

Abstract
HLA-donor specific antibodies (DSA) binding to vascular endothelial cells of the allograft trigger inflammation, vessel injury and antibody-mediated rejection (AMR). Accumulation of intragraft recipient macrophages is a histological characteristic of AMR, which portends worse outcome. HLA class I (HLA I) DSA enhance monocyte recruitment by activating endothelial cells and engaging FcγRs, but the DSA-activated donor endothelial influence on macrophage differentiation is unknown. In this study, we explored the consequence of DSA-activated endothelium on infiltrating monocyte differentiation. Here we show that cardiac allografts from murine recipients treated with MHC I DSA upregulated genes related to monocyte transmigration and Fc receptor stimulation. Human monocytes co-cultured with HLA I intact IgG- or F(ab’)2-stimulated primary human endothelium promoted monocyte differentiation into CD68+CD206+CD163+ macrophages (M(HLA I IgG)), while HLA I F(ab’)2-stimulated ECs solely induced higher CD206 (M(HLA I F(ab’)2)). Both macrophage subtypes exhibited significant changes in discrete cytokines/chemokines and unique gene expression profiles. Cross-comparison of gene transcripts between murine DSA-treated cardiac allografts and human co-cultured macrophages identified overlapping genes. These findings uncover the role of HLA I DSA-activated endothelium in monocyte differentiation, and point to a novel, remodeling phenotype of infiltrating macrophages that may contribute to vascular injury.
1. Introduction
In antibody-mediated autoimmune and inflammatory vascular diseases, antibodies binding to lumenal endothelial cells (EC) trigger inflammatory signaling contributing to autoimmune vasculitis 1–3, atherosclerosis4,5 and rejection of solid organ transplants. The production of donor specific major histocompatibility complex (MHC/HLA) antibodies (DSA) after transplantation of kidney, heart, liver and lung allografts occurs in as many as 50% of patients by 10 years 6,7. DSA mediate acute and chronic antibody-mediated rejection (AMR) and are associated with lower long-term graft survival 8. AMR manifests as endothelial injury and vascular inflammation consisting predominantly of macrophages 9, and evidence of complement deposition in the vessels of the graft 10. Chronic exposure of the allograft to DSA and repeated injury can culminate in transplant vasculopathy (TV), which is a leading cause of allograft loss and patient death 11–14.
Anti-endothelial cell antibodies contribute to vascular inflammation by activating endothelium directly, and through FcγR engagement on effector immune cells3. The binding of HLA I antibodies to donor endothelium also causes injury by multiple concurrent mechanisms, including Fc-independent agonistic HLA signaling 15,16, and IgG Fc-dependent activation of the classical complement cascade and recruitment of leukocyte effector cells 17–20. Antibody ligation of HLA I triggers Weibel Palade Body (WPB) exocytosis and induces P-selectin expression, which in turn promotes recipient platelet, monocyte, NK cell and neutrophil capture and infiltration into the graft 18,21. P-selectin-induced monocyte tethering is further enhanced through secondary interactions between the Fc portion of IgG and Fcγ receptors (FcγRs) on myeloid and lymphoid cells 17–19, promoting Mac-1-mediated firm adhesion to ICAM-1 on EC. These in vitro findings are supported by clinical and experimental evidence that allografts undergoing AMR exhibit characteristic macrophage infiltrates that promote graft injury and graft dysfunction 16,18,22. Collectively, these data support the critical role for macrophages in the pathogenicity of AMR.
Macrophages mature into heterogeneous subtypes with distinct effector functions, and play important roles in tissue homeostasis, host defense to pathogens, and disease pathogenesis 23,24. The effector functions of macrophages develop after transendothelial cell migration into the tissue and interaction with activating stimuli such as cytokines, chemokines, growth factors, immunoglobulin Fcγ receptors and Toll-like receptor (TLR) agonists 24,25. Macrophages are associated with both rejection and allograft fibrosis. M1-like macrophages were described to promote acute cellular rejection 26,27, while the M2-like subtype may dampen alloimmune responses, and contribute to tissue repair, remodeling and chronic rejection 28–31. Although macrophages are the principal component of AMR lesions, no studies have attempted to define their functional and phenotypic characteristics, especially in the context of HLA DSA.
In the current study, we sought to understand if antibody-stimulated endothelium could induce monocyte differentiation toward a specialized macrophage phenotype. Using a mouse model of acute AMR and an in vitro transwell co-culture model with human primary cells, we identified two novel macrophage subtypes, one polarized through direct contact with HLA I antibody-activated endothelium: M(HLA I F(ab’)2)—with increased CD68+CD206+ expression; and M(HLA I IgG) receiving a second activating signal via FcγR engagement that prompted an increase in CD163+ expression. This work dissects the Fc-independent and Fc-dependent effects of endothelium and donor specific HLA antibodies on macrophage differentiation, processes that are relevant to both acute and chronic AMR.
2. Materials and Methods
2.1. Murine Cardiac Allografts
B10.A (H-2a) hearts (Kk, Dd, Ld, IEk, IAk ) were transplanted heterotopically to immune deficient C57BL/6 (B6 Rag1−/−; H-2b) mice as previously described 32. One week post-operative, a mixture of IgG1 (AF3–12.1.3), IgG2a (16.1.2N), and IgG2b (15.1.5P) antibodies to H-2a (BioXCell, West Lebanon, NH) was transferred intraperitoneally at a dose of 100 mg for each antibody. Control animals received the same dose of isotype control antibodies (MOPC-21, C1.18, and MPC-11; BioXCell). Antibody injections were administered 4 times on alternate days and the mice were euthanized 1 hour after the fourth injection.
2.2. Immunohistochemistry and Nanostring for Cardiac Allografts
Samples containing full cross-sections through the cardiac grafts were immediately fixed in acid methanol and processed for immunohistochemistry as detailed in Supplemental Methods. RNA was isolated from homogenates of 3 experimental and 3 control allografts for gene expression measurements by NanoString. RNA was hybridized to the Mouse PanCancer Immune Profiling Panel supplemented with 10 genes.
2.3. HLA Antibodies
MAbs against HLA-A2 and A3 were derived from human hybridomas 33. F(ab’)2 fragments of mAbs were generated using Ides/FABricator 19. Anti-CD105 was from Millipore (Billerica, MA). Isotype control was from Sigma-Aldrich (St. Louis, MO).
2.4. Cells and culture
Primary human ECs were isolated from aortic rings as previously described 15,19. Human primary monocytes were isolated from healthy donors as previously reported 17 and detailed in Supplemental Methods. Methods for monocyte:endothelial cell co-culture are provided in the Supplement.
2.5. Flow cytometric analysis of macrophage subtypes
Macrophage phenotypes were assessed by flow cytometry using the fluorochrome-labeled antibodies described in the Supplement, acquired on an LSRFortessa™ (BD Biosciences, San Jose, CA) and the data were analyzed using FlowJo version 10 (Ashland, OR). (Fig. S6).
2.6. Cytokine/chemokine detection by multiplex assay
Cytokine/chemokine measurements were performed using human 38-plex kits (Millipore, HCYTMAG-60K-PX38) and analyzed on a Luminex 200 instrument as detailed in the Supplemental Methods 34.
2.7. Gene expression analysis of human macrophages
RNA expression of 594 genes was measured using the nCounter® Human Immunology v2 panel (Supplemental Methods).
2.8. In vitro macrophage and DC differentiation
In vitro induction of polarized macrophages and DC from human monocytes was performed as previously described (Supplemental Methods) 35–39.
2.9. Statistical analysis
Data are shown by box and whiskers graphs with range of min to max and all plots, or presented as mean ± SEM or mean ± SD. Statistical differences between groups were determined using paired t-tests or repeated measures one-way ANOVA followed by multiple correction (Dunnett’s, Sidak’s or Fisher’s LSD as recommended by the GraphPad Prism software). P-values < 0.05 were considered significant. For hierarchical clustering, individual genes or cytokines were normalized by Z-score, and then euclidean distance with average linkage was used. Analyses were performed in R or nSolver Analysis Software 3.0 (nanoString Technologies).
