

Abstract
The ability of DNA to produce a functional protein even after transfer to a foreign host is of fundamental importance in both evolutionary biology and biotechnology, enabling horizontal gene transfer in the wild and heterologous expression in the lab. However, the influence of genetic particulars on DNA’s functionality in a new host remains poorly understood, as do the evolutionary mechanisms of assimilation and refinement. Here, we describe an automation-enabled large-scale experiment wherein Escherichia coli strains were evolved in parallel after replacement of genes pgi or tpiA with orthologous DNA from donor species spanning all domains of life, from humans to hyperthermophilic archaea. We show via analysis of hundreds of clones evolved for 50,000+ cumulative generations across dozens of independent lineages that orthogene-upregulating mutations can completely mitigate fitness defects resulting from initial nonfunctionality, with coding sequence changes unnecessary. Gene target, donor species, and genomic location of the swap all influenced outcomes – both the nature of adaptive mutations (often synonymous) and the frequency with which strains successfully evolved to assimilate the foreign DNA. Additionally, time series DNA sequencing and replay evolution experiments revealed transient copy number expansions, the contingency of lineage outcome on first-step mutations, and the ability for strains to escape from sub-optimal local fitness maxima. Overall, this study establishes the influence of various DNA and protein features on cross-species genetic interchangeability and evolutionary outcomes, with implications for both horizontal gene transfer and rational strain design.
Horizontal Gene Transfer (HGT), the non-reproductive transmission of genetic material that can transcend species boundaries, is possible due to shared mechanisms of DNA decoding machinery inherited by all known life following descent from the Last Universal Common Ancestor1. In addition to shaping Earth’s web of life, HGT has both clinical and industrial importance due to influencing the spread of antimicrobial resistance2 and facilitating the engineering of organisms with desired phenotypes3, respectively. Understanding how HGT content assimilates into a new genome is thus of both basic and applied interest. The mechanistic basis for gene transfer has been increasingly uncovered4 and replacement studies have established the impressive extent to which many genes retain cross-species functionality despite billions of years of phylogenetic divergence5–7. However, studies on the functionality of a gene replacement immediately post-transfer are insufficient to reveal how organisms can adapt to utilize foreign DNA that initially provides no fitness benefit.
Several studies to-date have paired gene-swaps with evolution, providing insight into the process of foreign gene assimilation. Lind et al. replaced ribosomal genes in S. typhimurium with microbial orthologs, and within a few hundred generations of laboratory evolution found gene amplifications aimed at increasing orthogene copy number as a way to ameliorate fitness defects8. Kacar et al. replaced the essential elongation factor tufB in E. coli with an ancestral variant, and evolution similarly selected for upregulation9. Bershtein et al. replaced the essential folA gene in E. coli with orthologs from 35 close bacterial relatives and found that fitness defects were frequently evolutionarily compensated by protease-deactivating mutations that increased intracellular orthogene levels10. Such studies establish that insufficient expression levels regularly hinder in vivo functionality of foreign genes, but the influence of particular variables remains difficult or impossible to deconvolute from existing data. Codon optimization was typically performed on the foreign genes before insertion, a critical change that limits applicability of observed results to natural HGT. Essential genes were also the main targets for replacement, precluding any outcome in which an initially non-functional swap could evolve functionality, and vastly limiting the pool of organisms from which an orthogene could be taken without inducing lethality in the new host. Additional confounding factors complicate mutational interpretation, such as the use of gene targets involved in protein-protein interactions and complex formation, or differences in plasmid vs. chromosomal insertion. Recent advances in automation11 enable experimental evolution of a heretofore infeasible scale and data resolution, providing an empirical means to address unresolved details on cross-species gene functionality, potential for evolutionary assimilation, and mutational mechanisms.
Here, we designed and constructed eight distinct gene-swapped Escherichia coli strains with orthologous donor DNA from four species spanning all domains of life: the γ- and α- proteobacteria Vibrio cholerae (Vch) and Brucella melitensis (Bme), the hyperthermophilic archaeum Pyrobaculum aerophilum (Pae), and the mammalian eukaryote Homo sapiens (Hsa) (Fig. 1a). Glycolytic isomerase genes pgi and tpiA were selected for swapping for a number of reasons – they are nonessential but crucial for fitness, highly studied with known post-knockout evolutionary outcomes12–14, and do not require cofactors or participate in protein complexes (Extended Data Fig. 1 and Supplementary Table 1). To further minimize potential confounding factors, strain construction involved scarless chromosomal replacement, from start to stop codon, with the coding sequence of the foreign ortholog not subjected to any codon optimization. Automated Adaptive Laboratory Evolution (ALE) systems15 allowed dozens of lineages to be evolved for growth rate improvements with real-time tracking of adaptive trajectories. Such high-throughput evolution studies essentially serve as empirical Monte Carlo sampling of the fitness landscape, providing insight into the influence on adaptive outcomes of chromosomal location, local sequence context, and nucleotide/amino acid features (Fig. 1b).
Fig. 1. Gene-swap strain construction and laboratory evolution.
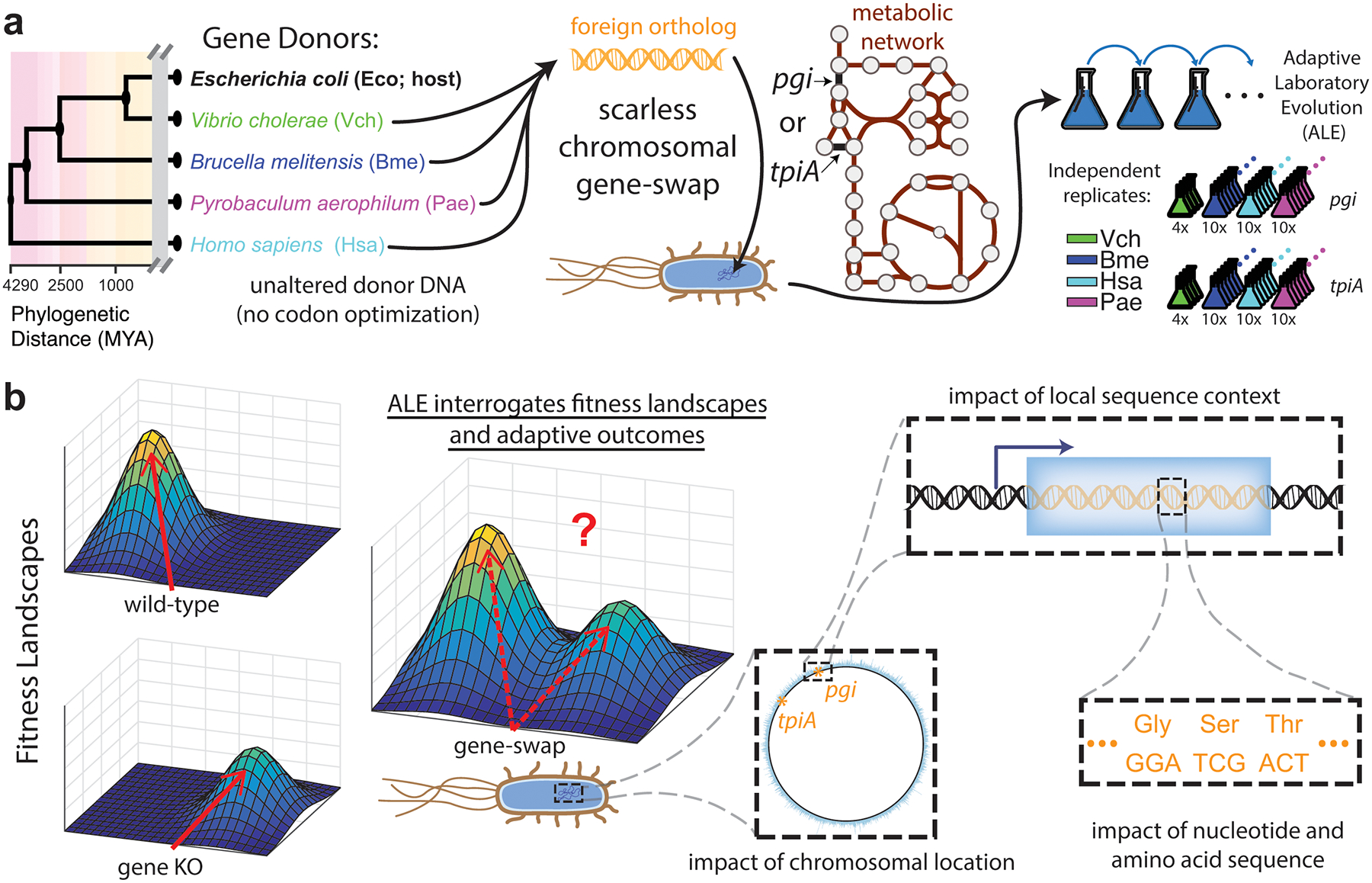
a, Schematic of experimental workflow. Native E. coli glycolytic isomerases pgi and tpiA were replaced with the coding sequence of foreign orthologs and subjected to laboratory evolution for improved exponential phase growth rate. b, Experimental evolution enables interrogation of the fitness landscape following gene replacement. The spectrum of different swaps and large replicate number provides insight into various factors influencing adaptive outcomes.
Results and Discussion
Gene-swapped strains exhibited an initial physiology that was consistent with phylogenetic distance from the donor species: Vch swaps did not show any phenotypic defects, Bme swaps grew slower than wild-type but faster than full knockouts, while Pae and Hsa swaps had the same slow growth rate as knockouts. The intrinsic growth rate gap between the wild-type and pgi or tpiA deficient strains, and their distinct evolutionary trajectories, enables fitness-based classification of gene-swap lineages into ‘success’ or ‘failure’ of orthogene assimilation (Fig. 2a). Across the 68 independently evolved gene-swap lineages we see significantly different gene- and organism-specific outcomes (Fig. 2b and Extended Data Fig. 2). While only a single tpiA-swap failure was found, pgi had less success with assimilation, with 40% of human swaps failing as well as 100% of archaeal swaps.
Fig. 2. Evolutionary outcomes.
