

Abstract
Motivation:
Trillions of bacteria in human body (human microbiota) affect human health and diseases by controlling host functions through small molecule metabolites. An accurate and comprehensive catalog of the metabolic output from human microbiota is critical for our deep understanding of how microbial metabolism contributes to human health. The large number of published biomedical research articles is a rich resource of microbiome studies. However, automatically extracting microbial metabolites from free-text documents and differentiating them from other human metabolites is a challenging task. Here we developed an integrated approach called Co-occurrence Metabolite Network Ranking (CoMNRank) by combining named entity extraction, network construction and topic sensitive network-based prioritization to extract and prioritize microbial metabolites from biomedical articles.
Methods:
The text data included 28,851,232 MEDLINE records. CoMNRank consists of three steps: (1) extraction of human metabolites from MEDLINE records; (2) construction of a weighted co-occurrence metabolite network (CoMN); (3) prioritization and differentiation of microbial metabolites from other human metabolites.
Results:
For the first step of CoMNRank, we extracted 11,846 human metabolites from MEDLINE articles, with a baseline performance of precision of 0.014, recall of 0.959 and F1 of 0.028. We then constructed a weighted CoMN of 6,996 nodes and 986,186 edges. CoMNRank effectively prioritized microbial metabolites: the precision of top ranked metabolites is 0.45, a 31-fold enrichment as compared to the overall precision of 0.014. Manual curation of top 100 metabolites showed a true precision of 0.67, among which 48% true positives are not captured by existing databases.
Conclusion:
Our study sets the foundation for future tasks of microbial entity and relationship extractions as well as data-driven studies of how microbial metabolism contributes to human health and diseases.
*Graphical Abstract
1. Introduction
Human microbiota is an important modifiable environmental factor and a part of the ecosystem of human body (Turnbaugh et al., 2007). These microbiota exists in symbiotic relation with human hosts by controlling both the metabolism and immune balance of the human body (Gill et al., 2006; Cho et al., 2012; Sommer et al., 2013; Gilbert et al., 2018). Strong evidence shows that the metabolism of human microbiota influences human health and well-being (Gill et al., 2006; Tremaroli et al., 2012; Nicholson et al., 2012; Trompette et al., 2014; Koppel et al., 2017; Tang et al., 2018). Recent studies showed that gut microbial metabolites are involved in many common complex diseases, including cardiovascular diseases (Tang et al., 2018; Brown et al., 2018), metabolic syndrome (Canfora et al., 2019), neuropsychiatric disorders (Hsiao et al., 2013; Smith., 2015; Valles-Colomer et al., 2019), cancers (Smith et al., 2013; Schwabe et al., 2013; Louis et al., 2014), and immune disorders (Rooks et al., 2016; Marino et al., 2017; Jangi et al., 2016; Haase et al., 2018).
We have recently developed data-driven computational approaches to understand how microbial metabolites contribute to human diseases, including colorectal cancer (Xu et al., 2015; Wang et al., 2018), Alzheimer’s disease (Xu et al., 2016), psoriasis (Wang et al., 2017), and rheumatoid arthritis (Wang et al., 2017, 2019). We used 172 known microbial metabolites from the Human Metabolome Database (HMDB), which is currently the most comprehensive human metabolome database of over 114,100 small molecule metabolites found in the human body (Wishart et al., 2013). Our studies have shown that the interactions of microbial metabolites with human host genetics have long-ranging systematic effects on intestine, joints, skin and brain. For example, our studies revealed strong mechanistic links between trimethylamine N-oxide (TMAO), a gut microbial metabolite of dietary meat and fat, and both colorectal cancer (Xu et al., 2015) and Alzheimer’s diseases (Xu et al., 2016). These computation-based findings were verified by other studies showing that patient plasma level of TMAO is positively associated with colorectal cancer risk (Bae et al., 2014) and that TMAO is elevated in the cerebrospinal fluid of Alzheimer’s disease patients (Vogt et al., 2018). Our previous data-driven studies critically depended on the coverage of microbial metabolites from HMDB.
