

Abstract
Background:
Approximately 4% of non-small cell lung cancers (NSCLC) harbor BRAF mutations, and ~50% of these are non-V600 mutations. Treatment of tumors harboring non-V600 mutations is challenging because of functional heterogeneity and lack of knowledge regarding their clinical significance and response to targeted agents.
Methods:
We conducted an integrative analysis of BRAF non-V600 mutations using genomic profiles of BRAF-mutant NSCLC from the Guardant360 database. BRAF mutations were categorized by clonality and class (1 and 2: RAS-independent; 3: RAS-dependent). Cell viability assays were performed in Ba/F3 models. Drug screens were performed in NSCLC cell lines.
Results:
305 unique BRAF mutations were identified. Missense mutations were most common (276, 90%), and 45% were variants of unknown significance (VUS). F468S and N581Y were identified as novel activating mutations. Class 1-3 mutations demonstrated higher clonality compared to mutations of unknown class (p<0.01). Three patients were treated with MEK +/− BRAF inhibitors. Patients harboring G469V and D594G mutations did not respond, while a patient with L597R mutation had durable response. Trametinib+/−dabrafenib, LXH254, and lifirafenib showed more potent inhibition of BRAF non-V600 mutant NSCLC cell lines compared to other MEK, BRAF, and ERK inhibitors, and comparable to inhibition of BRAF V600E cell line.
Conclusions:
In BRAF-mutant NSCLC, clonality is higher in known functional mutations and may allow identification of VUS more likely to be oncogenic drivers. Our data indicate certain non-V600 mutations are responsive to MEK and BRAF inhibitors. This integration of genomic profiling and drug sensitivity may guide treatment for BRAF-mutant NSCLC.
Keywords: Non-small cell lung cancer, BRAF, targeted therapy, cell-free DNA
Introduction
Characterization of the landscape of somatic mutations in cancer has led to identification of novel targetable mutations in several malignancies, particularly in non-small cell lung cancer (NSCLC)1-6. Among these oncogenic mutations are somatic alterations in the BRAF gene, a serine/ threonine protein kinase, which are present in ~7% of solid tumors and ~4% of NSCLC7,8. These mutations can increase activation of the RAF-MEK-ERK (MAP kinase, MAPK) pathway to drive cell proliferation and growth9. V600X mutations are the most common BRAF mutation and occur in ~50% of cutaneous melanomas10, and in ~2% of NSCLCs11-14 The Food and Drug Administration has approved the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) in NSCLC harboring a BRAF V600E mutation due to its efficacy in first and second-lines15,16. However, this success is tempered by the fact that ~50% of BRAF mutations in NSCLC are non-V600, and the responsiveness of these different mutations to BRAF +/− MEK inhibitors is not well characterized13,17.
The challenge of targeting BRAF mutations lies in the heterogeneous mutational repertoire of the BRAF gene. BRAF mutations are typically classified into three classes: class 1 mutations signal as RAS-independent active monomers (e.g. V600E); class 2 mutations are constitutively active RAS-independent dimers; and class 3 mutations have low/absent kinase activity and require additional upstream signaling through RAS or receptor tyrosine kinase mutations18-20. Combination therapy of a BRAF inhibitor with a MEK inhibitor is currently used to effectively block the MAPK pathway in tumors with class 1 mutations. Dual therapy may have activity in non-V600 BRAF mutations as well, as demonstrated by recent reports in melanoma21. In addition, novel MAPK pathway inhibitors may be effective in targeting class 2 and 3 mutations22-25.
Though significant progress has been made for treatment of BRAF V600E mutation positive cancers, two important challenges remain: first, BRAF non-V600 mutations have heterogeneous functionality7,19,26; second, many BRAF mutations remain variants of unknown significance (VUS). Therefore, an assessment of the genomic landscape of BRAF is needed to guide treatment decisions and trial development.
Cell-free DNA (cfDNA) is a blood-based method of assessing tumor DNA mutational status using next generation sequencing (NGS). Because sample acquisition is less invasive compared to tumor tissue, cfDNA can be a powerful tool to perform large scale molecular profiling across different tumor types. Cell-free DNA analysis compares favorably to tissue profiling for identification of targetable alterations and for assessing tumor genetic heterogeneity in patients with advanced-stage malignancies27,28.
In this report, we characterize the landscape of somatic mutations for a large cohort of patients harboring BRAF-mutant advanced-stage NSCLC using a clinical cfDNA assay. We also characterize BRAF non-V600 mutations through cell viability and pharmacologic assays to determine potential treatment strategies.
Methods
Genomic data collection
The Guardant Health Clinical Laboratory database was queried for cfDNA tests from patients with a diagnosis of NSCLC in which a BRAF mutation was identified. Synonymous mutations were excluded. This research is approved by Quorum Institutional Review Board for the generation of de-identified datasets for research purposes.
Cell-free DNA analysis
Samples were shipped to a Clinical Laboratory Improvement Act (CLIA)-certified, College of American Pathologists (CAP)–accredited laboratory (Guardant Health, Redwood City, California USA). Cell-free DNA was extracted from whole blood collected in 10-mL Streck cfDNA BCT®. After double centrifugation, 5–30 ng of cfDNA was isolated for digital targeted panel sequencing of up to 73 genes (Supplemental Figure S1A-D), as previously described (Guardant360®)29. Non-synonymous mutations and focal copy number alterations were further processed with R statistical computing program (version 3.3). All non-synonymous BRAF mutations and focal amplification were included in the analysis. Mutations previously reported as associated with clonal hematopoiesis were excluded30.
Clonality Analysis
BRAF mutations were defined as clonal or subclonal as previously reported5. In brief, we calculated variant allele frequency (VAF) for the BRAF mutation and for the variant with highest allele frequency in that sample. VAF underwent copy number (CN) normalization using VAF/log2(CN) calculation. The estimated mutation clonality was calculated by dividing each BRAF mutation adjusted VAF by the adjusted maximum somatic VAF present in the given sample. Mutations with clonality > 0.9 were considered clonal, and subclonal if < 0.1.
Cell culture and medium
LentiX-293T cells (Clontech) were cultured in DMEM (with high glucose, glutamine and sodium pyruvate) with 5% FBS and 1× non-essential amino acid. Growth medium for Ba/F3 cells was RPMI with 5% FBS and 1 ng/ml mouse IL-3. Assay medium was Advanced RPMI with 5% FBS and 1× GlutaMAX.
