

Abstract
Nicotine, the primary psychoactive component in tobacco, plays a major role in the initiation and maintenance of tobacco dependence and addiction, a leading cause of preventable death worldwide. An essential need thus exists for more effective pharmacotherapies for nicotine-use cessation. Previous reports suggest that pharmacological and genetic blockade of CB1 receptors attenuate nicotine reinforcement and reward; while exogenous agonists enhanced these abuse-related behaviors. In this study, we utilized complementary genetic and pharmacologic approaches to test the hypothesis that increasing the levels of the endocannabinoid 2-arachindonoylglycerol (2-AG), will enhance nicotine reward by stimulating neuronal CB1 receptors. Contrary to our hypothesis, we found that inhibition of monoacylglycerol lipase (MAGL), the primary catabolic enzyme of 2-AG, attenuates nicotine conditioned place preference (CPP) in mice, through a non-CB1 receptor-mediated mechanism. MAGL inhibition did not alter palatable food reward or Lithium Chloride (LiCl) aversion. In support of our findings, repeated MAGL inhibition did not induce a reduction in CB1 brain receptor levels or hinder function. To explore the potential mechanism of action, we investigated if MAGL inhibition affected other fatty acid levels in our CPP paradigm. Indeed, MAGL inhibition caused a concomitant decrease in arachidonic acid (AA) levels in various brain regions of interest, suggesting an AA cascade-dependent mechanism. This idea is supported by dose-dependent attenuation of nicotine preference by the selective COX-2 inhibitors valdecoxib and LM-4131. Collectively, these findings, along with our reported studies on nicotine withdrawal, suggest that inhibition of MAGL represents a promising new target for the development of pharmacotherapies to treat nicotine dependence.
Keywords: Nicotine, Nicotine Reward, Endocannabinoids, Monoacylglycerol lipase (MAGL), 2-Arachidonylglycerol (2-AG), Conditioned Place Preference
1. Introduction
Tobacco is one of the most widely abused drugs and the leading cause of preventable mortality and morbidity worldwide (World Health Organization, 2019). Nicotine, the main psychoactive component in tobacco, plays a major role in the initiation and maintenance of tobacco addiction. Over the past decade, a variety of nicotine cessation therapies have been developed including nicotine replacement therapies such as nicotine gums and patches, the antidepressant bupropion (Zyban®), and the partial α4β2* nicotinic agonist varenicline (Chantix®) (Cummings and Mahoney, 2008). Unfortunately, the efficacy of these treatments remain quite modest with only 20% of patients remaining abstinent after one year (Prado et al., 2011). Consequently, there remains an essential need for more effective pharmacotherapies than current existing treatments. Increased understanding of the neurobiological systems involved in nicotine intake and reward may uncover new and more efficacious therapies for nicotine cessation.
The endocannabinoid (EC) system contains several components that have been demonstrated to modulate nicotine reward and dependence in laboratory animal models; including the Cannabinoid Type 1 (CB1) and Type 2 (CB2) receptors, as well as endogenous ligands that bind and activate these receptors. For example, genetic deletion or antagonism of CB1 (Foll et al., 2008) or CB2 (Ignatowska-Jankowska et al., 2013; Navarrete et al., 2013) receptors reduces nicotine reward. The two best characterized endogenous ligands, N-arachidonoylethanolamine (anandamide; AEA; Devane et al., 1992) and 2-arachindonoylglycerol (2-AG; Mechoulam et al., 1995; Sugiura et al., 1995) are tightly regulated by serine hydrolases which include their respective primary catabolic enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) (Blankman and Cravatt, 2013; Clapper et al., 2009). Notably, CB1 receptors and nicotinic receptors overlap in many brain areas associated with the rewarding effects of various drugs of abuse including nicotine, such as the hippocampus (HIPP), and the mesolimbic dopamine system (Herkenham et al., 1990; Matsuda et al., 1993; Picciotto et al., 2000; Tsou et al., 1998). In addition, Valjent et al., 2002, demonstrated that combination of low dose nicotine and delta-9-tetrahydocannabinol (THC), the primary active constituent of marijuana, significantly enhanced nicotine CPP. Furthermore, WIN 55,212–2, a synthetic full CB1 receptor agonist, increased nicotine seeking behavior in a progressive ratio (PR) intravenous (i.v.) nicotine self-administration paradigm (Gamaleddin et al., 2012). Conversely, rimonabant, a CB1 receptor antagonist, decreased nicotine i.v. self-administration and CPP in rodents, similar to CB1 knockout (KO) mice (Castañé et al., 2005; Cohen et al., 2002; Le Foll and Goldberg, 2004; Merritt et al., 2008). Consistent with these findings, rimonabant blocked nicotine-induced dopamine release in the NAc (Cohen et al., 2002). A growing body of evidence indicates that elevating endogenous levels of AEA or 2-AG through the blockade of their primary metabolic enzymes elicit potential therapeutic effects in rodent models of pain and drug abuse, without eliciting the side effects associated with direct acting CB1 receptor agonists, such as THC (Ahn et al., 2009, 2008; Cravatt et al., 1996; Justinova et al., 2015, 2008; Long et al., 2009a, 2009b; Solinas et al., 2007). While we and others reported that FAAH inhibition augments both nicotine CPP and nicotine-induced accumbal dopamine release in mice (Merritt et al., 2008; Pavon et al., 2018), other groups found that FAAH inhibitors reduce nicotine intake and nicotine-induced dopamine release via a PPAR-α nuclear receptor mechanism in rats (Luchicchi et al., 2010; Melis et al., 2008; Scherma et al., 2008). However, little work has focused on the effects of MAGL inhibition in these models. Whereas MAGL inhibitors attenuate both precipitated and spontaneous somatic withdrawal signs as well as aversive withdrawal signs in nicotine-dependent mice (Muldoon et al., 2015), its role in nicotine reward remains largely unknown. Importantly, 2-AG is approximately 200-fold more abundant then AEA in the brain and acts as a full agonist at both CB1 and CB2 receptors (Gonsiorek et al., 2000; Savinainen et al., 2001). Moreover, cannabinoids unique retrograde signaling in the brain depends on 2-AG but not AEA (Pan et al., 2009) suggesting the importance of 2-AG in regulating the EC pathway.
Here, we used complementary genetic and pharmacologic approaches to test the hypothesis that MAGL inhibition enhances the rewarding effects of nicotine in mice by increasing 2-AG stimulation of CB1 receptors in the mesolimbic system. Accordingly, we tested whether administration of the MAGL enzyme inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate JZL184 (Long et al., 2009a, 2009b) in wild type mice or MAGL KO mice would display augmented nicotine CPP, and if so, are these effects mediated via a CB1 receptor-dependent mechanism. Also, we examined whether the JZL184 dosing regimen used in the nicotine CPP studies would affect receptor expression and function using radioligand binding and agonist-stimulated [35S] GTPγS binding assays, because prolonged MAGL blockade has been shown to lead to CB1 receptor down-regulation and desensitization. We then conducted a series of studies to explore the potential mechanism of MAGL inhibition on nicotine reward. Specifically, we examined changes in 2-AG levels within different brain regions implicated in nicotine reward, as well as evaluating whether JZL184 would affect other types of place conditioning, including CPP to palatable food and a conditioned place aversion (CPA) to lithium chloride (LiCl).
Finally, we tested the effects of two selective cyclooxygenase-2 (COX-2) inhibitors, valdecoxib and LM-4131, on nicotine reward. The COX enzyme is the rate-limiting enzyme in the synthesis of prostanoids from AA; which are not only found in the periphery but in the brain where they are shown to mediate synaptic transmission (Williams, 1996). Since MAGL is the rate-limiting synthetic enzyme in the production of arachidonic acid (AA) and its metabolites, and genetic deletion or repeated pharmacological inhibition of MAGL leads not only to increases in 2-AG but concomitant decreases in AA levels in the brain (Long et al., 2009a; Nomura et al., 2011, 2008; Schlosburg et al., 2010); we assessed whether inhibiting the COX enzyme would also result in nicotine CPP augmentation; thereby suggesting an AA-dependent cascade for JZL184’s effects.
2. Materials and Methods
2.1. Drugs and chemicals
(−)-Nicotine hydrogen tartrate salt, and lithium chloride were purchased from Sigma-Aldrich (St Louis, MO). The CB1 receptor antagonist rimonabant [N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide HCl] was provided by the National Institute on Drug Abuse Drug Supply Program (Bethesda, MD). JZL184 [2-[1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl]-1-(4-morpholinyl)-ethanone] was synthesized as previously described (Long et al., 2009a) by Organix, Inc. (Woburn, MA). Valdecoxib [4-(5-methyl-3-phenyl-4-isoxazolyl)-benzenesulfonamide], a selective COX-2 inhibitor, was provided by IronWood Pharmaceuticals (Cambridge, MA); and LM-4131, another selective COX-2 inhibitor was purchased from Cayman Chemicals (Ann Arbor, MI). JZL184, valdecoxib, LM-4131 and rimonabant were dissolved in ethanol, followed by addition of Emulphor-620 (Solvay S.A., Brussels, Belgium), and diluted with 0.9% saline to form a vehicle mixture of ethanol-emulphor-saline in a ratio of 1:1:18. The final concentration of ethanol in the vehicle is 5% and displays no evidence for neurotoxicity at the concentration used in the vehicle. The 1:1:18 preparation is a common vehicle and has been used in several published articles across many various species in the preparation of suspensions suitable for parenteral administration of cannabinergics that are not water soluble and therefore require organic solvents as carriers (Baumann et al., 2017; Ignatowska-Jankowska et al., 2013; Muldoon et al., 2015; Schlosburg et al., 2010; Wise et al., 2012). Nicotine, and lithium chloride were dissolved in physiological saline (0.9% sodium chloride). Nicotine was administered via subcutaneous injection (s.c.), whereas lithium chloride, JZL184, valdecoxib, LM-4131 and rimonabant were given via intraperitoneal (i.p.) injection. All doses are expressed as the free base of the drug and were administered at a volume of 10mL/kg. GTPγS, adenosine deaminase and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St Louis, MO).
[35S] GTPγS (1250 Ci mmol–1) and [3H] SR141716A (44.0 Ci mmol–1) were obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). Budget Solve (scintillation fluid) from Research Products Int. Corp. (Mount Prospect, IL) and Whatman GF/B glass fiber filters were obtained from Brandel (Gaithersburg, MD). Teklad LM-485 chow pellets were purchased from Harlan Laboratories (Frederick, MD). Palatable food was Kraft Philadelphia New York Style Cheesecake distributed from US Food Groups (Salem, VA) and purchased from Shockoe Expresso and Café (Richmond, VA).