The field of microbiome research is fast moving with an increasing number of microbial metabolites being identified and published in the literature. During our previous studies, we found that many microbial metabolites have been reported in biomedical literature, but not captured by HMDB. For example, the sentence “The dietary treatment also boosted serum concentrations of bacterial metabolites, including choline, glycerophosphorylcholine, dimethylamine, trimethylamine, trimethylamine-N-oxide, lactate, and succinate.” (PMID 29563518). The phrase ‘bacterial metabolites’ clearly states that all seven metabolites in the sentence are originated in microbes. Though HMDB include all these metabolites as these metabolites are indeed present in human body, three of them (choline, glycerophosphorylcholine, and lactate) are not classified as microbial metabolites. Another example is the sentence “We further investigate the bioactivity of the confirmed metabolites, and identify two microbiota-generated metabolites 5-hydroxy-L-tryptophan and salicylate as activators of the aryl hydrocarbon receptor” (PMID 25411059). HMDB includes both 5-hydroxy-L-tryptophan and salicylate but did not classify them as microbial metabolites. Consequently, our previous studies missed these five microbial metabolites, even though they are shown to be involved in important biologic process and human diseases (Hinz et al., 2012; Pincemail, 1995). Therefore, an accurate and comprehensive catalog of microbial metabolites that are also present in human body (“human microbial metabolites”) is the key to data-driven studies of how human microbial metabolism affects human health.
2. Approach
Microbial metabolites are defined as metabolites produced, but not necessarily exclusively, by microbes. While some microbial metabolites are exclusively produced by microbes, many others are produced by both human hosts and microbes. The large number of published biomedical research articles is a rich resource for building a catalog of human microbial metabolites as shown by above two examples. Automatic extraction of biomedical entities and relationships from free-text biomedical documents is in general a difficult task(Xu et al., 2013; Wang et al., 2013; Xu et al., 2014; Wang et al., 2018). Compared other biomedical natural language processing (NLP) tasks, extraction of machine-understandable knowledge of microbiome in human diseases from published biomedical research articles is a challenging task and less explored (Badal et al., 2019). Recently, researchers developed natural language processing and text mining techniques to extract disease-microbe (bacteria) relationship from published biomedical literature (Ma et al., 2019; Janssens et al., 2019). However, currently no research efforts have been devoted to extract microbial metabolites from biomedical literature. Compared to other biomedical entity extractions, extracting human microbial metabolites from free-text faces special challenges. First, metabolites found in human body can originate from many different resources, including human hosts, plants, foods, microbes, toxins, pollutants, cosmetics, drugs, among others. Metabolites originated from human microbiota constitute only a very small portion of human metabolome. For example, the list of 172 microbial metabolites in HMDB constitutes only 0.15% of 114,100 human metabolites. Second, the majority of metabolites from biomedical articles are not of microbial origin. Consequently, automatically extracting microbial metabolites from a large number of biomedical articles and differentiating them from other human metabolites is a challenging “finding a needle in haystack” task.
The goal of this study is to automatically prioritize microbial metabolites among all human metabolites in HMDB by leveraging their occurrence in biomedical literature. In this study, we developed an integrated approach called Co-occurrence Metabolite Network Ranking (CoMNRank) by combining named entity extraction (NER), network construction and topic sensitive network-based signal prioritization to extract and prioritize microbial metabolites from over 28 million MEDLINE reicords. As shown in the above two examples (PMID 29563518, PMID 25411059), the five missed microbial metabolites are included in HMDB indicating that they are present in human body, but they are not classified as microbial origin. We leveraged the large list of human metabolites from HMDB and performed a dictionary-based NER to extract tens of thousands of human metabolites from MEDLINE records. Though this baseline NER had low precision, it had a high recall. Importantly, this human metabolome-based NER step ensures extraction of only metabolites present in human body and exclusion of metabolites only present in soil, oceans, among others (“Human Metabolites vs Other”).
Since the majority of human metabolites extracted from published biomedical articles are not microbial metabolites, we then constructed a co-occurrence metabolite network (CoMN) and developed a topic sensitive network-based approach to further differentiate microbial metabolites from other metabolites (“Human Microbial Metabolites vs Other”). The CoMN-based ranking algorithm is based on three hypotheses: (1) if an article contains a known microbial metabolite (e.g., butyric acid, trimethylamine n-oxide), then the article is likely related to microbial metabolism (“Implicit Text Classification”); (2) other metabolites extracted from microbial metabolism-related articles are likely microbial metabolites (“Implicit Microbial Metabolite Classification”); and (3) if a metabolite co-occurs with multiple known microbial metabolites many times, the metabolite is more likely a microbial metabolite than others co-occurring with none or few known microbial metabolites few times in the entire MEDLINE collection (“Prioritization”). For example, the sentence “The dietary treatment also boosted serum concentrations of bacterial metabolites, including choline, glycerophosphorylcholine, dimethylamine, trimethylamine, trimethylamine-N-oxide, lactate, and succinate.” (PMID 29563518) contains a well-known microbial metabolite “trimethylamine-N-oxide”. Based on our assumption, this sentence is likely from a microbial metabolism-related study. Other metabolites (choline, glycerophosphorylcholine, dimethylamine, trimethylamine, lactate, and succinate) from this sentence are likely microbial metabolites. Indeed, all seven metabolites from this sentence are of microbial origin and only 4 are captured in HMDB as microbial metabolites.
