

Abstract
Bicuspid aortic valve (BAV) is the most common congenital heart defect, found in up to 2% of the population and associated with a 30% lifetime risk of complications. BAV is inherited as an autosomal dominant trait with incomplete penetrance and variable expressivity due to a complex genetic architecture that involves many interacting genes. In this review, we highlight the current state of knowledge about BAV genetics, principles and methods for BAV gene discovery, clinical applications of BAV genetics, and important future directions.
Keywords: bicuspid aortic valve, genetics, aortic aneurysm, congenital heart disease
INTRODUCTION
Bicuspid aortic valve (BAV) is a heterogeneous disorder that is primarily inherited in an autosomal dominant pattern with incomplete penetrance and variable expressivity1, 2. BAV is a feature of some genetic syndromes, including Turner syndrome and Loeys-Dietz syndrome, as well as complex congenital heart defects that disrupt the left ventricular outflow tract (LVOT). In non-syndromic cases, BAV inheritance is best explained by a complex genetic architecture involving many different interacting genes. Thus, fully elucidating the genetic and epigenetic networks involved in the complex pathophysiology of BAV will be crucial for the development of personalized risk stratification approaches3. In this review we highlight the current state of knowledge about BAV genetics, principles and methods for BAV gene discovery, clinical applications, and potential future directions.
GENETIC SYNDROMES WITH BAV
1. Turner syndrome
Turner syndrome (TS) features the highest prevalence of BAV among all genetic syndromes (Table 1)4. TS is caused by partial or complete absence of one X chromosome in women. BAV appears in more than 30% of women with TS, and the prevalence of associated coarctation of the aorta (CoA) and thoracic aortic aneurysm (TAA) exceeds non-syndromic BAV cases5. In combination with the higher prevalence of BAV in 46,X,Y men (3:1), this observation led to the hypothesis that copy number reduction of X chromosome genes may predispose to BAV formation2, 6, 7. Specific dosage-sensitive genes on the short arm of the X chromosome, including KDM6A and TIMP1, were implicated by recent genetic studies8, 9. In addition, genetic and non-genetic modifiers appear to influence the prevalence of BAV in TS. Impaired fetal thoracic lymphatic drainage causing LVOT obstruction during cardiac development may predispose individuals with TS and a history of central lymphedema to BAV and anomalous pulmonary venous drainage10. Potential autosomal genetic modifiers include polymorphic genomic duplications of 12p13.31 (SLC2A3, SLC2A14, NANOGP1) and rare deleterious variants of the TIMP3 gene in 22q, which are significantly associated with BAV, CoA and aortic dissections in TS, independently of karyotype7. These data imply that copy variation of X chromosome gene(s) and other genetic variants may interact to modify susceptibility to BAV.
Table 1.
Syndromes with BAV
| Syndrome | Gene(s) | Other features |
|---|---|---|
| Loeys-Dietz | TGFBR1, TGFBR2, TGFB2, TGFB3, SMAD3 | Bifid uvula HTAD |
| Multisystemic Smooth Muscle Dysfunction | ACTA2 | Mydriasis HTAD |
| Down | 21 duplication | Atrioventricular septal defects |
| Turner | X monosomy | Short stature Coarctation of the aorta |
| Velocardiofacial | 22q11.2 del | Truncus arteriosus Tetralogy of Fallot |
HTAD: hereditary thoracic aortic aneurysm and dissection.
2. Loeys-Dietz syndrome
Approximately 10% of patients with Loeys-Dietz syndrome (LDS) have BAV, making it the second most highly associated genetic syndrome after TS11. LDS is caused by dominantly inherited mutations of TGF-β pathway genes, including ligands (TGFB2 and TGFB3), receptors (TGFBR1 and TGFBR2) and downstream effectors (SMAD3), Pathogenic mutations in TGFBR1 or TGFBR2 are also rare causes of heritable non-syndromic thoracic aortic aneurysms and dissections12–18. While there are no specific anatomic features that distinguish BAV in LDS from BAV in non-syndromic cases, valvular disease tends to manifest at younger ages in patients with LDS, who frequently present with aortic regurgitation due to proximal aortic dilation, and may have other cardiac and vascular malformations or conduction abnormalities19. In LDS patients with TGFBR1 mutations, male sex and increased arterial tortuosity are more highly predictive of dissection and death than syndromic features20. However, the burden of rare variants in LDS genes is not increased in non-syndromic BAV patients, even among those with aortic dilatation21.
3. Velocardiofacial syndrome (VCFS)
BAV and aortic dilation are also more prevalent in VCFS22. VCFS patients have recognizable syndromic features, including learning disabilities, hypoparathyroidism, autoimmune problems and conotruncal heart malformations, including ventricular septal defects (60%), interrupted aortic arch (40%) and patent ductus arteriosus (30%)4. Large genomic deletions of 22q11.2 cause VCFS, and haploinsufficiency of TBX1 is strongly associated with cardiac abnormalities23. However, other genes in the commonly deleted VCFS region, including HIRA, UFD1L, CRKL and DGCR6, have been shown to regulate cardiac development and probably interact to explain the burden of conotruncal defects in VCFS patients24, 25. Mutations of single VCFS genes do not appear to be prevalent in non-syndromic BAV cases26, but smaller distal deletions adjacent to the commonly deleted VCFS region are evident in some patients with BAV and overlapping clinical features. The same polymorphic 12p13.31 duplications that are associated with BAV in TS are also associated with heart defects in VCFS, reinforcing the hypothesis that multiple genetic lesions interact to cause BAV7.
4. Genetic syndromes with less frequent BAV
Other genetic disorders that include BAV as an occasional feature include Down syndrome, caused by trisomy of chromosome 21, Alagille syndrome, caused by mutation of the NOTCH ligands JAG1 or JAG2, Kabuki syndrome, caused by mutation of the epigenetic regulators KMT2D or KDM6A, and FLNA mutations that cause periventricular nodular heterotopia and mitral valve malformations4, 27, 28. All of these disorders are associated with distinctive extracardiac features and other left-sided obstructive cardiac lesions.
DEVELOPMENTAL ORIGINS OF BAV
1. Defective endocardial endothelial to mesenchymal transition (EndMT) during embryonic development
The aortic valve develops in the atrioventricular canal and outflow tract of the primitive heart tube, when signals from the myocardium induce endocardial-mesenchymal transition (EndMT) to create the endocardial cushions, in which proliferative mesenchymal cells are embedded in a loose extracellular matrix (cardiac jelly)29. This is followed by a complex sequence of cell proliferation, differentiation, migration, adhesion and apoptosis that results in elongation and remodeling of the cushions into the distinct layers of the mature valves29–32. Valve remodeling is dynamic and reciprocally regulated by blood flow and hemodynamic shear stress33, 34.
EndMT is also required for late stages of aortic valve development, and defects in EndMT cause LVOT abnormalities and BAV (Figure 1). As demonstrated in mouse embryos, NOTCH1 is highly expressed in the LVOT mesenchyme and endocardium at the location of the nascent valve cusps.35 Homozygous NOTCH1 mutations cause premature death due to vascular endothelial defects, but haploinsufficiency causes BAV and TAA, which is accentuated on a nitric oxide synthase (Nos3)-null background30, 36–38. BAV and TAA are highly correlated with impairment of EndMT in NOTCH1-deficient vascular cells35, 36, 38–40.
Figure 1. Cellular contributions to the formation of the outflow tract.