2.2. Animals
Naive male 8–10-week-old ICR mice (Harlan Laboratories; Indianapolis, IN) served as subjects for the study. They were housed five per cage in a 21° C humidity-controlled facility with ad libitum access to food and water. Male and female littermate MAGL wild-type (WT) and knock-out (KO) mice were on a mixed 129SvEv/C57BL/6J background from the Scripps Research Institute (La Jolla, CA) (see Schlosburg et al., 2010, for information regarding initial breeders). The animal facility was accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. Experiments were performed during the light cycle and were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee.
2.3. Behaviour testing
2.3.1. Nicotine conditioned place preference
An unbiased CPP paradigm was used in this study (Kota et al., 2007). In brief, place conditioning chambers consisted of two distinct compartments separated by a smaller intermediate compartment with openings that allowed access to either side of the chamber. In the preconditioning phase (day 1), animals were confined to the intermediate compartment for a 5-min habituation period, and then allowed to move freely between compartments for 15 minutes. Time spent in each compartment was recorded in seconds (baseline measurements). These data were used to separate the animals into groups of approximately equal bias. Days 2 to 4 were the conditioning phase during which the saline group received saline in both compartments and drug groups received nicotine (0.1 or 0.5 mg/kg s.c.) in one compartment and saline in the opposite compartment. Drug-paired compartments were randomized among all groups. AM and PM sessions were 4 hours apart. Mice were counterbalanced in terms of which side was paired with drug and order of treatment (saline versus nicotine at first injection), allowing exposure to nicotine in the drug-paired side only once per day over the course of 3 conditioning days (Merritt et al., 2008). In this and all place conditioning studies, mice were given an i.p. injection of vehicle or JZL184 (16, 24 and 40 mg/kg) 2 hours before AM conditioning. JZL184 was only injected in the AM session due to its long-lasting duration of action (~7 h) (Long et al., 2009b). In a separate experiment, before both AM and PM conditioning, rimonabant (0.3 mg/kg i.p.) was administered 30 minutes before being placed in preference chambers. We used this dose of rimonabant because at higher doses, rimonabant has been shown to reduce nicotine CPP and self-administration in mice (Cohen et al., 2002; Merritt et al., 2008). Consequently, we chose a dose which shows minimal effects on nicotine CPP and self-administration.
On day 5 (test day), mice did not receive any drug. Activity counts and time spent on each side were recorded via photo sensors using Med Associates interface and software. Data are expressed as time spent on the drug-paired side minus time spent on the saline-paired side. A positive number indicates a preference for the drug-paired side, whereas a negative number indicates an aversion to the drug-paired side. A number at or near zero indicates no preference for either side.
We also assessed the effect of genetic deletion of MAGL on nicotine CPP. MAGL WT and KO mice were conditioned with nicotine (0.1 or 0.5 mg/kg, s.c.) or saline in the CPP test as described above. In another separate experiment, we performed a nicotine CPP dose response, where mice were given an i.p. injection of vehicle or JZL184 (24 mg/kg) 2 hours before each AM conditioning session followed by a s.c. injection of nicotine (0.1, 0.5, or 1 mg/kg) and subsequently subjected to the CPP test. Lastly, we examined the effect of COX-2 inhibition on nicotine CPP in a separate cohort of mice. Following the same CPP paradigm as above, mice were given an i.p. injection of either vehicle or valdecoxib in one experiment, and either vehicle or LM-4131 in a separate experiment 30 min before each AM conditioning session followed by a s.c. injection of nicotine (0.1, 0.5, or 1 mg/kg).
2.3.2. Palatable food CPP
On day 1, animals were confined to the intermediate compartment for a 5-min habituation period, and then they were allowed to move freely between compartments for 15 minutes. Time spent in each compartment was recorded. These data were used to separate the animals into groups of approximately equal bias. Immediately after baseline, mice were exposed to palatable food for 4–6 hours. Days 2 to 5 were the conditioning days during which the group exposed to chow pellet received a pellet in both compartments, while the palatable food groups received cheesecake in one compartment and pellet in the opposite compartment. Chow pellets were removed from the home cage 4 hours prior to conditioning. Palatable-food-paired compartments were randomized among all groups. 2 hours prior to the AM conditioning session, mice were given an i.p. injection of JZL184 (24 mg/kg) and then placed in a paired compartment containing either chow pellet or palatable food. Between each session mice had access to food for 1 hour at the end of the session. On day 6, animals were confined to the intermediate compartment for a 5-min habituation period, and then they were allowed to move freely between compartments for 15 minutes.
2.3.3. Lithium chloride induced conditioned place aversion (CPA)
Following the same CPP paradigm, mice were conditioned on days 2 to 4, during which the saline group received saline in both compartments and drug group received lithium chloride (LiCl) (150 mg/kg; i.p.) in one compartment and saline in the opposite compartment. Drug-paired compartments were randomized among all groups. Again, like other place conditioning assays, mice were either administered an i.p. injection of vehicle or JZL184 (24 mg/kg) 2 hours before AM conditioning. On day 5, animals were confined to the intermediate compartment for a 5-min habituation period, and then they were allowed to move freely between compartments for 15 minutes.
2.4. CP55, 940- Stimulated [35S] GTPγS binding in Membrane
Using a similar protocol as described for the place conditioning studies, mice were given an i.p. injection of JZL184 (24mg/kg) or vehicle once a day for three days. On day 3, 2 hours after the final injection, the mice underwent decapitation. Immediately following decapitation, the Prefrontal Cortex (PFC), Nucleus Accumbens (NAc), Hippocampus (HIPP) and Ventral Tegmental Area (VTA) were collected, snap frozen in liquid nitrogen and stored at −80°C until further processing. Each brain region was homogenized in a cold membrane buffer (50mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA, and 100 mM NaCl, pH 7.4) and centrifuged at 48,000g for 10 min at 4°C. Pellets were re-suspended in membrane buffer, and then centrifuged again. Pellets from second centrifugation were homogenized in membrane buffer and preincubated for 10 minutes at 30°C in 0.004 U/mL adenosine deaminase (240 U/mg protein). The assay was conducted at 30°C for 2 hours in membrane buffer including 5–10 ug (microgram) of membrane protein with 0.1% bovine serum albumin (BSA), 30 uM (micromolar) GDP (Sigma), various concentration of CP55, 940 – 300 nM, 100 nM, 30 nM, 10 nM, 3 nM and 1 nM and 0.10 nM [35S]GTPγS in a final volume of 0.5 mL. Nonspecific binding was determined in the absence of agonist and the presence of 30 uM (micromolar) unlabeled GTPγS. Reactions were terminated by rapid filtration under vacuum through Whatman GF/B glass fiber filters (Brandel, Gaithersburg, MD), followed by three washes with cold Tris HCl buffer, pH 7.4. Bound radioactivity was determined by liquid scintillation spectrophotometry at 50% efficiency for 35S after 1 hour shaking of the filters in 4 mL of Budget Solve scintillation fluid (Sim-Selley et al., 2006).
2.5. (CPP)SR141716A Binding in Membranes
As described above, mice were administered JZL184 (24 mg/kg) or vehicle once a day for 3 days and decapitated for brain harvesting 2 hours after the final injection on day 3. Immediately following decapitation, the Prefrontal Cortex (PFC), Nucleus Accumbens (NAc), Hippocampus (HIPP) and Ventral Tegmental Area (VTA) were collected, snap frozen in liquid nitrogen and then stored at −80° C until further processing. Membranes were incubated for 90 min at 30°C in buffer A with 0.5% BSA and varying concentrations of [3H] SR141716A in a total volume of 0.5 ml. Nonspecific binding was assessed in the presence of 2 uM unlabeled SR141716A. Incubations were terminated by vacuum filtration through Whatman GF/B glass fiber filters and washed three times with ice-cold Tris buffer with 0.25% BSA Tris-HCl, pH 7.4. Bound radioactivity was determined by liquid scintillation spectrophotometry at 45% efficiency for 3H after extraction of the filters in scintillation fluid (Sim-Selley et al., 2006).
2.6. Extraction and quantification of brain endocannabinoid levels with liquid chromatography-MS
In one experiment, mice were conditioned with either nicotine (0.5 mg/kg s.c.) or vehicle given once a day for 3 days (conditioning days 2–4); then, on the test day were euthanized via decapitation after the final drug administration and immediately following the CPP test. Immediately after decapitation, the Prefrontal Cortex (PFC), Striatum (STR), Hippocampus (HIPP) and Thalamus (THAL) were collected. Brain samples from each mouse were placed in individual vials and snap frozen in liquid nitrogen and stored at −80 °C until further processing for analysis of 2-AG levels using LC-MS. In a separate experiment, we used a similar CPP drug dosing protocol where mice were given either nicotine (0.5 mg/kg s.c.), i.p. injection of JZL184 (24 mg/kg), JZL184 (24 mg/kg i.p.) + nicotine (0.5 mg/kg s.c.) or vehicle once a day for 3 days. On day 3, 20 minutes after the final injection, the mice were euthanized via decapitation. Immediately following decapitation, the Prefrontal Cortex (PFC), Striatum (STR), Hippocampus (HIPP) and Thalamus (THAL) were collected. Brain samples from each mouse were placed in individual vials and snap frozen in liquid nitrogen and stored at −80 °C until further processing for analysis of 2-AG and AA levels using LC-MS.
On the day of processing, tissues were weighed and homogenized with 1.4 mL of chloroform-methanol (2:1, v/v; containing 0.0348 mg of phenylmethylsulfonyl fluoride/mL) after the addition of internal standards to each sample (1 nmol of 2-AG-d8, and 1 nmol AA-d8; Cayman Chemical, Ann Arbor, MI). Homogenates were then mixed with 0.3 mL of 0.73% w/v NaCl, vortexed, and then centrifuged for 10 min at 4000 rpm (4°C). The aqueous phase was collected and extracted two more times with 0.8 mL of chloroform. The organic phases from the three extractions were pooled, and the organic solvents were evaporated under nitrogen gas. Dried samples were reconstituted with 0.1 ml of chloroform and mixed with 1 mL of ice-cold acetone. The mixtures were then centrifuged for 5 min at 3000 rpm at 4°C. The upper layer of each mixture was collected and evaporated under nitrogen. Dried samples were reconstituted with 0.1 mL of methanol and placed in auto sample vials for analysis. The mobile phase consisted of water-methanol (10:90) with 0.1% ammonium acetate and 0.1% formic acid. The column used was a Discovery HS C18, 4.6 × 15 cm, 3-μm column (Supelco, Bellefonte, PA). Ions were analyzed in multiple reaction monitoring mode and the following transitions were monitored in positive mode: (379>287) and (279>269) for 2-AG; (387>96) for 2AG-d8; in negative mode: (303>259) and (303>59) for AA and (311>267) for AA-d8. A calibration curve was constructed for each assay on the basis of linear regression using the peak area ratios of the calibrators. The extracted standard curves ranged from 0.0625 to 64 nmol for 2-AG and from 1 to 32 nmol for AA.