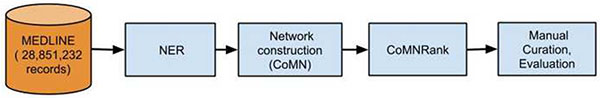
CoMNRank consists of three steps: NER, network construction and prioritization. After the baseline NER step (human metabolite extraction of low precision and high recall), we built a weighted co-occurrence metabolite network (CoMN) where nodes are metabolites that have appeared in MEDLINE, and edges represent the number of MEDLINE articles where any two metabolites co-occurred. We then used known microbial metabolites as seeds to prioritize other microbial metabolites from the weighted CoMN. We demonstrated the robustness of this approach by investigating how different seeds affected the overall performance. We manually curated top ranked metabolites and built a list of true microbial metabolites.
In summary, CoMNRank will set the strong foundation for future tasks of microbial metabolite entity and relationship extractions. A comprehensive list of human microbial metabolites will greatly facilitate data-driven studies of how human microbial metabolism contributes to human health and diseases as demonstrated in our study.
3. Methods
CoMNRank consists of three steps: (1) human metabolite extraction; (2) construction of Co-occurrence Metabolite Network (coMN); and (3) priorization of microbial metabolites from CoMN. We evaluated the comparative performance of different algorithm configurations for both metabolite extraction and prioritization using known microbial metabolites. We performed manual curation to calculate the true precision of top ranked metabolites.
3.1. Human metabolite extraction and evaluation
3.1.1. Human metabolite extraction
A total of 28,851,232 MEDLINE records (published up to July, 2018) were downloaded from the National Library of Medicine (MEDLINE., 2018). The MEDLINE fields of Title, Abstract, MeshHeadings, Keywords, and Chemicals, separately or combined, were used for metabolite extraction. We performed dictionary-based NER to extract human metabolites and to exclude metabolites present in soil, oceans and others. We built a lexicon consisting of all human metabolites from HMDB (Wishart et al., 2013), including preferred names (e.g., butyric acid) and their synonyms (e.g., 1-butanoate, butyrate). We experimented with two versions of HMDB: the year 2017 version and the most updated year 2018 version. The lexicon based on the 2017 version comprised of 42,003 unique metabolite concepts, 188,153 synonyms and 365,632 synonym→concept mappings. The lexicon based on the 2018 version is significantly larger and contains 114,100 unique metabolites, 510,603 synonyms and 11,131,600 synonym→concept mappings. Both versions of metabolite lexicons were used to recognize metabolites and their synonyms from MEDLINE records. Extracted entities were normalized by mapping synonyms to their preferred names (e.g., butyrate→butyric acid).
3.1.2. Evaluation of NER
We evaluated the performance of the baseline NER using the 172 known microbial metabolites from HMDB. Standard measures of precision (fraction of recognized entities as positive that are truly positive), recall (true positive rate) and F1 (harmonic average of the precision and recall) were calculated and compared. Since many microbial metabolites are reported in literature but not captured by the list of 172 microbial metabolites from HMDB, the calculated precision significantly underestimated the true precision. Therefore, these 172 known microbial metabolites were used to compare relative performances of different approaches (not for true precision calculation). We investigated which fields of MEDLINE records and which version of the HMDB-based metabolite lexicons to be used for microbial metabolite extraction. We evaluated the coverage of 172 known microbial metabolites from HMDB in each of the MEDLINE field. The fields with maximal coverage were used for subsequent NER, CoMN construction and prioritization. For example, the field “MeshHeadings” of all 28,851,232 MEDLINE records captured 31.5% of the 172 known microbial metabolites. The field “Chemicals” captured 70.9% and the combined all five fields captured as much as 95% of known microbial metabolites. We also compared the coverage of NER using the two versions of HMDB-based metabolite lexicons and the one with better performance was used subsequently.
3.2. Construct co-occurrence metabolite network (CoMN)
We built a weighted co-occurrence metabolite network (CoMN). Nodes on CoMN represent metabolites that appeared in MEDLINE records. The edge between two metabolites represents the number of MELDINE records in which these two metabolites appeared together. To investigate how the edge weighting affects subsequent prioritization, we also constructed a unweighted CoMN, where the edge (with weight of 1) represents the fact that two metabolites co-occurred at least once.
