

Abstract
Objective:
Subgroup analyses of clinical trial data can be an important tool for understanding when treatment effects differ across populations. That said, even effect estimates from pre-specified subgroups in well-conducted trials may not apply to corresponding subgroups in the source population. While this divergence may simply reflect statistical imprecision, there has been less discussion of systematic or structural sources of misleading subgroup estimates.
Study Design/Setting:
We use directed acyclic graphs to show how selection bias caused by associations between effect measure modifiers and trial selection, whether explicit (e.g. eligibility criteria) or implicit (e.g. self-selection based on race), can result in subgroup estimates that do not correspond to subgroup effects in the source population. To demonstrate this point, we provide a hypothetical example illustrating the sorts of erroneous conclusions that can result, as well as their potential consequences. We also provide a tool for readers to explore additional cases.
Conclusion:
Treating subgroups within a trial essentially as random samples of the corresponding subgroups in the wider population can be misleading, even when analyses are conducted rigorously and all findings are internally valid. Researchers should carefully examine associations between (and consider adjusting for) variables when attempting to identify heterogeneous treatment effects.
Keywords: Subgroups, external validity, selection bias, causal graphs
Background/Aims
A common sight in the published results of a randomized controlled trial is a forest plot showing the treatment effect in trial participants separately for two or more groups: for example, for men and women, or for those over and under the median age of participants, or for smokers and non-smokers. The expectation is that the treatment effect within each subgroup shows the effect in that type of participant, because randomization is maintained within subgroups.1, 2
Unfortunately, these simple analyses can lead to inaccurate conclusions about the benefits to patients in the corresponding subgroups in the source population.3–6 One subtle fact is that subgroup estimates can become misleading when trial participation depends on multiple factors, even when not all of these factors are associated with the outcome in the source population. This occurs whether factors are explicitly associated with participation (e.g. through eligibility criteria) or implicitly by virtue of self-selection (e.g. through lower participation of older patients or people of color).
Here, we use causal graphs to help explain this problem, and walk through an example of a potentially misleading subgroup analysis. We also provide a tool for researchers to construct and examine additional scenarios that may be relevant to their specific area of study and show that appropriately adjusted subgroup analyses can yield more informative results.
Methods
Theoretical Framework
Suppose we are planning a randomized controlled trial of a treatment X on an outcome Y (say, mortality). We are interested in examining whether the effect of X differs depending on the level of another categorical variable, V1, in the source population: that is, not just in the sample in which our study is conducted, but in the population which gave rise to our study sample (sometimes known as the target population).7 For example, if V1 is self-reported race, then our question is whether the effect of treatment is the same in each racial group. In this example, we assume that there is no effect of V1 on Y, no common causes of V1 and Y, and no effect measure modification of the treatment effect by V1 in our target population; i.e., that the effect of X on Y on the scale of interest is the same at both levels of V1. If the probability of participation for those with V1 is equal to the probability of participation for those without V1, each subgroup defined by V1 will yield an unbiased estimate of the treatment effect in the corresponding portion of the source population in expectation.
However, when V1 and another variable V2 (say, severity of illness) are both associated with study participation a problem can arise. Consider the directed acyclic graphs in Figure 1.8 The arrows from V1 and V2 into participation on the left represent the fact that altering either of the variables changes the chances of trial participation. This might be the case if Black adults and adults with more severe illness were less likely to participate in the trial.9 Variable V1 and V2 also share no common causes-meaning that there are no other variables, measured or unmeasured, that cause both V1 and V2. Because of this, there is only one causal path from V1 to Y in the source population in Figure 1A: the path from V1 -> Participation <- V2 -> Y. This path is blocked, however, because participation is a collider (so-called because the arrows from V1 and V2 “collide” at participation).10 As a result, we expect to see no association between V1 and Y in the full population and V1 should not modify the effect of X on Y.11
Figure 1.