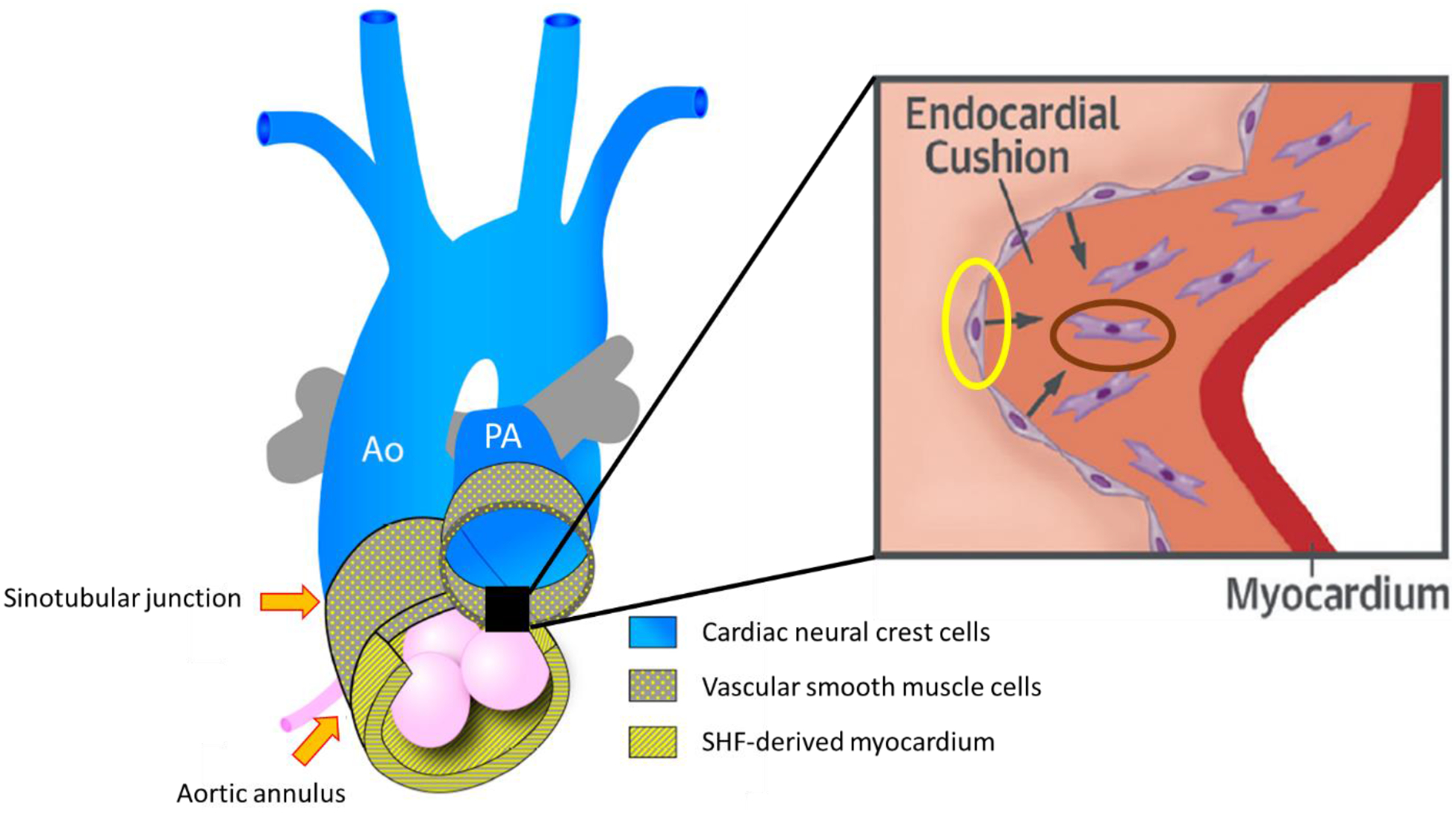
The outflow tract is formed by three cell lines: cardiac neural crest cells (blue), vascular smooth muscle cells (dotted yellow) and second heart field-derived myocardium (striped yellow). Inset shows endothelial (yellow oval) to mesenchymal (dark oval) transition in the endocardial cushions. Ao, aorta; PA, pulmonary artery; SHF, second heart field. Adapted from Martin et al and Kovacic et al99, 126.
2. Association between left-sided obstructive lesions and BAV
Due to the common embryologic origin of the aortic valve, LVOT and proximal aorta, BAV frequently co-exists with other left-sided congenital heart lesions, such as coarctation (CoA), Shone complex and hypoplastic left heart syndrome (HLHS). Approximately 50 to 85% of patients with CoA have BAV41. CoA consists of a discrete stenosis or hypoplastic segment located most often immediately after the origin of the left subclavian artery42. In humans, each subclavian artery derives from the 7th intersegmental artery from the right and left dorsal aortae immediately before they fuse into a single common aorta. Defective migration of differentiation of cardiac neural crest cells into 4th pharyngeal pouch and 4th branchial arch derivatives that develop into the endocardial cushions, proximal aorta and aortic isthmus may explain this association43. Some human teratogens that affect neural crest derivatives, such as maternal phenylketonuria, have also been implicated in the development of aortic arch (including CoA) and conotruncal malformations44.
Shone complex, characterized by mitral valve stenosis, LVOT obstruction and CoA, present with BAV in 71% of patients45. Additionally, up to 17% of patients with hypoplastic left heart syndrome (HLHS) have BAV41, 46, suggesting an underlying common genetic defect between these conditions. This is supported by family-based genome-wide linkage analysis, where recurrence risk ratios of BAV in HLHS families was similar to that in BAV families47.
Left ventricular noncompaction cardiomyopathy (LVNC) is an important cause of dilated cardiomyopathy due to impaired compaction of myocardial fibers during endomyocardial morphogenesis48. LVNC may be genetically linked to BAV through the Notch pathway, which is also essential to promote myocardial compaction and ventricular septation37. LVNC is found in conjunction with congenital heart defects in more than 10% of cases, predominately LVOT abnormalities including BAV49. In addition, LVNC may be more common in BAV patients than in the general population50, and BAV patients with LVNC may develop earlier onset aortic valve disease or TAA with a more malignant course requiring surgical intervention50, 51. There is limited evidence that the same genetic mutations that cause isolated LVNC can also cause BAV, but the genetic causes of most LVNC cases remain unknown.
NON-SYNDROMIC BAV
Most people with BAV do not have syndromic features, but may have other congenital heart and vascular abnormalities with variable disease severity. The frequency of other left-sided lesions such as CoA (7%), patent ductus arteriosus (8.5%), mitral valve abnormalities (11%), ventricular septal defects (14%) and TAA (50%) are all significantly increased in BAV52, 53. Valvular aortic stenosis or regurgitation may eventually necessitate aortic valve replacement in up to 50% of BAV patients54.
Complex inheritance is present in large families with non-syndromic BAV. The prevalence of BAV in first-degree family members is 10-fold higher than the general population55, 56. Inheritance is observed in more than half of the families if associated nonvalvular complications such as CoA, TAA, mitral valve or ventricular septal defects are included1. The heritability of BAV has been estimated to be as high as 90%, and multiple alleles can interact to cause BAV or other congenital heart defects without BAV in the same family1, 57, 58. With this basis, echocardiographic screening of first-degree family members is recommended in current guidelines59, 60.
BAV is more frequent in first-degree relatives of patients with severe left-sided lesions, which include aortic valve stenosis, CoA, mitral valve stenosis, interrupted aortic arch type A, HLHS and Shone complex61. There is greater than 10 and 5.5-fold excess of BAV in first-degree relatives of patients with HLHS62 and LVOT obstruction63, respectively. Family-based genome-wide linkage analysis have found that recurrence risk ratios of BAV in HLHS families are similar to that in BAV families, providing evidence that some HLHS and BAV are genetically related47. In these families, BAV likely represents a mild manifestation of the disease trait and thus implicates genetic pleiotropism, whereby environmental or stochastic factors play a significant role in the phenotypic expression64.
Imaging studies show that first-degree relatives of BAV probands who have tricuspid aortic valves frequently have subtle root and ascending aortic dilation or valvular defects causing perturbed outflow65. These observations support the hypothesis that tricuspid aortic valve stenosis and other subclinical features may represent an underrecognized ‘form fruste’ of BAV66.
IDENTIFICATION OF BAV GENES: PRINCIPLES
1. Distinguishing causation from phenotypic modification
For most Mendelian disorders, a single genetic mutation is sufficient to cause disease. The discovery of one rare pathogenic variant that segregates with disease in a family with clear and consistent inheritance of the phenotype usually provides adequate evidence to identify the causal gene67. In complex or polygenic disorders like BAV, a single genetic variant may increase the risk to develop disease, but is not sufficient to cause disease by itself. In many cases, environmental factors may interact with multiple genetic variants to modify the timing or penetrance of disease68. Thus, complex traits characteristically show reduced penetrance (same genetic variant but no disease) and variable expressivity (same genetic variant with different manifestations of disease). For example, interactions between genetic variants that cause BAV, genetic variants that accelerate aortic valve calcification and established clinical risk factors such as hypertension, dyslipidemia and smoking may increase the rate of progression of aortic stenosis in BAV patients2, 69, 70.