Transforming potential assay in Ba/F3 cells
BRAF wild-type and mutant constructs were made with pHAGE-PURO vector by HiTMMoB technique as previously described31. BRAF wild-type and mutant vectors were transduced and expressed in Ba/F3 cells by lentivirus approach as described previously32. Briefly, lentivirus was generated by transfecting the LentiX-293T cells with reagent. Ba/F3 cells were transduced by spinoculation (1,000× g for 3 hours) in the presence of 8 μg/ml polybrene and re-suspended in assay medium after spinning. Cell viability of transduced Ba/F3 cells was measured by Cell Titer-Glo assay (Promega) after 1 week.
Cell viability for each of the mutant Ba/F3 cell lines was scaled for comparison with BRAF wild type (scale of 1), and known activating mutations BRAF V600E and K601N (both scale of 100). Mutations were called activating if they fulfilled two criteria: 1) mean growth of mutant cell line is higher than the mean + 3 standard deviations (SD) of the universal negative controls (i.e. mCherry/GFP/Luc); 2) mean growth of mutant cell line is higher than the mean + 3 SD of BRAF wild type, and 10-fold higher than that of BRAF wild type. Mutations that did not fulfill these criteria were called not activating.
Drug Screen Assay
Human NSCLC cell lines were authenticated via DNA fingerprinting, routinely tested for the presence of Mycoplasma species, and maintained as described previously33. NSCLC cell lines were incubated with DMSO (vehicle control), BRAF inhibitors (dabrafenib, encorafenib, lifirafenib, LXH254, TAK580, vemurafenib), MEK inhibitors (binimetinib, cobimetinib, trametinib), BRAF + MEK inhibitor combinations (dabrafenib+trametinib, encorafenib+binimetinib, vemurafenib+cobimetinib), or ERK inhibitors (LY3214996, MK-8353, ravoxertinib, ulixertinib) for 72 hours at seven distinct concentrations. Concentrations for MEK inhibitors ranged from 0.064 nM to 1 uM by 5-fold dilution, and concentration for BRAF and ERK inhibitors ranged from 0.64 nM to 10 uM by 5-fold dilution. A CellTiter-Glo Luminescent Cell Viability Assay (Promega, San Luis Obispo, CA, USA) was performed as per the manufacturer's specifications. For both assays, three replicates were tested at each concentration, and each experiment was completed three separate times. We compared the efficacy of MEK and BRAF inhibitors for BRAF mutant cell lines where MEK+/−BRAF inhibitors showed activity (H2087, H1755, HCC364) with BRAF wild type (H1648) to calculate relative IC50s (average IC50 for BRAF mutant cell lines divided by IC50 for BRAF wild type) in order to estimate the potency of BRAF mutant cell line inhibition compared to wild type.
Clinical outcomes
Patients with NSCLC harboring a BRAF non-V600 mutation treated with a MEK +/− BRAF inhibitor were analyzed for clinical outcomes. Molecular profiling was performed in CLIA-certified laboratories and included different tissue-based assays: FoundationOne – Foundation Medicine Inc.; Molecular Diagnostics Laboratory 50-gene panel – MD Anderson Cancer Center; and single gene testing using PCR and Ion Torrent sequencer. Details on treatments, outcomes, and molecular profiling results were obtained from the local physicians as per local Institutional Review Board guidelines. The study was conducted in accordance with the Declaration of Helsinki and U.S. Common Rule.
Statistical analysis
Categorical variables were compared across groups through Chi-Square test or Fisher’s Exact-Test where applicable. Significance was determined as two-sided p-value < 0.05. Drug effects were estimated using the best-fit dose–response model. Statistical analyses were performed in GraphPad Prism version 8.1 (La Jolla, California, USA).
Results
Genomic cohort characteristics
From July/2014-June/2017, 1589 samples from 1515 unique patients were identified to have BRAF alterations according to results from the Guardant360 panel (Supplemental Figure S1A-D). The median number of samples collected per unique patient was 1 (range: 1-6). A total of 857 non-synonymous BRAF mutations were identified, with 305 unique mutations identified overall. Missense mutations accounted for the majority of mutations (276, 90%), while nonsense and splice-site mutations accounted for 5% each (n=14 and 15, respectively). These results are summarized in the CONSORT diagram in Figure 1.
Figure 1.
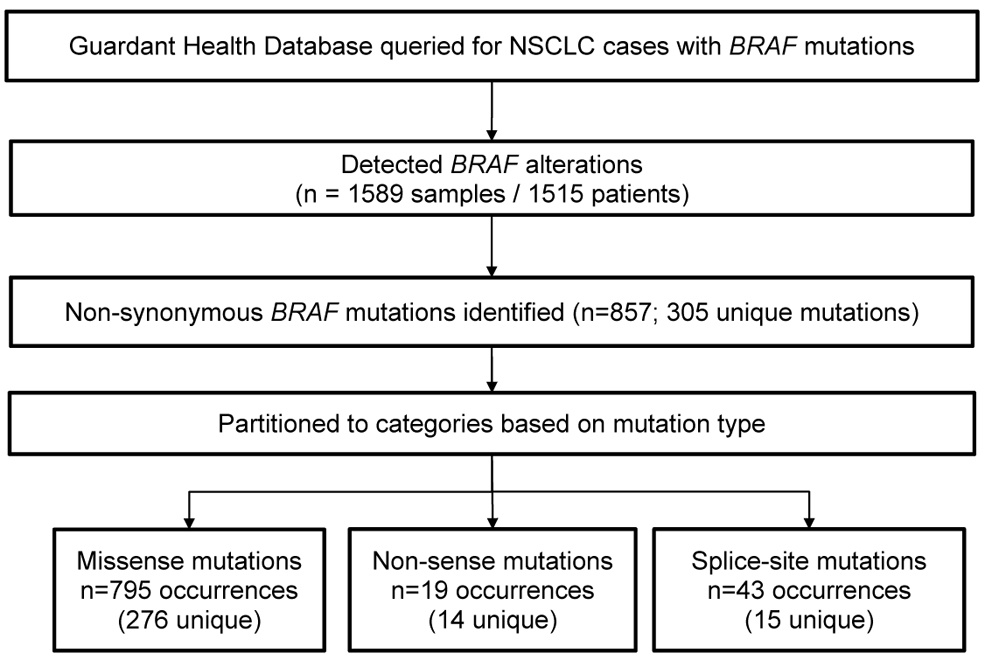
CONSORT diagram of patients in the Guardant Health Database harboring a BRAF mutation.