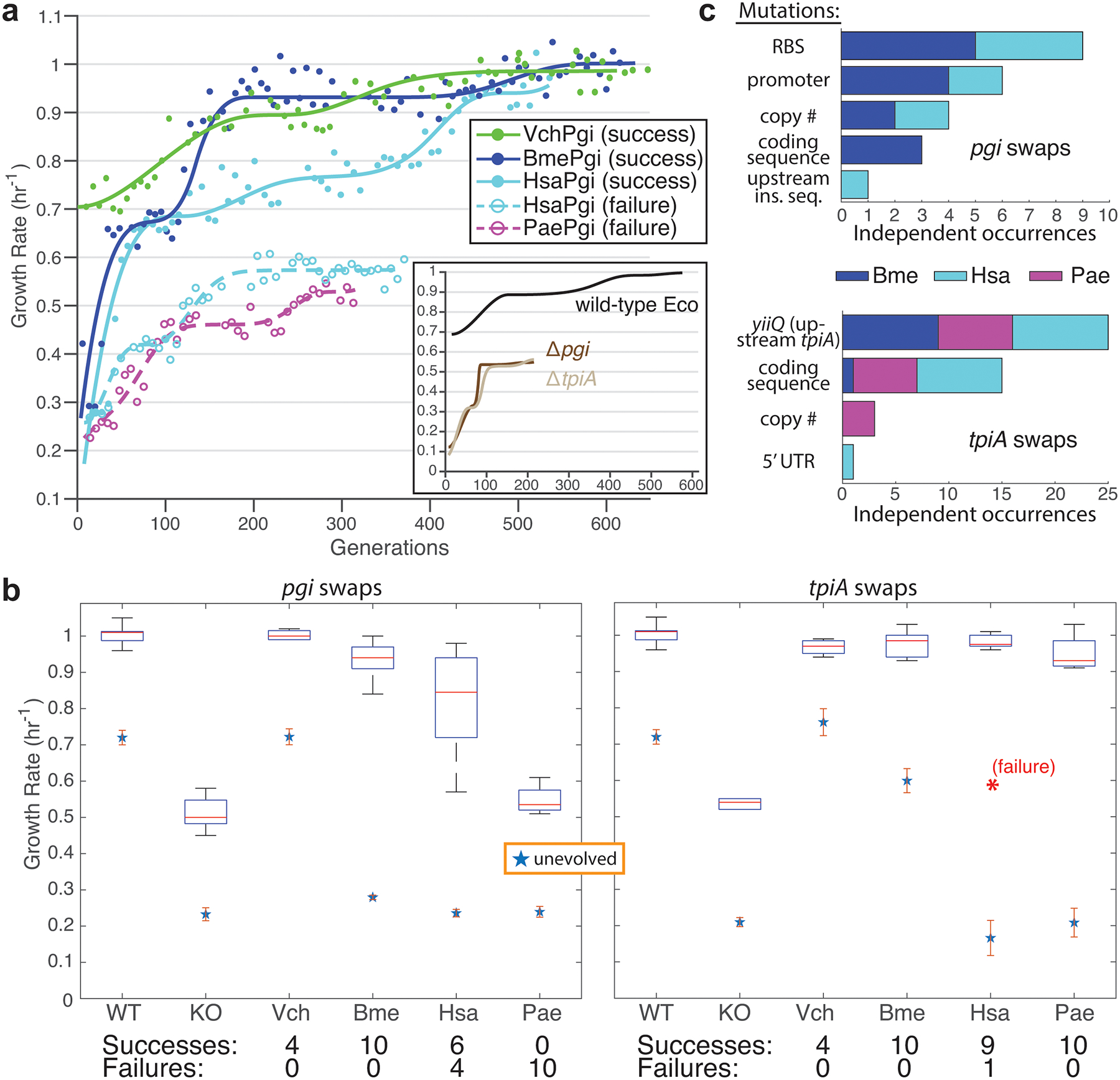
a, Robotic systems allowed growth rate tracking over the course of evolution (points represent growth rates from successive culture tubes, with lines being cubic interpolating splines fit to the points). Example trajectories demonstrate various outcomes; gene-swapped strains could evolve like the wild-type, successfully escape from knockout-like fitness levels, or fail to evolve any differently than knockout controls. b, Box plots (showing first and third quartile around median red line, with whiskers to extreme values) of fitness outcomes for evolved population endpoints vs. starting strains, with statistics on success and failure frequency. Error bars on starting strains represent standard deviation from quadruplicate measurements. c, Mutations found in or around orthogenes in all successful lineage endpoints. Vch swaps did not require orthogene mutations to reach high growth rates, while failure lineages never acquired any.
Evolved endpoint clones were isolated and whole genome sequenced to determine genetic mechanisms of adaptation, and mutational results provided striking reinforcement of the conclusions drawn from initial physiology and adaptive outcomes. All ‘failure’ lineages (not reaching a growth rate above 0.75/hr) lacked mutations in or around the foreign DNA, while every single ‘successful’ lineage acquired one or more mutations to this region, barring Vch swaps which did not require any orthogene changes to enable functionality in E. coli (Fig. 2c and Fig. 3a,b). Promoter and RBS mutations were the dominant adaptive mechanisms for pgi swaps, while tpiA swaps acquired the same synonymous L179L SNP in the upstream gene yiiQ more than 20 times independently. In most cases, fitness improvement to levels on par with evolved wild-type strains did not require any changes to the foreign coding sequence.
Fig. 3. Evolved strain mutations.
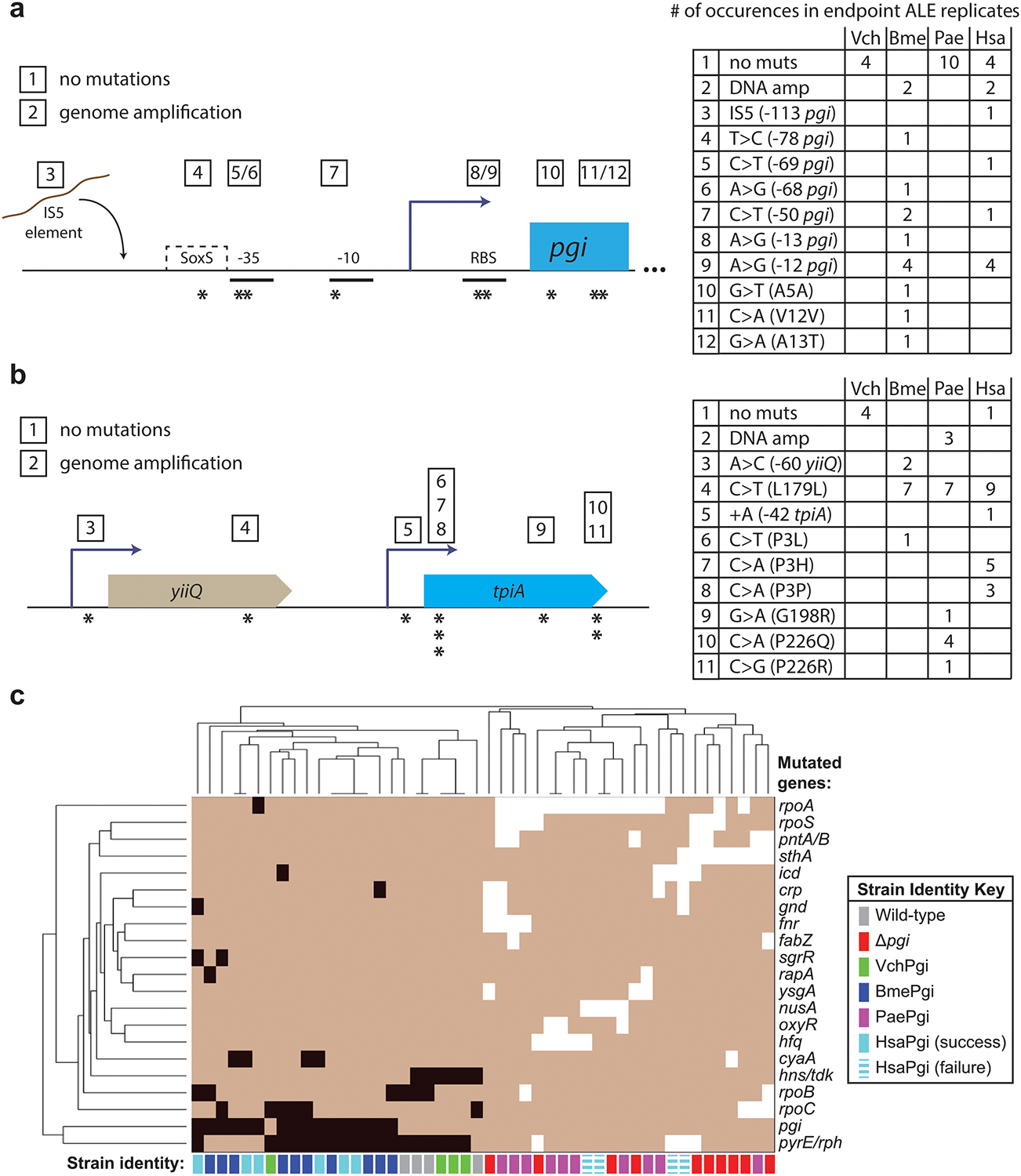
a, pgi-proximal mutations found in gene-swapped endpoint ALE strains. b, tpiA-proximal mutations found in gene-swapped endpoint ALE strains. c, Hierarchical clustering of strains based on mutations (indicated by black or white squares) found in evolved endpoints, considering only genes that mutated independently two or more times. Wild-type and successful lineages cluster together based on shared characteristic gene alterations (left), as do knock-out and failure lineages (right). All VchPgi and BmePgi strains were successful, while all PaePgi strains were failures.
We performed several assays to verify that fitness improvement in ‘successful’ lineages was due to mutations increasing orthogene levels. Knocking out the orthologous gene from various endpoint strains caused significant drops in growth rate for successes but had no impact on failures (Extended Data Fig. 3a). Enzyme assays on these same endpoint strains and their unevolved post-swap ancestors were consistent with these results, revealing enzymatic activity levels that increased following evolution, but only in successes (Extended Data Fig. 3b). Finally, we expressed the Homo sapiens orthogenes (the only donor species that led to failure and success lineages for both swapped genes) from inducible promoters inserted into Δpgi and ΔtpiA strains, demonstrating that growth rate increased with increasing induction level.
Failure lineages, in addition to never acquiring mutations within or proximal to the orthogenes (in any sequenced clones or populations), also had mutations highly characteristic of KO control evolutions. For example, the gene sthA did not mutate in any successful pgi swaps, but did in two of the failures (1 HsaPgi, 1 PaePgi) and in multiple Δpgi controls12. Lineages mirroring wild-type control evolutions likewise shared characteristic mutations – three of four evolved VchPgi strains acquired hns/tdk IS element mutations, a causal mechanism only observed for wild-type glucose evolutions16,17, highlighting the negligible influence of the Vch swap. Considering only genes that mutated two or more times independently across the replicates, hierarchical clustering cleanly discriminates between pgi successes and failures (Fig. 3c). The single unsuccessful HsaTpi lineage likewise had unique adaptive signatures characteristic of ΔtpiA evolutions – ptsG, galR, and nemR all mutated13. The large-scale nature of our study resulted in numerous genes targeted repeatedly for alteration, from which structural insights can be drawn and cross-study comparisons made that reveal mutational hotspots (Extended Data Fig. 4a and Supplementary Table 2).
Given the significant fitness defect resulting from absent or low-level pgi or tpiA flux, orthogene mutations (Fig. 2c) would be expected to improve fitness in one of two ways: by increasing expression level of the gene product through regulatory changes, or by enhancing specific enzyme activity through coding sequence alterations. While many of the observed mutations can be easily interpreted as expression-increasing (promoter/RBS SNPs and copy number expansions for upregulation), others require more detailed analysis. We find that the widespread yiiQ L179L SNP achieves orthogene upregulation by creating a new promoter 179 basepairs upstream of the tpiA start codon. Analysis with promoter prediction tools18 reveals the increased chance for RNA polymerase binding that the C→T mutation creates (Fig. 4a). This mechanism was documented in a prior study19, and demonstrates the strong impact of local sequence context on adaptive outcomes.
Fig. 4. Mutational mechanisms of orthogene assimilation.