2. Evidence framework to prove pathogenicity
According to the evidence framework developed by the Clinical Genome Resource71, requirements to prove that a genetic variant is pathogenic, or disease-causing, include: 1) segregation, or co-inheritance of the variant with disease in a family72; 2) at least two independent observations of a loss of function variant in cases, in a gene that has no loss of function variants in normal controls; 3) multiple independent observations of rare missense variants in cases, in a gene that is highly invariant in controls (i.e., ‘burden’ tests of variants in cases vs controls must be significant in at least two independent cohorts); or 4) direct functional studies to prove that the genetic variant alters the function or expression of the encoded protein. Most of the genetic variants that were discovered in cohorts of BAV patients do not meet these standards. Few demonstrate familial segregation, and most were not replicated in independent populations or validated using functional genomic studies. Therefore, significant validation will be required before this genetic data can be used to develop genetic tests for BAV or make decisions about the clinical treatment of BAV patients based on genetic information.
3. Family-based vs population-based gene identification methods
In family-based strategies, the inheritance pattern of a disease is determined by careful phenotypic and genetic analysis of affected and unaffected relatives (Figure 2). Segregation of genetic mutations with disease traits in families remains the most effective approach to identify causative genes73. Population-based studies are based on the hypothesis that a few common genetic variants are shared by groups of people with the disease. Population-based genetic strategies are designed to compare the frequencies of common variants in a large sample of individuals with the disease to a control group. Instead of directly identifying causal genetic variants, population-based approaches detect indirect associations between polymorphic genetic markers (single-nucleotide polymorphisms -SNPs-) and disease. Common genetic variants that are more frequent in disease populations tend to have a modest and indirect effect on the likelihood of developing disease, but are more likely to modify the timing or severity of disease74, 75. Genetic studies of selected populations with ‘extreme trait’ characteristics that are more likely to have a genetic cause, such as early onset or severe manifestations of disease, may increase the power of association studies to identify clinically relevant genetic variants76.
Figure 2. Family-based analysis.
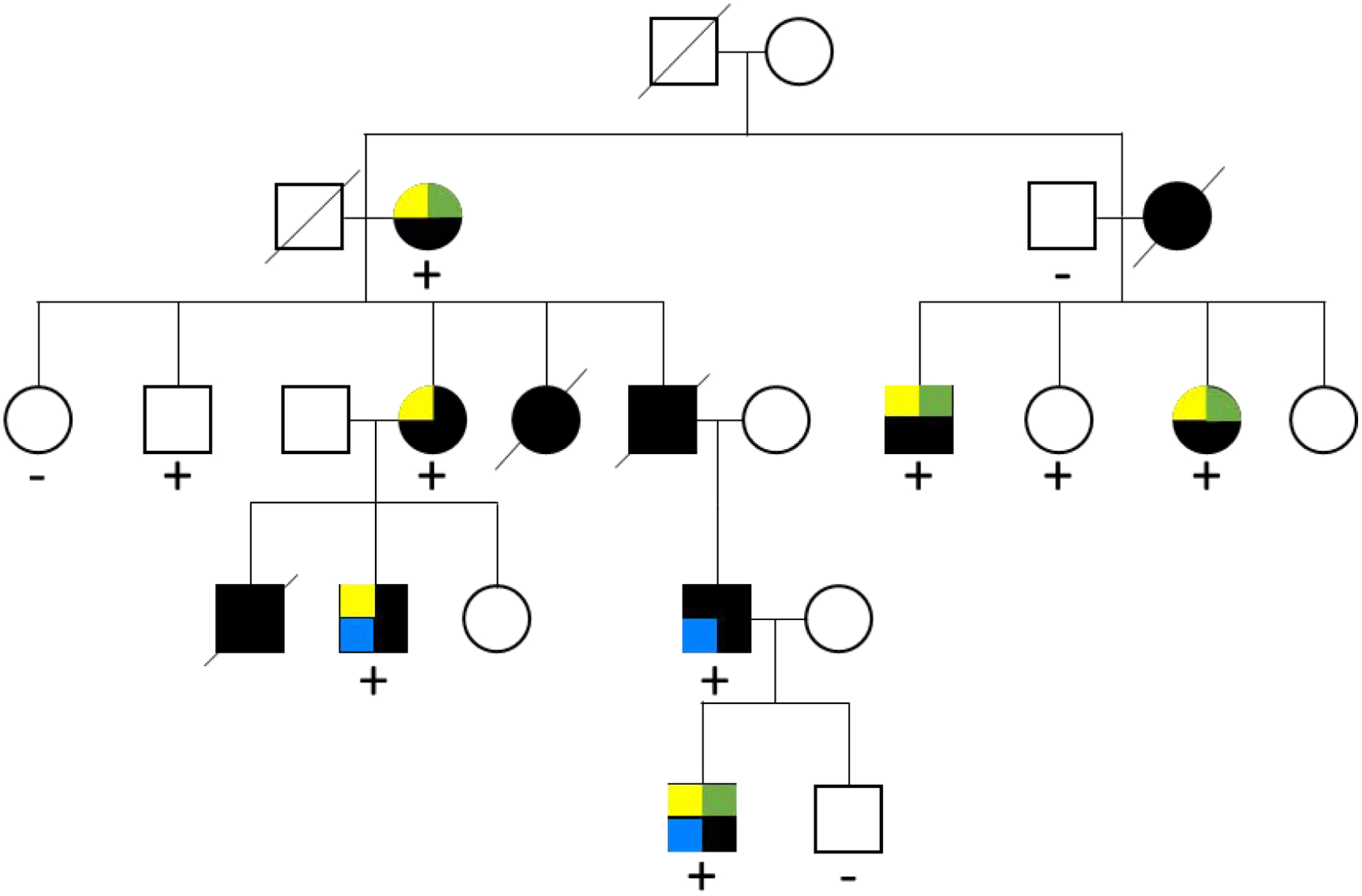
Kindred with five generations affected by bicuspid aortic valve (yellow square), coarctation of the aorta (green square) and thoracic aortic aneurysm (blue square) demonstrates reduced penetrance and variable expressivity that is characteristic of BAV pedigrees. Squares, males; circles, females; slashed, deceased family members; dark filled, affected; +, presence of mutation; −, absence of mutation. Adapted from Garg et al35.
IDENTIFICATION OF BAV GENES: METHODS
1. Genome-wide association studies (GWAS)
Currently available ‘chip’ genotyping technologies capture SNP variants that are evenly distributed throughout the genome and detect common (present in >1% of the population) genetic variants that modify disease risk or increase susceptibility to disease, but in general do not cause disease77. Thousands of individuals must be genotyped in large GWAS studies to detect the relatively modest effect of most SNPs75.
Several GWAS studies have focused on BAV, with variable results (Table 2). In 466 BAV patients who attended a cardiothoracic surgery clinic (83% TAA), GWAS identified more common noncoding and missense variants around the GATA4 gene in BAV cases than in tricuspid controls78. In a separate cohort of 480 non-syndromic BAV patients, no individual gene attained genome-wide significance, but a pathway-based analysis identified variants in genes that regulate cilia. Functional studies in zebrafish confirmed that disruption of primary cilia can cause BAV and other left-sided cardiac lesions79. Other smaller GWAS studies were underpowered to find significant associations80, 81.
Table 2.
Genes associated with BAV
| Gene | Locus | OMIM disease | Function | Size and Type of Cohort | References |
|---|---|---|---|---|---|
| NOTCH1 | 9q34.3 | Adams-Oliver syndrome 5, Aortic valve disease 1 | Endocardial cushion development, aortic valve calcification | Family-based genome-wide scan, including 14 BAV individuals | 35 |
| SMAD6 | 15q22.31 | Aortic valve disease 2 | Cardiac valves development and outflow tract septation | Targeted resequencing in 441 BAV/TAA cohort | 89 |
| GATA4 | 8p23.1 | Atrial septal defect 2, atrioventricular septal defect 4, tetralogy of Fallot, ventricular septal defect 1 | Myocardial differentiation and function | Sequencing of 150 nonsyndromic BAV individuals | 31 |
| GATA5 | 15q25-q26.1, 20q13.13 | Congenital heart defects, multiple types, 5 | Extracellular matrix remodeling and morphogenesis of valve leaflets | Sequencing of 110 nonsyndromic BAV individuals | 87 |
| GATA6 | 18q11.2 | Tetralogy of Fallot, patent ductus arteriosus, atrial septal defect 9, atrioventricular septal defect 5 | Outflow tract and subpulmonary myocardial development | Family-based sequencing study including 152 BAV individuals | 32 |
| ROBO4 | 11q24.2 | Aortic valve disease 8 | Outflow tract development and integrity of ascending aorta | Family-based targeted sequencing including 10 BAV individuals | 86 |
| MAT2A | 2p11.2 | Smooth muscle cell function and development | Family-based whole exome sequencing including 8 individuals with TAA with or without BAV | 85 | |
| ADAMTS19 | 5q23.3 | Perturbs shear stress signaling in valvular endothelial cells, increasing cellularity and proteoglycan deposition | Family-based exome sequencing, including 8 affected individuals with early-onset valvular heart disease | 127 |
BAV: bicuspid aortic valve; TAA: thoracic aortic aneurysm.