2.7. Statistical analysis
For most data, statistical analyses were performed using StatView® software (SAS, Cary, NC), with the exception of data on the genetic deletion of MAGL, nicotine CPP DRC, 2AG/AA levels and COX-2 inhibition by LM4131, which were analyzed using the GraphPad Prism software, version 8.1.3 (GraphPad Software, Inc., La Jolla, CA). For data analyzed using StatView®, a one-way analyses of variance (ANOVA) was used to determine the overall interaction of drug treatment (nicotine/JZL184 in the nicotine CPP study, nicotine/Rimonabant in the Rimonabant study and nicotine/valdecoxib in the Cox-2 inhibitor studies). Values of p < 0.05 were considered to be statistically significant. Significant results were further analyzed using the Neuman-Keuls post hoc test. For data analyzed using GraphPad Prism, one-way or two-way ANOVA were performed where appropriate to determine the overall interaction of drug treatment (nicotine in the genetic study, and nicotine/JZL184 in the dose response study). Values of p < 0.05 were considered statistically significant. Significant results were further analyzed using either the appropriate Dunnett’s or Sidak post hoc test. To determine if there was a nicotine effect in the LM-4131 CPP study, data for the nicotine control treatment groups were compared to the saline treatment groups using a two-tailed Welch’s t-test.
3. Results
3.1. Pharmacological inhibition or genetic deletion of MAGL attenuates nicotine conditioned place preference
First, we tested whether administration of the MAGL enzyme inhibitor JZL184 in wild type mice would display augmented nicotine CPP. For our experiments, we chose a dose of 0.5. mg/kg nicotine in the CPP test because it produces moderately robust preference in mice (Grabus et al., 2006; Merritt et al., 2008). As shown in Figure 1, mice conditioned with nicotine (0.5 mg/kg, s.c.) displayed significantly increased CPP compared to vehicle-treated mice. However, JZL184 significantly blocked the development of nicotine preference in a dose-related fashion [F (7,63) = 7.218; p < 0.000, Figure 1]. At doses ≥ 24 mg/kg, JZL184 completely blocked the development of nicotine preference. Next, we assessed if genetic deletion of MAGL would augment nicotine CPP. Similarly, we find that MAGL KO mice failed to develop a nicotine CPP compared to MAGL WT (Figure 2). Although, Two-way ANOVA showed no significant interaction between strain and dose of nicotine tested, there was a main effect of both nicotine dose [F (2,55) = 5.682; p = 0.0057] and strain [F (1,55) = 5.429; p = 0.0235]. A Sidak post hoc analysis revealed a significant difference in preference scores between MAGL WT and MAGL KO at the highest dose of nicotine (0.5 mg/kg) tested (p = 0.0165). As expected, mice pretreated with vehicle and conditioned with saline did not show a preference for either side.
Figure 1. Nicotine conditioned place preference is significantly blocked by JZL184.
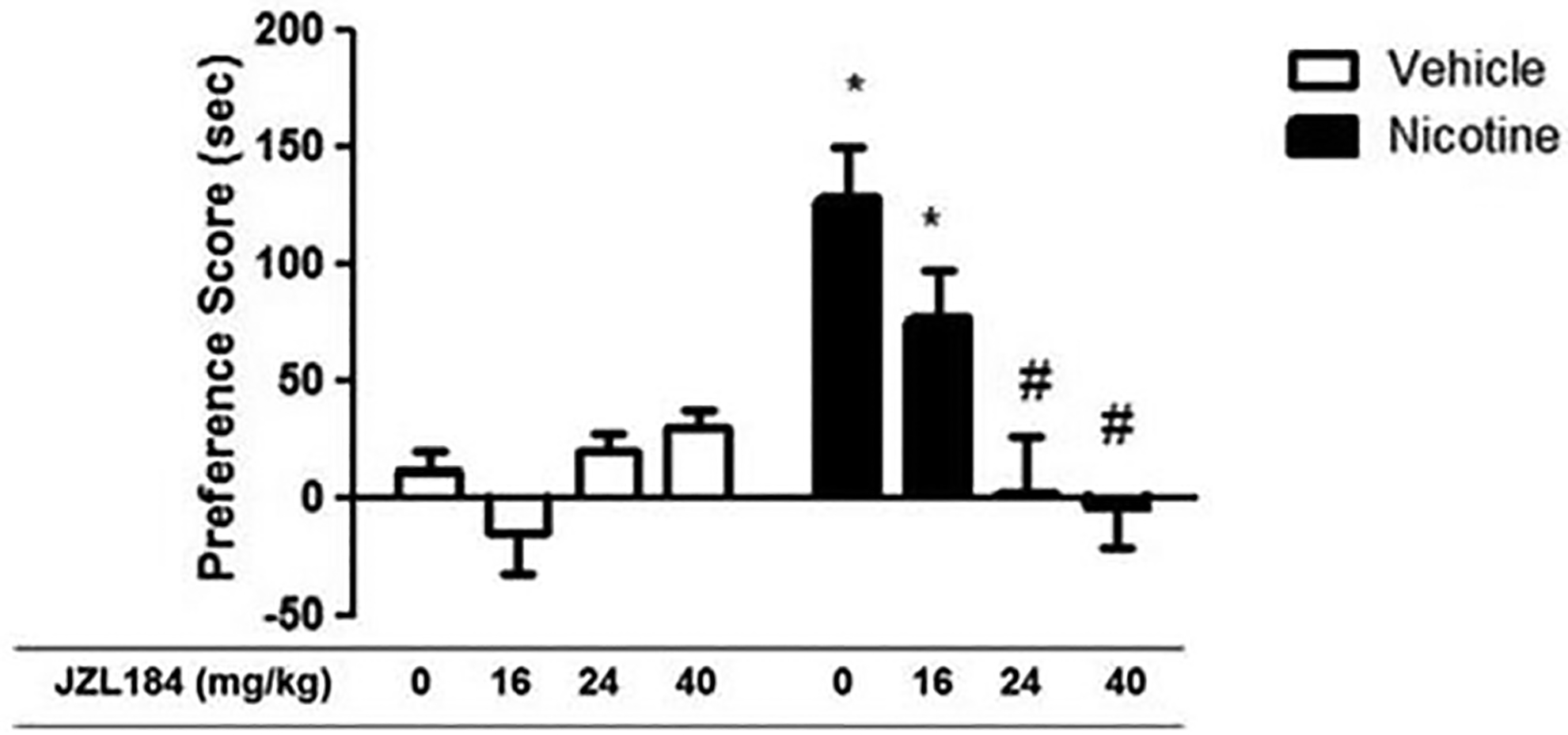
Mice were pretreated with either vehicle (0.9% saline, i.p.) or JZL184 (16, 24 and 40mg/kg, i.p.), and then injected with either vehicle (0.9% saline, i.p.) or nicotine (0.5 mg/kg, i.p.). Mice treated with nicotine produced significant place preference. Development of preference for nicotine was blocked by pretreatment of JZL184 at all tested doses (16, 24, and 40 mg/kg i.p.). Data are expressed as mean ± S.E.M. of 8–10 mice/group. *p<0.05 from vehicle control; #p<0.05 from nicotine control and nicotine + JZL184 (16mg/kg).
Figure 2. Genetic deletion of MAGL blocks nicotine preference.
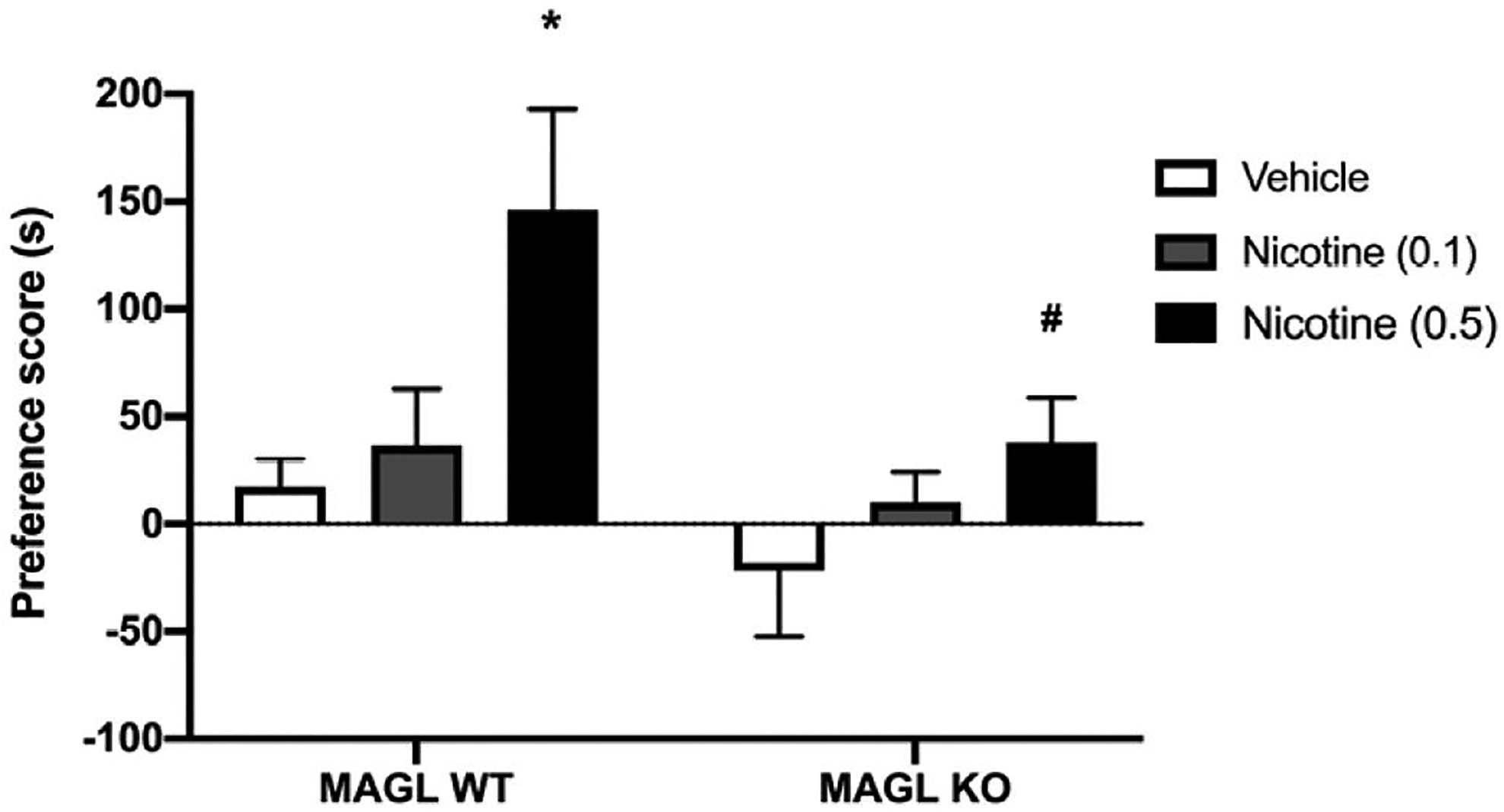
MAGL KO and WT mice were treated with nicotine (0.1 and 0.5 mg/kg s.c.). Nicotine preference is blocked in KO mice versus WT mice. Data are expressed as mean + S.E.M. of n = 9–14 mice/group. *p<0.05 from vehicle controls #p< 0.05 versus respective WT controls.
