

Abstract
Ferroptosis is considered a distinctive form of cell death compared to other types of death such as apoptosis. It is known to result from iron-dependent accumulation of lipid peroxides rather than caspase activation. However, we reported recently that ferroptosis interplays with apoptosis. In this study, we investigated a possible mechanism of this interplay between ferroptosis and apoptosis. Results from our studies reveal that combined treatment of the ferroptotic agent erastin and the apoptotic agent TRAIL effectively disrupted mitochondrial membrane potential (ΔΨm) and subsequently promoted caspase activation. The alterations of mitochondrial membrane potential are probably due to an increase in oligomerization of BAX and its accumulation at the mitochondria during treatment with erastin and TRAIL. Interestingly, the combined treatment-promoted apoptosis was effectively inhibited in BAX-deficient HCT116 cells, but not BAK-deficient cells. These results indicate that the BAX-associated mitochondria-dependent pathway plays a pivotal role in erastin-enhanced TRAIL-induced apoptosis.
Keywords: ferroptosis, apoptosis, crosstalk, BAX, mitochondria-dependent pathway
Introduction
Ferroptosis is a well-known form of iron-dependent, lipid hydroperoxide-associated, programmed cell death (1). Iron-dependent generation of reactive oxygen species, accumulation of lipid peroxidation, and depletion of plasma membrane polyunsaturated fatty acids have been well known to result in this lethal event (2, 3). Although considered different from autophagy, necroptosis, necrosis, apoptosis, and cell death of other types, recent studies reveal an interplay between ferroptosis and apoptosis through the endoplasmic reticulum (ER) stress signaling pathway (4). Interestingly, the interaction between apoptosis and ferroptosis promotes apoptosis, but not ferroptosis (4). Nevertheless, the mechanisms of crosstalk between ferroptosis and apoptosis remain obscure.
Apoptotic cell death occurs through two major pathways: the death receptor (DR) signaling pathway and the mitochondrial signaling pathway. In the extrinsic pathway (which refers to the death receptor signaling pathway), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) binds to death receptors such as death receptor-4 (DR4) and −5 (DR5) on the cell surface (5, 6). Once TRAIL has bound to death receptors, the death-inducing signaling complex (DISC) forms and leads to recruitment of caspase-8, followed by activation of executioners such as caspases-7, −6, and −3, culminating in apoptotic death (7). Unlike the extrinsic pathway, the intrinsic pathway (which refers to the mitochondrial signaling pathway) stress signal causes oligomerization of cytoplasmic Bcl-2-associated X protein (BAX)/Bcl-2 homologous antagonist killer (BAK); BAK then binds to mitochondria's outer membrane (8, 9). The BAK/BAK complex causes mitochondrial membrane permeabilization by pore formation and leads to release of cytochrome c into the cytosol. In the cytosol, released cytochrome c enables the formation of apoptosome, activating the inactive procaspase-9, which subsequently activates the executioner caspase-3 (10, 11).
In the current study, we reported that the apoptotic agent TRAIL in combination with the ferroptotic agent erastin remarkably enhanced TRAIL-induced apoptosis. The combined treatment enhanced TRAIL-induced oligomerization of BAX and disruption of mitochondrial membrane potential. These results suggest a novel mechanism by which the combined treatment of erastin and TRAIL induces promotion of apoptosis through the mitochondrial pathway.
Materials and Methods
Cell Culture
Human pancreatic adenocarcinoma BxPC3 cells, human colorectal carcinoma HCT116 cells, and human breast adenocarcinoma MCF7 cells were cultured in RPMI, McCoy's 5A, and DMEM, respectively, with 10% FBS. Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. Single knockout BAK (BAK−/−), single knockout BAX (BAX−/−), and double knockout BAX and BAK (BAX−/−BAK−/−) HCT116 cells were obtained from Dr. B. Vogelstein (Johns Hopkins University, Baltimore, MD). BID−/− HCT116 cells were obtained from Dr. X. Luo (University of Nebraska, Omaha, NE). MCF7/vector or MCF7/casp3 cell lines were provided by Dr. C. J. Froelich (Northwestern University, Chicago, IL).