As these results illustrate, the significant genetic heterogeneity of BAV makes it difficult to detect the causal effect of individual genes82. Methods to reduce heterogeneity include limiting the analysis to strictly defined BAV subtypes based on anatomic classifications or disease presentations, where the effect of single gene mutations may be amplified, or a hybrid approach in which familial segregation is used to prioritize rare variants73. GWAS is also more likely to succeed in comparisons that identify more common variants that modify BAV disease presentation or complications. For example, genetic variants that influence vascular calcification may increase the risk for bicuspid aortic stenosis83.
2. Next generation sequencing (NGS)
Current NGS technologies permit whole exome (WES) or whole genome (WGS) sequencing for approximately $100084. NGS comprehensively detects unique or rare variants that are not accessible to other methods and can accelerate gene discovery. WES studies identified rare missense variants in MAT2A85 and missense and loss of function variants in ROBO486 that segregate with non-syndromic BAV and TAA in families. In contrast, WES of unrelated, non-syndromic BAV cases has been less successful83. Rare loss of function variants in GATA4, GATA5 and GATA6 were each identified in more than one non-syndromic BAV cohort.31, 32, 87. More recent observations of rare, non-synonymous variants in MIB1, ADAMTS5 and ADAMTS19 have yet to be replicated88. Sequencing a panel of candidate genes based on prior information has been frequently used to validate their contributions to BAV in different contexts. In 441 non-syndromic BAV patients with TAA, targeted sequencing of 22 candidate genes identified recurrent loss of function mutations in SMAD6 that account for 2% of cases89. These studies are much less expensive than genome or exome-wide approaches, but are much more limited in scope and generally lack data to corroborate the pathogenicity of candidate variants. For example, rare missense variants of candidate genes are generally not well correlated with specific valve or aortic phenotypes90, 91.To date, SMAD6 and GATA6 have the most cumulative and consistent evidence for contribution to non-syndromic BAV based on recurrent deleterious mutations in multiple cohorts.
3. Rare genomic Copy Number Variants (CNVs)
A CNV is a contiguous genomic DNA segment that may be duplicated (≥3 copies) or deleted (≤1 copy) in comparison with the reference genome. While most CNVs are considered to be benign, more than 40 CNV loci are implicated in human diseases92. CNVs may account for up to 10% of BAV cases and are more likely to be found in patients that present at younger ages or with more severe valve or aortic complications93. Rare pathogenic CNVs in BAV patients affect a subset of genes that are mutated in syndromic and non-syndromic cases. A similar enrichment of rare CNVs was also observed in patients with related congenital lesions such as Tetralogy of Fallot and hypoplastic left heart syndrome94, 95. We identified rare and recurrent CNVs at 2q37.3 and 22q11.21 in patients with early onset BAV and TAA that are absent or extremely rare in controls and involve candidate genes that interact with each other during heart and vascular development93.
GENOMIC LANDSCAPE OF BAV: RARE OR UNIQUE VARIANTS IN MANY GENES
Cumulative data demonstrate that BAV is caused by rare or private mutations in many different genes that each contribute to a small proportion of cases. Many BAV genes also regulate LVOT and aortic development and can influence cardiac and aortic traits besides - Tiif to do BAV, leading to reduced penetrance and variable expressivity of BAV phenotypes86, 96. In addition, epigenetic and common variation of other genes that are not directly involved in BAV development can influence the rate of disease progression, but do not cause disease97, 98.
The power to identify BAV genes depends on the study population and approach. For example, rare or unique variants may be readily identified in single families where they may segregate with BAV. In contrast, large cohorts of unrelated probands may be needed to detect the relatively weaker associations between more common genetic variants and BAV. Thus, family-based exome and genome sequencing studies may be more appropriate for BAV gene discovery than population-based or GWAS approaches, which may identify disease modifiers rather than causal genes. The effect of single gene mutations may be more evident in pediatric, young adult and surgical cohorts with earlier onset and more aggressive valve or aortic complications than in older adults with later onset and more slowly progressive disease.
SINGLE GENES IN NON-SYNDROMIC BAV
Table 3 summarizes genes with the most extensive evidence for causative mutations in BAV patients. Many more potential candidate genes exist based on model organism data that have not been corroborated in human genetic studies.
Table 3.
Genome-wide association and next-generation sequencing studies of BAV
| Author | Type | Cases | Controls | Findings | Reference |
|---|---|---|---|---|---|
| Hanchard | GWAS | 778 non-syndromic left-sided lesions cases | 2756 patients without left-sided lesions from the high-density SNP association analysis of melanoma: case-control and outcomes investigation and from the GWAS of Parkinson disease: genes and environment | Locus 20q11 associated with BAV, MYH7B and MIR499A as candidate genes | 61 |
| Yang | GWAS | 466 non-syndromic BAV (83% TAA) attending cardiac surgery clinic at the University of Michigan Frankel Cardiovascular Center | 4660 age, sex and ethnicity matched controls from the Michigan Genomics Initiative | Noncoding variants near GATA4 associated with BAV | 78 |
| Fulmer | GWAS | 2131 non-syndromic BAV cases | 2728 patients without BAV from the Framingham Heart Study cohort | 15 SNPs associated with BAV, including EXOC4 | 79 |
| Helgadotir | GWAS | 208 non-syndromic BAV (Iceland, Sweden, USA) | 25139 controls (Iceland deCODE database, Stockholm POLCA/Olivia study, Michigan Genomics Initiative, Framingham Heart Study and National Institute of Neurological Disorders and Stroke) | PALMD intergenic and TEX41 intronic variants associated with BAV | 128 |
| Bjornsson | GWAS | 120 Icelandic non-syndromic CoA cases (75% with BAV) | 355166 disease-free individuals randomly selected from Icelandic genealogical databases at deCODE | Rare missense mutations of MYH6 in BAV cases | 81 |
| LeMaire | GWAS (3 stages) | 765 non-syndromic TAD, 385 non-syndromic TAD (192 BAV), 163 non-syndromic TAD (157 BAV) | 1355, 159 and 476 controls from Wellcome T rust Case-Control Consortium 1958 Birth Cohort and US National Institute of Neurological Disorders and Stroke | Common FBN1 variants associated with TAD, with or without BAV | 124 |
| Wooten | Modified GWAS | 68 non-syndromic BAV | 830 controls from Illumina iControlDB and 7 BAV negative familial controls | Rare AXIN1/PDIA2 haplotype associated with BAV | 80 |
| Gago-Dlaz | Population-based NGS | 565 Spanish non-syndromic BAV cases (none with cardiac or ascending aortic surgery) | 484 controls attending primary health care centers in Galicia and from Plataforma en Red Banco Nacional de ADN Carlos III | No significant associations with BAV | 83 |
| Guo | Family-based NGS | 34 family members with TAA (BAV 47%) | MAT2A mutations segregate with BAV and HTAD | 85 | |
| Gould | Family-based NGS | 286 family members with BAV/TAA | 193 unrelated controls | ROBO4 mutations segregate with BAV and HTAD and are enriched in non-syndromic cases. | 86 |
BAV: bicuspid aortic valve; GWAS: genome-wide association study; NGS: next generation sequencing; SNP: single-nucleotide polymorphism; TAA: thoracic aortic aneurysm; TAD: thoracic aortic aneurysm and dissection.