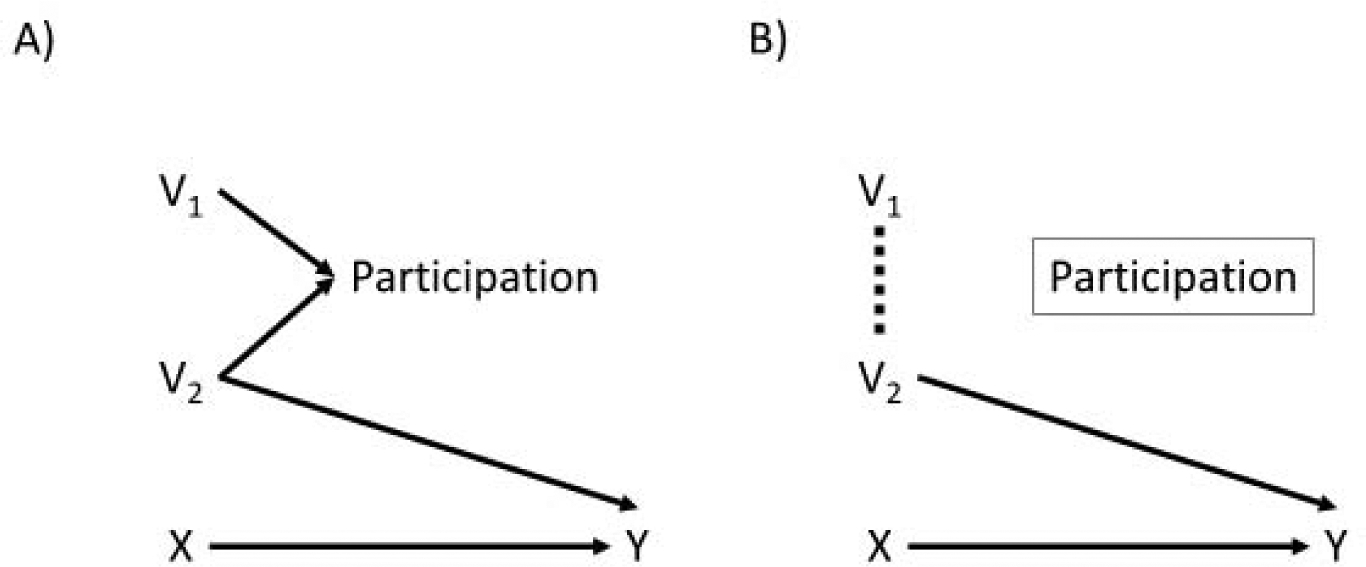
Two directed acyclic graphs. Panel A shows the causal relationships between V1, V2, X, participation, and Y in the source population; since there is no open path from V1 to Y in panel A, V1 will not modify the treatment effect of X on Y. Panel B shows the causal relationships between the same variables in the study population. Because we are conditioning on study participation, which depends on V1 and V2, there is now an open path from V1 to Y that can make V1 act as an effect modifier in the study population.
Of course, when we conduct a trial, the trial population naturally restricts to those who were recruited into the study - we cannot study people who were not recruited or did not participate. This leads to the situation shown in Figure 1B. Conditioning on the participation collider opens up an association between its parents, V1 and V2, in at least one stratum of participation. This means that we can expect V1 to be associated (via V2) with the outcome in the trial; if we look for variation of the effect of X on Y by levels of V1 within the trial participants we might well find it and, indeed, will find it on at least one of the relative (i.e. risk ratio) and absolute (i.e. risk difference) scales.11, 12 We put numbers to an example scenario.
Hypothetical Example
Suppose researchers are conducting a placebo-controlled trial of a novel therapeutic agent, Fakeium (X), that may reduce the 5-year risk of all-cause mortality (Y) after myocardial infarction when added to standard treatment. To achieve power to address a safety concern that has been seen in other drugs in the class, they had to recruit 89,375 patients from a source population of 1,000,000. Knowing that minority populations and people of color are often underrepresented in clinical trials and unsure about whether they may benefit more or less from treatment,13, 14 they pre-specified subgroup analyses of those who identified as Black and those who did not (they anticipated being underpowered for any analyses in smaller ethnic groups).