We then filtered the 276 unique missense mutations for those observed in this cohort at least four times. We observed that ~11% of unique mutations (31/276), which were present in 66% of the samples (526/795), accounted for the majority of BRAF missense mutations. Class 1 mutations corresponded exclusively to the V600E mutation, which was detected in 167 patients. Additionally, seven unique class 2 and nine unique class 3 mutations were detected in our cohort, in 136 and 116 samples respectively. BRAF G469A and K601E were the most common class 2 mutations, and G466V and N581S were the most common class 3 mutations. The total number of unique BRAF missense mutations of unknown class was 14, and the total number of occurrences was 107. These results are summarized in Table 1.
Table 1.
Clonality of BRAF missense mutations.
| BRAF Missense Mutations | ||||
|---|---|---|---|---|
| Class 1 | Class 2 | Class 3 | Unknown | |
| Unique mutationsa - N (%) | 1 (3) | 7 (23) | 9 (29) | 14 (45) |
| Occurrences – N (%) | ||||
| Total | 167 (100) | 136 (100) | 116 (100) | 107 (100) |
| Defined clonality | 106 (63) | 90 (66) | 64 (55) | 68 (64) |
| Undefined clonality | 61 (37) | 46 (34) | 52 (45) | 39 (36) |
| Clonality – N (%) | ||||
| Clonal | 93 (88) | 74 (82) | 52 (81) | 41 (60) |
| Subclonal | 13 (12) | 16 (18) | 12 (19) | 27 (40) |
with ≥4 occurrences.
We also observed BRAF focal amplification in 161 unique samples. In 150 samples (93.2%), BRAF focal amplification was the only BRAF alteration, while in 11 samples (6.8%) focal amplification co-occurred with BRAF missense mutations.
Cell line models identify novel activating BRAF mutations
Among the most frequent unique missense mutations, ~45% were VUS (Table 1 and Figure 2A – Class row - gray). Using Ba/F3 cell line models, we sought to test the functionality of the BRAF mutations identified in the Guardant Health database with cell viability assays.
Figure 2.

A) Clonality, class and functionality of BRAF mutations; B) Clonality by mutation class; C) Clonality by mutation functionality.
All class 1 and 2 mutations, with the exception of G469R, which is known to have intermediate kinase activity19, were found to be activating in the cell viability assays (Figure 2A – Class row – red and green represent classes 1 and 2, respectively). However, class 3 mutations showed heterogeneity, with only 4/9 (36%) mutations found to be activating (Figure 2A – Class row – blue represents class 3).
We also performed cell viability assays to test the functionality of the 14 unique VUS detected in the Guardant Health database with a frequency greater than or equal to 4. Among these, F468S and N581Y mutations were found to be activating (Figure 2A – Functionality row – orange represents activating mutations).
Clonality is associated with functionality of BRAF mutations in NSCLC
Next, we examined the genomic landscape of the BRAF mutations by characterizing their clonal and subclonal distribution. Class 1 (Figure 2A – Class row – red), class 2 (Figure 2A – Class row – green), and class 3 (Figure 2A Class row – blue) mutations were all associated with higher clonality compared to mutations of unknown class (Figure 2A – Class row – grey) (class 1 88% vs class 2 82% vs class 3 81% vs unknown class 60%, respectively, p<0.01) (Figure 2B). Classes 1, 2, and 3 had similar clonality, and there were no significant differences in clonality rates between these classes (1 vs 2: p=0.31; 1 vs 3: p=0.27; 2 vs 3: p=0.99) (Table 1 and Figure 2B). We also observed that mutations found to be activating in the cell viability assays (Figure 2A – Functionality row – orange) had higher clonality compared to mutations that were not activating (84% vs 71%, p<0.01) (Figure 2C). The clonal and subclonal relationship for each of the most common BRAF mutations (≥4 occurrences) is summarized in Supplemental Figure S2A and S2B.
TP53, EGFR, KRAS, and NF1 are the most commonly co-mutated genes in BRAF-mutant NSCLC
We next evaluated which genes are more frequently co-mutated in BRAF-mutant NSCLC. The four most commonly co-mutated genes were TP53 (57%), EGFR (26%), KRAS (15%) and NF1 (15%). Other commonly co-occurring cancer gene mutations included ARID1A (14%) and APC (11%) (Figure 3 – cBioPortal Oncoprinter34,35). BRAF Class 1, 2, and 3 mutations co-occurred with EGFR activating mutations exon 21 L858R and exon 19 deletion in 10% of samples (43/419) (Supplemental Table). In two samples, the BRAF mutation was observed at a VAF that was clonal. In all other samples, either the EGFR mutation was clonal (N=29), or the two variants were observed at similar VAF (N=12). In an exploratory analysis, we tested the association between KRAS and BRAF mutations, and found that class 3 BRAF mutations are more likely to have KRAS co-mutations compared to class 1 and 2 mutations (1: 6.0%; 2: 12.6%; 3: 23.5%, p<0.01). This finding is consistent with recently published work showing a requirement for upstream RAS signaling for optimal oncogenic output of class 3 BRAF mutations23. In addition, we observed BRAF class 2 mutations were more commonly associated with NF1 mutations when compared to BRAF class 1 mutations (1: 4.2%; 2: 11.8%; 3: 7.8%, p=0.05).
Figure 3.

Oncoprint of the most commonly co-mutated genes in BRAF-mutant and BRAF focally amplified lung cancers.
In addition, we evaluated the frequency of co-mutations in tumors with BRAF focal amplification. The frequency of mutations in canonical EGFR, KRAS and NF1 in samples with BRAF focal amplification were respectively 24.2% (39/161), 20.5% (33/161), and 13.0% (21/161) (Figure 3).
Advanced-stage NSCLC harboring BRAF non-V600 mutations show distinct patterns of sensitivity to MEK inhibition
To illustrate the importance of classifying BRAF mutations, we evaluated three patients with advanced-stage NSCLC harboring BRAF non-V600 mutations that received treatment with MEK +/− BRAF inhibitors.