1. NOTCH1
NOTCH1 encodes a single-pass transmembrane receptor and functions in a highly conserved pathway that promotes the endothelial to mesenchymal transition and plays a critical role in cardiac valve development35, 99, 100. The NOTCH1 pathway is also required to maintain vascular integrity by regulation of endothelial and vascular smooth muscle cell differentiation and proliferation101–103. Moreover, NOTCH1-dependent pathways can accelerate aortic valve calcification104, which develops early in the course of BAV105. Familial studies demonstrate that loss of function mutations in NOTCH1 are responsible for less than 1 WES identified rare and novel protein-altering variants in Notch pathway genes (NOTCH1, ARHGAP31, MAML1, SMARCA4, JARID2, JAG1) that co-segregate with LVOT obstructive phenotypes, including BAV and CoA, in French-Canadian families106. Rare variants of JARID2, a regulator of NOTCH1 expression, were found in WES of 4593 individuals with left-sided obstructive lesions and in families with BAV and aortic dilatation89, 107. To date, rare variants of NOTCH1 are the most commonly reported genetic variant in non-syndromic BAV cohorts6, 108, but in most cases familial segregation or functional studies confirming the pathogenicity of these variants is lacking13,86,91.
2. GATA factors
GATA (GATA binding protein) genes 4–6 encode zinc finger transcription factors that regulate early cardiac gene expression and cardiac cell lineage differentiation109, 110. GATA4 is required for the early stages of heart development, and rare variants of GATA4 are enriched in patients with Tetralogy of Fallot and isolated ventricular septal defects111. GATA5 is expressed in the endocardium and deletion of GATA5 in mice causes partially penetrant BAV112. GATA6 is more directly required for aortic valve development by regulating a conserved semaphorin-plexin pathway in cardiac neural crest cells113. Common variants of GATA4 are associated with BAV, and rare variants of GATA4, GATA5 and GATA6 were identified in candidate gene studies of BAV cohorts and in some families with autosomal dominant inheritance32, 78, 114, 115.
3. SMAD factors
Smad (Mothers against decapentaplegic homolog) proteins are intracellular mediators of signal transduction by TGF-β and bone morphogenetic protein ligands116. Mutations in SMAD3 cause a subtype of Loeys-Dietz syndrome that features a relatively late onset presentation with TAA, BAV and osteoarthritis. Recurrent rare missense and loss of function variants in SMAD4 and SMAD6 were also identified in non-syndromic probands with BAV and TAA52,100,117.
4. ROBO4 in patients with BAV with TAA
WES and familial studies identified recurrent rare variants of the ROBO4 (Roundabout homolog 4) gene in non-syndromic patients who were ascertained due to BAV and TAA. ROBO4 is expressed in endothelial cells, and mutation or targeted silencing of ROBO4 results in vascular defects and EndoMT in animal models86. Rare TBX20 and ADAMTS19 variants were identified in the same cohort, but each potentially accounts for less than 1% of cases26.
5. Familial TAA without syndromic features
Mutations of the ACTA2 gene, which encodes smooth muscle alpha-actin, are the most common cause (10–20%) of heritable non-syndromic TAA118, 119. ACTA2 is required for actin filament assembly and smooth muscle cell contraction120. The prevalence of BAV (3%) is increased in ACTA2 families, who may also manifest various features of a systemic smooth muscle dysfunction syndrome120. However, as with LDS genetic variants, ACTA2 mutations are not frequent in non-syndromic BAV cases121.
6. FBN1
The prevalence of BAV (4%) is also increased in Marfan syndrome, which is caused by mutation of the FBN1 gene. FBN1 encodes an extracellular glycoprotein (fibrillin-1) that is secreted by vascular smooth muscle cells and regulates the structural integrity of the aortic media. FBN1 mutations disrupt smooth muscle cell attachments to arterial elastic laminae, resulting in progressive destruction of the media by matrix metalloproteinases and eventual TAA122, 123. In addition, common variation of FBN1 is associated with non-syndromic TAA124, and rare FBN1 variants were identified in BAV patients who presented with aortic root aneurysms but had no features of Marfan syndrome125. Pathologic analysis of ascending aortic tissue from BAV patients demonstrated deficiency of fibrillin-containing microfibrils compared to tricuspid controls.
Current single gene mutations explain less than 10% of BAV cases. Several potential confounding factors may account for the missing heritability of BAV: 1) Multiple genetic variants in the same individual, affecting more than one candidate gene (compound heterozygotes) could explain up to 20% of BAV cases, based on recent analyses of other exome data; 2) Other types of variants (CNVs > noncoding > epigenetic) that are outside the exome may be discoverable by different technologies such as methylation arrays and whole genome sequencing; 3) The diversity of BAV phenotypes may cause the prevalence of BAV to be underestimated, if some affected individuals are not recognized.
CLINICAL APPLICATIONS OF BAV GENETIC INFORMATION
Family-based screening:
Based on the heritability of BAV in families, the current thoracic aortic disease guidelines recommend echocardiographic screening of all first-degree relatives of BAV probands59, 60. Affected relatives should receive a comprehensive clinical evaluation that includes complete imaging of the heart and thoracic aorta. Genetic tests may be appropriate for BAV patients when recognizable features of single-gene disorders or syndromes are present. If no external features of a genetic syndrome are present, genetic testing is generally reserved for BAV patients with high-risk clinical or imaging features, such as other congenital heart lesions, aneurysms or dissections beyond the proximal aorta, or a family history of dissection or sudden death. If genetic testing is contemplated, genetic counselors can facilitate screening, testing, clinical follow up and surveillance imaging of relatives.
Development of clinical genetic testing for BAV
When referring BAV patients for genetic counseling and testing, accurate phenotypic analysis of all available family members is essential for gene discovery. The variable expressivity of valve and aortic phenotypes in BAV families means that parents who have affected children and are assumed to harbor causative genetic mutations may not have BAV themselves or may have other cardiovascular abnormalities. Lifetime follow up of affected individuals is also essential, because the timing of BAV-related complications such as aortic valve disease or TAD may be very different between individuals, even within the same family.
Table 2 highlights genes that could potentially be included in a commercially available genetic test for BAV, based on current high-quality evidence. Those with familial BAV and TAD, as well as those with early onset complications of BAV, are most likely to have mutations in currently known TAD genes and therefore may represent the subset with the greatest potential benefit from genetic testing. Due to the frequency of rare CNVs in BAV cohorts, it may be reasonable to include SNP or chromosomal microarray analysis with single gene testing. The potential benefits of this approach to reduce healthcare costs by eliminating surveillance imaging of patients who test negative will need to be evaluated in future studies.
CONCLUSION
BAV is a complex disorder that is primarily inherited in an autosomal dominant pattern with incomplete penetrance and variable expressivity. Although BAV is a feature of some genetic syndromes and complex congenital heart defects, most cases are isolated and non-syndromic. Currently known single gene mutations do not explain most non-syndromic BAV cases. Alternative approaches to gene discovery using methods that account for multiple variants or interrogate the noncoding genome, family-based cohorts, and populations with highly penetrant disease may be necessary to discover new BAV genes. Until then, genetic testing should be considered for BAV patients who have features of genetic syndromes or heritable TAD.
ACKNOWLEDGMENTS
This work was supported in part by R01HL137028 (SP).