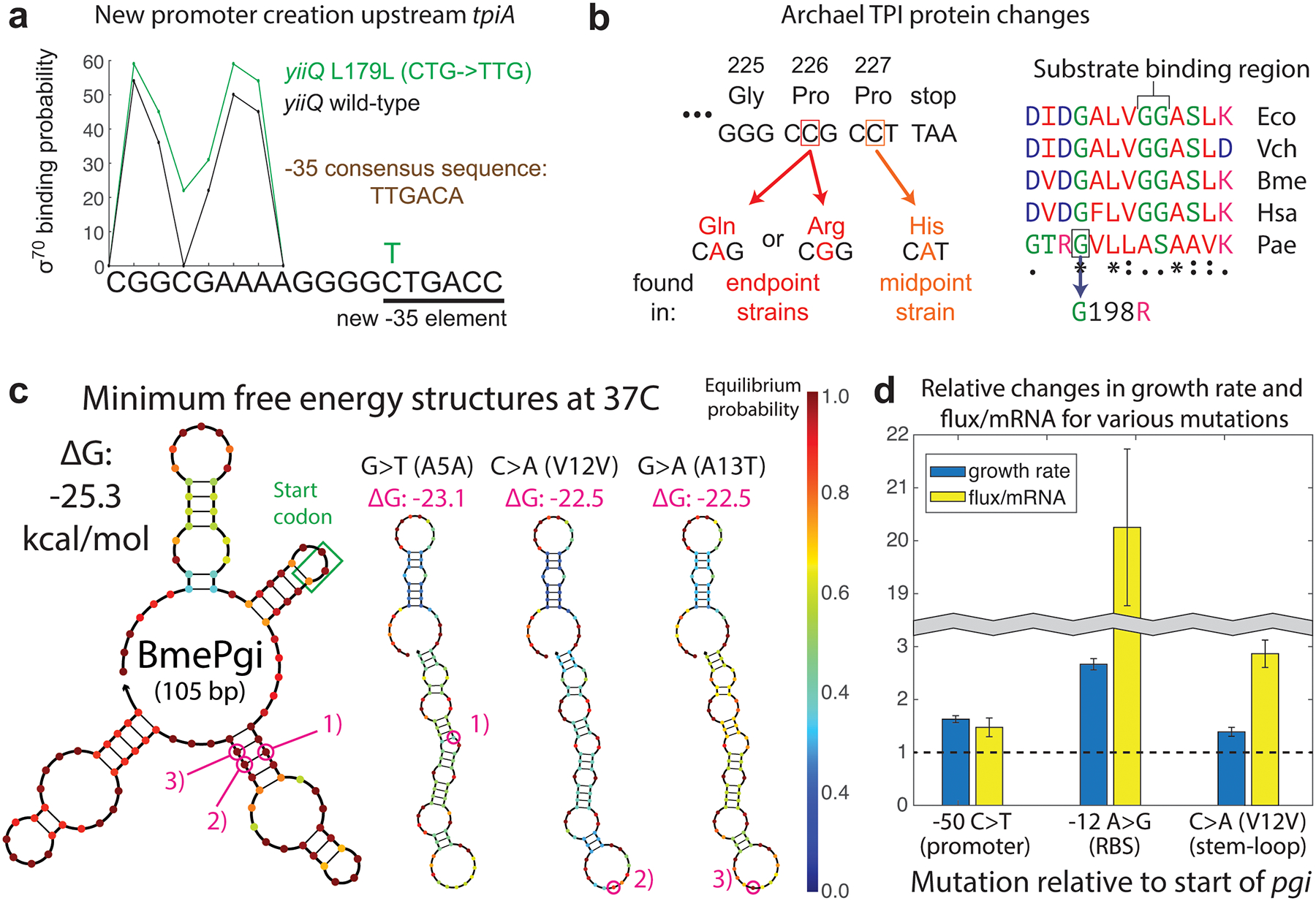
a, A promoter-creating synonymous SNP in the gene upstream of tpiA occurred independently more than 20 times. b, Missense SNPs in the archaeal TPI ameliorated polyproline ribosome stalling (left), and in one instance likely modified enzyme properties (right). c, N-terminal SNPs, frequently synonymous, targeted prohibitively strong stem-loops in mRNA secondary structure for destabilization. d, Various upregulating mutations had different impacts on the ratio of orthogene enzyme flux to mRNA level. Each pair of bars reflects growth, enzyme, and qPCR assays performed on two different strains – one lacking the mutation in question and one genetically identical except for the addition of the listed mutation. Error bars represent standard deviation from quadruplicate measurements.
Coding sequence changes to the orthogenes were less common than cis-regulatory alterations and fell into two types: C-terminal missense SNPs in the archaeal TPI, and a number of N-terminal, mostly synonymous SNPs across a variety of swapped strains. PaeTpi swaps repeatedly acquired SNPs to the penultimate amino acid, consistent with a mechanism of increasing expression by reducing polyproline ribosomal stalling20. This mechanism explains the occurrence of growth-improved clones stemming from changes to either of the final two proline residues (Fig. 4b), although it’s possible that the prolines hinder folding of the protein within E. coli rather than causing ribosomal stalling. The archael tpiA swap is also characterized by the only observed orthogene mutation that likely alters enzyme activity rather than expression level – residue 198 is adjacent to the conserved substrate-binding region of the enzyme.
Our observed N-terminal orthogene SNPs were frequently synonymous, and though earlier experimental evolution studies were unable to determine the particular causal mechanism for fitness improvement21,22, recent work identified mRNA secondary structure as the target for such SNPs in the case of a gene knockout which selected for native promiscuous enzyme upregulation23. Here we find that this mechanism of expression increase extends to orthogene assimilation, with both synonymous and nonsynonymous SNPs targeting tightly-bound stem-loops in mRNA secondary structure for destabilization. This opens up the transcript for increased ribosomal readthrough, most strikingly in the cases of BmePgi (Fig. 4c) and HsaTpi (Extended Data Fig. 4b), leading to more protein per mRNA. We empirically validated this mechanism with pairwise characterizations of pgi expression level and enzymatic activity in strains genetically identical except for single mutations of interest, thus enabling causal establishment (Fig. 4d). Moreover, we surveyed ~1,000 whole genome sequenced E. coli strains and found that SNPs preferentially accumulated in the strongest stem-loop region (Extended Data Fig. 5a). Synonymous SNPs of this type may be underappreciated drivers of adaptation rather than neutral signatures of drift, potentially overlooked in previous evolution experiments or even contributing to speciation (Extended Data Fig. 5b).
We next probed the evolutionary dynamics governing adaptive outcomes by sequencing multiple midpoint strains from every ALE lineage, and performed ‘continuation ALEs’ for a number of failure endpoints as well as ‘replay ALEs’ for selected clones of interest isolated from various lineages. We found that the first mutation fixed in an evolving population strongly constrained ultimate lineage outcome, exemplified by the single tpiA failure lineage (Fig. 5a). In 10/10 ΔtpiA controls the first adaptive step was knockout of ptsG, mirrored by the HsaTpi failure, but additional evolutionary time allowed this lineage to undo the ptsG nonsense SNP it had acquired and ultimately achieve success. Had the failure lineage knocked out ptsG via some other mechanism, such as a frameshifting indel, ORF restoration would have been prevented and escape from ‘failure’ might not have been possible. However, some lineages in the initial ALE were found to have achieved success despite first acquiring knockout-characteristic mutations (Extended Data Fig. 6), and added ALE time for failures normally enabled success, though not for any archaeal pgi swaps (Extended Data Fig. 7).
Fig. 5. Adaptive dynamics.
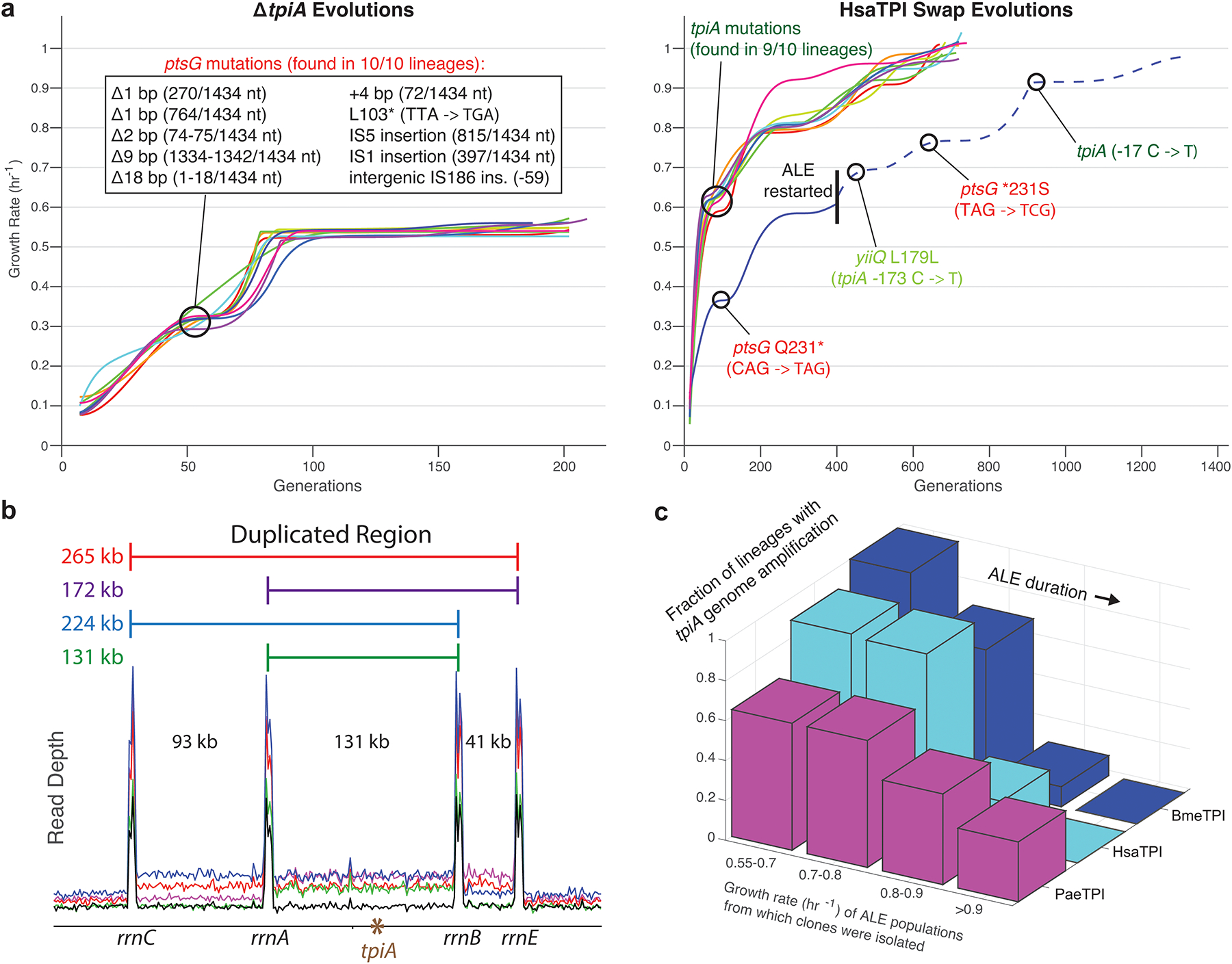
a, Knockout of ptsG was highly characteristic of ΔtpiA control evolutions. The single failure tpiA swap lineage took an initial step down this suboptimal knockout-like adaptive path, but when provided additional evolutionary time ultimately restored ptsG and successfully assimilated the orthogene. b, Copy number increases in tpiA came in four distinct types, driven by homology between flanking rrn operons. c, Increases in tpiA copy number were rampant at the start of evolution, but precipitously decreased in frequency as the experiment progressed.
Our midpoint sequencing also revealed that, for tpiA swaps, orthogene copy number expansions were much more widespread than indicated by endpoint clones. While pgi’s chromosomal location left it little flanking homology with which to facilitate genome amplifications (Extended Data Fig. 8a), tpiA fell between several rRNA operons which could cross over in different pairings that led to four distinct types of copy number expansions (Fig. 5b). The increased frequency with which such homology-facilitated expansions occur is a likely reason for the greater success rate of pgi vs. tpiA swaps, further emphasizing the importance of a gene’s chromosomal location on adaptive outcomes. Extent of amplified region had drastic changes in relative fitness for different growth environments (Extended Data Fig. 8b), and more than 80% of clones isolated across Bme/Hsa/PaeTpi lineages after their first jump in growth rate contained such expansions. As the ALE experiment progressed these tpiA-amplified strains were ultimately outcompeted by more cellular resource-parsimonious, i.e. efficient, methods of upregulation (e.g., yiiQ L179L and mRNA stem-loop SNPs), leaving only three PaeTpi endpoints with a persistent amplification (Fig. 5c). Replay ALEs confirmed that amplified regions of the genome could further increase in copy number, remain stable, or collapse back to single copy as evolution progressed (Extended Data Fig. 9).