Patient 1 is a 48-year-old male, never smoker diagnosed with BRAF G469V (class 2 mutation) stage IV lung adenocarcinoma. In addition to the BRAF mutation, the tumor harbored APC R1040fs*16 and CHD2 L1383* mutations, and NFKBIA and NKX2-1 amplifications as determined by tissue-based NGS platform (Foundation Medicine). The patient initially received radiation to a left frontal lobe lesion and to a left lung lesion followed by four cycles of carboplatin with pemetrexed. He was subsequently started on dabrafenib 150mg orally twice daily and trametinib 2mg orally daily (Figure 4A). After nine weeks of treatment, the patient had both systemic and intracranial disease progression (Figure 4B). He received palliative radiation to bone lesions and stereotactic radiation to the brain metastasis, and went on to receive two additional lines of cytotoxic chemotherapy (re-exposure to carboplatin and pemetrexed, and docetaxel with ramucirumab). The patient had further disease progression and clinical deterioration, and was transitioned to hospice.
Figure 4.

Outcomes of three patients with BRAF non-V600E mutant NSCLC treated with MEK +/− BRAF inhibitors. A) Patient 1: G469V mutation pre-treatment with dabrafenib + trametinib; B) Patient 1: G469V mutation post-treatment with dabrafenib + trametinib (PET-CT after 5 weeks and MRI after 9 weeks of treatment); C) Patient 2: L597R mutation pre-treatment with dabrafenib + trametinib; D) Patient 2: L597R mutation 3 months post-treatment with dabrafenib + trametinib; E) Patient 3: D594G mutation pre-treatment with trametinib; F) Patient 3: D594G mutation 4 months post-treatment with trametinib.
Patient 2 is a 63-year-old woman with a 40 pack-year smoking history diagnosed with stage IV lung adenocarcinoma whose tumor harbored a BRAF L597R mutation (class 2 mutation). Targeted mutational profiling (Ion Torrent sequencing) found no additional alterations. The patient was initiated on treatment with palliative radiation to a left hilar lesion followed by two cycles of carboplatin and pemetrexed, with a mixed tumor response (Figure 4C). Treatment was then switched to dabrafenib 150mg orally twice daily and trametinib 2mg orally daily. The patient had significant clinical benefit, and a CT scan performed at 13 weeks demonstrated resolution of mediastinal lymphadenopathy and left hepatic metastasis, all consistent with tumor response to treatment (Figure 4D). At 12 months of follow-up, the patient remains on treatment with dabrafenib and trametinib without disease progression.
Patient 3 is a 60-year-old male, former smoker (40 pack-year history), with stage IV lung adenocarcinoma harboring a BRAF D594G mutation (class 3 mutation). Tissue-based targeted panel sequencing (MD Anderson MDL 50-gene panel) found a co-occurring TP53 H193L mutation. The patient received two lines of cytotoxic chemotherapy (carboplatin plus pemetrexed and bevacizumab; carboplatin plus gemcitabine) and one line of immunotherapy (PD-1 inhibitor nivolumab) with disease progression (Figure 4E). He was subsequently started on single-agent trametinib. On his first restaging scan at two months, the patient was found to have stable disease and continued on treatment. At four months, the patient was admitted with hemoptysis and was diagnosed with disease progression in the mediastinal lymph nodes (Figure 4F). He underwent argon plasma coagulation and palliative radiation to the chest with resolution of hemoptysis. The patient developed progressive worsening of performance status at which point hospice was recommended. The patient passed away two months later.
BRAF non-V600 mutant cell lines are sensitive to MEK and BRAF inhibition
Because of the distinct patterns of response observed in the three patients with advanced-stage NSCLC treated with MEK +/− BRAF inhibition, we tested the sensitivity of five patient-derived lung adenocarcinoma cell lines harboring endogenous BRAF mutations to BRAF, MEK and ERK inhibitors: HCC364 (V600E mutation) and H1648 (BRAF wild-type), which served respectively as positive and negative controls; H2087 (L597V mutation – the same codon mutated as Patient 2), H1395 and H1755 (G469A mutation - the same codon mutated as Patient 1).
Pharmacologic screens in H2087 (L597V) and H1755 (G469A) cell lines showed that trametinib +/− dabrafenib (MEK inhibitor IC50: 2.5-15.2nM), lifirafenib (BRAF inhibitor IC50: 3.5-25.7nM) and LXH254 (BRAF inhibitor IC50: 2.0-17.2nM) were the most effective agents. The combinations of binimetinib +/− encorafenib and cobimetinib +/− vemurafenib inhibited cell growth albeit at higher drug concentrations in the H2087 (L597V) (MEK inhibitor IC50: 87.8-554.4nM) and H1755 (G469A) (MEK inhibitor IC50: 38.8-465.4nM) cell lines. H2087 (L597V) and H1755 (G469A) cell lines were resistant to single-agent dabrafenib, encorafenib, vemurafenib, and TAK580 (BRAF inhibitor IC50: >1μM) (Figure 5A-5B). The H1395 cell line (G469A) was resistant to all tested MEK +/− BRAF inhibitors (IC50 >1μM) (Figure 5C).
Figure 5.




Drug screen for MEK and BRAF inhibitors. A) H2087 (BRAF L597V mutation); B) H1755 (BRAF G469A mutation); C) H1395 (BRAF G469A mutation); D) HCC364 (BRAF V600E mutation); E) H1648 (BRAF wild-type); F) Relative IC50 of MEK and BRAF inhibitors for comparison between BRAF mutant and BRAF wild type cell lines.
The HCC364 (V600E) cell line was the most sensitive to MEK and BRAF inhibition. All regimens showed significant activity in this cell line (IC50: <1.0-27.0nM) with the exception of binimetinib (MEK inhibitor IC50: 115.6nM) and vemurafenib (BRAF inhibitor IC50: 423.2nM) (Figure 5D). As expected, MEK and BRAF inhibitors were ineffective in the H1648 cell line (BRAF wild-type), with lifirafenib, LXH254, and TAK580 showing the lowest IC50 (BRAF inhibitor IC50: 556.4-839.6nM) (Figure 5E). Of note, IC50s for HCC364 (BRAF V600E) cell line were similar to those of H2087 (BRAF L597V) and H1755 (BRAF G469A) cell lines for trametinib +/− dabrafenib, LXH254 and lifirafenib (Figures 5A, 5B, 5D).