3. Results
3.1. DSA induces acute endothelial and macrophage responses in cardiac allografts
To test the effects of donor specific antibody (DSA) on cardiac allografts in the absence of T cells, we transplanted B10.A (H-2a) hearts to immune deficient C57BL/6 (B6 Rag1−/−; H-2b) mice. A week after transplantation when perioperative inflammation had subsided, a mixture of IgG1, −2a, and −2b monoclonal antibodies to H-2a was injected intraperitoneal. Control mice received isotype matched control IgG. The antibody injections were administered 4 times on alternate days and the mice were euthanized 1 hour after the fourth injection. Immunohistology confirmed that DSA induced features characteristic of acute antibody-mediated rejection, including diffuse C4d deposition on capillary endothelium and increased numbers of intravascular and perivascular macrophages (Fig. 1). Antibody induced injury was manifested by capillary dilatation and endothelial cell swelling (Fig. 1, insets). Macrophage activation was indicated by more stellate morphology in grafts from DSA treated recipients compared to the quiescent fusiform macrophages in control grafts.
Fig. 1.

Immunohistology for C4d and Mac 2. C4d deposits are diffuse in capillaries, arteries and veins of graft from DSA treated mouse (A), but no C4d was detected in graft from isotype control treated mouse (B). Higher magnification demonstrates capillaries with C4d deposits are dilated and EC are swollen (inset A). Mac2+ macrophages are more numerous in the graft from DSA treated mouse (C) than in graft from isotype control treated mouse (D). Higher magnification demonstrates qualitative differences in Mac2 staining of intra- and pericapillary macrophages (Insets of C and D).
3.2. DSA upregulates gene expression indicative of monocyte stimulation by Fc receptors and transmigration through activated endothelium
DSA induced changes in gene expression in cardiac allograft tissue identified 27 differentially expressed genes (DEGs) between DSA treated and isotype treated recipient allografts at a p<0.1 significance level (n=3). All DEGs had increased expression in DSA treated recipient allografts compared to isotype control allografts. Nine of the top 10 upregulated genes were related to monocyte and macrophage function (Table 1, Table S1). Six of these genes code for chemotactic proteins (CXCL1, CCL2, CCL7 and CCL12) or receptors (CX3CR1 and GPR183). Two of the monocyte chemotactic proteins (CCL2/MCP-1 and CCL7/MCP-3) have been implicated in transmigration of human monocytes through activated EC in vitro 40. In addition, production of MCP-1 and CXCL1 (KC) were found to be increased when macrophages bind to DSA on EC in vitro 41. These findings prompted us to investigate in detail DSA induced transmigration of human monocytes, by employing an in vitro model with human primary monocytes and EC.
Table 1.
The 27 differentially expressed genes (DEGs) in DSA vs isotype treated cardiac allografts
| Gene Symbol | p | FC | Gene Symbol | p | FC |
|---|---|---|---|---|---|
| Complement related | Adhesion molecules | ||||
| CD55 | 0.007 | 1.5 | EMR1 | 0.063 | 1.6 |
| C7 | 0.071 | 2.6 | SELE | 0.073 | 3.6 |
| C1S1 | 0.097 | 1.4 | Signaling and transcription factors | ||
| Costimulatory molecules | TBP | 0.05 | 1.2 | ||
| CD79B | 0.063 | 1.4 | FOS | 0.056 | 1.6 |
| Immune/cellular response | POU2F2 | 0.08 | 1.4 | ||
| IL1RN | 0.002 | 2.2 | EGR2 | 0.067 | 2.4 |
| GPR183 | 0.034 | 2.0 | Chemokines/cytokines | ||
| PTGS2 | 0.073 | 2.6 | CCL7 | 0.0118 | 3.2 |
| CD200 | 0.073 | 1.4 | CXCL1 | 0.041 | 1.6 |
| CD9 | 0.093 | 1.6 | CCL12 | 0.052 | 1.7 |
| TMEM173 | 0.097 | 1.5 | CX3CR1 | 0.056 | 1.7 |
| ITGA2B | 0.099 | 17.3 | CCL2 | 0.06 | 3.7 |
| LRP1 | 0.075 | 1.2 | PPBP | 0.076 | 17.0 |
| Phagocytosis | CSF1 | 0.09 | 1.3 | ||
| CLEC5A | 0.091 | 1.5 | CXCL14 | 0.09 | 1.4 |
GPR183 (EBI2) expressed on macrophages leads to calcium mobilization and to directed cell migration.
FOS (cFos) suppresses the expression of inducible nitric oxide synthase (iNOS) and pro-inflammatory cytokines.
Data analyzed by student T-test of gene expression counts (p<0.1) between HLA I DSA treated mice allograft recipients and isotype treated controls (n=3). FC, fold change.
3.3. HLA I antibody-activated ECs promote the differentiation of human peripheral blood monocytes into CD68+CD206+CD163+ macrophages through Fc-dependent and Fc-independent mechanisms
Because the in vivo findings indicated that DSA induces genes implicated in Fcγ receptor activation of monocytes and transmigration of monocytes, we tested whether HLA I antibody binding to EC altered the function of interacting human peripheral blood monocytes. To mimic the process of monocyte transendothelial cell extravasation into the allograft during AMR, we developed an in vitro EC:monocyte co-culture system in which monocytes adhere to HLA I antibody-activated EC, transmigrate, and are then allowed to differentiate into macrophages over a period of 5 days (Fig. 2A). To determine the effect of HLA I antibody-activated endothelium on macrophage polarization, ECs were pretreated with HLA I IgG before the addition of purified monocytes. Both intact and the F(ab’)2 fragment of HLA I IgG stimulation significantly increased the numbers of transmigrated cells compared to untreated and hIgG treated ECs (Fig. 2B, S1). Expression of CD68, CD206 and CD163 on the transmigrated macrophages was significantly increased after co-culture with HLA I IgG-activated ECs, but not on those co-cultured with irrelevant human IgG-pretreated ECs (Fig. 3A, Fig. S2). There was no significant difference in expression of CD64, CD86 or CD1a. Given CD68 is a pan-macrophage marker, these data suggest that the HLA I IgG activated ECs increased monocyte transmigration and induced their differentiation. Activation of endothelium with TNF-α, a pro-inflammatory stimulus, increased the expression of CD206, but not CD163 and CD68 (Fig. 3A). To address the role of the Fc region in monocyte differentiation, we stimulated ECs with an HLA I F(ab’)2. There was no difference in CD206 expression between macrophages induced by intact HLA I IgG versus F(ab’)2 (Fig. 3A, Fig. S2). However, CD163 was significantly lower on M(HLA I F(ab’)2) (Fig. 3A, Fig. S2), implying that CD163 expression relies on the monocyte FcγR engagement by HLA I IgG Fc region, while CD206 does not.
Fig. 2.

HLA I IgG stimulation increased the transmigration of human monocytes. (A) ECs were seeded in the upper chamber on day −1 and grown to a confluent monolayer. On day 0, the EC were washed and starved for four hours, and then left unstimulated (UT) or stimulated with 1 μg/mL HLA I antibody (intact or F(ab’)2) or hIgG for an additional hour. For TNF-α stimulation, 10 ng/mL TNF-α was added at the beginning of the EC starvation. The unbound antibodies and TNF-α were extensively washed away before adding the monocytes. The cells were co-cultured for five days before phenotype or gene expression assessments. During the co-culture, half of the medium was replaced every two days, before which the supernatants were collected for cytokine detection. (B) Transmigrated mononuclear cells were imaged on day 2 before medium replacement and counted. Fold changes in mean numbers of transmigrated mononuclear cells co-cultured with unstimulated (UT), HLA IgG or HLA-I F(ab’)2 stimulated EC are shown by box and whiskers graph. Data were from two independent experiments using two monocyte donors cultured with ECs from three donors. *p < 0.05 was analyzed by repeated measures one-way ANOVA with Sidak’s tests.
Fig. 3.