Activity counts were also measured for each animal during the CPP test. Statistical analysis from two-way ANOVA revealed no significant differences in activity counts on post-conditioning days among any of the test groups, suggesting that the results were not due to alteration of locomotor effects (Table 1). Furthermore, due to the inverted U-shaped nature of the nicotine dose-response relationship in the CPP test (Grabus et al., 2006), it is possible that JZL184 elicited a rightward or leftward shift in the nicotine dose-response curve. To assess this possibility, we tested the effects of 24 mg/kg JZL184 on the nicotine dose response curve in the CPP test at 0.1, 0.5 and 1 mg/kg nicotine. Figure 3 demonstrates that JZL184 significantly blocked nicotine CPP at each of its active doses with no leftward or rightward shift in the nicotine DRC (significant interaction between JZL184 and nicotine dose; F (3, 56) = 3.031; p = 0.037). Thus, further experiments focused on the 24 mg/kg dose of JZL184 because it was the lowest active dose that blocked the development of nicotine preference (Figure 1), and because it did not significantly affect activity counts or elicit a shift in the nicotine dose-response curve in the CPP test.
Table 1.
Summary table of activity counts measured during nicotine CPP test. JZL184 does not alter locomotor activity on test day compared to vehicle. Data are expressed as mean ± S.E.M. of 6 – 8 mice/group.
| CPP Activity Counts (sec) | ||
|---|---|---|
| Saline | Nicotine (0.5 mg/kg) | |
| V ehicle | 1321 ± 177 | 1380 ± 122 |
| JZL184 (16 mg/kg) | 1338 ± 140 | 1232 ± 92 |
| JZL184 (24 mg/kg) | 1104 ± 199 | 1234 ± 127 |
| JZL184 (40 mg/kg) | 893 ± 168 | 1075 ± 58 |
Figure 3. Nicotine DRC in the CPP Test After JZL-184 administration.
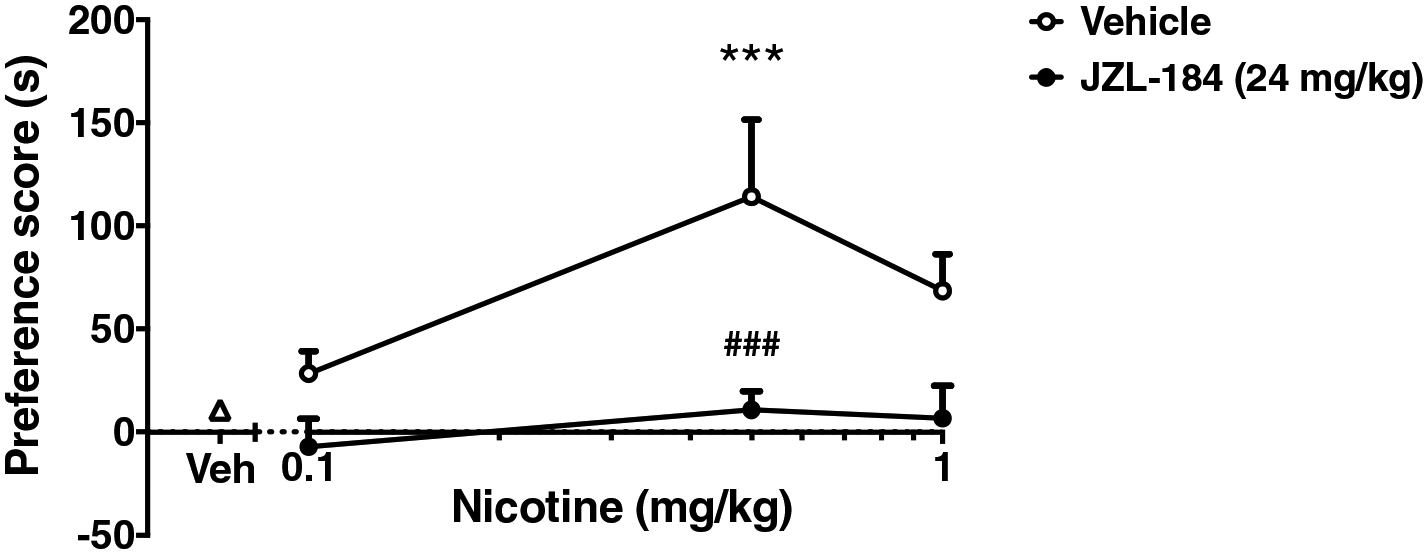
JZL dose-dependently attenuates preference in the nicotine CPP paradigm with no leftward or rightward shift in the nicotine DRC. Mice were pretreated with JZL184 (24mg/kg i.p.) or vehicle (0.9% saline, i.p.) 2 hours before AM conditioning, followed by treatment with nicotine. A dose response to nicotine at 0.1, 0.5, and 1 mg/kg was assessed for preference in the CPP test. Data are expressed as mean + S.E.M. of 8 mice/group. ***p<0.001 from nicotine vehicle (triangle) ###p<0.001 from JZL184 vehicle (circle).
3.2. MAGL inhibition does not interfere with either food reward or LiCl aversion
To assess if MAGL inhibition interferes with place conditioning to other stimuli, we tested if JZL184 would also affect place conditioning to palatable food-induced CPP and LiCl-induced conditioned place aversion. As shown in Figure 4B, JZL184 (24mg/kg i.p.) did not affect place preference scores for the context associated with palatable food F (3, 25) = 4.207; p < 0.02). Similarly, JZL184 (24mg/kg i.p.) did not significantly alter LiCl-induced CPA compared to vehicle group [(Treatment F (2, 11) = 4.162; p < 0.05); Figure 4B].
Figure 4. MAGL inhibition does not alter LiCl induced aversion, or palatable-food reward.
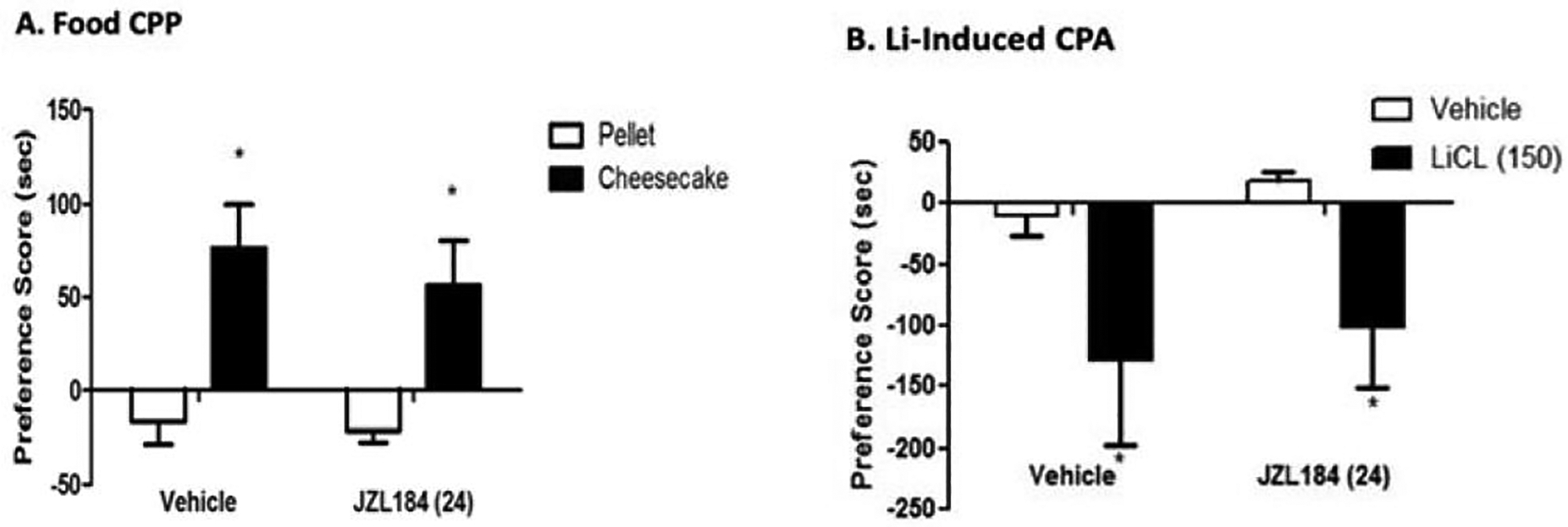
(A) Food CPP; mice were treated either with pellet or cheesecake. Mice treated with palatable food (cheesecake) showed significant preference score compared to their pellet counterparts. Pretreatment with JZL184 (24 mg/kg i.p.) did not alter food reward. (B) LiCl-induced CPP; mice were treated either with vehicle or LiCl (150 mg/kg i.p.). Mice treated with LiCl showed significant aversion score compared to their vehicle counterparts. Pretreatment with JZL184 (24 mg/kg i.p.) did not alter LiCl aversion. Data are expressed as mean ± S.E.M. of 8–10 mice/group. *p<0.05 vs all treatment groups.