All cell lines tested were found to be resistant to single-agent ERK inhibitors (Supplemental Figure S3A-E).
Next, we compared the IC50s of MEK and BRAF inhibitors for BRAF-mutant cell lines where these compounds showed activity and BRAF wild type cell line by calculating relative IC50 ratios. We observed that trametinib, trametinib+dabrafenib and LXH254 showed the lowest IC50 ratios (relative IC50 < 0.01), while lifirafenib, binimetinib+encorafenib, cobimetinib+vemurafenib, and cobimetinib showed intermediate IC50 ratios (relative IC50 0.1-0.01). Encorafenib, binimetinib, dabrafenib, vemurafenib, and TAK580 had the highest IC50 ratios (>0.1) (Figure 5F).
Discussion
The development of treatment strategies for non-V600 BRAF-mutant NSCLC has been hindered by the diversity and functional heterogeneity of these mutations as well as an incomplete understanding of which mutations are oncogenic drivers and targetable with available therapies. To address these issues we conducted an integrative analysis of the molecular landscape and function of BRAF mutant NSCLC.
We initially characterized the largest cohort described to date of BRAF mutations as well as additional co-occurring genetic alterations detected in the cfDNA from 1515 patients with NSCLC. We confirmed that TP53, EGFR, KRAS, and NF1 are among the most commonly co-mutated genes in BRAF mutant and BRAF focally amplified tumors, consistent with prior reports36. In addition, we found that KRAS mutations are enriched in cases showing class 3 BRAF mutations, which is also consistent with previous reports13,37 and pre-clinical data highlighting that these kinase impaired alterations require additional RAS activation (eg. KRAS mutations) or receptor tyrosine kinase activation (eg. EGFR mutations) to promote cell growth and survival19,20. We also found that there was a significant association between NF1 mutations and class 2 BRAF mutations compared to class 1. Finally, we found no significant difference amongst the BRAF mutation class and EGFR mutations. One limitation of this analysis is that the genomic dataset does not include detailed clinical history (e.g. prior treatment history). Therefore, clinical interpretation of co-occurring mutation status and clonality, particularly for co-occurring EGFR mutations, is limited.
Additionally, we found that mutational functionality is correlated with mutational clonality. Specifically, we demonstrated that class 1, 2, and 3 mutations were more often clonal compared to VUS in BRAF. This is relevant when diverse mutations are present in a given oncogene such as the cfDNA cohort in question, where ~45% of tumors harbored BRAF VUS. This novel correlation between clonality and functionality should be further investigated and validated in other tumor types and for other oncogenes, and may prove to be a broadly useful approach for identifying candidate mutations that are more likely to be true oncogenic drivers instead of passenger mutations. We leveraged Ba/F3 cell line models to evaluate the functionality of these mutations and characterized two novel VUS to be activating: F468S and N581Y. Despite these mutations having been described previously in other cancer types, such as colorectal, gastric, prostate, and oral cavity38-44, in vitro functional data were lacking. We recognize the limitations of the Ba/F3 cell model, which are two-fold. First, for activating mutations, IL-3 independence of Ba/F3 cells does not allow us to characterize their BRAF mutational class (1, 2, or 3). Second, BRAF mutations found to be not activating may still promote MAPK pathway signaling as class 3 kinase mutations, but lack the capacity to do so without a co-existing alteration.
Next, we analyzed the efficacy of MEK and BRAF inhibitors in clinical cases of NSCLC. We show that BRAF non-V600 mutations have heterogeneous patterns of sensitivity to these agents. Clinically, the patient with a tumor harboring BRAF class 2 L597R mutation was sensitive to trametinib and dabrafenib with tumor shrinkage and durable response at one year. To our knowledge, this is the first report of response to MEK and BRAF inhibition in a NSCLC patient bearing this mutation although activity was observed for L597X-mutant tumors in a melanoma patient treated with this combination and for a NSCLC patient treated with an ERK inhibitor 21,25. Conversely, the G469V mutation was resistant to trametinib and dabrafenib, and the patient developed rapid disease progression in two months. It is possible that the more complex genomic profile of the tumor harboring the G469V BRAF mutation may have contributed to this poor outcome (APC R1040fs*16, CHD2 L1383*, NFKBIA amplification, NKX2-1 amplification). Increased molecular complexity resulting in shorter benefit from tyrosine kinase inhibitors has been previously reported in other oncogene driven lung cancers, such as those harboring EGFR mutations1,45.
To better understand these differences in clinical outcome, we leveraged lung cancer cell lines to test the efficacy of BRAF, MEK, and ERK inhibitors. Trametinib, with or without dabrafenib, lifirafenib, and LXH254 were effective in inhibiting cell growth in the H2087 (L597V) and H1755 (G469A) cell lines. The cobimetinib + vemurafenib and binimetinib + encorafenib combinations had higher IC50s and were less effective for targeting these mutations. In addition, we found that single-agent binimetinib had limited activity across all BRAF cell lines, which is consistent with recent work in cell lines harboring class 2 BRAF mutations from histologies other than NSCLC showing that binimetinib had the least activity among tested MEK inhibitors26. Among the BRAF inhibitors, TAK580, dabrafenib, encorafenib, and vemurafenib all demonstrated similar single agent activity in the non-V600 setting, while lifirafenib and LXH254 demonstrated lower IC50s for H2087 (L597V) and H1755 (G469A). When comparing efficacy among BRAF mutant and BRAF wild type cell lines, we found that trametinib, trametinib+dabrafenib and LXH254 had the lowest IC50 ratios. This suggests that these regimens may have the best trade-off for targeting non-V600 mutant BRAF while sparing wild type BRAF. Although it is known that trametinib +/− dabrafenib are clinically well tolerated15,16, further pre-clinical and clinical evaluation of LXH254 is warranted particularly as this drug moves forward in clinical development. Notably, we identified different sensitivities to trametinib +/− dabrafenib, lifirafenib, and LHX254 between H1755 and H1395 cell lines, both of which harbor the same BRAF G469A mutation. These differences in sensitivity highlight our clinical finding that the G469V mutation was resistant to combination trametinib and dabrafenib, and prior pre-clinical findings suggesting that other tumor characteristics, such as co-mutations, can impact response to BRAF-targeted treatments26,44,46,47. Finally we found that novel ERK inhibitors failed to inhibit growth as single agents in all lung cancer cell lines tested. The reasons for this remain unclear, but nonetheless suggest that these compounds as single-agents are not effective for inhibition of BRAF signaling. This is consistent with phase I clinical data from single-agent MK-8353 where response rate in BRAF V600-mutant melanoma was only 20% (3/15 pts)48. Another explanation is that ERK inhibitors may require co-inhibition with other agents, such as BRAF inhibitors, to effectively block BRAF signaling. It is also possible that ERK inhibition acts on a context dependent manner because the efficacy of these agents has been more clearly described in melanoma and colorectal cancer pre-clinical models49,50.