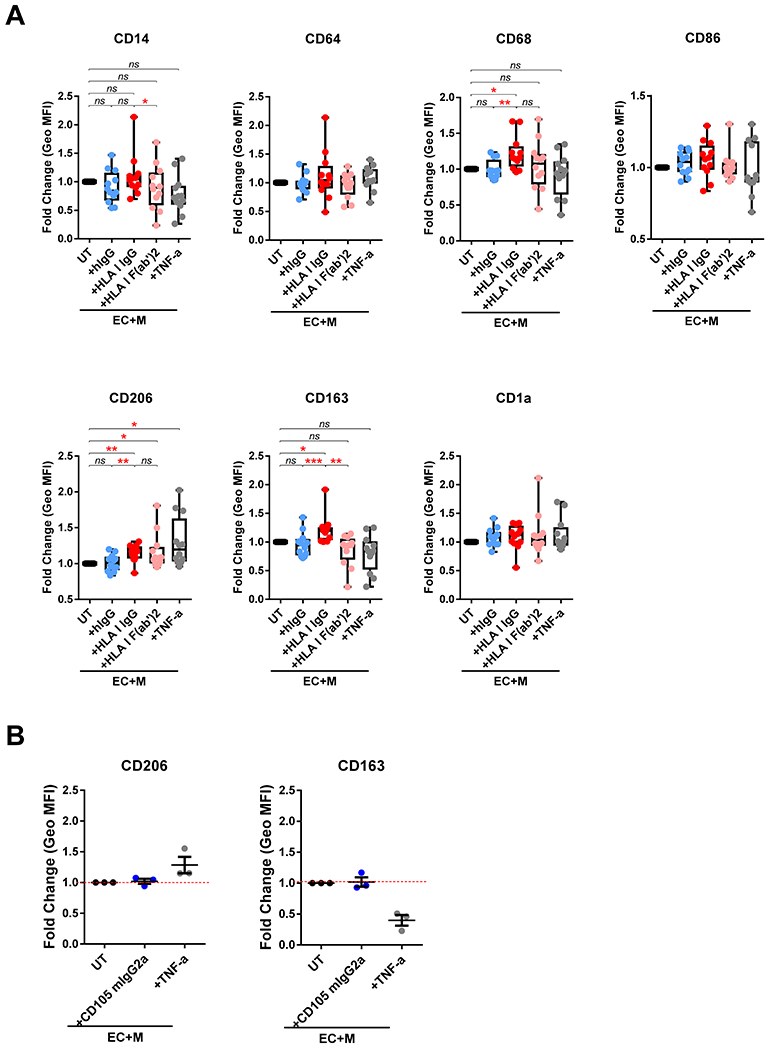
HLA I IgG activated EC-induced differentiation of monocytes into CD68+CD206+CD163+ macrophages. (A) Monocytes were co-cultured with unstimulated (UT), or HLA I IgG, HLA I F(ab’), hIgG, TNF-α stimulated ECs as in Figure 1. On day five, the macrophages in the lower chamber were collected and the assessments of surface markers expression were performed using flow cytometry. Results are expressed as fold change of Geo MFI value against EC+M+UT by box and whiskers graph. * p < 0.05, ** p < 0.01 and *** p < 0.001 were analyzed by repeated measures one-way ANOVA with Sidak’s tests. (B) Before the addition of monocytes, ECs were left untreated (UT) or treated with CD105 mIgG2a at 1 μg/mL (+CD105 mIgG2a) or TNF-α (10 ng/mL) (+TNF-α) and then co-cultured for 5 days. The cells in lower chamber were collected and assessed for the expression of CD206 and CD163 using flow cytometry. Data are expressed as mean ± SEM Fold change of Geo MFI value against EC+M+UT. The data in (A) represent four independent experiments with four monocyte donors against ECs from three donors. Data in (B) were from one experiment using one monocyte donor against ECs from three donors.
We previously found that antibody crosslinking of HLA I was sufficient to increase adhesion of monocytes in a P-selectin-dependent manner. To determine if monocyte differentiation was specifically induced by HLA I antibody-mediated EC activation, or required FcγRs, we co-cultured monocytes with anti-CD105 mIgG2a-stimulated ECs. Anti-CD105 mIgG2a binds to endoglin on EC surface and to FcγRs on human monocytes 42, but does not trigger EC exocytosis 17. Control CD105 mIgG2a treated EC did not increase the expression of either CD206 or CD163 on monocytes compared to untreated EC (Fig. 3B), indicating that M(HLA I IgG) differentiation is dependent on the HLA I antibody induced EC activation, even in the presence of Fc-FcγR ligation.
3.4. HLA I antibody-activated ECs increased cytokine/chemokine production by M(HLA I IgG)
To investigate the functional capacity of M(HLA I IgG), we collected co-culture supernatants on day 4 and examined the production of 38 cytokines and chemokines. Eight analytes were detected above the lower limit of quantitation (LLOQ). Cytokines/chemokines clustered into 3 main groups; control, HLA I antibody and TNF-α stimulated (Fig. 4A). ECs alone, monocytes alone or untreated/IgG-treated co-cultures produced few detectable cytokines and chemokines. In contrast, HLA I antibody (intact IgG and F(ab’)2 and TNF-α treatment induced high expression of cytokines and chemokines in EC-monocyte co-cultures. IL-6, IL-8/CXCL8, IL-10, MIP-1β/CCL4 and GROα/CXCL1 were significantly increased in HLA I IgG stimulated co-cultures compared to untreated and isotype control-treated co-cultures (Fig. 4B), while the differences in MDC/CCL22 and MIP-1β/CCL4 were not significant. There was no difference between M(HLA I IgG) and M(HLA I F(ab’)2) co-cultures, except for a trend towards increased IL-10 in M(HLA I IgG) compared to M(HLA I F(ab’)2) co-cultures (p = 0.1801) (Fig. 4B).
Fig. 4.
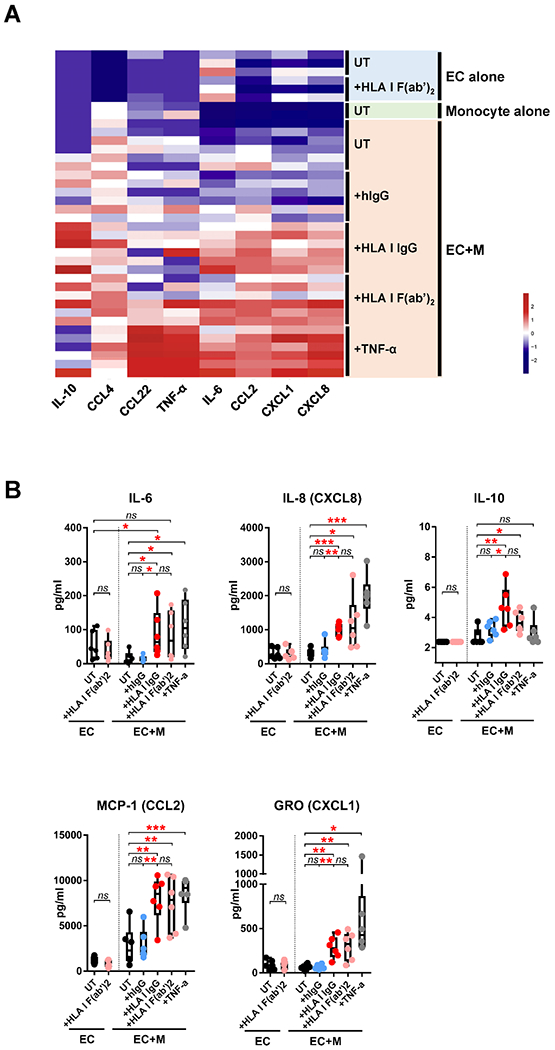
HLA I antibody-activated ECs increased cytokine/chemokine production by M(HLA I IgG). (A) Co-culture supernatants were collected on day 4 before medium replacement for cytokine detection. The data were Z-scaled and normalized by cytokine, and hierarchical clustering performed. (B) Statistical analysis of the data shown in (A). The data in (B) are represented by box and whiskers plots and analyzed by repeated measures one-way ANOVA with Fisher’s LSD tests. * p < 0.05, ** p < 0.01 and *** p < 0.001. The data represent two independent experiments using two monocyte donors against ECs from three donors.
We did not observe changes in cytokine production from HLA antibody stimulated endothelium alone (Fig. 4A, 4B), while positive control TNF-α induced the secretion of numerous cytokines and chemokines as expected (Fig. S3). These results indicate that interaction of activated macrophages with HLA I antibody-activated endothelium is necessary to induce cytokine/chemokine production.