One-quarter of patients in the source population are Black (V1), half of the myocardial infarctions (MIs) in the source population are less severe and half were more severe (V2), and race and MI severity are independent. Severity modifies treatment effect in that Fakeium is effective only in those with less severe MIs: these patients had a 30% chance of death in the placebo arm and a 25% chance of death in the Fakeium arm, while higher severity patients have a 40% chance of death regardless of treatment status. Race, however, does not modify treatment effect, and is not associated with the outcome. In the study, patients with severe myocardial infarctions had a 2% chance of being recruited if Black and a 10% chance of being recruited if non-Black. If the infarction was less severe, patients were more likely to participate regardless of race, with participation rates of 11% in non-Black and 8% in Black participants.
Table 1 shows the expected distributions of race and myocardial infarction severity in the source population and the trial population. While the odds for mild infarction are identical in Black and non-Black participants in the source population, they differ substantially in the trial because both race and MI severity have an effect on study sampling. This has direct consequences for the trial subgroup analyses. Table 2 presents the expected results of 1) an imaginary study and subgroup analyses conducted on the source population and 2) a study conducted in the trial population.
Table 1:
Expected distributions of race and myocardial infarction severity in the 1,000,000 patients in the source population and the 89,375 patients in the trial.
| Population | Severe infarctions | Mild infarctions | Odds of mild infarction | Odds ratio of severe vs mild infarction |
|---|---|---|---|---|
| Source population | ||||
| Non-Black race | 375,000 | 375,000 | 1:1 | Ref. |
| Black race | 125,000 | 125,000 | 1:1 | 1.00 |
| Trial population | ||||
| Non-Black race | 37,500 | 41,250 | 1:1.1 | Ref. |
| Black race | 2,500 | 10,000 | 1:4 | 3.63 |
Table 2:
Expected results of two randomized studies, one conducted in the source population if we did not have to recruit participants and one conducted in the trial population under our participation criteria above.
| Analysis and treatment arm | Participants | Deaths | Risk of five-year mortality | Risk difference for five-year mortality (95% CI) |
|---|---|---|---|---|
| Source population | ||||
| Placebo | 500,000 | 175,000 | 35.00% | Ref. |
| Fakeium | 500,000 | 160,000 | 32.00% | −3.00% (−3.18%, −2.82%) |
| Source population Black subgroup | ||||
| Placebo | 125,000 | 43,750 | 35.00% | Ref. |
| Fakeium | 125,000 | 40,000 | 32.00% | −3.00% (−3.37%, −2.63%) |
| Source population non-Black subgroup | ||||
| Placebo | 375,000 | 131,250 | 35.00% | Ref. |
| Fakeium | 375,000 | 120,000 | 32.00% | −3.00% (−3.21%, −2.79%) |
| Trial population | ||||
| Placebo | 45,625 | 15,687 | 34.38% | Ref. |
| Fakeium | 45,625 | 14,150 | 31.01% | −3.37% (−3.98%, −2.76%) |
| Trial Black subgroup | ||||
| Placebo | 6,250 | 2000 | 32.00% | Ref. |
| Fakeium | 6,250 | 1700 | 27.20% | −4.80% (−6.40%, −3.20%) |
| Trial non-Black subgroup | ||||
| Placebo | 39,375 | 13,687 | 34.76% | Ref. |
| Fakeium | 39,375 | 12,450 | 31.62% | −3.14% (−3.80%, −2.48%) |
Two conclusions might be drawn from the trial results. First, that Fakeium protects against mortality in the overall trial population (number needed to treat, NNT, of 30). Second, that Fakeium has a substantially more potent protective effect in those that identify as Black than those that do not (NNT of 21 in those patients versus NNT of 32 in the remaining patients). The p-value for heterogeneity between the two groups is 0.06. With these results Fakeium might be used more often in Black patients than non-Black patients, especially if it is expensive or has a burdensome side effect profile.
Of course, as discussed above and shown in the full population data, Fakeium is equally effective in Blacks and non-Blacks in the source population (the p value for interaction there is 1.00); when we do not condition on trial participation, only myocardial infarction severity modifies the effect of Fakeium.