Taken together, our laboratory evolution study of orthogene assimilation and refinement reveals mechanisms underlying cross-species gene functionality and adaptive outcomes, such as mRNA stem-loop formation, cis-regulation, and chromosomal copy number stability. Adjustments to enzyme abundance were much more common than alterations to specific enzyme activity, emphasizing that systems biology may be key to understanding adaptive evolutionary processes. Adaptive synonymous mutations were also found to be surprisingly widespread – one replay lineage even acquired two successive synonymous orthogene SNPs as its only coding sequence alterations. Calculations of adaptive protein substitution rates are known to be sensitive to even weak selection for synonymous mutations24, thus our results raise concerns about the applicability of Ka/Ks in evolutionary biology and the accuracy of existing genetic clock studies.
The collection of hundreds of evolved, whole genome sequenced strains generated in this study also highlights several interesting or unexpected features. Firstly, real-time tracking of culture growth rates with automated systems, at least in cases where individual mutations can cause large fitness jumps, reliably enables the isolation of strains identical except for single genetic alterations. We are able to capture all types of ‘quantum step’ in evolution, i.e. the smallest possible units of genetic change – from SNPs to indels to genome rearrangements. Strain pairs differing by such quantum steps in genome sequence space allow validation of mutational mechanisms without necessitating the creation of knock-ins (Fig. 4d). Importantly, genome rearrangements often underlie adaptive events but cannot be perfectly reproduced with current genome engineering techniques25, yet our strain collection gives us the ability to assign causal influence to various of these rearrangements (Extended Data Fig. 8b). Second, we see a shocking extent of mutational reproducibility, with the same exact DNA basepair change occurring independently more than 20 times across three distinct strains (Fig. 3b). This demonstrates that evolutionary outcomes can be (probabilistically) predicted to the single basepair level, something not observed in higher organisms to-date26. Finally, the non-orthogene mutations appearing in our strains fell predominantly within genes of the RNA Polymerase complex, where we catalogued more than 90 unique mutations. Although RNAP mutations are a common occurrence in evolution experiments27, the fact that identical SNPs appear repeatedly following both our gene-swaps and 5 entirely distinct metabolic gene knockouts28 (Supplementary Table 2) is unexpected. With evolution-generated strain collections such as the one herein paired with new analytical techniques29, we are on the path to elucidating RNAP’s role as a master regulator of transcriptional networks.
Methods
Strain Design and Engineering
DNA sequences for gene replacement were ordered from Gene Universal Inc. For the human genes, the coding sequence (introns removed) of the annotated main isoform was used. Strains were constructed in two different ways - pgi swaps with a modified gene gorging protocol30 as depicted in Extended Data Fig. 10, and tpiA swaps with a similar method but using CRISPR-induced double-stranded DNA breaks on the native E. coli sequence as the method of counter-selection, thus not requiring an antibiotic cassette. Orthologous gene knockouts (Extended Data Fig. 3a) were generated via P1 phage transduction31 from Δpgi and ΔtpiA strains. Strain construction was checked for compositional and locational accuracy with both whole-genome and Sanger sequencing.
Protein similarity scores (Supplementary Table 1) were obtained using EMBOSS Needle pairwise sequence alignment32. Codon adaptation index (Supplementary Table 1) was calculated with the tool from Biologics International Corp (https://www.biologicscorp.com/tools/CAICalculator).
Adaptive Laboratory Evolution
Strains were maintained in exponential-phase growth and evolved via batch culture serial propagation of 100 ul volumes into 15 mL (working volume) tubes of 4 g/L glucose M9 minimal media kept at 37 C and aerated via magnetic stirring, exactly as described previously33. Cultures were propagated for 30 days, or until measured population growth rate reached within 10% of the maximum known to occur for the growth conditions, so that fitness trajectories would be dominated by sequential selective sweeps of large-effect beneficial mutations34. Stopped cultures were designated ‘endpoints’ and, due to the stochastic timing with which populations experienced jumps in growth rate, total evolution time in generations varied across the cultures. Each independent ALE replicate was started from a unique pre-culture inoculated with a clone isolated from an LB plate, so as to prevent standing variation from influencing mutational independence across replicates.
DNA Sequencing and Analysis
Genomic DNA was isolated using bead agitation in 96-well plates as outlined previously35. Paired-end whole genome DNA sequencing libraries were generated with a Kapa HyperPlus library prep kit (Kapa Biosystems) and run on an Illumina HiSeq 4000 platform with a HiSeq SBS kit, 150 bp reads. The generated DNA sequencing fastq files were quality controlled with AfterQC 0.9.736 then processed with the breseq computational pipeline37 following standard procedures (https://barricklab.org/twiki/pub/Lab/ToolsBacterialGenomeResequencing/documentation/) and aligned to the E. coli genome (NCBI accession NC_000913.3) to identify mutations. Genome amplifications were identified with a custom script that identified discontinuities in read depth, and all read depth coverage plots and marginal mutation calls were also manually inspected.
mRNA Structural Analysis
Structures for mRNA transcripts were evaluated using the NUPACK computational tool38. Annotated transcription start sites served as the 5’ end for evaluated structures, and a range of transcript lengths in 5 bp intervals were analyzed to ensure robustness of results to region chosen.
Strain Characterizations
Strains were assayed for growth rate via serial propagation and OD sampling conditions identical to the ALE experiments, with given values an average over at minimum 4 independent growth tubes. Strains were assayed for pgi expression level via RT-qPCR as follows: total RNA was purified with the Qiagen mRNeasy kit, assessed for quality on an Agilent Bioanalyzer, and quantified with a Nanodrop. RNA was converted to cDNA with the NEB LunaScript RT Kit, then quantified with the NEB Luna Universal qPCR kit. A panel of 5 housekeeping genes were assayed along with pgi to allow for normalization. Strains were assayed for enzymatic flux with a PGI activity colorimetric assay kit from BioVision (#K775) as per manufacturer protocols. Flux/mRNA values (Fig. 4d) were obtained by taking the ratio of colorimetric assay results to qPCR results. For both expression level and flux measurements, cultures were first flash frozen (both biological and technical duplicates) in liquid nitrogen at the same OD in mid-exponential growth phase.
Statistical Analysis
Quantitative values presented (i.e. growth rates, qPCR mRNA levels, enzyme flux assays) are averages from quadruplicate measurements, with error bars representing standard deviation.
Data Availability
The genome sequence data that support the findings of this study are available from ALEdb (https://aledb.org) under project name ‘SvNS.’
Code Availability
AfterQC, the software used to trim and filter DNAseq reads, is available at https://github.com/OpenGene/AfterQC. Breseq, the software used to identify mutations, is available at https://github.com/barricklab/breseq. Co, the software used to edit genome references (https://github.com/SBRG/svns_refseq), is available at https://github.com/biosustain/co.
Extended Data
Extended Data Fig. 1. Orthogene properties.
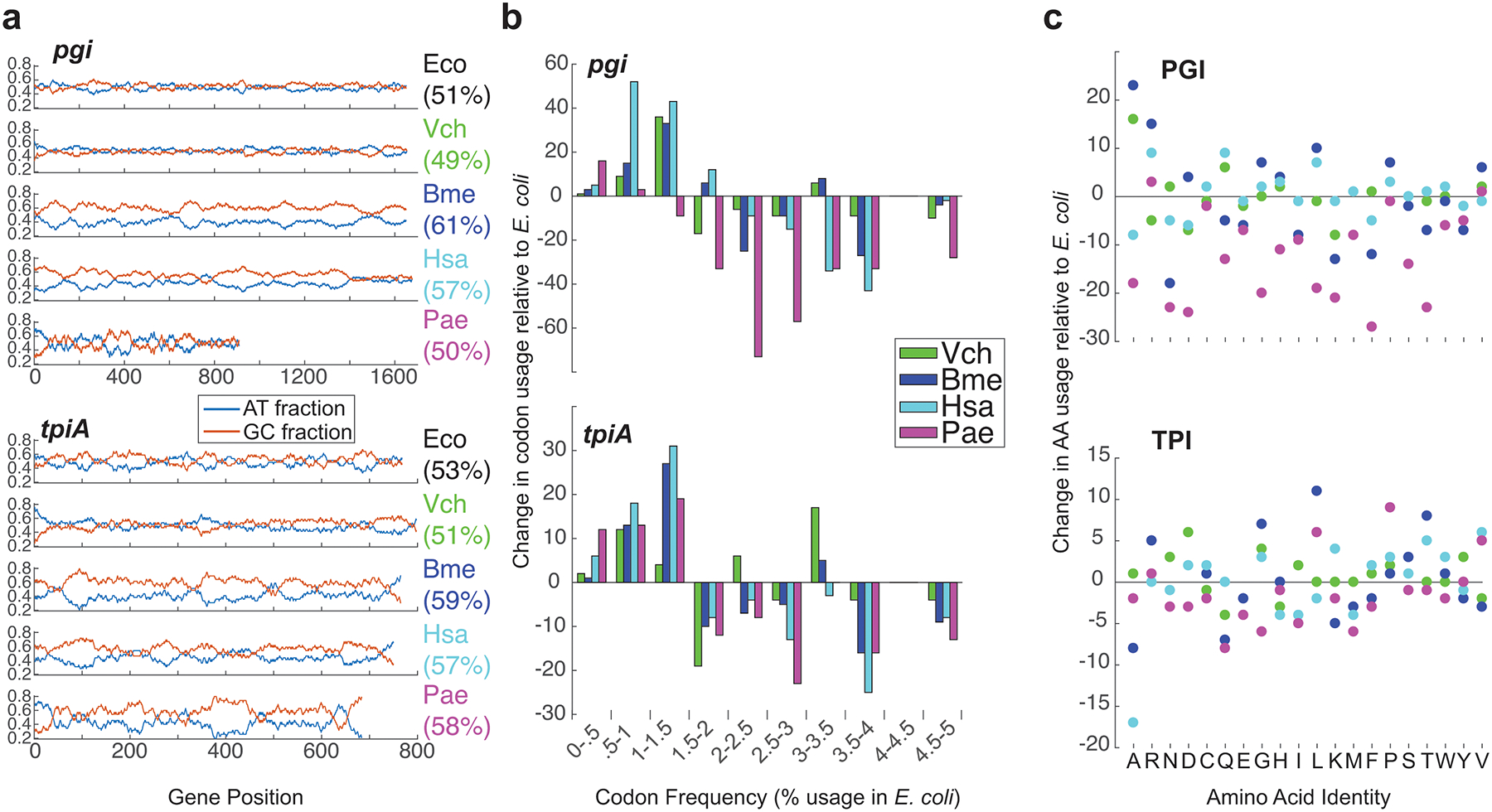
a, GC content of native and donor gene sequences (GC% total in parentheses). b, Histogram of the change in codon usage resulting from replacement of native E. coli sequences with foreign versions. c, Change in protein’s amino acid usage resulting from replacement of native sequences with foreign versions.
Extended Data Fig. 2. Evolutionary trajectories.