3.5. Co-culture with HLA I antibody-stimulated ECs dramatically changes the macrophage transcriptional profile
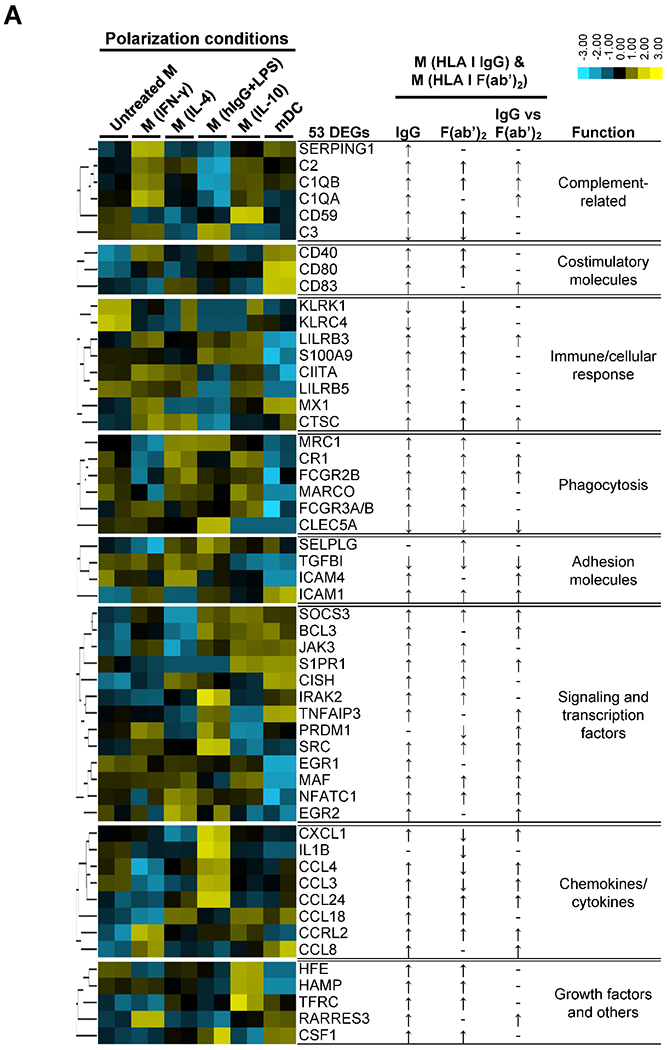
To further explore macrophage functional changes induced by ECs stimulated with intact and F(ab’)2 HLA antibody, we analyzed gene expression in transmigrated monocytes after 5 days using Nanostring Human Immunology Panel. The volcano plots (Fig. 5A) compare gene expression among M(EC), M(HLA I IgG) and M(HLA I F(ab’)2. Compared to M(EC), 43 genes were differentially expressed (38 increased and 5 decreased) in M(HLA I IgG) and 13 DEGs (11 increased, 2 decreased) in M(HLA I F(ab’)2). Eleven genes increased in both M(HLA I IgG) and M(HLA I F(ab’)2) compared with M(EC) (Table 2). In addition, 14 genes were differentially expressed comparing M(HLA I IgG) to M(HLA I F(ab’)2), suggesting that the Fc component of HLA antibody promotes additional gene expression. As a result, a total of 53 DEGs from the three comparisons were identified (Table 2, Table S2).
Fig. 5.

Volcano plots of the NanoString data and heatmap of the 53 differentially expressed genes (DEGs) in HLA I antibody-induced macrophages. After five days co-culture with unstimulated (UT), or HLA I IgG, HLA I F(ab’)2, hIgG stimulated ECs, macrophages in lower chambers were collected and lysed in RLT buffer for NanoString analysis. (A) Volcano plots of log2 (fold change, FC) on x-axis vs -log10 (p-value) on y-axis from the comparisons of the gene expression in each two of the HLA I IgG, HLA I F(ab’)2 and UT groups. Each circle represents one gene. For each comparison, genes with a p < 0.01 and absolute FC > 1.5 were considered DEGs and highlighted in gray . (B) Hierarchical clustering and heatmap of 53 DEGs, Z-scaled and normalized by gene. Group 1, 2 and 3 indicate the three gene clusters with differential expression patterns across the three conditions. Data were obtained coculturing two monocyte donors against the ECs from three donors.
Table 2.
The 53 differentially expressed genes (DEGs) in co-cultured macrophages
| Gene Symbol | IgG vs UT | F(ab’)2 vs UT | IgG vs F(ab’)2 | Gene Symbol | IgG vs UT | F(ab’)2 vs UT | IgG vs F(ab’)2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p | FC | p | FC | p | FC | p | FC | p | FC | p | FC | ||
| Complement-related | SELPLG | ns | - | *** | 2.37 | ns | - | ||||||
| C1QA | ** | 2.32 | ns | - | ** | 2.19 | TGFBI | *** | 0.62 | **** | 0.75 | * | 0.83 |
| C1QB | ** | 3.40 | ** | 1.55 | ** | 2.10 | Signaling and transcription factors | ||||||
| C2 | **** | 1.79 | ** | 1.32 | ** | 1.37 | BCL3 | ** | 1.50 | ns | - | * | 1.30 |
| C3 | ** | 0.61 | * | 0.68 | ns | - | CISH | ** | 11.13 | * | 13.74 | ns | - |
| CD59 | ** | 1.62 | * | 1.33 | ns | - | EGR1 | ** | 1.25 | ns | - | *** | 1.57 |
| SERPING1 | *** | 2.38 | ns | - | ns | - | EGR2 | ** | 1.74 | ns | - | *** | 1.73 |
| Costimulatory molecules | IRAK2 | ** | 2.37 | ** | 2.33 | ns | - | ||||||
| CD40 | *** | 3.09 | * | 2.44 | ns | - | JAK3 | ** | 8.24 | ** | 6.25 | ns | - |
| CD80 | ** | 7.36 | * | 4.89 | ns | - | MAF | ** | 1.67 | * | 1.27 | ** | 1.30 |
| CD83 | ** | 1.68 | ns | - | ** | 1.64 | NFATC1 | ** | 1.94 | *** | 1.35 | * | 1.40 |
| Immune/cellular response | PRDM1 | ns | - | ** | 0.65 | * | 1.49 | ||||||
| CIITA | ** | 2.97 | * | 2.67 | ns | - | S1PR1 | *** | 5.87 | * | 3.44 | * | 1.72 |
| CTSC | *** | 1.51 | ** | 1.29 | ** | 1.18 | SOCS3 | *** | 4.56 | ** | 2.34 | ** | 1.97 |
| KLRC4 | ** | 0.45 | * | 0.55 | ns | - | SRC | *** | 1.60 | * | 1.30 | *** | 1.24 |
| KLRK1 | ** | 0.52 | * | 0.59 | ns | - | TNFAIP3 | * | 1.42 | ns | - | ** | 1.67 |
| LILRB3 | ** | 1.59 | ** | 1.35 | * | 1.18 | Chemokines/cytokines | ||||||
| LILRB5 | ** | 1.74 | ns | - | ns | - | CCL18 | ** | 3.64 | * | 3.13 | ns | - |
| MX1 | ** | 1.65 | * | 2.04 | ns | - | CCL24 | ** | 32.47 | * | 10.91 | ** | 2.52 |
| S100A9 | ** | 1.58 | * | 1.50 | ns | - | CCL3 | * | 1.30 | ** | 0.76 | *** | 1.76 |
| Phagocytosis | CCL4 | * | 1.39 | ** | 0.72 | ** | 2.03 | ||||||
| CLEC5A | ** | 0.28 | * | 0.60 | * | 0.46 | CCL8 | ** | 1.59 | ns | - | * | 1.52 |
| CR1 | **** | 2.08 | *** | 1.67 | * | 1.25 | CCRL2 | *** | 1.81 | *** | 1.30 | *** | 1.39 |
| MARCO | ** | 2.62 | ** | 2.01 | ns | - | CXCL1 | * | 1.30 | ** | 0.76 | ** | 1.75 |
| MRC1 | ** | 1.68 | * | 1.51 | ns | - | IL1B | ns | - | ** | 0.62 | ns | - |
| Fcγ receptors | Growth/differentiation factors/others | ||||||||||||
| FCGR2B | ** | 2.15 | ** | 1.38 | ** | 1.56 | CSF1 | * | 8.90 | ** | 8.89 | ns | - |
| FCGR3A/B | ** | 2.16 | **** | 1.67 | ns | - | HAMP | *** | 1.97 | * | 2.02 | ns | - |
| Adhesion molecules | HFE | ** | 1.82 | ** | 1.60 | ns | - | ||||||
| ICAM1 | ** | 1.74 | * | 1.19 | * | 1.47 | TFRC | *** | 2.25 | ** | 1.84 | ns | - |
| ICAM4 | ** | 2.22 | ns | - | *** | 2.18 | RARRES3 | * | 4.61 | ns | - | ** | 1.93 |
UT, Ab and F(ab’)2 represent monocytes co-cultured with unstimulated, HLA-I antibody (intact or F(ab’)2) stimulated endothelial cells, respectively. ns p > 0.05,
p < 0.05,
p < 0.01,
p < 0.001 and
p < 0.0001 were analyzed by repeated measures one-way ANOVA with Fisher’s LSD tests of gene expression counts. FC, fold change. -, not applicable. The data were from two independent experiments using two monocyte donors against the ECs from three donors.