This finding is not the result of statistical noise or hunting for significant p values but instead the result of conditioning on a collider inflating the prevalence of lower severity myocardial infarctions among black trial participants. Even if we repeated the trial an infinite number of times, or tripled the trial enrollment, estimated benefits in Blacks would still be exaggerated in this analysis in the trial population; the problem arises from the causal structure of selection from the source population into the trial, not from random chance.
Generally, this type of non-random selection requires external data from the source population to recover unbiased effects in said source population.15, 16 Here, however, we can account for this non-random selection by performing a subgroup analysis that adjusts for both race (V1) and MI severity (V2) by fitting a multivariable identity-link linear regression with a term for treatment, a term for race, a term for MI severity, and an interaction term between race and treatment.
Because MI severity is now being held constant, the interaction between race and treatment will appear to be null (even though the lack of an interaction term between MI severity and treatment means the model is technically misspecified). If we instead fit a regression that includes a term for treatment, a term for MI severity, an interaction term between the two, and a term for race, we can see that the treatment effect varies across MI severity even if race is held constant. Including these predictors of the outcome in the model also has the advantage of helping reduce the impact of chance imbalances between study arms and between subgroups.17 By looking at the full constellation of variables that might impact treatment effect at once (the trees, collectively) instead of just looking at them one by one (the forest), it can become much clearer which variables are associated directly with better or worse treatment effects in the trial.
Discussion
In our sample case, taking marginal subgroup analyses at face value was dangerous even when the analysis was pre-specified. Considering this finding through the lens of collider bias-a problem much discussed as a threat to internal validity10, 18-helped clarify both why it occurred and how it might be resolved. This potential bias extends to many other scenarios where multiple variables are associated with trial participation, which we explored with the help of an Excel spreadsheet (eSupplement 1) that readers can use themselves as well. These associations can be implicit (via cultural or institutional norms related to trial participation) or explicit (via inclusion and exclusion criteria).
This work is not meant to discourage researchers from conducting subgroup analyses or attempting to identify heterogeneity of treatment effect across different groups; identifying and understanding interaction, particularly additive-scale interaction, is vital for public health.7, 19 Rather, it seeks to encourage researchers to move beyond simple stratification and to think deeply about the relationships between various subgroups, trial participation, and the outcome. They can then conduct adjusted subgroup analyses that inform later research using statistical methods (like outcome modeling or weighting) to generalize or transport treatment effects to other target populations.20, 21
From experimentation with the spreadsheet, the magnitude of the V1-V2 association that is created by conditioning on participation varies heavily depending on the strength of the individual V1 and V2 causal effects on participation, as well as whether they interact with one another with respect to participation. In fact, if their effects on participation are perfectly multiplicative and V1 and V2 are independent, no V1-V2 association is present within the trial sample (though one is created in the unsampled individuals). For the most part, this type of exact mathematical cancellation (called unfaithfulness in the world of causal diagrams) is not likely to occur naturally.22, 23
If there are multiple explicit inclusion criteria that can overlap, such as requiring at least one of hypertension and diabetes after myocardial infarction in cardiovascular trials, the bias is present in the subgroups that do not possess one of the inclusion criteria (e.g. those without hypertension and those without diabetes). This occurs because hypertension only has a causal effect on participation in those without diabetes and diabetes only has a causal effect on participation in those without hypertension; the result is that the collider-generated association is only present in those strata. When the associations are due to implicit factors, as in our example, subgroups across levels of V1 are all potentially misleading.
Notably, even if V1 does modify the treatment effect (Figure 2), the subgroup effects in the trial can still become a biased estimate of the effects in the source population. Additionally, any subgroup analyses for V2 will now differ as well. The collider-induced association could push the difference between groups either further from or closer to the null, depending on the direction of the modification and the specific relationships between the variables and participation. Changes in subgroup composition and participation across attempts to replicate the subgroup results could result in failure to reject null hypotheses of homogeneity, even when real effect measure modification does exist.
Figure 2.