ABBREVIATIONS
- BAV
bicuspid aortic valve
- CoA
coarctation of the aorta
- CNVs
copy-number variants
- EndMT
endothelial to mesenchymal transition
- GWAS
genome-wide association study
- HLHS
hypoplastic left heart syndrome
- LDS
Loeys-Dietz syndrome
- LVNC
left ventricular noncompaction cardiomyopathy
- LVOT
left ventricular outflow tract
- NGS
next generation sequencing
- SNP
single-nucleotide polymorphism
- TAA
thoracic aortic aneurysm
- TAD
thoracic aortic aneurysm and dissection
- TS
Turner syndrome
- VCFS
Velocardiofacial syndrome
- WES
whole exome sequencing
- WGS
whole genome sequencing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: None
REFERENCES
- 1.Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004;44:138–143. [DOI] [PubMed] [Google Scholar]
- 2.Prakash SK, Bosse Y, Muehlschlegel JD, et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: insights from the International BAVCon (Bicuspid Aortic Valve Consortium). Journal of the American College of Cardiology. 2014;64:832–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balistreri CR, Cavarretta E, Sciarretta S, Frati G. Light on the molecular and cellular mechanisms of bicuspid aortic valve to unveil phenotypic heterogeneity. Journal of molecular and cellular cardiology. 2019;133:113–114. [DOI] [PubMed] [Google Scholar]
- 4.Niaz T, Poterucha JT, Olson TM, et al. Characteristic Morphologies of the Bicuspid Aortic Valve in Patients with Genetic Syndromes. J Am Soc Echocardiogr. 2018;31:194–200. [DOI] [PubMed] [Google Scholar]
- 5.Carlson M, Silberbach M. Dissection of the aorta in Turner syndrome: two cases and review of 85 cases in the literature. BMJ case reports. 2009;2009:bcr0620091998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andreassi MG, Della Corte A. Genetics of bicuspid aortic valve aortopathy. Current opinion in cardiology. 2016;31:585–592. [DOI] [PubMed] [Google Scholar]
- 7.Prakash SK, Bondy CA, Maslen CL, et al. Autosomal and X chromosome structural variants are associated with congenital heart defects in Turner syndrome: The NHLBI GenTAC registry. Am J Med Genet A. 2016;170:3157–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corbitt H, Gutierrez J, Silberbach M, Maslen CL. The genetic basis of Turner syndrome aortopathy. American journal of medical genetics. Part C, Seminars in medical genetics 2019;181:117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corbitt H, Morris SA, Gravholt CH, et al. TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS genetics. 2018;14:e1007692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark EB. Neck web and congenital heart defects: a pathogenic association in 45 X-O Turner syndrome? Teratology. 1984;29:355–361. [DOI] [PubMed] [Google Scholar]
- 11.Patel ND, Crawford T, Magruder JT, et al. Cardiovascular operations for Loeys-Dietz syndrome: Intermediate-term results. The Journal of thoracic and cardiovascular surgery. 2017;153:406–412. [DOI] [PubMed] [Google Scholar]
- 12.Girdauskas E, Schulz S, Borger MA, Mierzwa M, Kuntze T. Transforming growth factor-beta receptor type II mutation in a patient with bicuspid aortic valve disease and intraoperative aortic dissection. The Annals of thoracic surgery. 2011;91:e70–71. [DOI] [PubMed] [Google Scholar]
- 13.Jondeau G, Ropers J, Regalado E, et al. International Registry of Patients Carrying TGFBR1 or TGFBR2 Mutations: Results of the MAC (Montalcino Aortic Consortium). Circ Cardiovasc Genet. 2016;9:548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andelfinger G, Loeys B, Dietz H. A Decade of Discovery in the Genetic Understanding of Thoracic Aortic Disease. The Canadian journal of cardiology. 2016;32:13–25. [DOI] [PubMed] [Google Scholar]
- 15.van de Laar IM, van der Linde D, Oei EH, et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. Journal of medical genetics. 2012;49:47–57. [DOI] [PubMed] [Google Scholar]
- 16.Schepers D, Tortora G, Morisaki H, et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Human mutation. 2018;39:621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. [DOI] [PubMed] [Google Scholar]
- 18.Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. The New England journal of medicine. 2006;355:788–798. [DOI] [PubMed] [Google Scholar]
- 20.Morris SA, Orbach DB, Geva T, Singh MN, Gauvreau K, Lacro RV. Increased vertebral artery tortuosity index is associated with adverse outcomes in children and young adults with connective tissue disorders. Circulation. 2011;124:388–396. [DOI] [PubMed] [Google Scholar]
- 21.Arrington CB, Sower CT, Chuckwuk N, et al. Absence of TGFBR1 and TGFBR2 mutations in patients with bicuspid aortic valve and aortic dilation. The American journal of cardiology. 2008;102:629–631. [DOI] [PubMed] [Google Scholar]
- 22.John AS, McDonald-McGinn DM, Zackai EH, Goldmuntz E. Aortic root dilation in patients with 22q11.2 deletion syndrome. American journal of medical genetics. Part A 2009;149A:939–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yagi H, Furutani Y, Hamada H, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. [DOI] [PubMed] [Google Scholar]
- 24.Gao W, Higaki T, Eguchi-Ishimae M, et al. DGCR6 at the proximal part of the DiGeorge critical region is involved in conotruncal heart defects. Human genome variation. 2015;2:15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Mierop LH, Kutsche LM. Cardiovascular anomalies in DiGeorge syndrome and importance of neural crest as a possible pathogenetic factor. The American journal of cardiology. 1986;58:133–137. [DOI] [PubMed] [Google Scholar]
- 26.Luyckx I, Kumar AA, Reyniers E, et al. Copy number variation analysis in bicuspid aortic valve-related aortopathy identifies TBX20 as a contributing gene. European journal of human genetics : EJHG. 2019;27:1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Digilio MC, Gnazzo M, Lepri F, et al. Congenital heart defects in molecularly proven Kabuki syndrome patients. American journal of medical genetics. Part A 2017;173:2912–2922. [DOI] [PubMed] [Google Scholar]
- 28.Meester JAN, Verstraeten A, Alaerts M, Schepers D, Van Laer L, Loeys BL. Overlapping but distinct roles for NOTCH receptors in human cardiovascular disease. Clinical genetics. 2019;95:85–94. [DOI] [PubMed] [Google Scholar]
- 29.Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annual review of physiology. 2011;73:29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Etiology and Morphogenesis of Congenital Heart Disease. In: Nakanishi TMR, Baldwin HS, Keller BB, Srivastava D, Yamagishi H, ed: Springer International Publishing AG, part of Springer Nature; 2018; 2016:371–376. [Google Scholar]
- 31.Li RG, Xu YJ, Wang J, et al. GATA4 Loss-of-Function Mutation and the Congenitally Bicuspid Aortic Valve. The American journal of cardiology. 2018;121:469–474. [DOI] [PubMed] [Google Scholar]
- 32.Xu YJ, Di RM, Qiao Q, et al. GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene. 2018;663:115–120. [DOI] [PubMed] [Google Scholar]
- 33.Pasipoularides A. Calcific Aortic Valve Disease: Part 1--Molecular Pathogenetic Aspects, Hemodynamics, and Adaptive Feedbacks. Journal of cardiovascular translational research. 2016;9:102–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasipoularides A Clinical-pathological correlations of BAV and the attendant thoracic aortopathies. Part 2: Pluridisciplinary perspective on their genetic and molecular origins. Journal of molecular and cellular cardiology. 2019;133:233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. [DOI] [PubMed] [Google Scholar]
- 36.Koenig SN, Bosse KM, Nadorlik HA, Lilly B, Garg V. Evidence of Aortopathy in Mice with Haploinsufficiency of Notch1 in Nos3-Null Background. Journal of cardiovascular development and disease. 2015;2:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Etiology and Morphogenesis of Congenital Heart Disease. In: Nakanishi TMR, Baldwin HS, Keller BB, Srivastava D, Yamagishi H, ed: Springer International Publishing AG, part of Springer Nature; 2018; 2016:103–114. [Google Scholar]
- 38.Kostina AS, Uspensky Vcapital Ie C, Irtyuga OB, et al. Notch-dependent EMT is attenuated in patients with aortic aneurysm and bicuspid aortic valve. Biochimica et biophysica acta. 2016;1862:733–740. [DOI] [PubMed] [Google Scholar]
- 39.Koenig SN, Lincoln J, Garg V. Genetic basis of aortic valvular disease. Current opinion in cardiology. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koenig SN, Bosse K, Majumdar U, Bonachea EM, Radtke F, Garg V. Endothelial Notch1 Is Required for Proper Development of the Semilunar Valves and Cardiac Outflow Tract. Journal of the American Heart Association. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quaegebeur JM, Jonas RA, Weinberg AD, Blackstone EH, Kirklin JW. Outcomes in seriously ill neonates with coarctation of the aorta. A multiinstitutional study. The Journal of thoracic and cardiovascular surgery. 1994;108:841–851; discussion 852–844. [PubMed] [Google Scholar]