Chemicals and Reagents
Production of recombinant human TRAIL was performed as previously described (Lee et al., 2001). The extracellular domain of TRAIL (amino acids 114-281) cDNA was cloned into a pET-23d plasmid (Novagen, Madison, WI). For purification of His-tagged TRAIL protein, the Qiagen express protein purification system (Qiagen, Valencia, CA) was used. ERA was purchased from Selleckchem (Houston, TX).
Cell Survival Assay
For determination of cell survival, trypan blue exclusion assay was used. In brief, cells were trypsinized and stained with trypan blue. Live cells and dead cells were counted using a hemocytometer under a light microscope.
Western Blot Analysis and Antibodies
Immunoblotting was carried out as previously described (12). Anti-PARP-1, anti-cleaved caspase-3, anti-cleaved caspase-8, anti-cleaved caspase-9, anti-BAX, anti-BAK, anti-BID, and anti-CHOP antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-actin, goat anti-rabbit IgG-horseradish peroxidase (HRP), and goat anti-mouse IgG-HRP antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Mitochondrial Fraction and Protein Crosslinking
For mitochondrial fraction, cells were lysed by Dounce homogenizer in homogenization buffer (HB) containing 10 mM EGTA, 10 mM HEPES (pH 7.4), and 0.25 M sucrose and complete protease inhibitors. The cell lysates were centrifuged for 15 min at 1000 g at 4°C to remove nuclei. The supernatants were centrifuged for 15 min at 10,000g at 4°C and the pellet was saved. The mitochondrial pellet was resuspended in HB. For protein crosslinking, 1 mM dithiobis (succinimidyl propionate) (Pierce, Rockford, IL, USA), an amine-reactive crosslinker, was added to mitochondria for 60 min at room temperature. The mitochondrial pellet was resuspended in native sample buffer without reducing agents and then analyzed using immunoblotting.
GFP-BAX and JC-1 Imaging
For GFP-BAX imaging, GFP-BAX-transfected-HCT116 cells were grown on coverslips. The cells were incubated for 20 min in 4% paraformaldehyde fixing solution at room temperature. After fixation, cells were washed with PBS and stained with DAPI (40,6-diamidino-2-phenylindole) (Cell Signaling Technology) and MitoTracker (Invitrogen) and then examined with an OlympusFluoview 1000 confocal microscope. To analyze mitochondrial membrane potential, cells were stained with JC-1 dye (5,50,6,60-tetrachloro-1,10,3,30-tetraethylbenzimidazol carbocyanineiodide) (Cayman Chemical, USA) and examined using fluorescence microscopy. Nuclei were stained with DAPI and examined with an OlympusFluoview 1000 confocal microscope.
Statistical Analysis
All values represent mean ± SD. Statistical analysis was performed using one-way analysis of variance followed by the Student t test as indicated using SigmaPlot software. P (calculated probability) values of less than 0.05 were defined as statistically significant. Significance of P value indicated as *P < 0.05; **P < 0.01; ***P < 0.001.
Results
Combined Treatment of Erastin and TRAIL-promoted Apoptosis is Mediated by Activation of Caspases
To examine whether the crosstalk between ferroptosis and apoptosis is mediated through activation of caspases, human colon cancer HCT116 cells and human pancreatic cancer BxPC3 cells were treated with erastin (ERA) in combination with TRAIL and activation of caspases was examined. Data from trypan blue exclusion assay and immunoblotting assay revealed that the combined treatment enhanced cytotoxicity and apoptosis (PARP-1 cleavage) (Figs. 1A and 1B), respectively. The enhanced apoptosis was probably due to increased activation (cleavage) of caspases (8, 9, and 3). To examine the role of caspases in the combined treatment, in particular executioner caspase-3, we treated cells with the caspase-3 inhibitor Z-DEVD-FMK prior to TRAIL treatment (Fig. 1C) or employed engineered human breast cancer MCF7 cells, which lack caspase-3 expression (Fig. 1D). Inhibition of caspase-3 completely protected cells from the combined treatment-enhanced apoptosis. Moreover, unlike caspase-3-deficient MCF7/Vector cells, caspase-3-reconstituted MCF7 cells (MCF7/Casp3) showed an increase in apoptosis during the combined treatment of ERA and TRAIL.