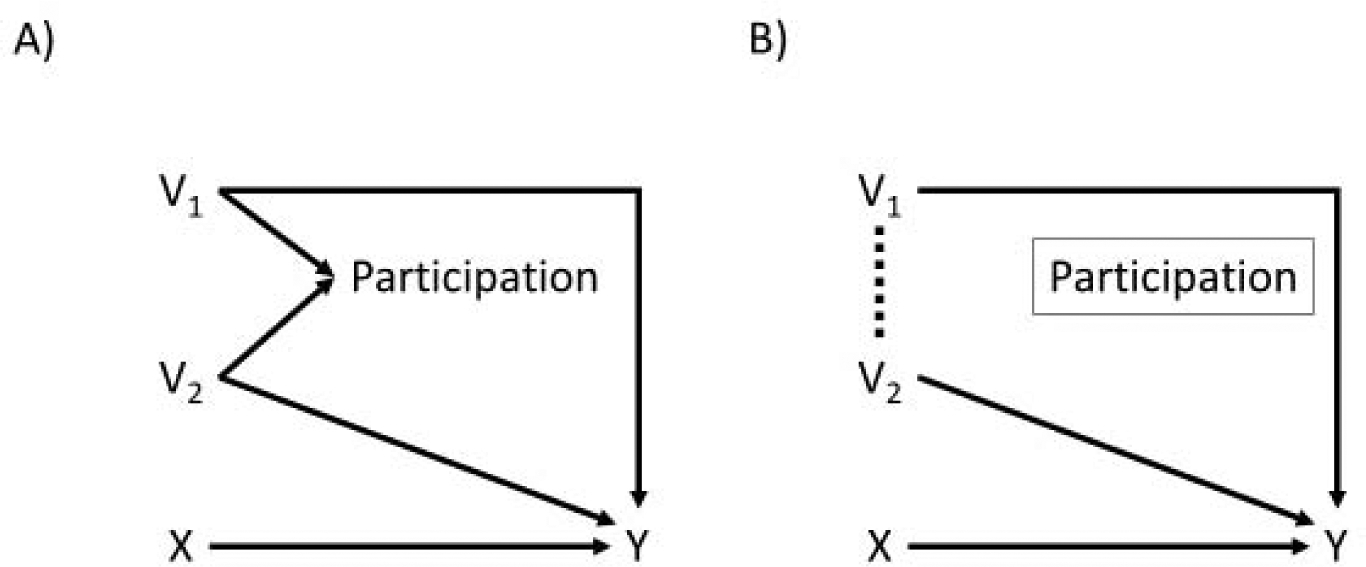
Directed acyclic graphs for when both V1 and V2 directly modify the treatment effect of X on Y. Panel A is the directed acyclic graph for the full population, while Panel B is the directed acyclic graph for the trial population. Both variables now have arrows indicating a causal effect on Y, and as a result both V1 and V2 are expected to have a different association with Y before and after conditioning on study participation.
We used a hypothetical example because that was the only way to know and tweak the input parameters; that said, we think this problem may occur frequently. With respect to explicit associations with participation, trials (especially cardiovascular trials like AFCAPS1 or the trials of direct-acting oral anticoagulants in atrial fibrillation24–26) often only enroll patients with a high enough risk based on some risk score, and then stratify by the components of the risk score in subsequent subgroup analyses. While it is more difficult to assess the prevalence of the implicit associations with participation, demographic factors such as sex, race, and age (which have historically been associated with trial participation)27 may impact participation differently in those with and without various chronic conditions. In either case, these risk factors or variables associated with the outcome will act as effect measure modifiers on the additive (i.e. risk difference) or relative (i.e. risk ratio) scale, if not both.12 The final result is misleading subgroup effects across the demographic factors, the chronic conditions, or (most likely) both.