- 42.Sinning C, Zengin E, Kozlik-Feldmann R, et al. Bicuspid aortic valve and aortic coarctation in congenital heart disease-important aspects for treatment with focus on aortic vasculopathy. Cardiovascular diagnosis and therapy. 2018;8:780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyabara S, Nakayama M, Suzumori K, Yonemitsu N, Sugihara H. Developmental analysis of cardiovascular system of 45,X fetuses with cystic hygroma. American journal of medical genetics. 1997;68:135–141. [DOI] [PubMed] [Google Scholar]
- 44.Seagraves NJ, McBride KL. Cardiac teratogenicity in mouse maternal phenylketonuria: defining phenotype parameters and genetic background influences. Molecular genetics and metabolism. 2012;107:650–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aslam S, Khairy P, Shohoudi A, et al. Shone Complex: An Under-recognized Congenital Heart Disease With Substantial Morbidity in Adulthood. The Canadian journal of cardiology. 2017;33:253–259. [DOI] [PubMed] [Google Scholar]
- 46.Hickey EJ, Caldarone CA, McCrindle BW. Left ventricular hypoplasia: a spectrum of disease involving the left ventricular outflow tract, aortic valve, and aorta. Journal of the American College of Cardiology. 2012;59:S43–54. [DOI] [PubMed] [Google Scholar]
- 47.Hinton RB, Martin LJ, Rame-Gowda S, Tabangin ME, Cripe LH, Benson DW. Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. Journal of the American College of Cardiology. 2009;53:1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 1990;82:507–513. [DOI] [PubMed] [Google Scholar]
- 49.Stahli BE, Gebhard C, Biaggi P, et al. Left ventricular non-compaction: prevalence in congenital heart disease. International journal of cardiology. 2013;167:2477–2481. [DOI] [PubMed] [Google Scholar]
- 50.Agarwal A, Khandheria BK, Paterick TE, Treiber SC, Bush M, Tajik AJ. Left ventricular noncompaction in patients with bicuspid aortic valve. Journal of the American Society of Echocardiography : official publication of the American Society of Echocardiography. 2013;26:1306–1313. [DOI] [PubMed] [Google Scholar]
- 51.Basu R, Hazra S, Shanks M, Paterson DI, Oudit GY. Novel mutation in exon 14 of the sarcomere gene MYH7 in familial left ventricular noncompaction with bicuspid aortic valve. Circulation. Heart failure 2014;7:1059–1062. [DOI] [PubMed] [Google Scholar]
- 52.Ciotti GR, Vlahos AP, Silverman NH. Morphology and function of the bicuspid aortic valve with and without coarctation of the aorta in the young. The American journal of cardiology. 2006;98:1096–1102. [DOI] [PubMed] [Google Scholar]
- 53.Verma S, Siu SC. Aortic dilatation in patients with bicuspid aortic valve. The New England journal of medicine. 2014;370:1920–1929. [DOI] [PubMed] [Google Scholar]
- 54.Michelena HI, Khanna AD, Mahoney D, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA. 2011;306:1104–1112. [DOI] [PubMed] [Google Scholar]
- 55.Siu SC, Silversides CK. Bicuspid aortic valve disease. Journal of the American College of Cardiology. 2010;55:2789–2800. [DOI] [PubMed] [Google Scholar]
- 56.Huntington K, Hunter AG, Chan KL. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J Am Coll Cardiol. 1997;30:1809–1812. [DOI] [PubMed] [Google Scholar]
- 57.Loscalzo ML, Goh DL, Loeys B, Kent KC, Spevak PJ, Dietz HC. Familial thoracic aortic dilation and bicommissural aortic valve: a prospective analysis of natural history and inheritance. Am J Med Genet A. 2007;143A:1960–1967. [DOI] [PubMed] [Google Scholar]
- 58.Longobardo L, Jain R, Carerj S, Zito C, Khandheria BK. Bicuspid Aortic Valve: Unlocking the Morphogenetic Puzzle. The American journal of medicine. 2016;129:796–805. [DOI] [PubMed] [Google Scholar]
- 59.Guntheroth WG. A critical review of the American College of Cardiology/American Heart Association practice guidelines on bicuspid aortic valve with dilated ascending aorta. Am J Cardiol. 2008;102:107–110. [DOI] [PubMed] [Google Scholar]
- 60.Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology,American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Journal of the American College of Cardiology. 2010;55:e27–e129. [DOI] [PubMed] [Google Scholar]
- 61.Hanchard NA, Swaminathan S, Bucasas K, et al. A genome-wide association study of congenital cardiovascular left-sided lesions shows association with a locus on chromosome 20. Human molecular genetics. 2016;25:2331–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hinton RB Jr., Martin LJ, Tabangin ME, Mazwi ML, Cripe LH, Benson DW. Hypoplastic left heart syndrome is heritable. Journal of the American College of Cardiology. 2007;50:1590–1595. [DOI] [PubMed] [Google Scholar]
- 63.Lewin MB, McBride KL, Pignatelli R, et al. Echocardiographic evaluation of asymptomatic parental and sibling cardiovascular anomalies associated with congenital left ventricular outflow tract lesions. Pediatrics. 2004;114:691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McBride KL, Pignatelli R, Lewin M, et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: Segregation, multiplex relative risk, and heritability. American journal of medical genetics. Part A 2005;134A:180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Galian-Gay L, Carro Hevia A, Teixido-Tura G, et al. Familial clustering of bicuspid aortic valve and its relationship with aortic dilation in first-degree relatives. Heart. 2019;105:603–608. [DOI] [PubMed] [Google Scholar]
- 66.Capoulade R, Schott JJ, Le Tourneau T. Familial bicuspid aortic valve disease: should we look more closely at the valve? Heart. 2019;105:584–586. [DOI] [PubMed] [Google Scholar]
- 67.Glazier AM, Nadeau JH, Aitman TJ. Finding genes that underlie complex traits. Science. 2002;298:2345–2349. [DOI] [PubMed] [Google Scholar]
- 68.Page GP, George V, Go RC, Page PZ, Allison DB. “Are we there yet?”: Deciding when one has demonstrated specific genetic causation in complex diseases and quantitative traits. American journal of human genetics. 2003;73:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Balistreri CR, Forte M, Greco E, et al. An overview of the molecular mechanisms underlying development and progression of bicuspid aortic valve disease. Journal of molecular and cellular cardiology. 2019;132:146–153. [DOI] [PubMed] [Google Scholar]
- 70.Rashedi N, Otto CM. Aortic Stenosis: Changing Disease Concepts. Journal of cardiovascular ultrasound. 2015;23:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Strande NT, Riggs ER, Buchanan AH, et al. Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. American journal of human genetics. 2017;100:895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic Bases of Bicuspid Aortic Valve: The Contribution of Traditional and High-Throughput Sequencing Approaches on Research and Diagnosis. Frontiers in physiology. 2017;8:612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schnell AWJ. Family-Based Study Designs. Molecular Epidemiology 2008. [Google Scholar]
- 74.Duggal P, Ladd-Acosta C, Ray D, Beaty TH. The Evolving Field of Genetic Epidemiology: From Familial Aggregation to Genomic Sequencing. American journal of epidemiology. 2019;188:2069–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. American journal of human genetics. 2014;95:5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hopper JL, Bishop DT, Easton DF. Population-based family studies in genetic epidemiology. Lancet. 2005;366:1397–1406. [DOI] [PubMed] [Google Scholar]
- 77.Bush WS, Moore JH. Chapter 11: Genome-wide association studies. PLoS computational biology. 2012;8:e1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang B, Zhou W, Jiao J, et al. Protein-altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nature communications. 2017;8:15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fulmer D, Toomer K, Guo L, et al. Defects in the Exocyst-Cilia Machinery Cause Bicuspid Aortic Valve Disease and Aortic Stenosis. Circulation. 2019;140:1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wooten EC, Iyer LK, Montefusco MC, et al. Application of gene network analysis techniques identifies AXIN1/PDIA2 and endoglin haplotypes associated with bicuspid aortic valve. PloS one. 2010;5:e8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bjornsson T, Thorolfsdottir RB, Sveinbjornsson G, et al. A rare missense mutation in MYH6 associates with non-syndromic coarctation of the aorta. European heart journal. 2018;39:3243–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dawn Teare M, Barrett JH. Genetic linkage studies. Lancet. 2005;366:1036–1044. [DOI] [PubMed] [Google Scholar]
- 83.Gago-Diaz M, Brion M, Gallego P, et al. The genetic component of bicuspid aortic valve and aortic dilation. An exome-wide association study. Journal of molecular and cellular cardiology. 2017;102:3–9. [DOI] [PubMed] [Google Scholar]
- 84.Muzzey D, Evans EA, Lieber C. Understanding the Basics of NGS: From Mechanism to Variant Calling. Current genetic medicine reports. 2015;3:158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo DC, Gong L, Regalado ES, et al. MAT2A mutations predispose individuals to thoracic aortic aneurysms. American journal of human genetics. 2015;96:170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gould RA, Aziz H, Woods CE, et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nature genetics. 2019;51:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shi LM, Tao JW, Qiu XB, et al. GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. International journal of molecular medicine. 2014;33:1219–1226. [DOI] [PubMed] [Google Scholar]