Figure 1. ERA enhanced TRAIL-induced apoptosis by promoting activation of caspases.
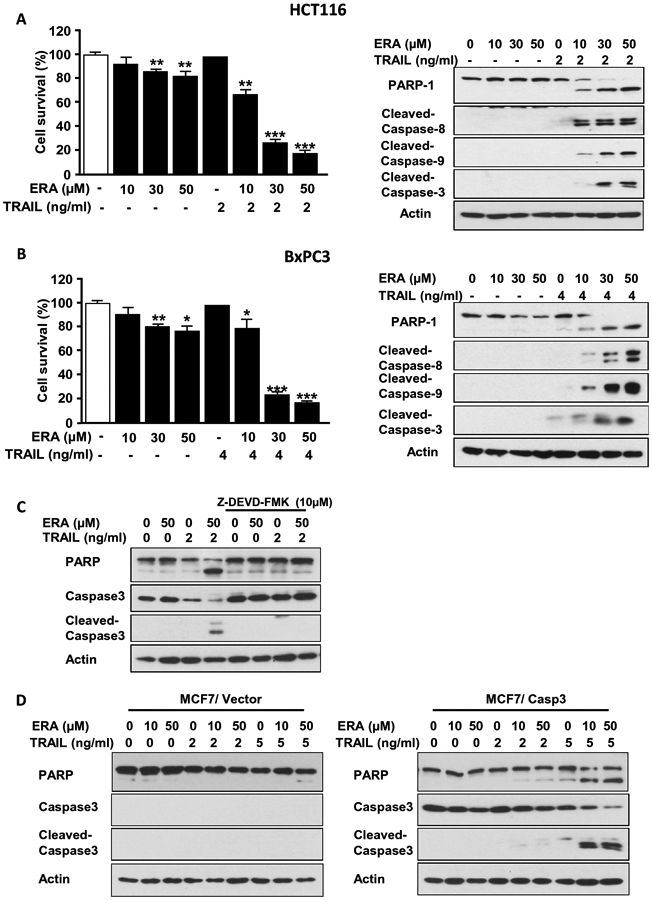
Human colorectal carcinoma HCT116 cells (A) and human pancreatic adenocarcinoma BxPC3 cells (B) were pretreated with ERA (10-50 μM) for 20 h and treated with TRAIL (2 ng or 4 ng/ml) for 4 h in the presence of ERA. Cell survival was analyzed using trypan blue exclusion assay (left panels). Western blotting was used to detect the expression of indicated proteins (right panels). (C) HCT116 cells were pretreated with ERA (50 μM) for 19 h and then pretreated with Z-DEVD-FMK (10 μM) for 1 h prior to TRAIL (2 ng/ml) treatment for 4 h in the presence of ERA and Z-DEVD-FMK. Western blotting was used to detect the expression of indicated proteins. (D) MCF7/Vector (left panel) and MCF7/Casp3 (right panel) cells were pretreated with ERA (10 or 50 μM) for 20 h and treated with TRAIL (2 or 5 ng/ml) for 4 h in the presence of ERA. Western blotting was used to detect the expression of indicated proteins. Error bars represent the mean ± SD from triplicate experiments. For statistical analysis, the Student t test (two-sided, paired) was used. P-values: ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001
The Intrinsic Mitochondrial Apoptotic Pathway Plays a Pivotal Role in Combined Treatment-enhanced Apoptosis
Since caspase-3 is activated through both the intrinsic and extrinsic pathways (13), we examined which pathway plays a pivotal role in enhancing apoptosis during the combined treatment. First, we studied the role of the intrinsic mitochondrial pathways in apoptosis (14). BAX and BAK are well known pro-apoptotic proteins. It is known that activation of BAX and BAK leads to oligomerization and subsequently pore formation on the mitochondrial membrane, causing apoptosis (14). We employed BAX or BAK single deficient HCT116 cells (BAX−/− or BAK−/−) or BAX and BAK double deficient HCT116 cells (BAX−/−/BAK−/−) and treated them with ERA in combination with TRAIL. As shown in Figure 2, BAX−/−/BAK−/− cells effectively suppressed the combined treatment-enhanced apoptosis. Data from single deficient cells demonstrate that BAX proteins were more effective than BAK proteins in the combined treatment-enhanced apoptosis.