Taken together, our clinical and pre-clinical data suggest that targeting BRAF non-V600 mutations with MEK and novel BRAF inhibitors is feasible, and there are ongoing trials to test these as well as other drug combinations. For example, trials evaluating binimetinib + encorafenib (NCT03839342), and second-generation BRAF inhibitors BGB-3245 (NCT04249843) and PLX8394 (NCT02428712) are ongoing. In addition, there are active early phase trials testing the safety of LXH254 (NCT02607813) and lifirafenib (NCT03905148). We feel that these trials as well as broader testing with these agents in combination with other inhibitors of the MAPK pathway will be critical to determine the best treatment strategy for patients whose tumors harbor BRAF non-V600 mutations.
In conclusion, we report the broader genetic landscape of advanced-stage BRAF mutant NSCLC in a large clinical cohort. We show that non-V600 BRAF mutations may be sensitive to MEK and novel BRAF inhibitors, with trametinib-based regimens, LXH254 and lifirafenib showing highest activity in the tested cell lines. In addition, we have shown the utility of combining functional and genomic approaches for classification of VUS to improve our ability to detect and characterize the diverse and heterogeneous BRAF non-V600 mutations. In doing so, we have improved our ability to understand the clinical contribution of these mutations, which ultimately may lead to novel therapies to enhance clinical outcomes for patients whose tumor harbor these mutations.
Supplementary Material
Acknowledgements.
The authors acknowledge funding support from Sheikh Khalifa Al Nahyan Ben Zayed Institute for Personalized Cancer Therapy, Cancer Prevention Research Institutive of Texas (CPRIT) Precision Oncology Decision Support Core RP150535 (to P.K.S.S. and F.M.B.); NIH / NCI U01CA217882, NIH / NCI U54CA224081, NIH / NCI R01CA204302, NIH / NCI R01CA211052, NIH / NCI R01CA169338, the Pew-Stewart Foundations (to T.G.B.); The University of Texas MD Anderson Lung Cancer Moonshots Program, Lung SPORE grant 5 P50 CA070907, and MD Anderson Cancer Center Support Grant P30 CA016672 (to J.V.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES:
Marcelo V. Negrao has nothing to disclose.
Victoria M. Raymond is an employee and shareholder at Guardant Health, during the conduct of the study.
Richard B. Lanman reports personal fees from Guardant Health, Inc., during the conduct of the study.
Jacqulyne P. Robichaux reports Royalties from licensed patent on EGFR/HER2 exon 20 from Spectrum Pharmaceuticals, outside the submitted work.
Junqin He has nothing to disclose.
Monique B. Nilsson reports Royalties from licensed patent on EGFR/HER2 exon 20 from Spectrum Pharmaceuticals, outside the submitted work.
Patrick K. S. Ng has nothing to disclose.
Bianca E. Amador has nothing to disclose.
Emily B. Roarty has nothing to disclose.
Rebecca J. Nagy is an employee and shareholder at Guardant Health, during the conduct of the study.
Kimberly C. Banks reports personal fees from Guardant Health, during the conduct of the study. Viola W. Zhu reports other from AstraZeneca, other from Roche-Foundation Medicine, other from Roche/Genentech, other from Takeda, other from TP Therapeutics, outside the submitted work.
Chun Ng has nothing to disclose.
Young Kwang Chae reports grants from Guardant Health, during the conduct of the study; grants from Abbvie, grants from BMS, grants from Biodesix, grants from Lexent Bio, grants from Freenome, personal fees from Roche/Genentech, personal fees from AstraZeneca, personal fees from Foundation Medicine, personal fees from Counsyl, personal fees from Neogenomics, personal fees from Boehringher Ingelheim, personal fees from Biodesix, personal fees from Immuneoncia, personal fees from Lilly Oncology, personal fees from Merck, personal fees from Takeda, outside the submitted work.
Jeffrey M. Clarke reports other from Guardant, during the conduct of the study; grants from Bristol-Myers Squibb, grants from Genentech, grants from Spectrum, grants from Adaptimmune, grants from Medpacto, grants from Bayer, grants from AbbVie, grants from Moderna, grants from Array, grants from Eli Lilly, other from Merck, other from AstraZeneca, other from Merck, other from Eli Lilly, other from Achilles Therapeutics, outside the submitted work.
Jeffrey A. Crawford reports grants from AstraZeneca, grants from Genentech, grants from Helsinn, personal fees from Amgen, personal fees from AstraZeneca, personal fees from Coherus, personal fees from Enzychem, personal fees from G1 Therapeutics, personal fees from GlaxoSmithKline, personal fees from Merck, personal fees from Pfizer, personal fees from Spectrum, personal fees from Beyong Spring, personal fees from Merrimack, personal fees from Mylan, personal fees from Roche, outside the submitted work.