3.3. Evaluation of CB1 receptors in the effects of JZL184 on nicotine CPP
As an initial step to investigate the possible role of the CB1 receptor in MAGL inhibition-induced nicotine preference attenuation. We tested the effects of rimonabant on JZL184-induced nicotine CPP attenuation. Mice were pretreated with rimonabant (0.3mg/kg i.p.) and JZL184 (24mg/kg i.p.) and conditioned with nicotine (0.5 mg/kg s.c.) in the CPP test. As discussed in the methods section, we chose this dose of rimonabant because it has been demonstrated to elicit minimal effects on nicotine CPP and self-administration; since rimonabant alone at higher doses attenuates nicotine CPP and self-administration in mice (Cohen et al., 2002; Merritt et al., 2008). As shown in Figure 5, 0.3 mg/kg rimonabant did not significantly affect JZL184’s attenuation of nicotine CPP as determined by a one-way ANOVA analysis [F (6, 55) = 5.860; p < 0.0001; Figure 5].
Figure 5. JZL184-mediated reduction in nicotine preference is not CB1 receptor-mediated.
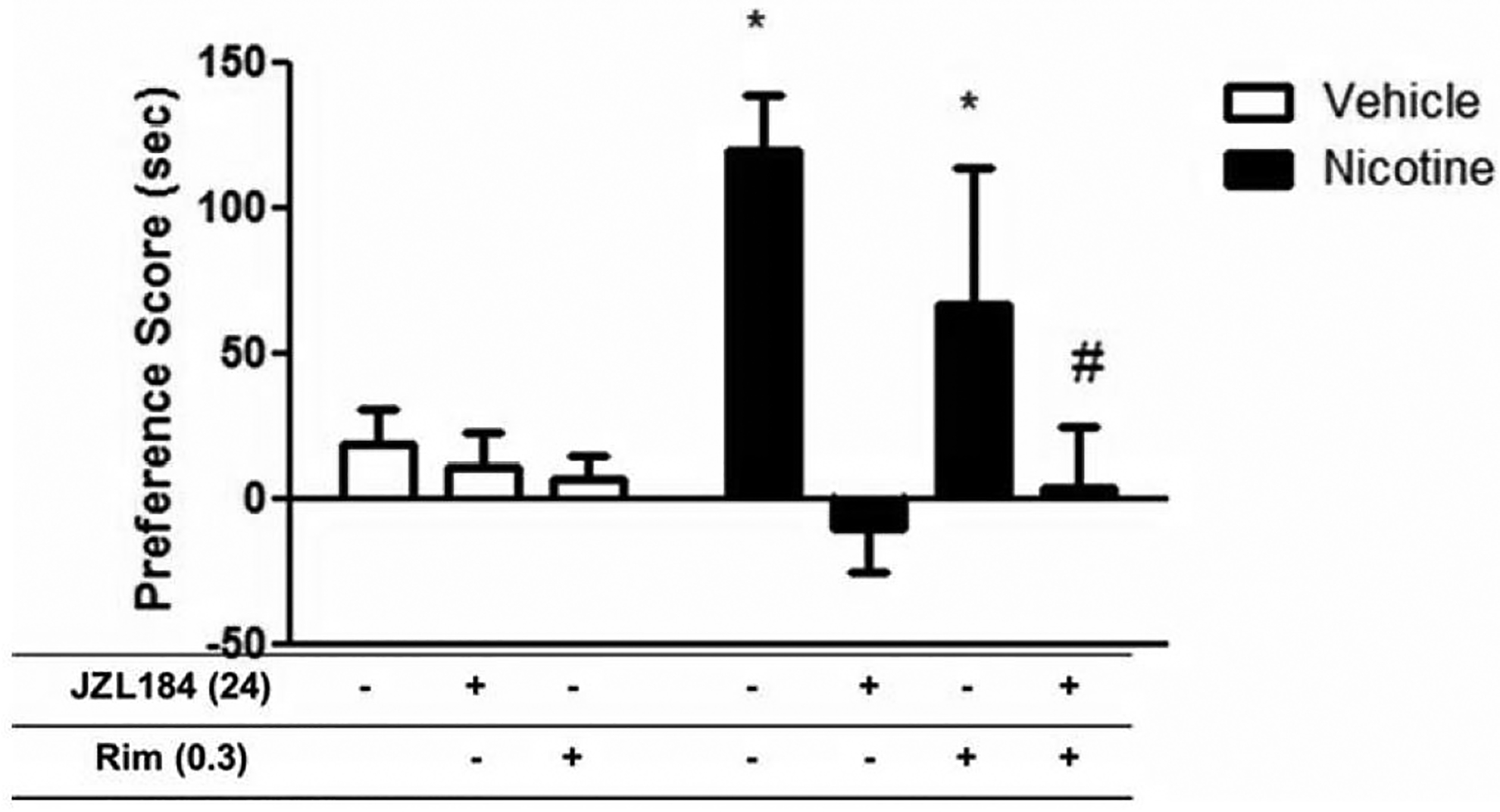
Mice were pretreated with JZL184 (24mg/kg i.p.) for 90 mins, followed by treatment with rimonabant (0.3 mg/kg i.p.) and tested in the CPP paradigm using 0.5 mg/kg nicotine administered s.c. Rimonabant did not attenuate JZL184’s effects on nicotine preference. Data are expressed as mean ± S.E.M. of 6–8 mice/group. *p<0.05 from vehicle controls; #p<0.05 vs Nicotine and Nicotine +Rim.
Next, we determined if the dosing regimen of repeated administration of JZL184 (24 mg/kg) which blocked nicotine CPP led to CB1 receptor downregulation and/or sensitization in brain areas of interest. As shown in Table 2, this JZL184 dosing regimen did not alter CB1 receptor levels in the PFC [F (1,4) = 2.063; p < 0.2], NAc [F (1,7) = 0.187; p < 0.7)], HIPP [F (1,4) = 0.24; p < 0.9], and the VTA [F (1,8) = 0.215; p < 0.7]. Furthermore, the same dosing regimen did not alter CB1 receptor function as assessed using the CP55,940-stimulated [35S] GTPγS binding assay in the PFC [F (1,4) = 0.003; p < 1.0], NAc [F (1,6) = 1.372; p < 0. 21], HIPP [F (1,4) = 0.004; p < 1.0], and the VTA [F (1,6) = 1.468; p < 0.3] (Table 2).
Table 2.
Effect of repeated JZL184 administration on CB1 receptor desensitization in mice. Repeated treatment with JZL184 does not produce CB1 receptor desensitization in PFC, NAc, HIPP, and VTA. Data are presented as means ± S.E.M. n = 6 tissue samples per group, run in separate experiments with each individual sample run in triplicate for [35 S] GTPγS binding, and each individual sample run in duplicate for receptor binding.
| Vehicle | JZL184 (24 mg/kg) | |
|---|---|---|
| Prefrontal Cortex | ||
| Bmax (pM) | 2.2 ± 0.2 | 1.8 ± 0.2 |
| Kd (nM) | 1.6 ± 0.5 | 1.3 ± 0.4 |
| Nucleus Accumbens | ||
| Bmax (pM) | 1.6 ± 0.6 | 1.2 ± 0.7 |
| Kd (nM) | 4.3 ± 2.5 | 1.8 ± 0.9 |
| Hippocampus | ||
| Bmax (pM) | 2.2 ± 0.6 | 2.1 ± 0.6 |
| Kd (nM) | 0.95 ± 0.25 | 1.85 ± 1.0 |
| Ventral Tegmental Area | ||
| Bmax (pM) | 0.53 ± 0.16 | 0.7 ± 0.2 |
| Kd (nM) | 0.99 ± 0.35 | 1.7 ± 1.2 |
| Vehicle | JZL184 (24 mg/kg) | |
|---|---|---|
| Prefrontal Cortex | ||
| Emax (% stim.) | 62.8 ± 20.2 | 64.5 ± 24.6 |
| EC50 (nM) | 6.0 ± 1.2 | 7.9 ± 1.9 |
| Nucleus Accumbens | ||
| Emax (% stim.) | 56.3 ± 3.8 | 45.5 ± 6.0 |
| EC50 (nM) | 15.11 ± 3.1 | 22.5 ± 4.5 |
| Hippocampus | ||
| Emax (% stim.) | 61.9 ± 12.4 | 63.0 ± 14.6 |
| EC50 (nM) | 8.5 ± 2.7 | 6.2 ± 0.3 |
| Ventral Tegmental Area | ||
| Emax (% stim.) | 51.54 ± 6.61 | 36.4 ± 10.6 |
| EC50 (nM) | 18.7 ± 8.7 | 19.4 ± 5.6 |
3.4. Nicotine CPP does not alter 2-AG levels on test day
Next, we examined changes in 2-AG levels within the brain in response to nicotine in the CPP paradigm. Mice were conditioned with either nicotine (0.5 mg/kg s.c.), or vehicle given once a day for 3 days (conditioning days 2–4). On test day (day 5), the PFC, STR, HIPP, and THAL were dissected for analysis of 2-AG levels immediately following the CPP test. As shown in table 3, mice treated with nicotine in the CPP paradigm did not show significant changes in 2-AG levels compared to vehicle in the PFC [F (1,22) = 4.2; p < 0.06], STR [F (1,21) = 0.3; p < 0.6], HIPP [F (1,22) = 0.8; p < 0.4], and THAL [F (1,14) = 0.9; p < 0.4].
Table 3.