Figure 2. The role of BAX and BAK in the combined treatment of ERA and TRAIL-induced apoptosis.
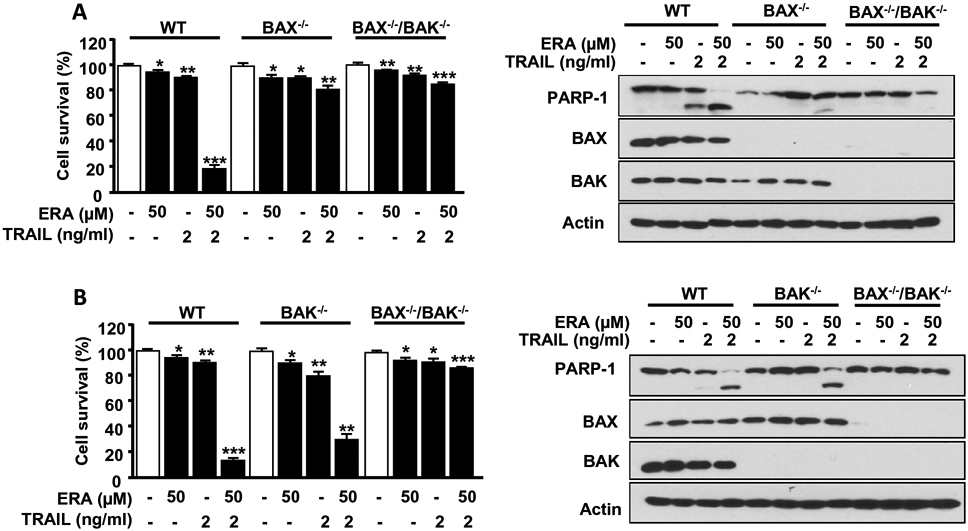
HCT116 WT, BAX−/− single knockout, or BAX−/−/BAK−/− double knockout cells (A) or HCT116 WT, BAK−/− or BAX−/−/BAK−/− cells (B) were pretreated with ERA (50 μM) for 20 h and then exposed to TRAIL (5 ng/ml) for an additional 4 h. Cell survival was analyzed using trypan blue exclusion assay (left panels). Whole-cell lysates were analyzed with immunoblotting assay using indicated antibodies (right panels). Error bars represent the mean ± SD from triplicate experiments. For statistical analysis, Student's t-test (two-sided, paired) was used. p-values: *P<0.05, **P < 0.01, ***P < 0.001.
Activation of caspases occurs under the control of BH3-only proteins as well as Bcl-2 family proteins (15). It is possible that BH3-only proteins like BID may play an important role in the activation of BAX during the combined treatment of ERA and TRAIL (15, 16). We further investigated this possibility using BID-deficient (BID−/−) HCT116 cells. Data from Figure 3A demonstrate that HCT116 BID−/− cells effectively suppressed the apoptosis enhanced by the combined treatment of ERA and TRAIL. Similar results were observed with another ferroptotic agent artesunate (ART) (Fig. 3B). Like the combined ERA and TRAIL treatment, the combined treatment of ART and TRAIL enhanced apoptosis in HCT116 wild-type (WT) cells. HCT116 BID−/− cells effectively suppressed the apoptosis enhanced by the combined treatment of ART and TRAIL.