There are several important limitations to this work. The extent of this problem can vary substantially depending on the underlying causal relationships between variables. The degree to which the results are misleading largely depends on whether and by how much V2 modifies the treatment effect of interest-if it only modifies the risk difference, the subgroup risk ratios can be unbiased, and vice versa (and unfortunately, whether a variable modifies the risk difference, risk ratio, or both cannot be derived from a directed acyclic graph).28 Another limitation is that we only explored binary variables; while these findings likely generalize to multilevel categorical or continuous variables, their magnitude and severity may differ. Our spreadsheet worked with relatively few potential effect modifiers, examining at most three modifiers simultaneously (V1, V2, V3). That said, with a larger covariate space, this phenomenon becomes more and more likely to occur.
Additionally, chance imbalances between treatment arms or between subgroups are not captured in these types of diagrams and can also result in biased estimates, particularly in smaller studies.17 Finally and perhaps most importantly, we merely demonstrated the existence of this potential problem; further work to understand its plausibility and impact in different types of trials using real data and more complex simulations is necessary to understand its impact.
Two final notes: first, this is not an internal validity problem. The estimates within each subgroup are valid estimates of treatment effect for that subgroup of trial participants. There is no graphical basis for adjustment for V1 and V2 when estimating these internal effects. But if subgroup analyses are being performed to help understand differences in treatment effects in larger (target) populations, they can easily mislead investigators, potentially with significant clinical consequences.7 Second, while we presented this through the lens of randomized trials because of their analytic simplicity, the same types of causal relationships can cause problems for observational data that undergoes an analogous selection process (e.g. prospective enrollment or restricting to those with specific insurance plans) if researchers simply split the study population after adjusting for confounding.
Conclusion
It is critical to identify heterogeneity in the effects of treatments in the source population of a study and not just in the study population. Subgroup analyses appear to be a simple method of doing so, but associations between subgroup strata and trial participation can lead to biased and potentially dangerous conclusions about treatment effects in subgroups in the source population. Moving beyond crude subgroup analyses is a crucial step towards the end goal of estimating treatment effects in routine clinical care; glances at forest plots are poor substitutes for in-depth and considered examinations of the trees.
Supplementary Material
What is new?
| Subgroup results can be misleading for reasons based on the factors underlying trial participation. |
| These results can result in incorrect beliefs about the efficacy of treatments in clinical populations. |
| Examining multiple potential modifiers of treatment effect at once, rather than conducting multiple crude subgroup analyses, can yield useful information about how and why variables are associated with subgroup differences in treatment effects. |
Acknowledgements:
The authors would like to thank the International Society of Pharmacoepidemiology and Drug Safety for accepting a related abstract for presentation at their 2019 conference.
Funding: MJF was supported by NIH/NHLBI R01HL118255.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: The authors have no competing interests to disclose.
References:
- 1.Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998; 279: 1615–22. [DOI] [PubMed] [Google Scholar]
- 2.Westreich D Epidemiology by Design: A Causal Approach to the Health Sciences. Oxford University Press, 2019. [Google Scholar]
- 3.Tanniou J, Tweel IV, Teerenstra S and Roes KC. Level of evidence for promising subgroup findings in an overall non-significant trial. Statistical Methods in Medical Research. 2016; 25: 2193–213. [DOI] [PubMed] [Google Scholar]
- 4.Bell S, Kivimäki M and Batty GD. Subgroup analysis as a source of spurious findings: an illustration using new data on alcohol intake and coronary heart disease. Addiction (Abingdon, England). 2015; 110: 183–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brookes ST, Whitley E, Peters TJ, Mulheran PA, Egger M and Davey Smith G. Subgroup analyses in randomised controlled trials: quantifying the risks of false-positives and false-negatives. Health Technology Assessment (Winchester, England). 2001; 5: 1–56. [DOI] [PubMed] [Google Scholar]