Effect of nicotine CPP on 2-AG levels in mice. 2-AG levels are not altered after nicotine treatment in the CPP model compared to vehicle. Data are expressed as mean ± S.E.M. of 6 mice/group.
| 2-AG Levels (nmol/g) | ||||
|---|---|---|---|---|
| Treatment | PFC | STR | HIPP | THAL |
| Vehicle | 7.72 ± 0.38 | 6.18 ± 1.31 | 10.18 ± 0.97 | 10.35 ± 0.72 |
| Nic (0.5 mg/kg) | 5.76 ± 0.65 | 6.95 ± 0.71 | 8.71 ± 1.06 | 9.82 ± 0.48 |
3.5. JZL184 selectively increases 2AG levels with concomitant decreases in AA levels in various brain regions
In a separate experiment, we also assessed changes in 2-AG levels within the brain in response to JZL treatment. Mice were treated with either vehicle, nicotine (0.5 mg/kg s.c.), JZL184 (24mg/kg i.p.) or JZL184 (24mg/kg i.p.) + nicotine (0.5 mg/kg, s.c.) for 3 days. On the 3rd day, 20 minutes after the last injection of JZL184, the PFC, STR, HIPP, and THAL were dissected for analysis of 2-AG levels. Overall, one-way ANOVA revealed significant differences between treatment groups in the PFC [F (3, 23) = 50.18; p < 0.0001], STR [F (3, 23) = 34.42; p < 0.0001], HIPP [F (3, 23) = 224.8; p < 0.0001], and THAL [F (3, 23) = 32.06; p < 0.0001] (Table 4). A Dunnett’s test post hoc analysis showed significantly increased 2-AG levels in the JZL184 and JZL184 + nicotine groups compared to the vehicle groups in all brain regions tested. However, there were no significant differences observed between the nicotine only treated group compared to the vehicle group.
Table 4.
Effect of JZL184 on 2-AG and free AA levels in the brain. JZL184 selectively increases 2AG levels with an associated decrease in AA levels in various brain regions. Data are expressed as mean ± S.E.M. of 6–7 mice/group.
| 2-AG Levels (nmol/g) | PFC | STR | HIPP | THAL |
|---|---|---|---|---|
| Vehicle | 7.51 ± 1.66 | 9.75 ± 1.75 | 11.53 ± 2.11 | 9.72 ± 2.85 |
| Nicotine (0.5 mg/kg) | 6.31 ± 1.82 | 10.57 ± 2.94 | 10.08 ± 1.91 | 9.04 ± 1.01 |
| JZL (24 mg/kg) | 196.72 ± 29.38* | 144.51 ± 9.88* | 140.66 ± 6.81* | 72.44 ± 5.26* |
| JZL + Nicotine | 212.76 ± 11.19* | 152.20 ± 24.07* | 165.31 ± 7.60* | 73.53 ± 1.23* |
| AA Levels (nmol/g) | PFC | STR | HIPP | THAL |
| Vehicle | 218.05 ± 16.46 | 149.85 ± 7.94 | 187.70 ± 7.59 | 89.40 ± 3.83 |
| Nicotine (0.5 mg/kg) | 224.92 ± 30.34 | 147.09 ± 25.93 | 172.22 ± 17.86 | 84.83 ± 9.46 |
| JZL (24 mg/kg) | 135.50 ± 20.20* | 83.18 ± 29.38* | 117.89 ± 10.20* | 48.07 ± 2.59* |
| JZL + Nicotine | 121.42 ± 19.88* | 76.32 ± 13.58* | 100.88 ± 8.53* | 40.64 ± 0.67* |
p<0.05 vs vehicle.
Furthermore, since the effects of MAGL inhibition on nicotine preference were not CB1 receptor mediated, and because MAGL is the rate-limiting synthetic enzyme in the production of arachidonic acid (AA) and its metabolites; we investigated if JZL184 was blocking nicotine’s effects by altering other fatty acids such as AA. Similarly, mice were treated with either vehicle, nicotine (0.5 mg/kg s.c.), JZL184 (24mg/kg i.p.) or JZL184 (24mg/kg i.p.) + nicotine (0.5 mg/kg, s.c.) for 3 days. On the 3rd day, 20 minutes after the last injection of JZL184, the PFC, STR, HIPP, and THAL were dissected for analysis of AA levels. We found that JZL184 treatment significantly decreased AA levels in the PFC [F (3, 23) = 6.028; p = 0.0035], STR [F (3, 23) = 5.089; p = 0.0076], HIPP [F (3, 23) = 11.92; p < 0.0001], and THAL [F (3, 23) = 8.213; p = 0.0007]. No significant differences were observed between the nicotine only treated group compared to the vehicle group. The magnitude to which JZL184 altered fatty acid levels are also displayed in Table 4. This decrease was almost uniform across regions (1.5 to 2-fold), while bigger differences were seen in 2-AG levels after JZL184 treatment. As previously reported, repeated MAGL inhibition not only caused an increase in 2-AG levels but a decrease in AA (Schlosburg et al., 2010).
3.6. AA Cascade-dependent Modulation of Nicotine Preference
Since CB1 receptors did not mediate the attenuation of nicotine CPP by JZL184, and because pharmacological inhibition of MAGL leads not only to increases in 2-AG but concomitant decreases in AA levels in the brain; we assessed whether the inhibition of cyclooxygenase, one of the major hydrolytic enzymes of AA would also prevent the development of nicotine CPP. As demonstrated in figure 6, both valdecoxib (0.5, 1, and 5 mg/kg; i.p.) [F (4, 33) = 3.8; p < 0.01], and LM-4131 (0.5, 1, and 5 mg/kg; i.p.) [F (3,32) = 5.102; p = 0.0053] dose-dependently blocked the development of nicotine preference in the CPP test; which may suggest an AA cascade-dependent mechanism for JZL184’s effects.
Figure 6. Selective COX-2 inhibitors valdecoxib and LM-4131, dose-dependently block development of nicotine preference.
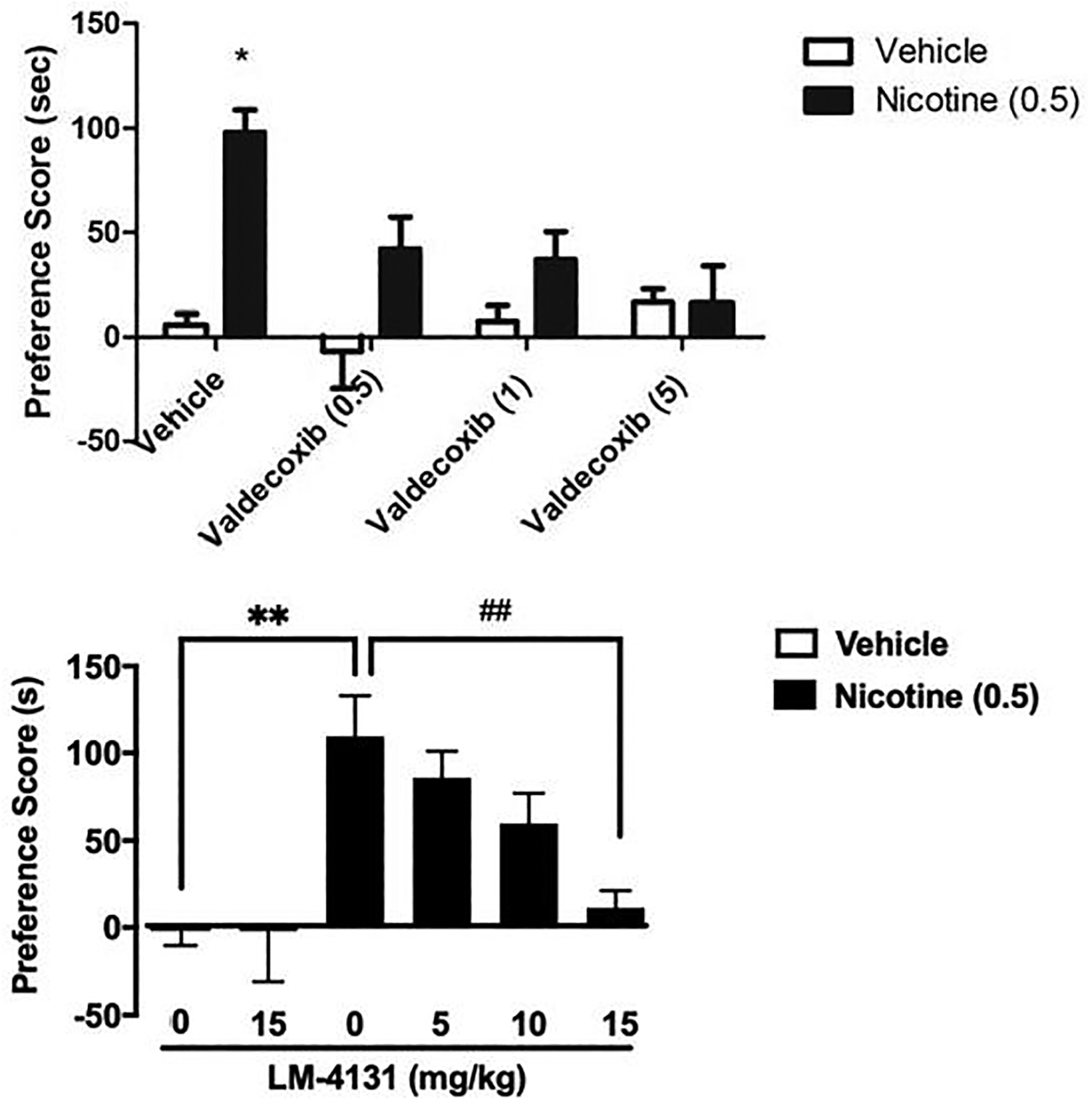
Mice treated with nicotine (0.5 mg/kg) produced significant place preference. Development of preference was blocked by (A) pretreatment of Valdecoxib (0.5, 1 or 5 mg/kg i.p.). Data are expressed as mean ± S.E.M. of 8–10 mice/group. *p<0.05 vs saline controls. (B) Pretreatment of LM-4131 (0, 5, 10, or 15 mg/kg). Data are expressed as mean ± S.E.M. of 8–10 mice/group. *p<0.05, **p<0.01 vs vehicle controls; ##p<0.01 vs nicotine control.
4. Discussion
In the present study, we report that genetic deletion of MAGL and pharmacological inhibition of MAGL by JZL184 reduces nicotine conditioned reward in the mouse CPP test. We find that JZL184’s effects are selective to nicotine; since MAGL inhibition did not alter LiCl aversion or palatable food reward. However, its effects are not CB1 mediated. In support of our findings, repeated MAGL inhibition did not induce a reduction in CB1 brain receptor levels or function. We also investigated if repeated MAGL inhibition in the CPP paradigm affected other fatty acids levels. Indeed, MAGL inhibition not only causes increases in 2-AG levels but also a concomitant decrease in AA levels in various brain regions of interest, suggesting an AA cascade dependent-mechanism. This idea is supported by a dose-dependent attenuation of nicotine preference by selective COX-2 inhibitors valdecoxib, and LM-4131.