Figure 3. The role of BID in the combined treatment of ERA/ART and TRAIL-induced apoptosis.
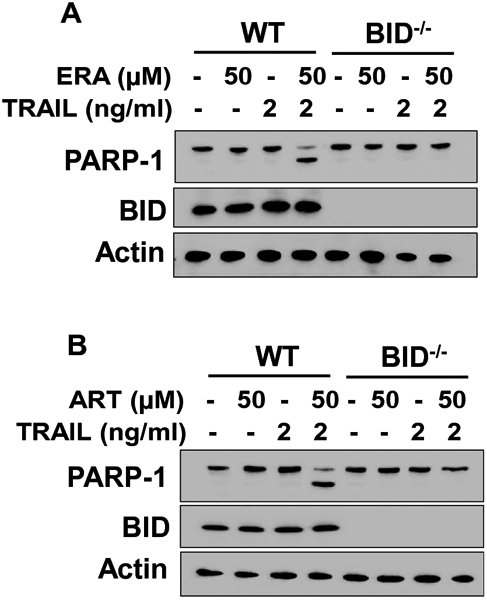
HCT116 WT or BID−/− cells were pretreated with ERA (50 μM) or artesunate (ART, 50 μM) for 20 h and then exposed to TRAIL (2 ng/ml) for an additional 4 h. Whole-cell lysates were analyzed with immunoblotting assay using indicated antibodies.
Next, we examined the role of BAX in the combined treatment-enhanced apoptosis. Figure 4A clearly demonstrates an increase in multi-oligomers of BAX during the combined treatment with ERA and TRAIL. As pro-apoptotic BAX oligomers are known to localize in mitochondria (17), we observed that BAX aggregates and trackers were co-localized during the combined treatment (Fig. 4B). Confocal microscopic analysis with JC-1-stained mitochondria confirmed that loss of mitochondrial membrane potential (ΔΨm) occurred during the combined treatment (Fig. 4C). These results imply that the combined treatment of TRAIL and ERA enhanced apoptosis through the BAX-associated mitochondria-dependent pathway.
Figure 4. BAX oligomerization and disruption of mitochondrial membrane potential during the combined treatment of ERA and TRAIL in HCT116 cells.
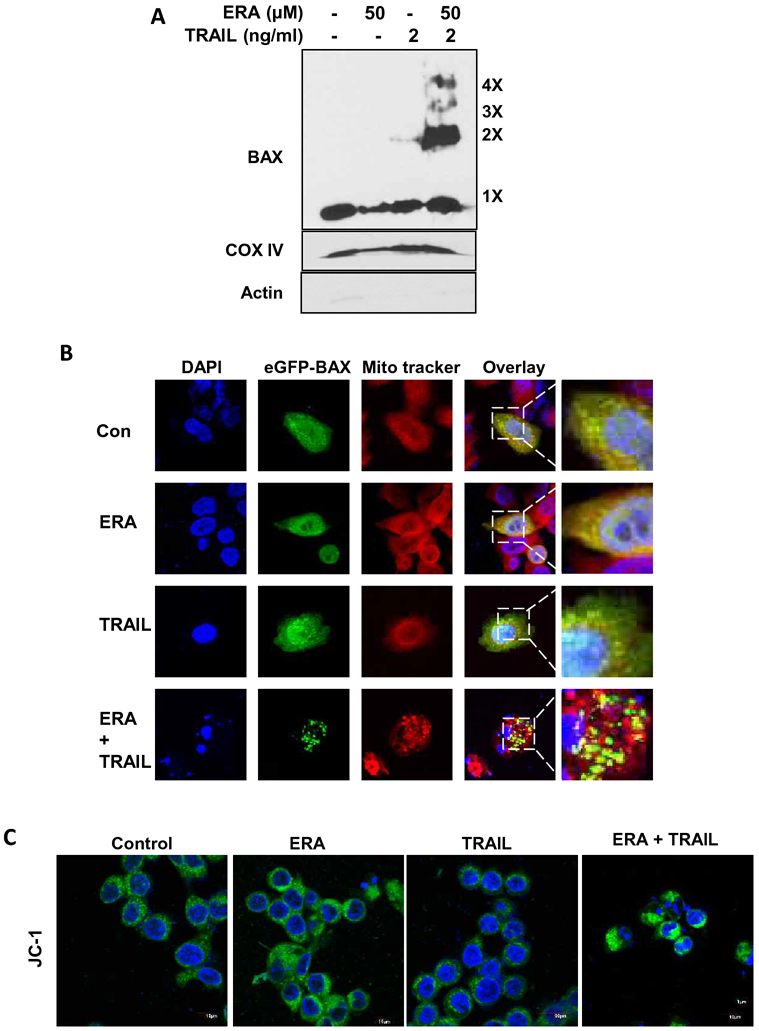
(A) Cells were pretreated with ERA (50 μM) for 20 h and treated with TRAIL (2 ng/ml) for 4 h in the presence of ERA. After treatment, mitochondrial fractions were isolated and cross-linked and then subjected to immunoblotting with an antibody to BAX. BAX monomers (1×) and multimers (2×–4×) are indicated. We used actin (cytosolic marker) and COX IV (mitochondrial marker as fractional markers. (B) Cells were transfected with GFP-BAX plasmids. The cells were pretreated with ERA (50 μM) for 20 h and treated with TRAIL (2 ng/ml) for an additional 4 h. After treatment, the cells were stained with MitoTracker Red. Co-localization of GFP-BAX puncta and MitoTracker Red was examined using a confocal microscope. (C) Cells were pretreated with ERA (50 μM) for 20 h and treated with TRAIL (2 ng/ml) for 4 h in the presence of ERA. After treatment, the cells were stained with JC-1 probe and detected using a confocal microscope. Green fluorescence represents the monomeric form of JC-1, indicating low mitochondrial membrane potential (ΔΨm).