Unsupervised hierarchical clustering of the expression patterns of the 53 DEGs (Fig. 5B, Fig. S4) revealed three groups. Compared to M(EC), 5 genes in group 1 DEGs were down-regulated in both M(HLA I IgG) and M(HLA I F(ab’)2), whereas 36 group 3 DEGs were up-regulated in both conditions. Twelve group 2 DEGs were upregulated in M(HLA I IgG), but down-regulated in M(HLA I F(ab’)2).
These DEGs encode complement-related, costimulatory, immune/cellular response, phagocytosis, adhesion, signaling and transcription factors, chemokines/cytokines, growth and differentiation related molecules (Table 2). Compared to M(EC), most of these DEGs were significantly increased in M(HLA I IgG) (Table 2, Fig. S4), such as: C1QA, C1QB, CD59, SERPING1, CTSC, CR1, JAK3, MAF, S1PR1, and TNFAIP3. Compared to M(HLA I IgG), complement-related (C1QA, C1QB, C2), costimulatory (CD83), adhesion (ICAM4), signaling and transcription factors (EGR1, EGR2, MAF, SOCS3, TNFAIP3), and chemokines/cytokines genes (eotaxin-2/CCL24, MIP-1α/CCL3, MIP-1β/CCL4, CCRL2, GROα/CXCL1) were lower than in M(HLA F(ab’)2). Chemokines/cytokines genes MIP-1α/CCL3, MIP-1β/CCL4, GROα/CXCL1 and IL1B were decreased in M(HLA I F(ab’)2) compared to the untreated M(EC) (Table 1, Fig. S4). These data suggest that, compared to HLA I F(ab’)2, HLA I IgG induces a broader spectrum of immunological functions in macrophages 38,43.
3.6. M(HLA I IgG) is a novel macrophage subset with distinct features compared to classically polarized macrophages
To understand whether HLA I antibody-activated EC poise transmigrated monocytes to unique, not-yet-described macrophage phenotypes, we generated cytokine-polarized control macrophage subsets to compare with M(HLA I IgG) and M(HLA I F(ab’)2). Consistent with previous studies 44–46, M(IFN-γ) were CD64hiCD15loCD206loCD163lo; M(IL-4) were CD206hiCD68hiCD14loCD64loCD163lo. M(IL-10) were CD163hiCD14hiCD68hiCD86lo. For M(hIgG+LPS), several markers, including CD64, CD68, CD163 and CD1a were significantly decreased, whereas no cell surface protein that we assessed was increased, as reported 46 (Fig. 6A, Fig. S4). mDC were CD1ahiCD86hiCD206hi 47. These phenotypes were distinct from that observed in M(HLA I IgG), which exhibited coexpression of CD163 and CD206.
Fig. 6.

Characterization of in vitro polarized macrophages and dendritic cells (DCs). (A) Freshly isolated human peripheral monocytes from 4 different donors were left unstimulated (untreated M), or stimulated with IFN-γ (50 ng/mL), IL-4 (40 ng/mL), hIgG (10 μg/mL, pre-coated) + LPS (100 ng/mL), IL-10 (50 ng/mL) or IL-4 (20 ng/mL) + GM-CSF (50 ng/mL) + LPS (100 ng/mL, on day 4) for five days to induce M(IFN-γ), M(IL-4), M(hIgG+LPS), M(IL-10) and mature DC (mDC), respectively. Stimuli and half of the culture medium were refreshed every two days. Expression of seven surface markers were measured by flow cytometry. Results are expressed as fold change of Geo MFI value against untreated M, and shown by box and whiskers plots. Statistical significance * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001 was analyzed by repeated measures one-way ANOVA with Dunnett’s tests compared to untreated M. (B) Cell culture supernatants from polarized macrophages were collected on day 4 before medium replacement, and assessed for cytokines using 38-plex Luminex. Cytokine concentrations are shown as mean ± SD. Measurement of IL-10, used as stimuli to generate M(IL-10), was not included in the analysis and indicated by “N/A”. Table 5B represents the cytokine levels from Figure 3. ↑indicates increased vs –unchanged cytokine expression in M(HLA I IgG) and M(HLA I F(ab’)2). Data were from four independent experiments using 3 ECs with four monocyte donors.
When comparing M(HLA I IgG) to these controls, we found the expression of GRO/CXCL1, MCP-1/CCL2, MDC/CCL22 and TNF-α was consistent with those of M(IL-10). However, M(HLA I IgG) also shared some properties with M(hIgG+LPS) as well as M(IFN-γ), such as IL-6, IL-8/CXCL8 and MCP-1/CCL2 (Fig. 6B), indicating a distinct subtype of M(HLA I IgG).
Finally, we compared the expression of the 53 DEGs identified in M(HLA I IgG) and M(HLA I F(ab’)2) with that of macrophages polarized by cytokines (Fig. 7A). Hierarchical clustering based on the overall expression pattern of these DEGs indicate that antibody-stimulated conditions were indeed different, and were more similar to M(IL-10), and secondly to M(IL-4), than to LPS-treated macrophages (M(hIgG+LPS) and M(IFN-γ)) (Fig. 7B). For M(HLA I IgG), 13 of the 53 DEGs were shared with M(IL-4), 20 with M(IL-10) and 11 with both (Fig. 6C). Notably, 4 of the unshared 9 DEGs in M(HLA I IgG) were also dramatically increased in M(hIgG+LPS), and 3 of them were pro-inflammatory genes, suggesting some similarity exists between the M(HLA I IgG) and the M(hIgG+LPS). M(HLA I F(ab’)2) shared 18 genes with M(IL-4), 16 with M(IL-10) and 14 with both (Fig. 7C). These data suggest that M(HLA I IgG) exhibit a distinct pattern of gene expression that is a unique phenotype not consistent with any of the canonical cytokine-stimulated conditions described to date.
Fig. 7.

HLA I antibody-polarized macrophages exhibit a unique transcriptional profile. (A) Heat map shows expression of the 53 DEGs in macrophages from two independent experiments using 2 monocyte donors polarized using canonical stimuli compared to expression of DEGs in M(HLA I IgG) and M(HLA I F(ab’)2) obtained by testing two monocyte donors against 3 ECs. ↑, ↓, and – indicate up-regulated, down-regulated and unchanged, respectively, refering to the data in Table 1. (B) Hierarchical clustering was used to compare the pattern of gene expression between the control cultures, M(HLA I IgG) and M(HLA I F(ab’)2). (C) For macrophages polarized using canonical stimuli DEGs were defined as an absolute fold change > 1.5. The radar chart shows the difference in the numbers of DEGs shared with between M(HLA I IgG) and M(HLA I F(ab’)2 and M(IL-4) and M(IL-10).
3.7. Human in vitro M(HLA I IgG) and M(HLA I F(ab’)2) polarized macrophages share similar DEGs to murine DSA treated cardiac grafts.
To identify if human in vitro co-culture macrophages contain DEGs analogous to murine cardiac allografts treated with MHC I DSA mixture, we performed a comparative analysis between DEGs from both parties. Overall, 11 DEGs overlapped between the human in vitro and the murine in vivo data (Table 3). The DEGs encoded immune/cellular responses, chemokines/cytokines, transcription factors, phagocytosis, and complement-related proteins. Both M(HLA I IgG) and M(HLA I F(ab’)2) independently shared 7 analogous DEGs compared with the DEGs from DSA treated grafts. Shared DEGs between M(HLA I IgG) and DSA treated grafts included genes related to immune responses (IL1RN), chemokines (CXCL1, CCL2, and CSF1), signaling transcriptions factors (EGR2 and POU2F2) and phagocytosis (CLEC5A). Shared DEGs between M(HLA I F(ab’)2) and DSA treated grafts also included genes related to immune/cellular response (GRP183, and PTGS2), cytokines/chemokines (CXCL1, CCL2, and CSF1), phagocytosis (CLEC5A) and complement (C1S1). Five DEGs (CXCL1, EGR2, PTGS2, CLEC5A, and TMEM173) were identified as significantly different between M(HLA I IgG) and M(HLA F(ab’)2. Finally, four DEGs were shared across all three groups: M(HLA I IgG), M(HLA I F(ab’)2), and MHC I DSA treated allografts. These four genes included chemokines/cytokines (CXCL1, CCL2, and CSF1), and phagocytosis (CLEC5a).