Our findings contradict our initial hypothesis that MAGL inhibition would enhance the rewarding effects of nicotine. Previous studies reported that CB1 agonists enhanced nicotine reward and reinforcement, while CB1 antagonists blocked it (Cohen et al., 2002; Gamaleddin et al., 2012; Le Foll and Goldberg, 2004; Merritt et al., 2008). In addition, Buczynski et al., (2016) showed that an inhibitor of 2-AG biosynthesis diacylglycerol lipase (DAGL), led to a significant reduction of neuronal 2-AG levels and a decrease in nicotine i.v. self-administration in rats. However, we show in our study that administration of JZL184 dose-dependently blocked the development of nicotine reward in the CPP test without disrupting conditional learning for food and aversion or motor activity. In support of our findings, another study demonstrated that JZL184 did not disrupt water maze performance in mice (Wise et al., 2012). In addition, JZL184 pretreatment shifted nicotine dose-response curve downward and blocked nicotine CPP at all doses. We therefore determined if JZL184’s receptor mechanism of action was due to activation of CB1 receptors. Rimonabant (0.3mg/kg) did not block the effect of the lowest active dose of JZL184 (24mg/kg i.p.) on nicotine preference, suggesting that JZL184’s effect is not CB1 mediated. We used this dose of rimonabant because rimonabant alone at higher doses has been shown to reduce nicotine CPP and self-administration in mice (Cohen et al., 2002; Merritt et al., 2008). Consequently, we chose a dose that had minimal effects on nicotine CPP and self-administration. While it is possible that the rimonabant dose used was insufficient to antagonize CB1 receptors, we recently reported that this dose was sufficient in blocking JZL184’s effect on nicotine precipitated withdrawal (Muldoon et al., 2015); and another study reported that this dose was sufficient to block THC drug discrimination in mice (McMahon et al., 2008). The lack of a CB1-mediated mechanism in the nicotine CPP test contrasts with JZL184’s blockade of nicotine withdrawal (Muldoon et al., 2015). One possible explanation of this duality in JZL184’s effect is the difference in endocannabinoid mechanisms involved in nicotine reward and withdrawal. For example, our previous results show that decreased 2-AG signaling may contribute to the severity of somatic symptoms of nicotine withdrawal (Muldoon et al., 2015), while no changes in 2-AG levels in multiple brain regions were observed in the CPP test in response to nicotine.
Consequently, since the effect of MAGL inhibition on nicotine preference were not CB1 receptor mediated, we investigated if JZL184 was altering other fatty acid levels in the brain such as AA; since MAGL is the rate-limiting synthetic enzyme in the production of arachidonic acid (AA) and its metabolites. We assessed whether the lowest active dose of JZL184 used in our studies was altering free AA levels in various brain regions implicated in nicotine’s behavioral effects (PFC, STR, HIPP, and THAL). Indeed, we found that compared to vehicle, we not only saw an increase in 2-AG levels but also an associated decrease of AA levels in these brain regions. This may suggest that JZL184’s effect is being mediated by an AA cascade-dependent mechanism. Our findings corroborate other studies that have found that genetic deletion or repeated pharmacological inhibition of MAGL by high dose of JZL184 leads not only to increases in 2-AG but concomitant decreases in AA levels in the brain (Long et al., 2009a; Nomura et al., 2011, 2008; Schlosburg et al., 2010).
In the AA cascade, the COX enzyme is the rate-limiting factor in the synthesis of prostanoids from AA. Prostanoids are found not only in the periphery but in the brain where they are shown to mediate synaptic transmission (Williams, 1996). Reports show that the acquisition of morphine preference in the morphine CPP test is blocked in mice co-administered the selective COX-2 inhibitor, celecoxib, during the conditioning phase (Ghahremani et al., 2006). Here, we found that valdecoxib and LM-4131, two selective COX-2 inhibitors, dose-dependently blocked the acquisition of nicotine preference in the CPP test. This is an interesting finding since it has been shown that prostanoids, a products of AA metabolism, play a regulatory role in neural plasticity involved with learning and memory (Teather, 2002), and in COX-2-mediated regulation of endocannabinoid metabolism as new emerging evidence finds (Hermanson et al., 2014; Martínez-Torres et al., 2019).
5. Conclusion
Collectively, these findings along with our reported studies on nicotine withdrawal (Muldoon et al., 2015), suggest that inhibition of MAGL represents a promising new target for the development of pharmacotherapies to treat nicotine dependence. Nicotine somatic and affective withdrawal symptoms, in addition to nicotine’s rewarding effects, have been stated as a major motivational factor for relapse (Epping-Jordan et al., 1998; S. Watkins, George F. Koob, Athina, 2000). One limitation to our study is the absence of experiments examining CB1 receptor desensitization with JZL184 in combination with nicotine. While we cannot discount potential nicotine and JZL184 interaction on CB1 receptor desensitization, studies in adult rodents were not found to have significant changes in CB1 receptors after chronic nicotine exposure (Gonzalez et al, 2002; Balerio et al., 2004). Another limitation is the use of only male mice. Animal studies suggest sex differences in endocannabinoid pharmacology (Craft et al., 2013). It will be important to examine in future studies if there are any sex differences associated with the effects of JZL184 on nicotine CPP. Nonetheless, our studies pose translational relevance since ABX-1431, a selective MAGL inhibitor, was well-tolerated and safe in phase 1 clinical studies and is currently in clinical phase 2 studies for the treatment of Gilles de la Tourette syndrome and other neurological disorders (Jiang and van der Stelt, 2018).
Highlights.
Inhibition of monoacylglycerol lipase (MAGL) attenuates nicotine conditioned place preference (CPP) in male mice, through a non-CB1 receptor-mediated mechanism.
JZL184 selectively increases 2AG levels with concomitant decreases in AA levels in various brain regions
Inhibition of MAGL represents a promising new target for the development of pharmacotherapies to treat nicotine dependence.
Acknowledgements
This research was supported by the National Institute on Drug Abuse of the National Institutes of Health under Awards Number P30 DA033934, DA005274 and DA032246 to MID.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no conflict of interest.
References
- Ahn K, Johnson DS, Cravatt BF, 2009. Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opin. Drug Discov 4, 763–784. 10.1517/17460440903018857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, McKinney MK, Cravatt BF, 2008. Enzymatic Pathways That Regulate Endocannabinoid Signaling in the Nervous System. Chem. Rev 108, 1687–1707. 10.1021/cr0782067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Glennon RA, Wiley JL, 2017. Neuropharmacology of New Psychoactive Substances (NPS), Current Topics in Behavioral Neurosciences, Current Topics in Behavioral Neurosciences. Springer International Publishing, Cham. 10.1007/978-3-319-52444-3 [DOI] [Google Scholar]
- Blankman JL, Cravatt BF, 2013. Chemical Probes of Endocannabinoid Metabolism. Pharmacol. Rev 65, 849–871. 10.1124/pr.112.006387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañé A, Berrendero F, Maldonado R, 2005. The role of the cannabinoid system in nicotine addiction, in: Pharmacology Biochemistry and Behavior. 10.1016/j.pbb.2005.01.025 [DOI] [PubMed] [Google Scholar]
- Clapper JR, Mangieri RA, Piomelli D, 2009. The endocannabinoid system as a target for the treatment of cannabis dependence. Neuropharmacology 56, 235–243. 10.1016/j.neuropharm.2008.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen C, Perrault G, Voltz C, Steinberg R, Soubrié P, 2002. SR141716, a central cannabinoid (CB1) receptor antagonist, blocks the motivational and dopamine-releasing effects of nicotine in rats. Behav. Pharmacol 13, 451–463. 10.1097/00008877-200209000-00018 [DOI] [PubMed] [Google Scholar]
- Craft RM, Marusich JA, Wiley JL, 2013. Sex differences in cannabinoid pharmacology: A reflection of differences in the endocannabinoid system? Life Sci. 92, 476–481. 10.1016/j.lfs.2012.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB, 1996. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384, 83–87. 10.1038/384083a0 [DOI] [PubMed] [Google Scholar]
- Cummings KM, Mahoney MC, 2008. Strategies for smoking cessation: what is new and what works? Expert Rev. Respir. Med 2, 201–213. 10.1586/17476348.2.2.201 [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanuš L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R, 1992. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science (80-.). 258, 1946–1949. 10.1126/science.1470919 [DOI] [PubMed] [Google Scholar]
- Epping-Jordan MP, Watkins SS, Koob GF, Markou A, 1998. Dramatic decreases in brain reward function during nicotine withdrawal. Nature 393, 76–79. 10.1038/30001 [DOI] [PubMed] [Google Scholar]
- Foll B. Le, Forget B, Aubin H-J, Goldberg SR, 2008. Blocking cannabinoid CB1 receptors for the treatment of nicotine dependence: insights from pre-clinical and clinical studies. Addict. Biol 13, 239–252. 10.1111/j.1369-1600.2008.00113.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamaleddin I, Wertheim C, Zhu AZX, Coen KM, Vemuri K, Makryannis A, Goldberg SR, Le Foll B, 2012. Cannabinoid receptor stimulation increases motivation for nicotine and nicotine seeking. Addict. Biol 17, 47–61. 10.1111/j.1369-1600.2011.00314.x [DOI] [PubMed] [Google Scholar]
- Ghahremani MH, Eghtesad E, Tahsili-Fahadan P, Sharifzadeh M, Amini M, Tootian Z, 2006. Inhibition of the cyclooxygenase pathway attenuates morphine-induced conditioned place preference in mice. Pharmacol. Biochem. Behav 85, 356–361. 10.1016/j.pbb.2006.09.002 [DOI] [PubMed] [Google Scholar]
- Gonsiorek W, Lunn C, Fan X, Narula S, Lundell D, Hipkin RW, 2000. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: Antagonism by anandamide. Mol. Pharmacol [PubMed] [Google Scholar]