Discussion
We can draw several conclusions based on the data presented in this study. The combined treatment of ERA and TRAIL enhanced apoptosis by promoting the activation of caspases, in particular, caspase-3. One way to activate caspase-3 is through the BAX-associated mitochondria-dependent intrinsic pathway.
We previously reported that ferroptotic agents such as ERA and ART induce ER stress (4). ER stress may activate BID through caspase-2; subsequently, truncated BID then translocates to the mitochondria and then activates BAX through oligomerization (16, 18). It is also well-known that the PERK/eIF2α/ATF4 pathway mediated by the ER stress response regulates expression of several target genes, i.e., CHOP (C/EBP homologous protein) (19). Previous studies show that ER stress triggers the association of CHOP with the promoter of PUMA (p53 upregulated modulator of apoptosis), a pro-apoptotic molecule, and consequently induces PUMA expression (20). PUMA is well known to interact with antiapoptotic Bcl-2 family members such as Mcl-1 (myeloid cell leukemia-1), Bcl-2 (B-cell lymphoma 2), and Bcl-xL (B-cell lymphoma-extra large), and subsequently activates multidomain proapoptotic proteins BAX and/or BAK via oligomerization. These oligomerized molecules bind to the mitochondrial membrane, which triggers mitochondrial dysfunction, cytochrome c release, and subsequently, caspase activation. It is possible that the ferroptotic agent ERA upregulates PUMA as well as anti-apoptotic Bcl-2 family proteins such as Mcl-1, which results in maintenance of the balance between them. However, the combined treatment of ERA and TRAIL triggers an imbalance between PUMA and Bcl-2 family proteins and consequently promotes apoptosis. This possibility needs to be further studied.
Acknowledgements
We thank Christine Burr (Department of Surgery, University of Pittsburgh) for her critical review of the manuscript.