Table 3.
The 11 differentially expressed genes (DEGs) similar across in vitro human co-cultured macrophages and DSA treated cardiac grafts.
| Mice allografts | IgG vs UT | F(ab)2 vs UT | IgG vs F(ab)2 | |||||
|---|---|---|---|---|---|---|---|---|
| Gene symbol | p | p | FC | p | FC | p | FC | Function |
| IL1RN | 0.002 | * | 2.42 | ns | 1.92 | ns | 1.23 | Immune/cellular response |
| GPR183 | 0.034 | ns | 1.00 | * | 0.84 | ns | 1.2 | Immune/cellular response |
| CXCL1 | 0.041 | * | 1.47 | ** | 0.87 | ** | 1.75 | Chemokines/cytokines |
| CCL2 | 0.060 | * | 1.78 | ** | 1.94 | ns | 0.97 | Chemokines/cytokines |
| EGR2 | 0.067 | ** | 1.74 | ns | 1.02 | *** | 1.73 | Signaling/ transcription |
| PTGS2 | 0.073 | ns | 1.46 | ** | 0.68 | * | 2.3 | Immune/cellular response |
| POU2F2 | 0.080 | * | 1.23 | ns | 1.09 | ns | 1.18 | Signaling/ transcription |
| CSF1 | 0.090 | * | 8.90 | ** | 8.89 | ns | 1.09 | Chemokines/cytokines |
| CLEC5A | 0.091 | ** | 0.28 | * | 0.60 | * | 0.46 | Phagocytosis |
| TMEM173 | 0.097 | ns | 1.23 | ns | 1.03 | * | 1.2 | Immune/cellular response |
| C1S1 | 0.097 | ns | 37.3 | * | 26.5 | ns | 1.2 | Complement-related |
Left: In vivo mouse cardiac allograft differentially expressed genes with p < 0.1 between HLA I DSA treated allograft recipients and isotype treated controls (n=3). Right: UT, Ab and F(ab’)2 represent monocytes co-cultured with unstimulated, HLA-I antibody (intact or F(ab’)2) stimulated endothelial cells, respectively. ns p > 0.05,
p < 0.05,
p < 0.01,
p < 0.001 and
p < 0.0001 were analyzed by repeated measures one-way ANOVA with Fisher’s LSD tests of gene expression counts. FC, fold change.
4. Discussion
Here we describe for the first time that ECs activated by HLA I IgG skew monocytes towards a novel macrophage polarization state through the synergistic actions of EC activation and FcγR ligation. Using an in vivo mouse model of acute AMR, we identified DSA upregulated genes related to monocyte stimulation by Fcγ receptors and transmigration through DSA-activated endothelium. Cardiac allografts from recipients treated with MHC I DSA had an increased expression of transcripts associated with macrophage recruitment, adhesion and immune/cellular response. To directly examine the mechanisms of MHC I DSA on macrophage polarization, we utilized an in vitro model of human peripheral blood monocytes transmigrating through HLA I IgG-activated endothelial monolayers. These macrophages had concurrent expression of CD163 and CD206, enhanced production of IL-10, IL-6, IL-8/CXCL8, MCP-1/CCL2 and GROα/CXCL1, and increased expression of transcripts involved in antigen presentation and regulation of complement cascades. In vitro co-cultured polarized macrophages and in vivo DSA-exposed allografts shared analogous DEGs involved in macrophage recruitment and immune cellular/response.
HLA I outside-in signaling promotes monocyte adhesion and recruitment by triggering mobilization of WPBs, induction of P-selectin, ICAM-1 clustering and subsequent leukocyte capture and firm adhesion to the activated endothelium, a process partially mediated by the Fc-FcγR interaction between the HLA I IgG and the monocytes 17,19,20. The question addressed in this work is whether the HLA I antibody modifies monocyte differentiation during recruitment and transendothelial migration.
Once monocytes transmigrate into the subendothelial connective tissue they differentiate into distinct phagocytes including macrophages and DCs. Historically, macrophages were classified into two major functional subgroups termed M1 and M2, with distinct functions 38,48. M1 or “classically activated” macrophages differentiate in response to interferon-gamma (IFN-γ) or TLR ligands, whereas M2 or “alternatively activated” macrophages develop in the presence of IL-10, IL-4 or IL-13 and mediate tissue repair and promote fibrosis 24,25,48. However, the current viewpoint is that the M1/M2 nomenclature underrepresents the actual functional heterogeneity of macrophage subtypes which are now classified based on their interaction with different immunological stimuli (i.e. growth factors, cytokines, bacterial products), rather than attributing them to distinct subsets 23,38. Indeed, monocyte co-culture even with unstimulated endothelium triggered phenotypic changes 49 and changes in gene expression 50.
Less is known about monocyte emigration and differentiation during antibody-mediated vascular inflammation. In the setting of AMR, HLA crosslinking by antibodies on the EC induces EC activation and intracellular signaling. Our data suggest this process is specific for HLA I and requires EC activation 17. Since the IgG subclass and FcγR polymorphisms regulate HLA I IgG-promoted monocyte adhesion 17,19, we used HLA I F(ab’)2 in parallel to see whether HLA I IgG Fc region took part in driving differential polarization. Our results indicate the critical role of antibody Fc region in further M(HLA I IgG) differentiation, as M(HLA I IgG) express CD206 and CD163 concurrently. Similarly, hemoglobin/haptoglobin stimulation promotes concomitant expression of CD206 and CD163 in macrophages, termed M(Hb) 51.
Several studies reported antibody ligation of HLA I molecules on EC directly triggered production of cytokines, chemokines 52,53 and growth factors , of which some have been shown to augment M2-like polarization of macrophages 54–57. In our model, stimulation with HLA I antibody did not induce cytokine production directly from ECs. Instead, cell:cell contact of monocytes with antibody-activated ECs was required for cytokine production. This inconsistency may be due to differences in EC vascular origin, passage number, and/or HLA antibody concentrations. However, recruited human monocytes trigger a regulatory pathway of cytokine-mediated signaling at the EC interface, substantiating cross talk between the monocyte and EC in vascular inflammation 58. The mechanism(s) underlying how HLA antibody-activated ECs instruct monocyte differentiation needs to be investigated.
Although endothelial-polarized macrophages shared some patterns of gene and cytokine expression with cytokine activated macrophages, there was no complete overlap with macrophages derived in the presence of prototypic stimuli. EC activated by HLA I crosslinking (HLA I F(ab’)2) induced expression of genes in macrophages typical of alternatively activated macrophages. When macrophages also received FcγR signaling, numerous genes characteristic of M(IFNγ) were enhanced. Many of these additional genes overlapped with M(hIgG+LPS), indicating the involvement of the HLA I IgG Fc region in monocyte differentiation. Thus, the present data suggest that the HLA I IgG enhanced monocyte differentiation occurs not only by activating ECs, but also through the action of the antibody Fc region. These features suggest the M(HLA I IgG) is a previously undescribed subtype of alternatively activated macrophage.
Our study also provides phenotypic markers for M(hIgG+LPS), which had remained elusive so far 45,46. Indeed, we found several surface markers (CD64, CD68, CD163, CD1a) that were significantly decreased in M(hIgG+LPS), which was to some extent consistent with the findings of others 59.