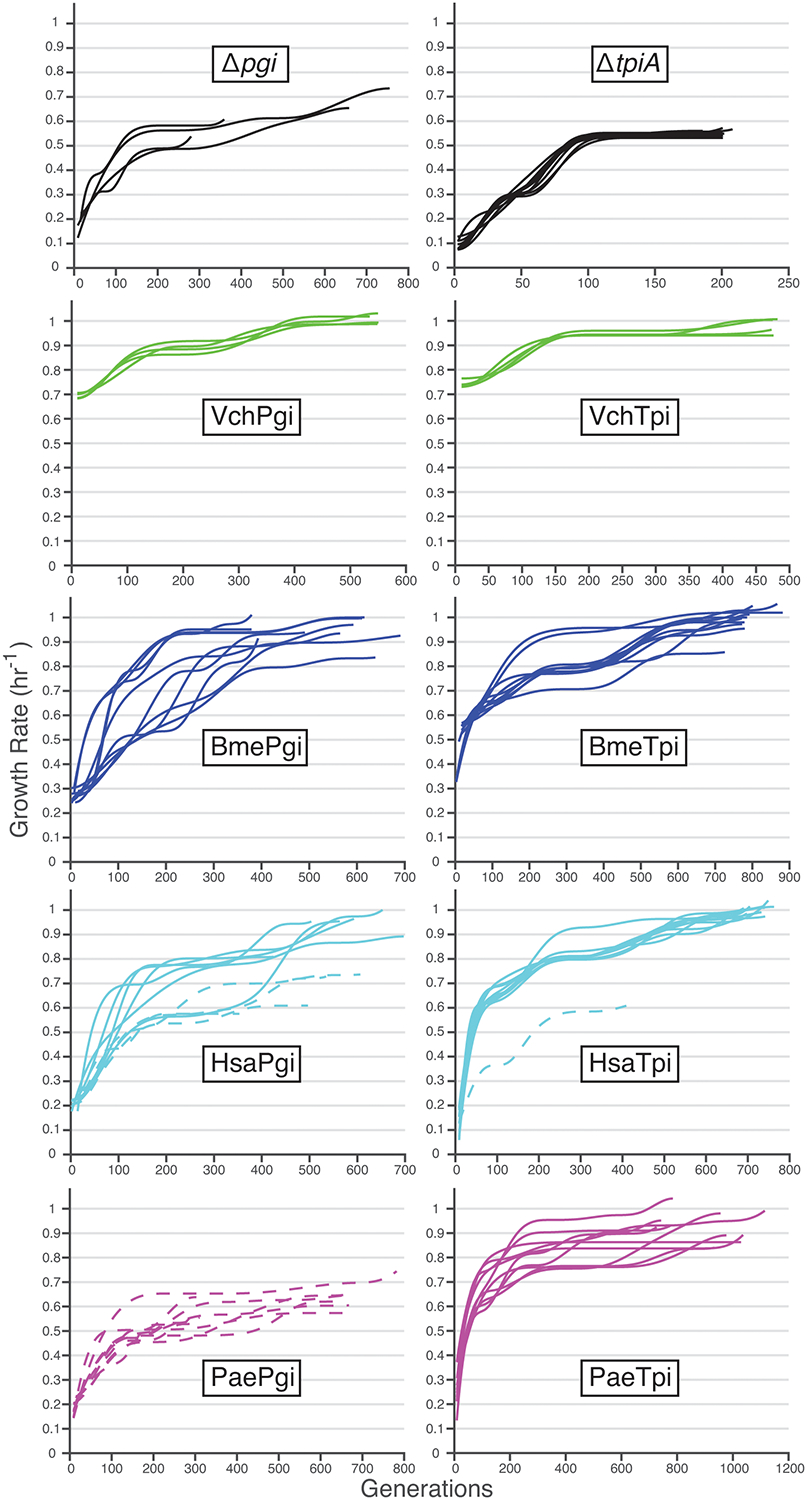
Fitness improvements over the course of evolution for knockout controls and gene-swapped strains, with failure lineages indicated by dotted lines. ΔtpiA controls were increased from four to ten to provide more comparison lineages for the single HsaTpi failure.
Extended Data Fig. 3. Orthogene impact on strain fitness.
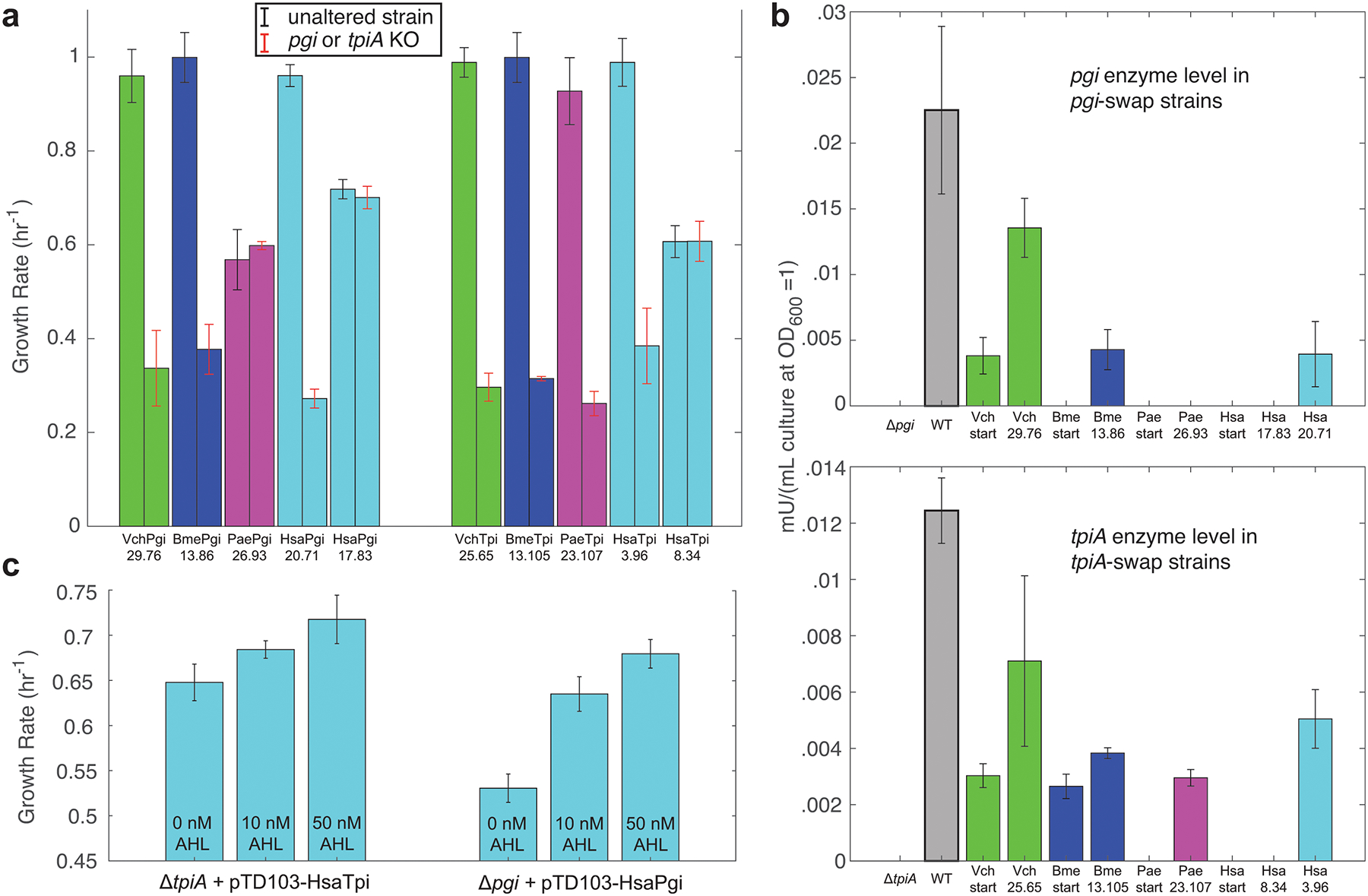
a, Growth rates of various pgi- and tpiA-swap evolved strains before and after knockout of the orthogene. b, Enzyme activity levels of various strains, determined by colorimetric assay. No bar indicates an activity level below detection of the assay. c, Growth rates at various AHL concentrations of pgi or tpiA knockout strains containing plasmids with AHL-inducible expression of the Homo sapiens pgi or tpiA. Growth rates higher than knockout levels even with no AHL induction may be due to leaky plasmid expression. In all panels strain names correspond with those given in the Supplementary Dataset containing DNA sequencing data, and error bars represent standard deviation from quadruplicate measurements. Orthogene-assimilation failures: PaePgi 26.93, HsaPgi 17.83, HsaTpi 8.34. Orthogene-assimilation successes: VchPgi 29.76, BmePgi 13.86, HsaPgi 20.71, VchTpi 25.65, BmeTpi 13.105, PaeTpi 23.107, HsaTpi 3.96.
Extended Data Fig. 4. Recurring mutations highlight regions under selection.
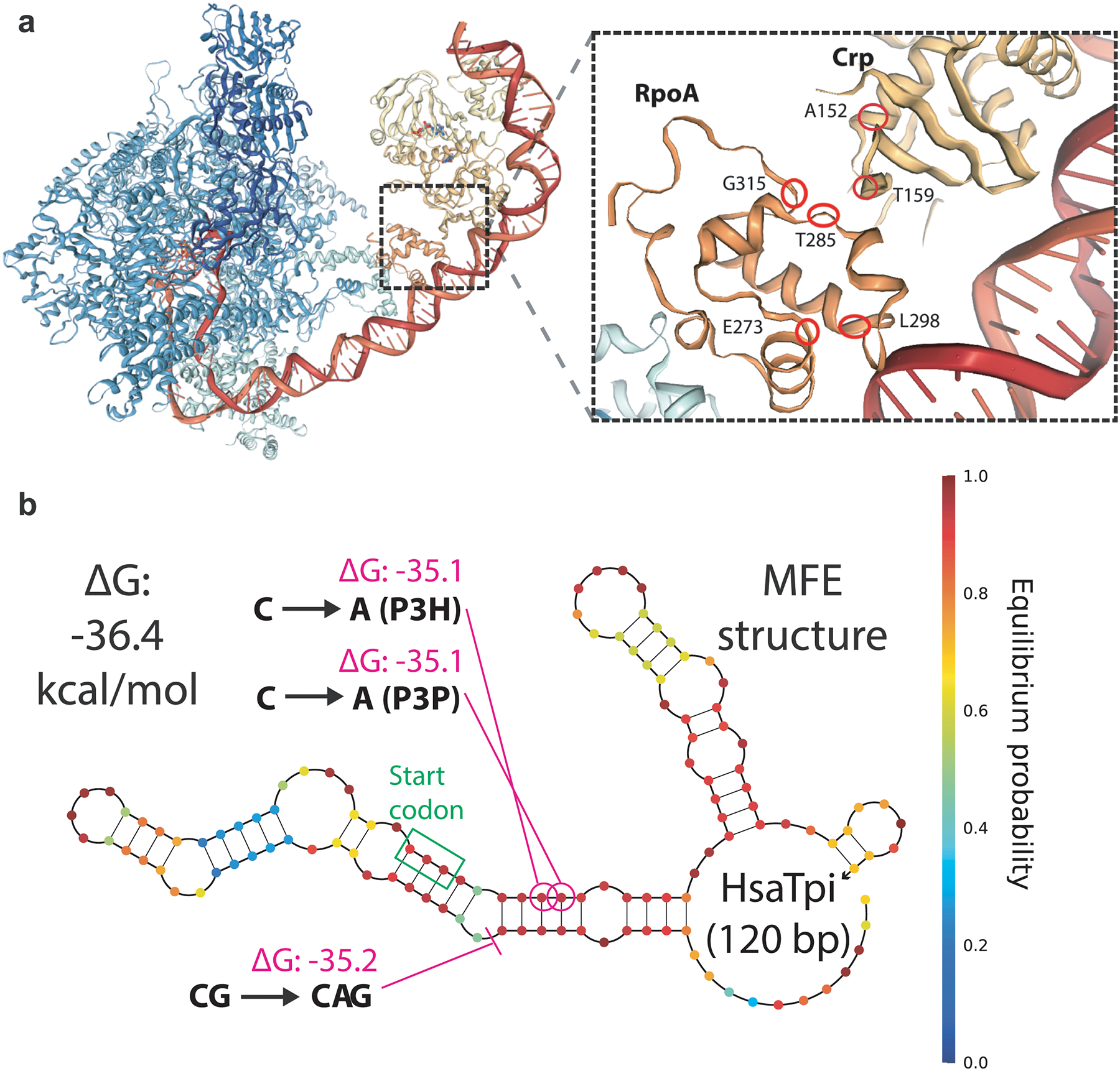
a, Mutations to crp and the C-terminus of rpoA were highly characteristic of pgi failures and knockout controls. Mapping to the cryoEM structure of the transcription activation complex (PDB ID: 6B6H) reveals that these characteristic mutations cluster in the same spatial region. b, Minimum free energy (MFE) structure at 37 °C of tpiA transcript for HsaTpi swap, with observed ALE endpoint mutations. The coding sequence changes destabilize G-C rungs of the strongest stem-loop, while the 5’-UTR +A insertion destabilizes this same stem-loop via increased stabilization of a stem-loop-adjacent unstructured region.