Grant sponsor: NCI R03CA205267, R03CA212125, and P30CA047904
Abbreviations used in this paper:
- ATF4
activating transcription factor 4
- BAK
Bcl-2 homologous antagonist killer
- BAX
Bcl-2–associated X protein
- Bcl-2
B-cell lymphoma 2
- Bcl-xL
B-cell lymphoma-extra large
- BH3
Bcl-2 homology
- BID
BH3 interacting-domain death agonist
- C/EBP
CCAAT-enhancer-binding proteins
- CHOP
CCAAT-enhancer-binding protein homologous protein
- DAPI
40,6-diamidino-2-phenylindole
- DISC
death-inducing signaling complex
- DR
death receptor
- DR4
death receptor 4
- DR5
death receptor 5
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- eIF2α
eukaryotic initiation factor 2α
- ER
endoplasmic reticulum
- ERA
erastin
- GFP
green fluorescent protein
- HB
homogenization buffer
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HRP
horseradish peroxidase
- JC-1
5,50,6,60-tetrachloro-1,10,3,30-tetraethylbenzimidazol carbocyanineiodid
- PARP-1
poly [ADP-ribose] polymerase 1
- PERK
protein kinase RNA-like endoplasmic reticulum kinase
- PUMA
p53 upregulated modulator of apoptosis
- SD
standard deviation
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- WT
wild-type
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest disclosure: The authors declare no competing financial interests.
References
- 1.Dixon SJ, Lemberg KM, Lamprecht MR, et al. (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magtanong L, Ko PJ, Dixon SJ. (2016) Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ 23:1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113:E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong SH, Lee DH, Lee YS, et al. (2017) Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget 8:115164–115178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillissen B, Richter A, Richter A, et al. (2017) Bax/Bak-independent mitochondrial depolarization and reactive oxygen species induction by sorafenib overcome resistance to apoptosis in renal cell carcinoma. J Biol Chem 292:6478–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li P, Nijhawan D, Budihardjo I, et al. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479–489. [DOI] [PubMed] [Google Scholar]
- 8.Kalkavan H, Green DR. (2018) MOMP, cell suicide as a BCL-2 family business. Cell Death Differ 25:46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganten TM, Haas TL, Sykora J, et al. (2004) Enhanced caspase-8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic drugs. Cell Death Differ 11 Suppl 1:S86–96. [DOI] [PubMed] [Google Scholar]
- 10.Yuan S, Akey CW. (2013) Apoptosome structure, assembly, and procaspase activation. Structure 21:501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baliga B, Kumar S. (2003) Apaf-1/cytochrome c apoptosome: an essential initiator of caspase activation or just a sideshow? Cell Death Differ 10:16–18. [DOI] [PubMed] [Google Scholar]
- 12.Lee YS, Lee DH, Jeong SY, et al. (2019) Ferroptosis-inducing agents enhance TRAIL-induced apoptosis through upregulation of death receptor 5. J Cell Biochem 120:928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krautwald S, Ziegler E, Rolver L, et al. (2010) Effective blockage of both the extrinsic and intrinsic pathways of apoptosis in mice by TAT-crmA. J Biol Chem 285:19997–20005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C, Youle RJ. (2009) The role of mitochondria in apoptosis*. Annu Rev Genet 43:95–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shamas-Din A, Kale J, Leber B, Andrews DW. (2013) Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol 5:a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei MC, Lindsten T, Mootha VK, et al. (2000) tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 17.Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. (1997) Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 139:1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elkholi R, Floros KV, Chipuk JE. (2011) The Role of BH3-Only Proteins in Tumor Cell Development, Signaling, and Treatment. Genes Cancer 2:523–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su N, Kilberg MS. (2008) C/EBP homology protein (CHOP) interacts with activating transcription factor 4 (ATF4) and negatively regulates the stress-dependent induction of the asparagine synthetase gene. J Biol Chem 283:35106–35117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh AP, Klocke BJ, Ballestas ME, Roth KA. (2012) CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS One 7:e39586. [DOI] [PMC free article] [PubMed] [Google Scholar]