MHC I DSA treated allografts shared overlapping genes with in vitro co-cultured human macrophages. Although it is difficult classify allograft DEGs to specific cell-types from bulk transcriptomes, many genes have been previously described to be associated with macrophage recruitment (e.g. CCL2 and CXCL1) 40,41, polarization (e.g. CSF1) 60, immune responses (GPR183) 61, and phagocytosis (CLEC5A) 62. . CXCL1 and CCL2 were also increased in vitro in co-culture supernatants from M(HLA I IgG) and M(HLA I F(ab’)2). Banff 2017 identified several transcripts (CX3CR1, FCGR3A, CD163, and CD206) in renal AMR that overlap with our M(HLA I) signature63. Moreover, CD68 is a component of the ISHLT working formulation to identify intravascular macrophages in heart allografts with AMR 64. None of the 27 DEGs are included in CD16a-inducible transcripts following in vitro cross-linking on purified NK cells that are also expressed during AMR of kidney grafts 65. Nonetheless, NK cell transcripts related to Fc receptor stimulation warrants further investigation. Overall, our in vitro data using human primary cells and in vivo murine studies further implicate DSA in the development of distinct macrophages specific to AMR. The clinical relevance and mechanisms by which these CD206+ and CD163+ macrophages may contribute to the processes of acute and chronic transplant rejection needs to be elucidated. Notably, CD163+ macrophages were identified within fibrotic areas of patient renal allografts28 and CD206+ macrophages were increased within murine renal allografts with chronic rejection 30. Studies are underway to validate these findings in biopsies from patients with AMR. This macrophage subset could be an attractive therapeutic target for patients with DSA at risk of TV.
The ECs used in the current study were isolated from human aortic rings which may not reflect the positional identity and heterogeneity of EC from different vascular beds. This study mainly probed the effect of HLA-I antibodies activated ECs on monocyte differentiation. Whether and how HLA II antibodies activate the ECs, and drive monocyte differentiation is a subject of ongoing study. Although our mouse model of AMR indicated macrophage polarization via Fc receptors and transmigration across DSA-activated endothelium, further transcriptome profiling of isolated graft infiltrating macrophages will be critical. We show that the HLA-I antibody activated ECs can drive monocyte differentiation, but it requires further study to determine what biological impact the macrophages, in turn, have on the intimal cells that contributes to vascular disease.
In conclusion, our findings demonstrate the ability of antibody-induced vascular inflammation promotes monocyte polarization to a unique CD68+CD206+CD163+ macrophage phenotype. The increase in genes indicative of monocyte stimulation by Fc receptors and transmigration through activated endothelium in murine cardiac allografts suggests a pathogenic role of these macrophages in tissue injury that remains to be elucidated.
Supplementary Material
Acknowledgments
The authors express their thanks to OneLegacy and all the organ and tissue donors and their families for giving the gift of life by their generous donation. The authors would like to acknowledge Dr. Nina Dvorina for her expert immunohistochemical stains, and the efforts of the CTOT Nanostring Core. This work was supported in part by NIH U19AL128913, PO1AI120944, U01AI124319, RO1AI042819 (to EFR), RO1AI135201 (to E.F.R and RLF); by the Jiangsu Provincial Medical Youth Talent QNRC2016739 and Science and Technology Development Program of Suzhou SYS201601, CXTDB2017009, GSWS2019033 (to X. W.); by the Jiangsu Provincial Key Medical Discipline ZDXKA2016012 (to J. H.); by the CTOT NanoString Core (UO1 AI063594 to R.L.F).
Abbreviations:
- Ab
antibody
- AMR
antibody-mediated rejection
- DSA
donor specific HLA antibodies
- EC
endothelial cell
- HLA
human leukocyte antigen
- M(HLA IgG)
macrophages differentiated by endothelium activated with intact HLA IgG
- M(HLA I F(ab’)2)
macrophages differentiated by endothelium activated with HLA IgG F(ab’)2 fragment
- TV
transplant vasculopathy
Footnotes
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Guilpain P, Mouthon L. Antiendothelial cells autoantibodies in vasculitis-associated systemic diseases. Clin Rev Allergy Immunol. 2008. 35(1-2): 59–65. [DOI] [PubMed] [Google Scholar]
- 2.Legendre P, Régent A, Thiebault M, Mouthon L. Anti-endothelial cell antibodies in vasculitis: A systematic review. Autoimmun Rev. 2017. 16(2): 146–153. [DOI] [PubMed] [Google Scholar]
- 3.Florey OJ, Johns M, Esho OO, Mason JC, Haskard DO. Antiendothelial cell antibodies mediate enhanced leukocyte adhesion to cytokine-activated endothelial cells through a novel mechanism requiring cooperation between Fc{gamma}RIIa and CXCR1/2. Blood. 2007. 109(9): 3881–9. [DOI] [PubMed] [Google Scholar]
- 4.Iseme RA, McEvoy M, Kelly B, et al. A role for autoantibodies in atherogenesis. Cardiovasc Res. 2017. 113(10): 1102–1112. [DOI] [PubMed] [Google Scholar]
- 5.Matsuura E, Atzeni F, Sarzi-Puttini P, Turiel M, Lopez LR, Nurmohamed MT. Is atherosclerosis an autoimmune disease. BMC Med. 2014. 12: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiebe C, Gibson IW, Blydt-Hansen TD, et al. Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant. 2012. 12(5): 1157–67. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Q, Hickey M, Drogalis-Kim D, et al. Understanding the correlation between DSA, complement activation and antibody mediated rejection in heart transplant recipients. Transplantation. 2018. 102(10): e431–e438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clerkin KJ, Farr MA, Restaino SW, et al. Donor-specific anti-HLA antibodies with antibody-mediated rejection and long-term outcomes following heart transplantation. J Heart Lung Transplant. 2017. 36(5): 540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valenzuela NM, Reed EF. Antibody-mediated rejection across solid organ transplants: manifestations, mechanisms, and therapies. J Clin Invest. 2017. 127(7): 2492–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loupy A, Toquet C, Rouvier P, et al. Late Failing Heart Allografts: Pathology of Cardiac Allograft Vasculopathy and Association With Antibody-Mediated Rejection. Am J Transplant. 2016. 16(1): 111–20. [DOI] [PubMed] [Google Scholar]
- 11.Roux A, Bendib Le Lan I, Holifanjaniaina S, et al. Antibody-Mediated Rejection in Lung Transplantation: Clinical Outcomes and Donor-Specific Antibody Characteristics. Am J Transplant. 2016. 16(4): 1216–28. [DOI] [PubMed] [Google Scholar]
- 12.Wong KL, Taner T, Smith BH, et al. Importance of Routine Antihuman/Leukocyte Antibody Monitoring: De Novo Donor Specific Antibodies Are Associated With Rejection and Allograft Vasculopathy After Heart Transplantation. Circulation. 2017. 136(14): 1350–1352. [DOI] [PubMed] [Google Scholar]
- 13.Tran A, Fixler D, Huang R, Meza T, Lacelle C, Das BB. Donor-specific HLA alloantibodies: Impact on cardiac allograft vasculopathy, rejection, and survival after pediatric heart transplantation. J Heart Lung Transplant. 2016. 35(1): 87–91. [DOI] [PubMed] [Google Scholar]
- 14.Loupy A, Vernerey D, Viglietti D, et al. Determinants and Outcomes of Accelerated Arteriosclerosis: Major Impact of Circulating Antibodies. Circ Res. 2015. 117(5): 470–82. [DOI] [PubMed] [Google Scholar]
- 15.Jin YP, Korin Y, Zhang X, Jindra PT, Rozengurt E, Reed EF. RNA interference elucidates the role of focal adhesion kinase in HLA class I-mediated focal adhesion complex formation and proliferation in human endothelial cells. J Immunol. 2007. 178(12): 7911–22. [DOI] [PubMed] [Google Scholar]
- 16.Jindra PT, Hsueh A, Hong L, et al. Anti-MHC class I antibody activation of proliferation and survival signaling in murine cardiac allografts. J Immunol. 2008. 180(4): 2214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valenzuela NM, Mulder A, Reed EF. HLA class I antibodies trigger increased adherence of monocytes to endothelial cells by eliciting an increase in endothelial P-selectin and, depending on subclass, by engaging FcγRs. J Immunol. 2013. 190(12): 6635–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valenzuela NM, Hong L, Shen XD, et al. Blockade of p-selectin is sufficient to reduce MHC I antibody-elicited monocyte recruitment in vitro and in vivo. Am J Transplant. 2013. 13(2): 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valenzuela NM, Trinh KR, Mulder A, Morrison SL, Reed EF. Monocyte recruitment by HLA IgG-activated endothelium: the relationship between IgG subclass and FcγRIIa polymorphisms. Am J Transplant. 2015. 15(6): 1502–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salehi S, Sosa RA, Jin YP, et al. Outside-in HLA class I signaling regulates ICAM-1 clustering and endothelial cell-monocyte interactions via mTOR in transplant antibody-mediated rejection. Am J Transplant. 2018. 18(5): 1096–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamakuchi M, Kirkiles-Smith NC, Ferlito M, et al. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc Natl Acad Sci U S A. 2007. 104(4): 1301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pilmore HL, Painter DM, Bishop GA, McCaughan GW, Eris JM. Early up-regulation of macrophages and myofibroblasts: a new marker for development of chronic renal allograft rejection. Transplantation. 2000. 69(12): 2658–62. [DOI] [PubMed] [Google Scholar]