Extended Data Fig. 5. Various pgi transcripts and mutations.
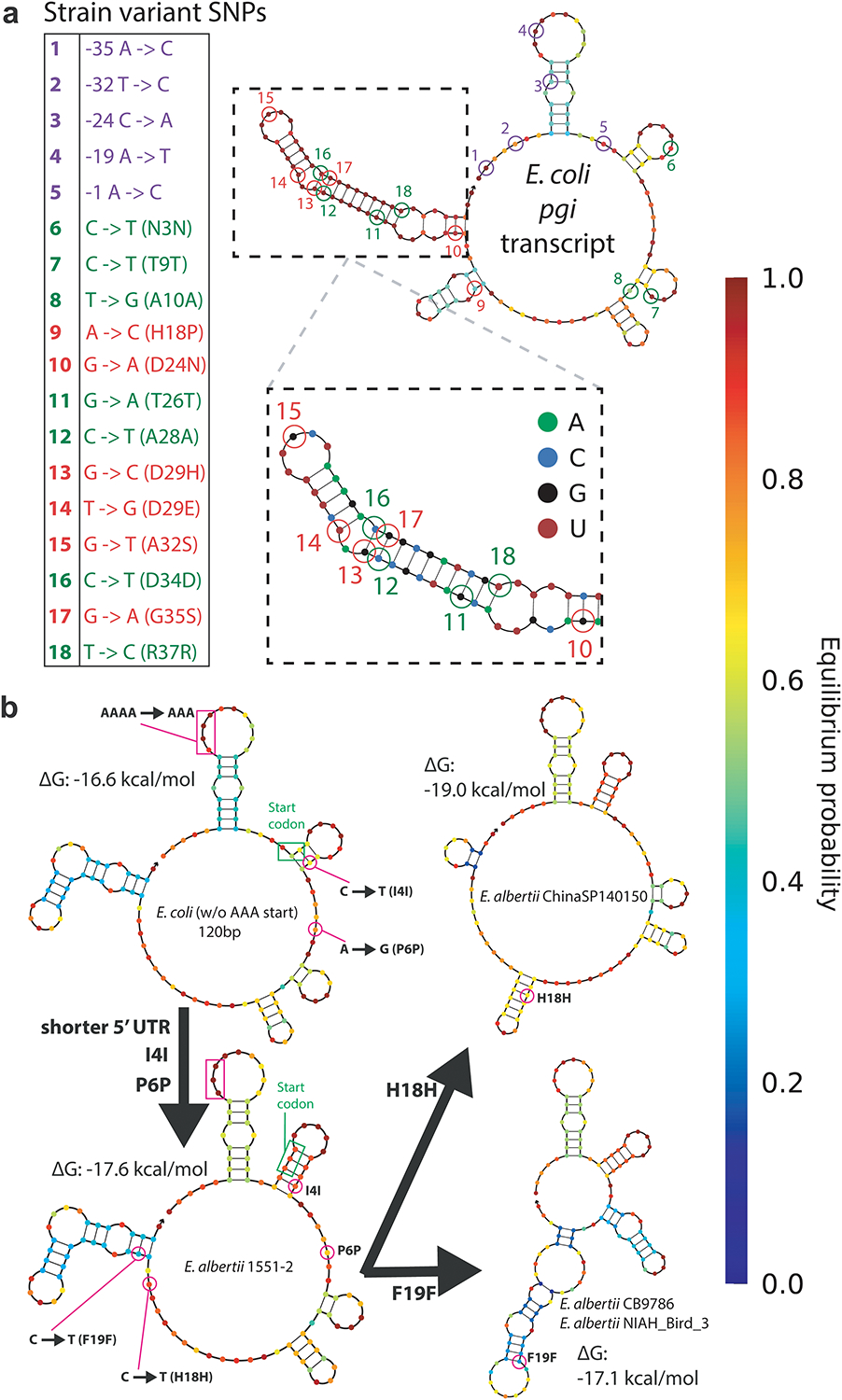
a, SNP accumulation in pgi across 924 Escherichia coli strain variants with whole genome sequences available. b, The only pgi differences in E. coli and various E. albertii strains within the first 120 bp of transcript are minor 5’-UTR changes and a number of synonymous mutations.
Extended Data Fig. 6. Orthogene assimilation in the presence of knockout-characteristic mutations.
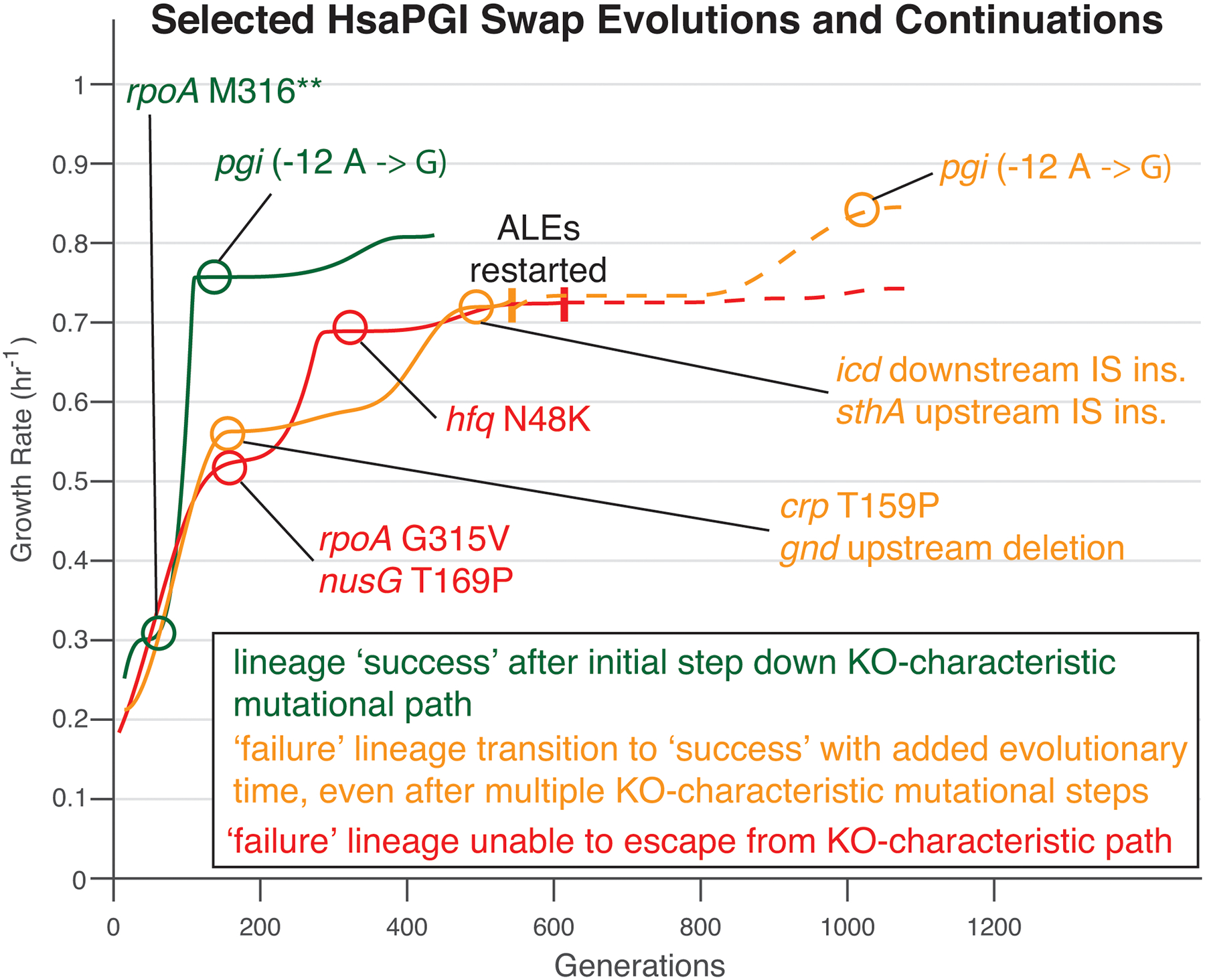
Even in the presence of one or more Δpgi-characteristic mutations a lineage could still successfully assimilate the orthogene, with added evolutionary time facilitating but not guaranteeing this outcome.
Extended Data Fig. 7. Continuation ALEs.
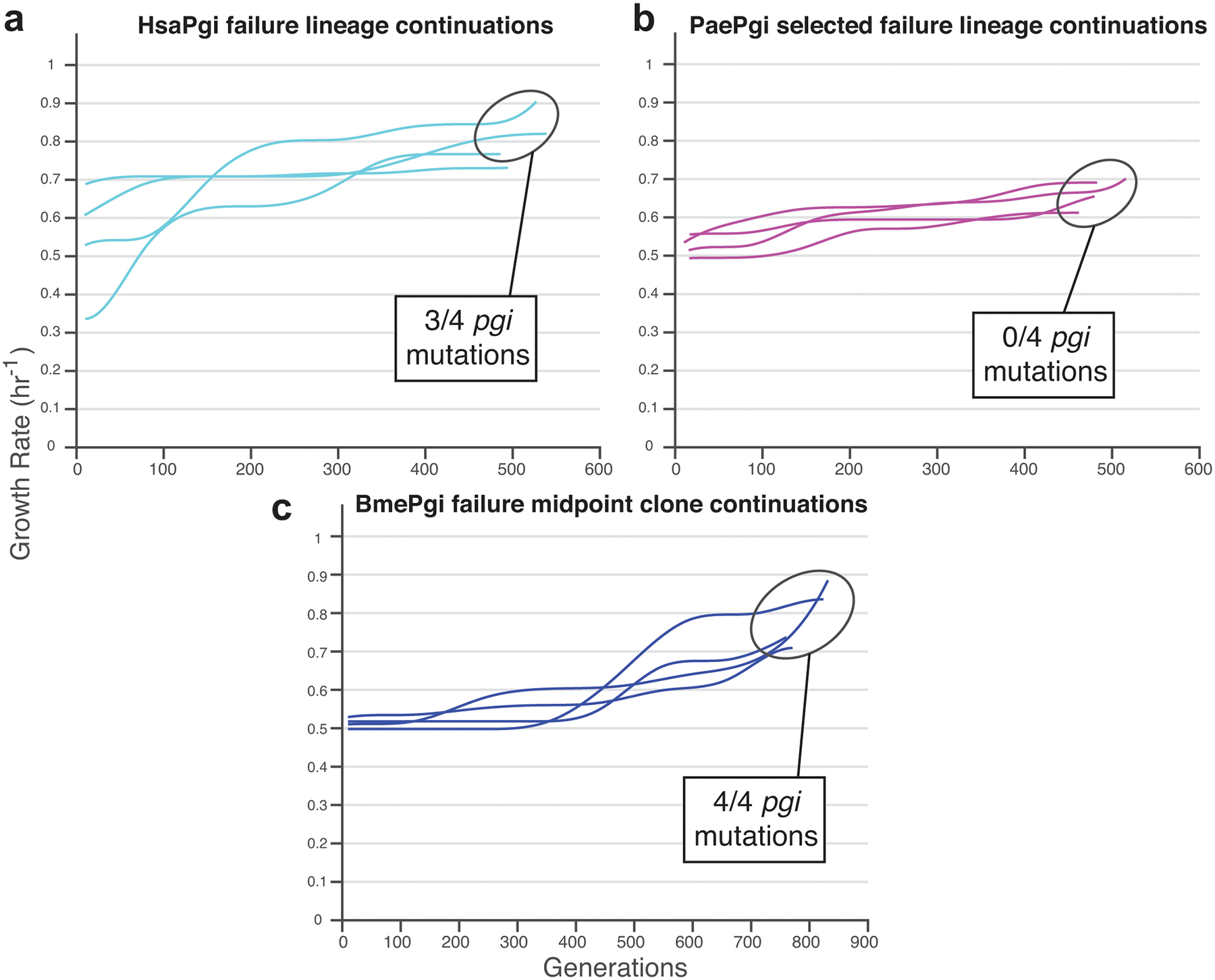
a, The only four failure HsaPgi lineages were given additional evolutionary time and reached success in most cases. b, Additional evolutionary time did not enable success for any of four continued PaePgi failure lineages. c, A single BmePgi midpoint clone was isolated with two knockout-characteristic mutations (to rpoA and nusA) and no orthogene changes. Independent lineages founded from this clone all eventually acquired orthogene mutations and higher growth rates.