Funda Meric-Bernstam reports grants and personal fees from eFFECTOR Therapeutics, personal fees from PACT Pharma, grants and personal fees from Zymeworks, personal fees from Jackson Laboratory, grants and personal fees from Genentech, personal fees from F. Hoffman-La Roche, personal fees from Paraxel International, grants and personal fees from Pfizer Inc., personal fees from IBM Watson, personal fees from Samsung Bioepis, personal fees from Aduro Biotech, personal fees from Kolon Life Sciences, personal fees from Origimed, personal fees from Sumitomo Dainippon Pharma, personal fees from Seattle Genetics, grants and personal fees from DebioPharm, personal fees from Dialectica, personal fees from Piers Pharmaceutical, personal fees from Xencor, personal fees from Chugai Biopharmaceuticals, grants and personal fees from Puma Biotechnology, personal fees from Mersana, personal fees from Immunomedics, personal fees from Silverback Therapeutics, personal fees from Inflection Biosciences, personal fees from Spectrum Pharamceuticals, grants and personal fees from Seattle Genetics, personal fees from Grail, personal fees from Darwin Health, personal fees from Clearlight Diagostics, grants from Aileron Therapeutics, grants from AstraZeneca, grants from Bayer Healthcare, grants from Calithera Biosciences, grants from Curis, grants from CytomX Therapeutics, grants from Daiichi Sankyo, grants from Guardant Health, grants from K Group, grants from Millennium Pharmaceuticals, grants from Novartis, grants from PPD Investigator Services , grants from Taiho Pharmaceutical , personal fees from Mayo Clinic, outside the submitted work.
Sai-Hong Ignatius Ou reports personal fees from Pfizer, personal fees from Merck, personal fees from Takeda/Ariad, personal fees from Astra Zeneca, personal fees from Roche/Genentech, personal fees from Foundation Medicine Inc., personal fees from Spectrum, other from Turning Point Therapeutics, outside the submitted work.
David R. Gandara reports personal fees from Guardant Health, during the conduct of the study; grants from Roche-Genentech, grants from Novartis, grants from Merck, personal fees from AstraZeneca, personal fees from Celgene, personal fees from CellMax, personal fees from FujiFilm, personal fees from Roche-Genentech, personal fees from Inivata, personal fees from IO Biotech, personal fees from Lilly, personal fees from Merck, personal fees from Samsung Bioepis, outside the submitted work.
John V. Heymach reports personal fees from Genentech, during the conduct of the study; personal fees and other from AstraZeneca, personal fees and other from GlaxoSmithKline, personal fees from Boehringer Ingelheim, personal fees from Exelixis, personal fees from Genentech, personal fees from Guardant Health, personal fees from Hengrui, personal fees from Lilly, personal fees from Novartis, personal fees and other from Spectrum, personal fees from EMD Serono, personal fees from Synta, other from Bayer, other from Takeda, other from Biotree, outside the submitted work.
Trever G. Bivona reports grants from NIH, during the conduct of the study; grants and other from Novartis, grants and other from Revolution Medicines, personal fees from Astrazeneca, personal fees from Takeda, personal fees from Strategia, personal fees from Springworks, personal fees from Array, Pfizer, personal fees from Rain , outside the submitted work.
Caroline E. McCoach reports personal fees from Novartis , grants from Revolution medicines, outside the submitted work.
References
- 1.Blakely CM, Watkins TBK, Wu W, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet. 2017;49(12):1693–1704. doi: 10.1038/ng.3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCoach CE, Blakely CM, Banks KC, et al. Clinical utility of cell-free DNA for the detection of ALK fusions and genomic mechanisms of ALK inhibitor resistance in non-small cell lung cancer. Clin Cancer Res. 2018;24(12):2758–2770. doi: 10.1158/1078-0432.CCR-17-2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nik-Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534(7605):47–54. doi: 10.1038/nature17676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toledo RA, Garralda E, Mitsi M, et al. Exome sequencing of plasma DNA portrays the mutation landscape of colorectal cancer and discovers mutated VEGFR2 receptors as modulators of antiangiogenic therapies. Clin Cancer Res. 2018;24(15):3550–3559. doi: 10.1158/1078-0432.CCR-18-0103 [DOI] [PubMed] [Google Scholar]
- 5.Zill OA, Banks KC, Fairclough SR, et al. The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced cancer patients. Clin Cancer Res. 2018;24(15):3528–3538. doi: 10.1158/1078-0432.CCR-17-3837 [DOI] [PubMed] [Google Scholar]
- 6.Strickler JH, Loree JM, Ahronian LG, et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov. 2018;8(2):164–173. doi: 10.1158/2159-8290.CD-17-1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yaeger R, Corcoran RB. Targeting alterations in the RAF–MEK pathway. Cancer Discov. 2019;9(3):329–341. doi: 10.1158/2159-8290.CD-18-1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7(6):596–609. doi: 10.1158/2159-8290.CD-16-1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin L, Asthana S, Chan E, et al. Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc Natl Acad Sci. 2014;111(7):E748–E757. doi: 10.1073/pnas.1320956111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akbani R, Akdemir KC, Aksoy BA, et al. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161(7):1681–1696. doi: 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- 12.Eric AC, Joshua DC, Angela NB, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tissot C, Couraud S, Tanguy R, Bringuier PP, Girard N, Souquet PJ. Clinical characteristics and outcome of patients with lung cancer harboring BRAF mutations. Lung Cancer. 2016;91:23–28. doi: 10.1016/j.lungcan.2015.11.006 [DOI] [PubMed] [Google Scholar]
- 14.Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer. 2015;121(3):448–456. doi: 10.1002/cncr.29042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Planchard D, Besse B, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. 2016;17(7):984–993. doi: 10.1016/S1470-2045(16)30146-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Planchard D, Smit EF, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–1316. doi: 10.1016/S1470-2045(17)30679-4 [DOI] [PubMed] [Google Scholar]
- 17.Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29(15):2046–2051. doi: 10.1200/JCO.2010.33.1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao Z, Torres NM, Tao A, et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell. 2015;28(3):370–383. doi: 10.1016/j.ccell.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao Z, Yaeger R, Rodrik-Outmezguine VS, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548(7666):234–238. doi: 10.1038/nature23291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell. 2010;140(2):209–221. doi: 10.1016/j.cell.2009.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowyer SE, Rao AD, Lyle M, et al. Activity of trametinib in K601E and L597Q BRAF mutation-positive metastatic melanoma. Melanoma Res. 2014;24(5):504–508. doi: 10.1097/CMR.0000000000000099 [DOI] [PubMed] [Google Scholar]