- 23.Liberale L, Dallegri F, Montecucco F, Carbone F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb Haemost. 2017. 117(1): 7–18. [DOI] [PubMed] [Google Scholar]
- 24.Das A, Sinha M, Datta S, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol. 2015. 185(10): 2596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013. 229(2): 176–85. [DOI] [PubMed] [Google Scholar]
- 26.Mannon RB. Macrophages: contributors to allograft dysfunction, repair, or innocent bystanders. Curr Opin Organ Transplant. 2012. 17(1): 20–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Azad TD, Donato M, Heylen L, et al. Inflammatory macrophage-associated 3-gene signature predicts subclinical allograft injury and graft survival. JCI Insight. 2018. 3(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikezumi Y, Suzuki T, Yamada T, et al. Alternatively activated macrophages in the pathogenesis of chronic kidney allograft injury. Pediatr Nephrol. 2015. 30(6): 1007–17. [DOI] [PubMed] [Google Scholar]
- 29.Toki D, Zhang W, Hor KL, et al. The role of macrophages in the development of human renal allograft fibrosis in the first year after transplantation. Am J Transplant. 2014. 14(9): 2126–36. [DOI] [PubMed] [Google Scholar]
- 30.Wu C, Zhao Y, Xiao X, et al. Graft-Infiltrating Macrophages Adopt an M2 Phenotype and Are Inhibited by Purinergic Receptor P2X7 Antagonist in Chronic Rejection. Am J Transplant. 2016. 16(9): 2563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, Chen S, Lan P, et al. Macrophage subpopulations and their impact on chronic allograft rejection versus graft acceptance in a mouse heart transplant model. Am J Transplant. 2018. 18(3): 604–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rahimi S, Qian Z, Layton J, Fox-Talbot K, Baldwin WM 3rd, Wasowska BA. Non-complement- and complement-activating antibodies synergize to cause rejection of cardiac allografts. Am J Transplant. 2004. 4(3): 326–34. [DOI] [PubMed] [Google Scholar]
- 33.Mulder A, Kardol MJ, Arn JS, et al. Human monoclonal HLA antibodies reveal interspecies crossreactive swine MHC class I epitopes relevant for xenotransplantation. Mol Immunol. 2010. 47(4): 809–15. [DOI] [PubMed] [Google Scholar]
- 34.Sosa RA, Zarrinpar A, Rossetti M, et al. Early cytokine signatures of ischemia/reperfusion injury in human orthotopic liver transplantation. JCI Insight. 2016. 1(20): e89679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008. 8(12): 958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008. 13: 453–61. [DOI] [PubMed] [Google Scholar]
- 37.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010. 32(5): 593–604. [DOI] [PubMed] [Google Scholar]
- 38.Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014. 41(1): 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rehman A, Hemmert KC, Ochi A, et al. Role of fatty-acid synthesis in dendritic cell generation and function. J Immunol. 2013. 190(9): 4640–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams MR, Sakurai Y, Zughaier SM, Eskin SG, McIntire LV. Transmigration across activated endothelium induces transcriptional changes, inhibits apoptosis, and decreases antimicrobial protein expression in human monocytes. J Leukoc Biol. 2009. 86(6): 1331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee CY, Lotfi-Emran S, Erdinc M, et al. The involvement of FcR mechanisms in antibody-mediated rejection. Transplantation. 2007. 84(10): 1324–34. [DOI] [PubMed] [Google Scholar]
- 42.Bruhns P Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012. 119(24): 5640–9. [DOI] [PubMed] [Google Scholar]
- 43.Fuentes-Duculan J, Suárez-Fariñas M, Zaba LC, et al. A subpopulation of CD163-positive macrophages is classically activated in psoriasis. J Invest Dermatol. 2010. 130(10): 2412–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004. 25(12): 677–86. [DOI] [PubMed] [Google Scholar]
- 45.Ambarus CA, Krausz S, van Eijk M, et al. Systematic validation of specific phenotypic markers for in vitro polarized human macrophages. J Immunol Methods. 2012. 375(1-2): 196–206. [DOI] [PubMed] [Google Scholar]
- 46.Ambarus CA, Santegoets KC, van Bon L, et al. Soluble immune complexes shift the TLR-induced cytokine production of distinct polarized human macrophage subsets towards IL-10. PLoS One. 2012. 7(4): e35994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia CQ, Kao KJ. Heparin induces differentiation of CD1a+ dendritic cells from monocytes: phenotypic and functional characterization. J Immunol. 2002. 168(3): 1131–8. [DOI] [PubMed] [Google Scholar]
- 48.Italiani P, Boraschi D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front Immunol. 2014. 5: 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tso C, Rye KA, Barter P. Phenotypic and functional changes in blood monocytes following adherence to endothelium. PLoS One. 2012. 7(5): e37091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas-Ecker S, Lindecke A, Hatzmann W, Kaltschmidt C, Zänker KS, Dittmar T. Alteration in the gene expression pattern of primary monocytes after adhesion to endothelial cells. Proc Natl Acad Sci U S A. 2007. 104(13): 5539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finn AV, Nakano M, Polavarapu R, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. 2012. 59(2): 166–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reyes-Vargas E, Pavlov IY, Martins TB, Schwartz JJ, Hill HR, Delgado JC. Binding of anti-HLA class I antibody to endothelial cells produce an inflammatory cytokine secretory pattern. J Clin Lab Anal. 2009. 23(3): 157–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naemi FM, Carter V, Kirby JA, Ali S. Anti-donor HLA class I antibodies: pathways to endothelial cell activation and cell-mediated allograft rejection. Transplantation. 2013. 96(3): 258–66. [DOI] [PubMed] [Google Scholar]
- 54.Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem. 2009. 284(49): 34342–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mauer J, Chaurasia B, Goldau J, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol. 2014. 15(5): 423–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sierra-Filardi E, Nieto C, Domínguez-Soto A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014. 192(8): 3858–67. [DOI] [PubMed] [Google Scholar]
- 57.Li N, Qin J, Lan L, et al. PTEN inhibits macrophage polarization from M1 to M2 through CCL2 and VEGF-A reduction and NHERF-1 synergism. Cancer Biol Ther. 2015. 16(2): 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chimen M, Yates CM, McGettrick HM, et al. Monocyte Subsets Coregulate Inflammatory Responses by Integrated Signaling through TNF and IL-6 at the Endothelial Cell Interface. J Immunol. 2017. 198(7): 2834–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008. 118(11): 3522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jones CV, Ricardo SD, Macrophages and CSF-1: implications for development and beyond. Organogenesis. 2013. 9(4): 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Preuss I, Ludwig MG, Baumgarten B, et al. Transcriptional regulation and functional characterization of the oxysterol/EBI2 system in primary human macrophages. Biochem. Biophys. Res. Commun. 2014. 446(3): 663–668. [DOI] [PubMed] [Google Scholar]
- 62.Xiong W, Wang H, Lu L, et al. The macrophage C-type lectin receptor CLEC5A (MDL-1) expression is associated with early plaque progression and promotes macrophage survival. J Transl Med. 2017. 15(1): 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haas M, Loupy A, Lefaucheur C, et al. The Banff 2017 Kidney Meeting Report: Revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant. 2018. 18(2):293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berry GJ, Burke MM, Andersen C, et al. The 2013 International Society for Heart and Lung Transplantation Working Formulation for the standardization of nomenclature in the pathologic diagnosis of antibody-mediated rejection in heart transplantation. J Heart Lung Transplant. 2013. 32(12):1147–1162. [DOI] [PubMed] [Google Scholar]
- 65.Parkes MD, Halloran PF, Hidalgo LG. Evidence for CD16a-Mediated NK Cell Stimulation in Antibody-Mediated Kidney Transplant Rejection. Transplantation. 2017101(4):e102–102e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.