Extended Data Fig. 8. Orthogene copy number expansions.
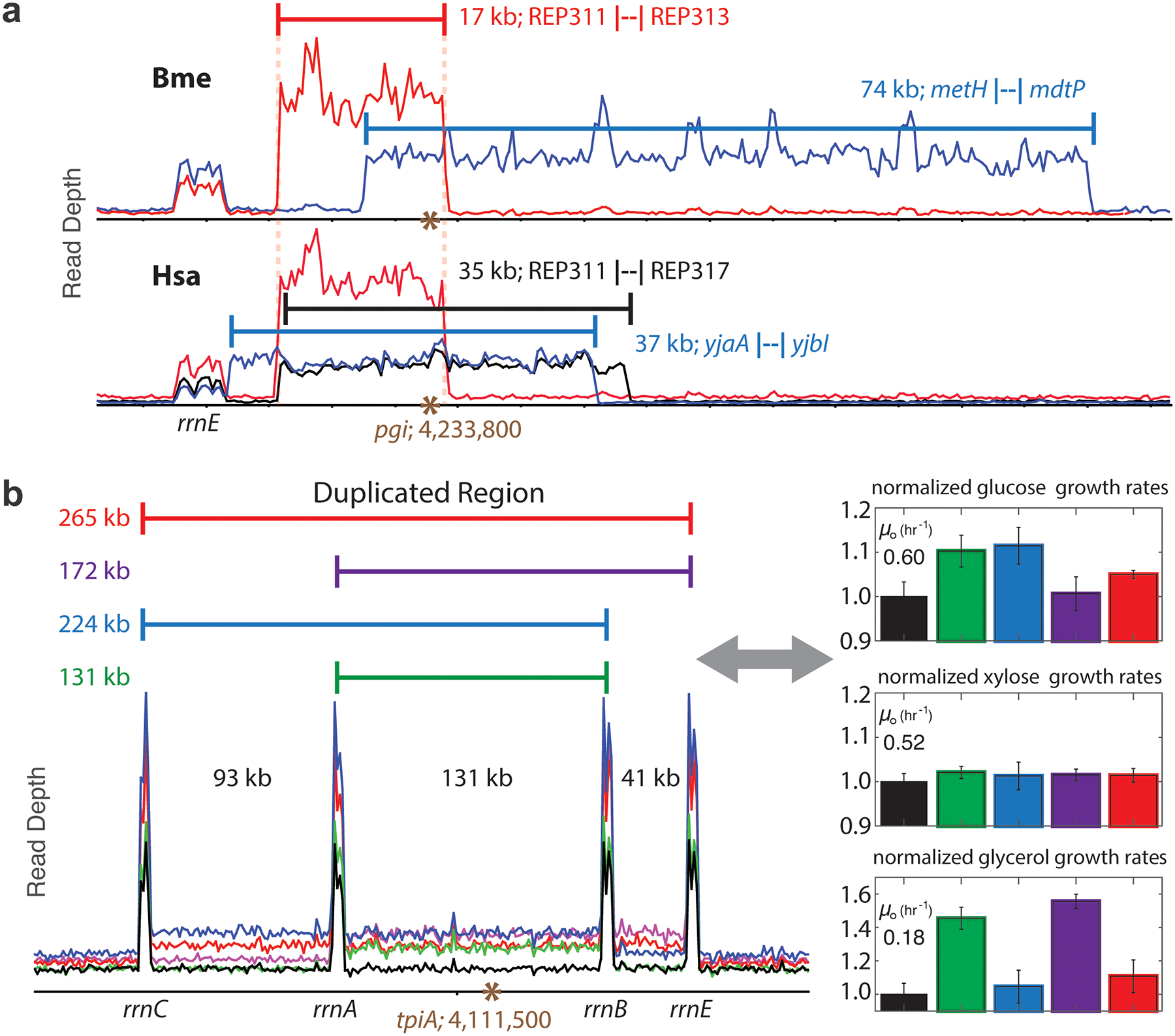
a, All orthogene copy number expansions found in pgi swap ALE strains. Homology between flanking genes or repetitive extragenic palindromic (REP) sequences facilitated the expansions. b, Relative growth rates on various carbon sources for HsaTpi strains genetically identical except for the size of tpiA duplication. Inclusion of the rrnB-rrnE region hindered growth on glucose, while the rrnC-rrnA expansion hindered glycerol growth. Error bars represent standard deviation from quadruplicate measurements.
Extended Data Fig. 9. Replay ALEs.
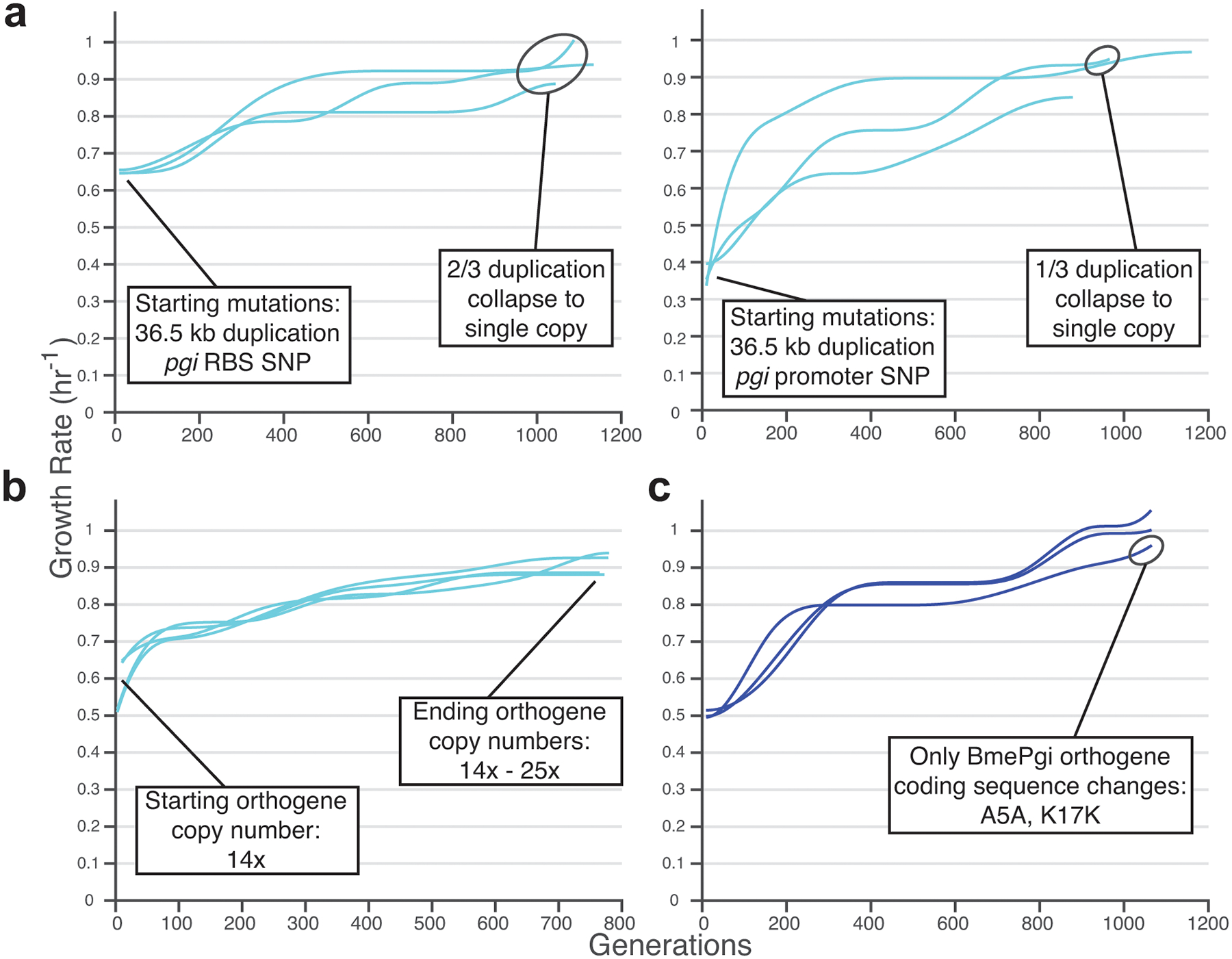
a, Two distinct HsaPgi strains with the same gnd- containing genome duplication but different orthogene mutations were split into lineages and evolved, which could lead to collapse of the gnd duplication. b, An HsaPgi strain that had acquired a genomic copy number increase of the orthogene was used to found four lineages. Evolution resulted in copy number remaining stable or increasing. c, A BmePgi strain with a synonymous orthogene SNP (A5A) was used to found three lineages. Evolution enabled further orthogene-upregulating mutations to increase growth rate, and one lineage acquired a second synonymous SNP as the only coding sequence changes to the foreign DNA.
Extended Data Fig. 10. Method of scarless strain construction.