- Grabus SD, Martin BR, Brown SE, Damaj MI, 2006. Nicotine place preference in the mouse: influences of prior handling, dose and strain and attenuation by nicotinic receptor antagonists. Psychopharmacology (Berl). 184, 456–463. 10.1007/s00213-006-0305-7 [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, De Costa BR, Rice KC, 1990. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. U. S. A 87, 1932–1936. 10.1073/pnas.87.5.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanson DJ, Gamble-George JC, Marnett LJ, Patel S, 2014. Substrate-selective COX-2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation. Trends Pharmacol. Sci 35, 358–367. 10.1016/j.tips.2014.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Muldoon PP, Lichtman AH, Damaj MI, 2013. The cannabinoid CB2 receptor is necessary for nicotine-conditioned place preference, but not other behavioral effects of nicotine in mice. Psychopharmacology (Berl). 229, 591–601. 10.1007/s00213-013-3117-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, van der Stelt M, 2018. Activity-Based Protein Profiling Delivers Selective Drug Candidate ABX-1431, a Monoacylglycerol Lipase Inhibitor, To Control Lipid Metabolism in Neurological Disorders. J. Med. Chem 61, 9059–9061. 10.1021/acs.jmedchem.8b01405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justinova Z, Mangieri RA, Bortolato M, Chefer SI, Mukhin AG, Clapper JR, King AR, Redhi GH, Yasar S, Piomelli D, Goldberg SR, 2008. Fatty Acid Amide Hydrolase Inhibition Heightens Anandamide Signaling Without Producing Reinforcing Effects in Primates. Biol. Psychiatry 64, 930–937. 10.1016/j.biopsych.2008.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justinova Z, Panlilio LV, Moreno-Sanz G, Redhi GH, Auber A, Secci ME, Mascia P, Bandiera T, Armirotti A, Bertorelli R, Chefer SI, Barnes C, Yasar S, Piomelli D, Goldberg SR, 2015. Effects of Fatty Acid Amide Hydrolase (FAAH) Inhibitors in Non-Human Primate Models of Nicotine Reward and Relapse. Neuropsychopharmacology 40, 2185–2197. 10.1038/npp.2015.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota D, Martin BR, Robinson SE, Damaj MI, 2007. Nicotine Dependence and Reward Differ between Adolescent and Adult Male Mice. J. Pharmacol. Exp. Ther 322, 399–407. 10.1124/jpet.107.121616 [DOI] [PubMed] [Google Scholar]
- Le Foll B, Goldberg SR, 2004. Rimonabant, a CB1 antagonist, blocks nicotine-conditioned place preferences. Neuroreport 15, 2139–2143. 10.1097/00001756-200409150-00028 [DOI] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF, 2009a. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol 5, 37–44. 10.1038/nchembio.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF, 2009b. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci 106, 20270–20275. 10.1073/pnas.0909411106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchicchi A, Lecca S, Carta S, Pillolla G, Muntoni AL, Yasar S, Goldberg SR, Pistis M, 2010. Effects of fatty acid amide hydrolase inhibition on neuronal responses to nicotine, cocaine and morphine in the nucleus accumbens shell and ventral tegmental area: involvement of PPAR-α nuclear receptors. Addict. Biol 15, 277–288. 10.1111/j.1369-1600.2010.00222.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Torres S, Cutando L, Pastor A, Kato A, Sakimura K, de la Torre R, Valjent E, Maldonado R, Kano M, Ozaita A, 2019. Monoacylglycerol lipase blockade impairs fine motor coordination and triggers cerebellar neuroinflammation through cyclooxygenase-2. Brain. Behav. Immun 81, 399–409. 10.1016/j.bbi.2019.06.036 [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Bonner TI, Lolait SJ, 1993. Localization of cannabinoid receptor mRNA in rat brain. J. Comp. Neurol 327, 535–50. 10.1002/cne.903270406 [DOI] [PubMed] [Google Scholar]
- McMahon LR, Ginsburg BC, Lamb RJ, 2008. Cannabinoid agonists differentially substitute for the discriminative stimulus effects of Δ9-tetrahydrocannabinol in C57BL/6J mice. Psychopharmacology (Berl). 198, 487–495. 10.1007/s00213-007-0900-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z, 1995. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol 50, 83–90. 10.1016/0006-2952(95)00109-D [DOI] [PubMed] [Google Scholar]
- Melis M, Pillolla G, Luchicchi A, Muntoni AL, Yasar S, Goldberg SR, Pistis M, 2008. Endogenous fatty acid ethanolamides suppress nicotine-induced activation of mesolimbic dopamine neurons through nuclear receptors. J. Neurosci 10.1523/JNEUROSCI.3221-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt LL, Martin BR, Walters C, Lichtman AH, Damaj MI, 2008. The Endogenous Cannabinoid System Modulates Nicotine Reward and Dependence. J. Pharmacol. Exp. Ther 326, 483–492. 10.1124/jpet.108.138321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muldoon PP, Chen J, Harenza JL, Abdullah RA, Sim-Selley LJ, Cravatt BF, Miles MF, Chen X, Lichtman AH, Damaj MI, Muldoon P, 2015. Inhibition of monoacylglycerol lipase reduces nicotine withdrawal. www.brjpharmacol.org Br. J. Pharmacol 172, 869 10.1111/bph.12948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete F, Rodríguez-Arias M, Martín-García E, Navarro D, García-Gutiérrez MS, Aguilar MA, Aracil-Fernández A, Berbel P, Miñarro J, Maldonado R, Manzanares J, 2013. Role of CB2 Cannabinoid Receptors in the Rewarding, Reinforcing, and Physical Effects of Nicotine. Neuropsychopharmacology 38, 2515–2524. 10.1038/npp.2013.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Hudak CSS, Ward AM, Burston JJ, Issa RS, Fisher KJ, Abood ME, Wiley JL, Lichtman AH, Casida JE, 2008. Monoacylglycerol lipase regulates 2-arachidonoylglycerol action and arachidonic acid levels. Bioorg. Med. Chem. Lett 18, 5875–5878. 10.1016/j.bmcl.2008.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ, Hoover HH, Cravatt BF, 2011. Monoacylglycerol Lipase Exerts Dual Control over Endocannabinoid and Fatty Acid Pathways to Support Prostate Cancer. Chem. Biol 18, 846–856. 10.1016/j.chembiol.2011.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu Q, 2009. Blockade of 2-Arachidonoylglycerol Hydrolysis by Selective Monoacylglycerol Lipase Inhibitor 4-Nitrophenyl 4-(Dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) Enhances Retrograde Endocannabinoid Signaling. J. Pharmacol. Exp. Ther 331, 591–597. 10.1124/jpet.109.158162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon FJ, Serrano A, Sidhpura N, Polis I, Stouffer D, de Fonseca FR, Cravatt BF, Martin-Fardon R, Parsons LH, 2018. Fatty acid amide hydrolase (FAAH) inactivation confers enhanced sensitivity to nicotine-induced dopamine release in the mouse nucleus accumbens. Addict. Biol 23, 723–734. 10.1111/adb.12531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Caldarone BJ, King SL, Zachariou V, 2000. Nicotinic receptors in the brain: Links between molecular biology and behavior. Neuropsychopharmacology. 10.1016/S0893-133X(99)00146-3 [DOI] [PubMed] [Google Scholar]
- Prado GF, Lombardi EMS, Bussacos MA, Arrabal-Fernandes FL, Terra-Filho M, Santos U. de P., 2011. A real-life study of the effectiveness of different pharmacological approaches to the treatment of smoking cessation: re-discussing the predictors of success. Clinics 66, 65–71. 10.1590/S1807-59322011000100012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins S, George F. Koob, Athina S, 2000. Neural mechanisms underlying nicotine addiction: acute positive reinforcement and withdrawal. Nicotine Tob. Res 2, 19–37. 10.1080/14622200050011277 [DOI] [PubMed] [Google Scholar]
- Savinainen JR, Järvinen T, Laine K, Laitinen JT, 2001. Despite substantial degradation, 2-arachidonoylglycerol is a potent full efficacy agonist mediating CB 1 receptor-dependent G-protein activation in rat cerebellar membranes. Br. J. Pharmacol 134, 664–672. 10.1038/sj.bjp.0704297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherma M, Panlilio LV, Fadda P, Fattore L, Gamaleddin I, Le Foll B, Justinová Z, Mikics E, Haller J, Medalie J, Stroik J, Barnes C, Yasar S, Tanda G, Piomelli D, Fratta W, Goldberg SR, 2008. Inhibition of Anandamide Hydrolysis by Cyclohexyl Carbamic Acid 3′-Carbamoyl-3-yl Ester (URB597) Reverses Abuse-Related Behavioral and Neurochemical Effects of Nicotine in Rats. J. Pharmacol. Exp. Ther 327, 482–490. 10.1124/jpet.108.142224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu Q, Lichtman AH, Cravatt BF, 2010. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci 13, 1113–1119. 10.1038/nn.2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim-Selley LJ, Schechter NS, Rorrer WK, Dalton GD, Hernandez J, Martin BR, Selley DE, 2006. Prolonged Recovery Rate of CB 1 Receptor Adaptation after Cessation of Long-Term Cannabinoid Administration. Mol. Pharmacol 70, 986–996. 10.1124/mol.105.019612 [DOI] [PubMed] [Google Scholar]
- Solinas M, Tanda G, Justinova Z, Wertheim CE, Yasar S, Piomelli D, Vadivel SK, Makriyannis A, Goldberg SR, 2007. The Endogenous Cannabinoid Anandamide Produces δ−9-Tetrahydrocannabinol-Like Discriminative and Neurochemical Effects That Are Enhanced by Inhibition of Fatty Acid Amide Hydrolase but Not by Inhibition of Anandamide Transport. J. Pharmacol. Exp. Ther 321, 370–380. 10.1124/jpet.106.114124 [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K, 1995. 2-arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun 215, 89–97. 10.1006/bbrc.1995.2437 [DOI] [PubMed] [Google Scholar]
- Teather LA, 2002. Post-Training Cyclooxygenase-2 (COX-2) Inhibition Impairs Memory Consolidation. Learn. Mem 9, 41–47. 10.1101/lm.43602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sañudo-Peña MC, Mackie K, Walker JM, 1998. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83, 393–411. 10.1016/S0306-4522(97)00436-3 [DOI] [PubMed] [Google Scholar]
- Williams JH, 1996. Retrograde messengers and long-term potentiation: A progress report. J. Lipid Mediat. Cell Signal 14, 331–339. 10.1016/0929-7855(96)00542-1 [DOI] [PubMed] [Google Scholar]
- Wise LE, Long KA, Abdullah RA, Long JZ, Cravatt BF, Lichtman AH, 2012. Dual Fatty Acid Amide Hydrolase and Monoacylglycerol Lipase Blockade Produces THC-Like Morris Water Maze Deficits in Mice. ACS Chem. Neurosci 3, 369–378. 10.1021/cn200130s [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization, 2019. Tobacco [WWW Document]. World Heal. Organ URL https://www.who.int/en/news-room/fact-sheets/detail/tobacco (accessed 8.31.19).