- 6.VanderWeele TJ and Knol MJ. Interpretation of subgroup analyses in randomized trials: heterogeneity versus secondary interventions. Annals of Internal Medicine. 2011; 154: 680–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Westreich D, Edwards JK, Lesko CR, Cole SR and Stuart EA. Target Validity and the Hierarchy of Study Designs. American Journal of Epidemiology. 2019; 188: 438–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenland S, Pearl J and Robins JM. Causal diagrams for epidemiologic research. Epidemiology (Cambridge, Mass) 1999; 10: 37–48. [PubMed] [Google Scholar]
- 9.Loree JM, Anand S, Dasari A, et al. Disparity of Race Reporting and Representation in Clinical Trials Leading to Cancer Drug Approvals From 2008 to 2018. JAMA Oncology. 2019: e191870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cole SR, Platt RW, Schisterman EF, et al. Illustrating bias due to conditioning on a collider. International Journal of Epidemiology. 2010; 39: 417–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.VanderWeele TJ and Robins JM. Four types of effect modification: a classification based on directed acyclic graphs. Epidemiology (Cambridge, Mass). 2007; 18: 561–8. [DOI] [PubMed] [Google Scholar]
- 12.Hernan MA. Invited Commentary: Selection Bias Without Colliders. American Journal of Epidemiology. 2017; 185: 1048–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen MS Jr., Lara PN, Dang JH, Paterniti DA and Kelly K. Twenty years post-NIH Revitalization Act: enhancing minority participation in clinical trials (EMPaCT): laying the groundwork for improving minority clinical trial accrual: renewing the case for enhancing minority participation in cancer clinical trials. Cancer. 2014; 120 Suppl 7: 1091–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park IU and Taylor AL. Race and ethnicity in trials of antihypertensive therapy to prevent cardiovascular outcomes: a systematic review. Annals of Family Medicine. 2007; 5: 444–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westreich D, Edwards JK, Lesko CR, Stuart E and Cole SR. Transportability of Trial Results Using Inverse Odds of Sampling Weights. American Journal of Epidemiology. 2017; 186: 1010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cole SR and Stuart EA. Generalizing evidence from randomized clinical trials to target populations: The ACTG 320 trial. American Journal of Epidemiology. 2010; 172: 107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenland S and Mansournia MA. Limitations of individual causal models, causal graphs, and ignorability assumptions, as illustrated by random confounding and design unfaithfulness. European Journal of Epidemiology. 2015; 30: 1101–10. [DOI] [PubMed] [Google Scholar]
- 18.Hernan MA, Hernandez-Diaz S and Robins JM. A structural approach to selection bias. Epidemiology (Cambridge, Mass). 2004; 15: 615–25. [DOI] [PubMed] [Google Scholar]
- 19.Lesko CR, Henderson NC and Varadhan R. Considerations when assessing heterogeneity of treatment effect in patient-centered outcomes research. Journal of Clinical Epidemiology. 2018; 100: 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westreich D, Edwards JK, Lesko CR, Stuart E and Cole SR. Transportability of trial results using inverse odds of sampling weights. American Journal of Epidemiology. 2017; 186: 1010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dahabreh IJ and Hernán MA. Extending inferences from a randomized trial to a target population. European Journal of Epidemiology. 2019: 1–4. [DOI] [PubMed] [Google Scholar]
- 22.Mansournia MA and Greenland S. The relation of collapsibility and confounding to faithfulness and stability. Epidemiology (Cambridge, Mass). 2015; 26: 466–72. [DOI] [PubMed] [Google Scholar]
- 23.Mansournia MA, Hernán MA and Greenland S. Matched designs and causal diagrams. International Journal of Epidemiology. 2013; 42: 860–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. The New England Journal of Medicine. 2009; 361: 1139–51. [DOI] [PubMed] [Google Scholar]
- 25.Patel MR, Mahaffey KW, Garg J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. The New England Journal of Medicine. 2011; 365: 883–91. [DOI] [PubMed] [Google Scholar]
- 26.Granger CB, Alexander JH, McMurray JJ, et al. Apixaban versus warfarin in patients with atrial fibrillation. The New England Journal of Medicine. 2011; 365: 981–92. [DOI] [PubMed] [Google Scholar]
- 27.Murthy VH, Krumholz HM and Gross CP. Participation in cancer clinical trials: race-, sex-, and age-based disparities. JAMA. 2004; 291: 2720–6. [DOI] [PubMed] [Google Scholar]
- 28.Weinberg CR. Can DAGs clarify effect modification? Epidemiology (Cambridge, Mass). 2007; 18: 569–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.