- 22.Okimoto RA, Lin L, Olivas V, et al. Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF -mutant lung cancer . Proc Natl Acad Sci. 2016;113(47):13456–13461. doi: 10.1073/pnas.1610456113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nichols RJ, Haderk F, Stahlhut C, et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol. 2018;20(9):1064–1073. doi: 10.1038/s41556-018-0169-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amir N, Philippe G, Sylvia DB, et al. Type II RAF inhibitor causes superior ERK pathway suppression compared to type I RAF inhibitor in cells expressing different BRAF mutant types recurrently found in lung cancer. Oncotarget. 2018;9(22):16110–16123. doi: 10.18632/oncotarget.24576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan RJ, Infante JR, Janku F, et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose-escalation and expansion study. Cancer Discov. 2018;8(2):184–195. doi: 10.1158/2159-8290.CD-17-1119 [DOI] [PubMed] [Google Scholar]
- 26.Dankner M, Lajoie M, Moldoveanu D, et al. Dual MAPK inhibition is an effective therapeutic strategy for a subset of class II BRAF mutant melanomas. Clin Cancer Res. 2018;24(24):6483–6494. doi: 10.1158/1078-0432.CCR-17-3384 [DOI] [PubMed] [Google Scholar]
- 27.Leighl NB, Page RD, Raymond VM, et al. Clinical Utility of Comprehensive Cell-Free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-Small Cell Lung Cancer. Clin Cancer Res. 2019:clincanres.0624.2019. doi: 10.1158/1078-0432.CCR-19-0624 [DOI] [PubMed] [Google Scholar]
- 28.Parseghian CM, Loree JM, Morris VK, et al. Anti-EGFR-resistant clones decay exponentially after progression: Implications for anti-EGFR re-challenge. Ann Oncol. 2019;30(2):243–249. doi: 10.1093/annonc/mdy509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Odegaard JI, Vincent JJ, Mortimer S, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res. 2018;24(15):3539–3549. doi: 10.1158/1078-0432.CCR-17-3831 [DOI] [PubMed] [Google Scholar]
- 30.Ptashkin RN, Mandelker DL, Coombs CC, et al. Prevalence of Clonal Hematopoiesis Mutations in Tumor-Only Clinical Genomic Profiling of Solid Tumors. JAMA Oncol. 2018;4(11):1589–1593. doi: 10.1001/jamaoncol.2018.2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsang YH, Dogruluk T, Tedeschi PM, et al. Functional annotation of rare gene aberration drivers of pancreatic cancer. Nat Commun. 2016;7:1–11. doi: 10.1038/ncomms10500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ng PKS, Li J, Jeong KJ, et al. Systematic Functional Annotation of Somatic Mutations in Cancer. Cancer Cell. 2018;33(3):450–462.e10. doi: 10.1016/j.ccell.2018.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferrarotto R, Goonatilake R, Yoo SY, et al. Epithelial-mesenchymal transition predicts polo-like kinase 1 inhibitor-mediated apoptosis in non-small cell lung cancer. Clin Cancer Res. 2016;22(7):1674–1686. doi: 10.1158/1078-0432.CCR-14-2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012;2(5):401 LP – 404. doi: 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal. 2013;6(269):pl1 LP–pl1. doi: 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheikine Y, Pavlick D, Klempner SJ, et al. BRAF in Lung Cancers: Analysis of Patient Cases Reveals Recurrent BRAF Mutations, Fusions, Kinase Duplications, and Concurrent Alterations. JCO Precis Oncol. 2018;(2):1–15. doi: 10.1200/PO.17.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dagogo-Jack I, Martinez P, Yeap BY, et al. Impact of BRAF Mutation Class on Disease Characteristics and Clinical Outcomes in BRAF-Mutant Lung Cancer. Clin Cancer Res. 2019;25(1):158–165. doi: 10.1158/1078-0432.CCR-18-2062 [DOI] [PubMed] [Google Scholar]
- 38.Belic J, Graf R, Bauernhofer T, et al. Genomic alterations in plasma DNA from patients with metastasized prostate cancer receiving abiraterone or enzalutamide. Int J Cancer. 2018;143(5):1236–1248. doi: 10.1002/ijc.31397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuboki Y, Yamashita S, Niwa T, et al. Comprehensive analyses using next-generation sequencing and immunohistochemistry enable precise treatment in advanced gastric cancer. Ann Oncol. 2016;27(1):127–133. doi: 10.1093/annonc/mdv508 [DOI] [PubMed] [Google Scholar]
- 40.Maitra A, Biswas NK, Amin K, et al. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat Commun. 2013;4. doi: 10.1038/ncomms3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mouradov D, Sloggett C, Jorissen RN, et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014;74(12):3238–3247. doi: 10.1158/0008-5472.CAN-14-0013 [DOI] [PubMed] [Google Scholar]
- 42.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–713. doi: 10.1038/nm.4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suehiro Y, Wong CW, Chirieac LR, et al. Epigenetic-genetic interactions in the APC/WNT, RAS/RAF, and P53 pathways in colorectal carcinoma. Clin Cancer Res. 2008;14(9):2560–2569. doi: 10.1158/1078-0432.CCR-07-1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krauthammer M, Kong Y, Bacchiocchi A, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet. 2015;47(9):996–1002. doi: 10.1038/ng.3361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Offin M, Rizvi H, Tenet M, et al. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant lung cancers. Clin Cancer Res. 2019;25(3):1063–1069. doi: 10.1158/1078-0432.CCR-18-1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74(8):2340–2350. doi: 10.1158/0008-5472.CAN-13-2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whittaker SR, Theurillat JP, Van Allen E, et al. A genome-Scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3(3):351–362. doi: 10.1158/2159-8290.CD-12-0470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moschos SJ, Sullivan RJ, Hwu W-J, et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight. 2018;3(4). doi: 10.1172/jci.insight.92352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kirouac D, Schaefer G, Chan J, et al. Clinical responses to ERK inhibitor (GDC-0994) treatment combinations predicted using a Quantitative Systems Pharmacology model of MAPK signaling in BRAF(V600E)-mutant colorectal cancer. Eur J Cancer. 2016;69:S20. doi: 10.1016/S0959-8049(16)32639-9 [DOI] [Google Scholar]
- 50.Bhagwat S V, McMillen WT, Cai S, et al. Abstract 4973: Discovery of LY3214996, a selective and novel ERK1/2 inhibitor with potent antitumor activities in cancer models with MAPK pathway alterations. Cancer Res. 2017;77(13 Supplement):4973LP – 4973. doi: 10.1158/1538-7445.AM2017-497328754668 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.