- 88.Lin X, Liu X, Wang L, et al. Targeted next-generation sequencing identified ADAMTS5 as novel genetic substrate in patients with bicuspid aortic valve. International journal of cardiology. 2018;252:150–155. [DOI] [PubMed] [Google Scholar]
- 89.Gillis E, Kumar AA, Luyckx I, et al. Candidate Gene Resequencing in a Large Bicuspid Aortic Valve-Associated Thoracic Aortic Aneurysm Cohort: SMAD6 as an Important Contributor. Frontiers in physiology. 2017;8:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bonachea EM, Zender G, White P, et al. Use of a targeted, combinatorial next-generation sequencing approach for the study of bicuspid aortic valve. BMC medical genomics. 2014;7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pileggi S, De Chiara B, Magnoli M, et al. Sequencing of NOTCH1 gene in an Italian population with bicuspid aortic valve: Preliminary results from the GISSI OUTLIERS VAR study. Gene. 2019;715:143970. [DOI] [PubMed] [Google Scholar]
- 92.Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annual review of genomics and human genetics. 2009;10:451–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Prakash S, Kuang SQ, Gen TACRI, Regalado E, Guo D, Milewicz D. Recurrent Rare Genomic Copy Number Variants and Bicuspid Aortic Valve Are Enriched in Early Onset Thoracic Aortic Aneurysms and Dissections. PloS one. 2016;11:e0153543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Greenway SC, Pereira AC, Lin JC, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nature genetics. 2009;41:931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Warburton D, Ronemus M, Kline J, et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Human genetics. 2014;133:11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hui DS, Bonow RO, Stolker JM, Braddock SR, Lee R. Discordant Aortic Valve Morphology in Monozygotic Twins: A Clinical Case Series. JAMA cardiology. 2016;1:1043–1047. [DOI] [PubMed] [Google Scholar]
- 97.Haunschild J, Schellinger IN, Barnard SJ, et al. Bicuspid aortic valve patients show specific epigenetic tissue signature increasing extracellular matrix destruction. Interactive cardiovascular and thoracic surgery. 2019;29:937–943. [DOI] [PubMed] [Google Scholar]
- 98.Tobin SW, Alibhai FJ, Lee MM, et al. Novel mediators of aneurysm progression in bicuspid aortic valve disease. Journal of molecular and cellular cardiology. 2019;132:71–83. [DOI] [PubMed] [Google Scholar]
- 99.Martin PS, Kloesel B, Norris RA, Lindsay M, Milan D, Body SC. Embryonic Development of the Bicuspid Aortic Valve. Journal of cardiovascular development and disease. 2015;2:248–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.MacGrogan D, Luna-Zurita L, de la Pompa JL. Notch signaling in cardiac valve development and disease. Birth defects research. Part A, Clinical and molecular teratology. 2011;91:449–459. [DOI] [PubMed] [Google Scholar]
- 101.Balistreri CR, Crapanzano F, Schirone L, et al. Deregulation of Notch1 pathway and circulating endothelial progenitor cell (EPC) number in patients with bicuspid aortic valve with and without ascending aorta aneurysm. Scientific reports. 2018;8:13834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee A, Wei S, Schwertani A. A Notch more: Molecular players in bicuspid aortic valve disease. Journal of molecular and cellular cardiology. 2019;134:62–68. [DOI] [PubMed] [Google Scholar]
- 103.Harrison OJ, Visan AC, Moorjani N, et al. Defective NOTCH signaling drives increased vascular smooth muscle cell apoptosis and contractile differentiation in bicuspid aortic valve aortopathy: A review of the evidence and future directions. Trends in cardiovascular medicine. 2019;29:61–68. [DOI] [PubMed] [Google Scholar]
- 104.Ducharme V, Guauque-Olarte S, Gaudreault N, Pibarot P, Mathieu P, Bosse Y. NOTCH1 genetic variants in patients with tricuspid calcific aortic valve stenosis. The Journal of heart valve disease. 2013;22:142–149. [PubMed] [Google Scholar]
- 105.Kostina A, Shishkova A, Ignatieva E, et al. Different Notch signaling in cells from calcified bicuspid and tricuspid aortic valves. Journal of molecular and cellular cardiology. 2018;114:211–219. [DOI] [PubMed] [Google Scholar]
- 106.Preuss C, Capredon M, Wunnemann F, et al. Family Based Whole Exome Sequencing Reveals the Multifaceted Role of Notch Signaling in Congenital Heart Disease. PLoS genetics. 2016;12:e1006335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li AH, Hanchard NA, Furthner D, et al. Whole exome sequencing in 342 congenital cardiac left sided lesion cases reveals extensive genetic heterogeneity and complex inheritance patterns. Genome medicine. 2017;9:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Foffa I, Ait Ali L, Panesi P, et al. Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC medical genetics. 2013;14:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mozzini C, Girelli D, Cominacini L, Soresi M. An Exploratory Look at Bicuspid Aortic Valve (Bav) Aortopathy: Focus on Molecular and Cellular Mechanisms. Current problems in cardiology. 2019:100425. [DOI] [PubMed] [Google Scholar]
- 110.Lentjes MH, Niessen HE, Akiyama Y, de Bruine AP, Melotte V, van Engeland M. The emerging role of GATA transcription factors in development and disease. Expert reviews in molecular medicine. 2016;18:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McCulley DJ, Black BL. Transcription factor pathways and congenital heart disease. Current topics in developmental biology. 2012;100:253–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. The Journal of clinical investigation. 2011;121:2876–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Peterkin T, Gibson A, Loose M, Patient R. The roles of GATA-4, −5 and −6 in vertebrate heart development. Seminars in cell & developmental biology. 2005;16:83–94. [DOI] [PubMed] [Google Scholar]
- 114.Alonso-Montes C, Martin M, Martinez-Arias L, et al. Variants in cardiac GATA genes associated with bicuspid aortic valve. European journal of clinical investigation. 2018;48:e13027. [DOI] [PubMed] [Google Scholar]
- 115.Gharibeh L, Komati H, Bosse Y, et al. GATA6 Regulates Aortic Valve Remodeling, and Its Haploinsufficiency Leads to Right-Left Type Bicuspid Aortic Valve. Circulation. 2018;138:1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Imamura T, Takase M, Nishihara A, et al. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997;389:622–626. [DOI] [PubMed] [Google Scholar]
- 117.Luyckx I, MacCarrick G, Kempers M, et al. Confirmation of the role of pathogenic SMAD6 variants in bicuspid aortic valve-related aortopathy. European journal of human genetics : EJHG. 2019;27:1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Guo DC, Papke CL, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. American journal of human genetics. 2009;84:617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Regalado ES, Guo DC, Prakash S, et al. Aortic Disease Presentation and Outcome Associated With ACTA2 Mutations. Circulation. Cardiovascular genetics 2015;8:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nature genetics. 2007;39:1488–1493. [DOI] [PubMed] [Google Scholar]
- 121.Tortora G, Wischmeijer A, Berretta P, et al. Search for genetic factors in bicuspid aortic valve disease: ACTA2 mutations do not play a major role. Interactive cardiovascular and thoracic surgery. 2017;25:813–817. [DOI] [PubMed] [Google Scholar]
- 122.Fedak PW, de Sa MP, Verma S, et al. Vascular matrix remodeling in patients with bicuspid aortic valve malformations: implications for aortic dilatation. The Journal of thoracic and cardiovascular surgery. 2003;126:797–806. [DOI] [PubMed] [Google Scholar]
- 123.Nistri S, Grande-Allen J, Noale M, et al. Aortic elasticity and size in bicuspid aortic valve syndrome. European heart journal. 2008;29:472–479. [DOI] [PubMed] [Google Scholar]
- 124.LeMaire SA, McDonald ML, Guo DC, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nature genetics. 2011;43:996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pepe G, Nistri S, Giusti B, et al. Identification of fibrillin 1 gene mutations in patients with bicuspid aortic valve (BAV) without Marfan syndrome. BMC medical genetics. 2014;15:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kovacic JC, Dimmeler S, Harvey RP, et al. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. Journal of the American College of Cardiology. 2019;73:190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wunnemann F, Ta-Shma A, Preuss C, et al. Loss of ADAMTS19 causes progressive non-syndromic heart valve disease. Nature genetics. 2020;52:40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Helgadottir A, Thorleifsson G, Gretarsdottir S, et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nature communications. 2018;9:987. [DOI] [PMC free article] [PubMed] [Google Scholar]