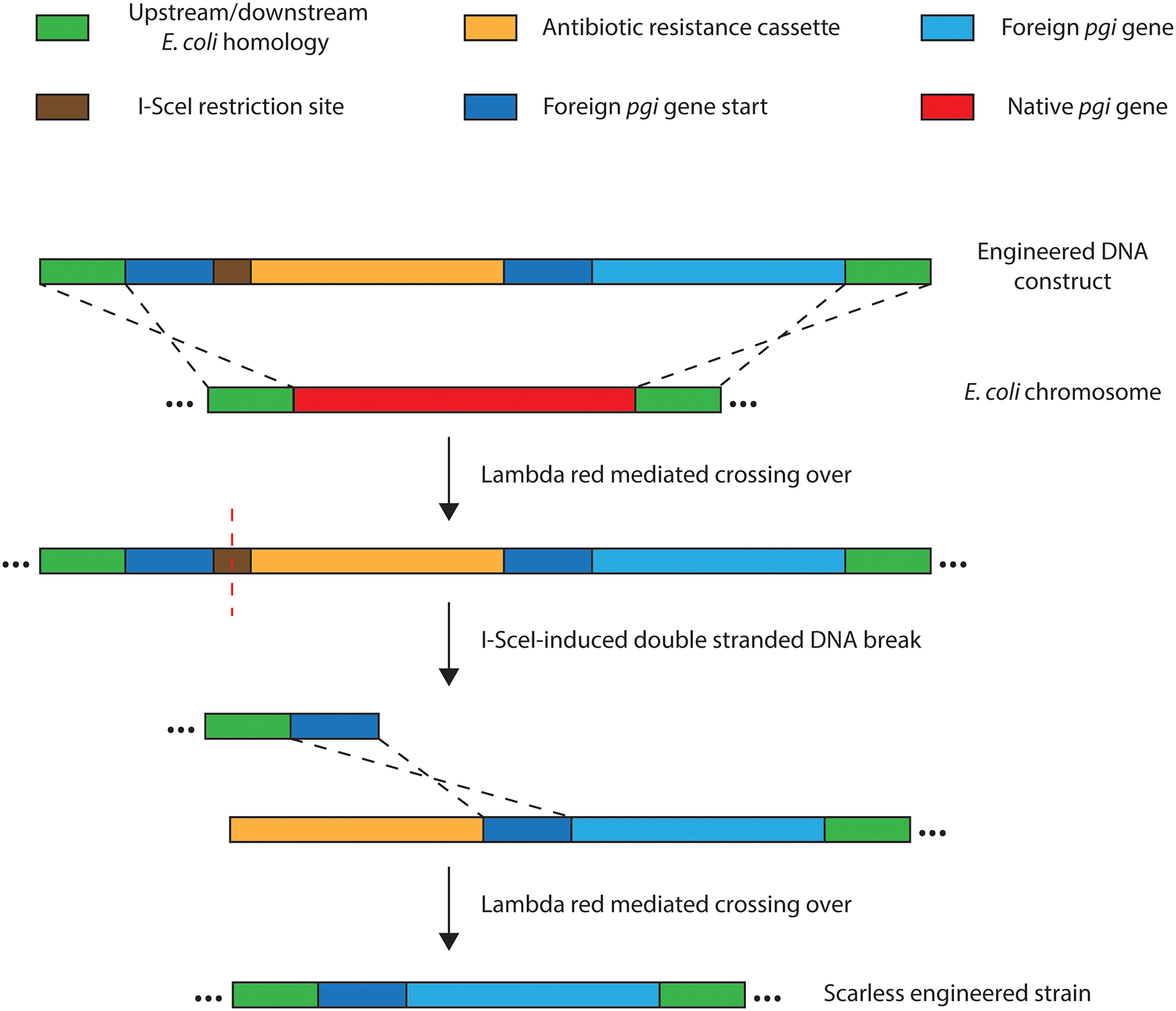
The pgi swap strains were constructed as shown. The tpiA swap strains used a construct lacking the foreign gene start homology, I-SceI site, and antibiotic cassette; double stranded DNA breaks were induced by CRISPR-targeting to native gene sequence, obviating the need for antibiotics.
Supplementary Material
Acknowledgments
This work was supported by the Novo Nordisk Foundation (Grant NNF10CC1016517) and in part by the NIH (R01GM057089). T.E.S was supported in part by the NSF Graduate Research Fellowship grant no. DGE-1144086. We thank Elizabeth Brunk, Edward Catoiu, M. Omar Din, Connor Olson, Muyao Wu, and Ying Hutchison for useful advice and discussions. We thank Adam Feist for making automated evolution machines available for the experiments performed in this study.
Footnotes
Competing Interests Statement
The authors declare no competing interests.
References
- 1.Soucy SM, Huang J & Gogarten JP Horizontal gene transfer: building the web of life. Nat Rev Genet 16, 472–482, doi: 10.1038/nrg3962 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Palmer KL, Kos VN & Gilmore MS Horizontal gene transfer and the genomics of enterococcal antibiotic resistance. Curr Opin Microbiol 13, 632–639, doi: 10.1016/j.mib.2010.08.004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Potvin G, Ahmad A & Zhang Z Bioprocess engineering aspects of heterologous protein production in Pichia pastoris: A review. Biochemical Engineering Journal 64, 91–105, doi: 10.1016/j.bej.2010.07.017 (2012). [DOI] [Google Scholar]
- 4.Chen J et al. Genome hypermobility by lateral transduction. Science 362, 207–212, doi: 10.1126/science.aat5867 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Kachroo AH et al. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348, 921–925, doi: 10.1126/science.aaa0769 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kachroo AH et al. Systematic bacterialization of yeast genes identifies a near-universally swappable pathway. Elife 6, doi: 10.7554/eLife.25093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kacar B, Garmendia E, Tuncbag N, Andersson DI & Hughes D Functional Constraints on Replacing an Essential Gene with Its Ancient and Modern Homologs. mBio 8, doi: 10.1128/mBio.01276-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lind PA, Tobin C, Berg OG, Kurland CG & Andersson DI Compensatory gene amplification restores fitness after inter-species gene replacements. Mol Microbiol 75, 1078–1089, doi: 10.1111/j.1365-2958.2009.07030.x (2010). [DOI] [PubMed] [Google Scholar]
- 9.Kacar B, Ge X, Sanyal S & Gaucher EA Experimental Evolution of Escherichia coli Harboring an Ancient Translation Protein. J Mol Evol 84, 69–84, doi: 10.1007/s00239-017-9781-0 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bershtein S et al. Protein Homeostasis Imposes a Barrier on Functional Integration of Horizontally Transferred Genes in Bacteria. PLoS Genet 11, e1005612, doi: 10.1371/journal.pgen.1005612 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandberg TE, Salazar MJ, Weng LL, Palsson BO & Feist AM The emergence of adaptive laboratory evolution as an efficient tool for biological discovery and industrial biotechnology. Metab Eng 56, 1–16, doi: 10.1016/j.ymben.2019.08.004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charusanti P et al. Genetic basis of growth adaptation of Escherichia coli after deletion of pgi, a major metabolic gene. PLoS genetics 6, e1001186, doi: 10.1371/journal.pgen.1001186 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCloskey D et al. Adaptation to the coupling of glycolysis to toxic methylglyoxal production in tpiA deletion strains of Escherichia coli requires synchronized and counterintuitive genetic changes. Metab Eng 48, 82–93, doi: 10.1016/j.ymben.2018.05.012 (2018). [DOI] [PubMed] [Google Scholar]
- 14.McCloskey D et al. Multiple optimal phenotypes overcome redox and glycolytic intermediate metabolite imbalances in Escherichia coli pgi knockout evolutions. Appl Environ Microbiol, doi: 10.1128/AEM.00823-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandberg TE et al. Evolution of Escherichia coli to 42 degrees C and subsequent genetic engineering reveals adaptive mechanisms and novel mutations. Mol Biol Evol 31, 2647–2662, doi: 10.1093/molbev/msu209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaCroix RA et al. Use of adaptive laboratory evolution to discover key mutations enabling rapid growth of Escherichia coli K-12 MG1655 on glucose minimal medium. Appl Environ Microbiol 81, 17–30, doi: 10.1128/AEM.02246-14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandberg TE et al. Evolution of E. coli on [U-13C]Glucose Reveals a Negligible Isotopic Influence on Metabolism and Physiology. PLoS One 11, e0151130, doi: 10.1371/journal.pone.0151130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Avila ESS, Notari DL, Neis FA, Ribeiro HG & Echeverrigaray S BacPP: a web-based tool for Gram-negative bacterial promoter prediction. Genet Mol Res 15, doi: 10.4238/gmr.15027973 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Kershner JP et al. A Synonymous Mutation Upstream of the Gene Encoding a Weak-Link Enzyme Causes an Ultrasensitive Response in Growth Rate. J Bacteriol 198, 2853–2863, doi: 10.1128/Jb.00262-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peil L et al. Distinct XPPX sequence motifs induce ribosome stalling, which is rescued by the translation elongation factor EF-P. Proc Natl Acad Sci U S A 110, 15265–15270, doi: 10.1073/pnas.1310642110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bailey SF, Hinz A & Kassen R Adaptive synonymous mutations in an experimentally evolved Pseudomonas fluorescens population. Nat Commun 5, 4076, doi: 10.1038/ncomms5076 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Agashe D et al. Large-Effect Beneficial Synonymous Mutations Mediate Rapid and Parallel Adaptation in a Bacterium. Mol Biol Evol 33, 1542–1553, doi: 10.1093/molbev/msw035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kristofich J et al. Synonymous mutations make dramatic contributions to fitness when growth is limited by a weak-link enzyme. PLoS Genet 14, e1007615, doi: 10.1371/journal.pgen.1007615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsumoto T, John A, Baeza-Centurion P, Li B & Akashi H Codon Usage Selection Can Bias Estimation of the Fraction of Adaptive Amino Acid Fixations. Mol Biol Evol 33, 1580–1589, doi: 10.1093/molbev/msw027 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Leon D, D’Alton S, Quandt EM & Barrick JE Innovation in an E. coli evolution experiment is contingent on maintaining adaptive potential until competition subsides. PLoS Genet 14, e1007348, doi: 10.1371/journal.pgen.1007348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Concha C et al. Interplay between Developmental Flexibility and Determinism in the Evolution of Mimetic Heliconius Wing Patterns. Current Biology 29, 3996–4009.e3994, doi: 10.1016/j.cub.2019.10.010 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Conrad TM et al. RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proceedings of the National Academy of Sciences of the United States of America 107, 20500–20505, doi: 10.1073/pnas.0911253107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wytock TP et al. Experimental evolution of diverse Escherichia coli metabolic mutants identifies genetic loci for convergent adaptation of growth rate. PLoS Genet 14, e1007284, doi: 10.1371/journal.pgen.1007284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sastry AV et al. The Escherichia coli transcriptome mostly consists of independently regulated modules. Nat Commun 10, 5536, doi: 10.1038/s41467-019-13483-w (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herring CD, Glasner JD & Blattner FR Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli. Gene 311, 153–163 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Thomason LC, Costantino N & Court DLE coli genome manipulation by P1 transduction. Curr Protoc Mol Biol Chapter 1, Unit 1 17, doi: 10.1002/0471142727.mb0117s79 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Li W et al. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43, W580–584, doi: 10.1093/nar/gkv279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sandberg TE, Lloyd CJ, Palsson BO & Feist AM Laboratory Evolution to Alternating Substrate Environments Yields Distinct Phenotypic and Genetic Adaptive Strategies. Appl Environ Microbiol, doi: 10.1128/AEM.00410-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenski RE Experimental evolution and the dynamics of adaptation and genome evolution in microbial populations. ISME J 11, 2181–2194, doi: 10.1038/ismej.2017.69 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marotz C et al. DNA extraction for streamlined metagenomics of diverse environmental samples. Biotechniques 62, 290–293, doi: 10.2144/000114559 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Chen S et al. AfterQC: automatic filtering, trimming, error removing and quality control for fastq data. BMC Bioinformatics 18, 80, doi: 10.1186/s12859-017-1469-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deatherage DE & Barrick JE Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol 1151, 165–188, doi: 10.1007/978-1-4939-0554-6_12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zadeh JN et al. NUPACK: Analysis and design of nucleic acid systems. J Comput Chem 32, 170–173, doi: 10.1002/jcc.21596 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genome sequence data that support the findings of this study are available from ALEdb (https://aledb.org) under project name ‘SvNS.’