3.3. CoMN-based prioritization and evaluation
The majority of metabolites extracted from MEDLINE records are not microbial metabolites, meaning that the baseline NER without further prioritization will have an extremely low precision. We developed a topic sensitive CoMN-based approach to prioritize microbial metabolites from extracted metabolites. We used known microbial metabolites as seeds (i.e.,”microbial metabolism topic”) to prioritize other microbial metabolites from CoMN.
3.3.1. CoMN-based prioritization
The input to the prioritization algorithm is a vector consisting of one or more known microbial metabolites. A probability of 1.0 is assigned to the input seed if the input vector consists of one metabolite and 0.25 if the input vector consists of 4 seeds. The output is a list of metabolites prioritized based on their co-occurrence relevance to the input seeds. We used the standard network-based ranking algorithms, which we previously applied to prioritize disease genes (Chen et al., 2015, 2017), drug candidates (Xu et al., 2015, 2016; Wang et al., 2018), drug targets (Zhou et al., 2018), and disease-microbial metabolite associations (Xu et al., 2015, 2016). Given an input vector consisting of known microbial metabolites, the ranking scores for all metabolites on the CoMN are iteratively updated by:
| (1) |
where S(k+1) is the score vector at step k + 1, S0 is the initial vector, and 1 – α is the restarting probability, M is the transition matrix of the CoMN, with normalized edge weights. We used the restarting probabilities α of 0.7, which was used in our previous studies. We also experimented with different restarting probabilities α and found that there were no significant difference on the overall performance. We also experimented with (1) different types of seeds (microbial metabolite seeds, non-microbial metabolites, and no seeds); (2) seeds with different MEDLINE frequency; (3) different number of microbial metabolites as seeds; and (4) weighted vs un-weighted CoMNs. The performances of CoMNRank with these different configurations were evaluated and compared.
3.3.2. Evaluation of prioritization
We used Precision-Recall (PR) curves (instead of receiver operating characteristic curve, or ROC curve) to evaluate and compare different prioritization methods. PR curves are often used to evaluate ranked results in information retrieval and classification (Manning et al., 2008; Davis et al., 2006). Since microbial metabolites constitutes only a tiny portion of all extracted human metabolites, PR curves provides a more informative and accurate measure of the algorithm performance than ROC curves for any highly skewed dataset (Davis et al., 2006). A PR space is defined as precision and recall as x and y axes, respectively. Using the 172 microbial metabolites from HMDB as the evaluation dataset, we calculated precisions at 11 different recall cutoffs (0.05, 0.1, 0.2, … 1.0) and plotted the PR curves for CoMNRank of different configurations. Since many microbial metabolites from literature are not captured by these 172 microbial metabolites, PR curves calculated with this evaluation dataset likely underestimate true performance of ranking algorithms. Therefore these 172 microbial metabolites were only used to compare relative performances of different approaches.
3.4. Manual curation and evaluation
We manually curated top 100 metabolites by the two authors reading the MEDLINE articles where the metabolites appear. The PubMed identifiers (PMIDs) of the MEDLINE articles with supporting evidence were extracted and associated with each identified microbial metabolites. The manual curation was labor-intensive requiring reading thousands of articles, therefore we only manually curated top 100 ranked metabolites. After we demonstrated that CoMNRank was able to enrich true positive among top as evaluated with the 172 known microbial metabolites, the goal of manual curation is to calculate the true precision of top ranked metabolites and to show that many true positives among top ranked metabolites are not captured by existing databases. We obtained a total of 220 microbial metabolites by combining 172 known microbial metabolites from HMDB with the additional 48 manually curated microbial metabolites. Manual curation data along with other data is publicly available at nlp.case.edu/public/data/CoMNRank.
4. Results
4.1. Distribution of known microbial metabolites in MEDLINE records and performance of the baseline extraction step
We investigated which MELDINE fields had the best coverage of known microbial metabolites. As expected, the best coverage was achieved when all five fields of MEDLINE records (MeshHeadings, Chemicals, Keywords, Title and Abstract) were used (Figure 1). A total of 22.0%,70.9% and 76.2% of the 172 known microbial metabolites appeared in the structured fields of MeshHeadings, Chemicals, and Keywords, respectively. When these three structured fields were combined, a coverage of 88.9% was achieved. When the free-text fields of Title and Abstract were used in addition to the structured fields, a coverage of 95.9% was achieved. This high coverage (or recall) demonstrates that these five fields of MELDLINE records captures the majority of known microbial metabolites and can serve as a rich resource for extracting not-yet-captured microbial metabolites.
Fig. 1.
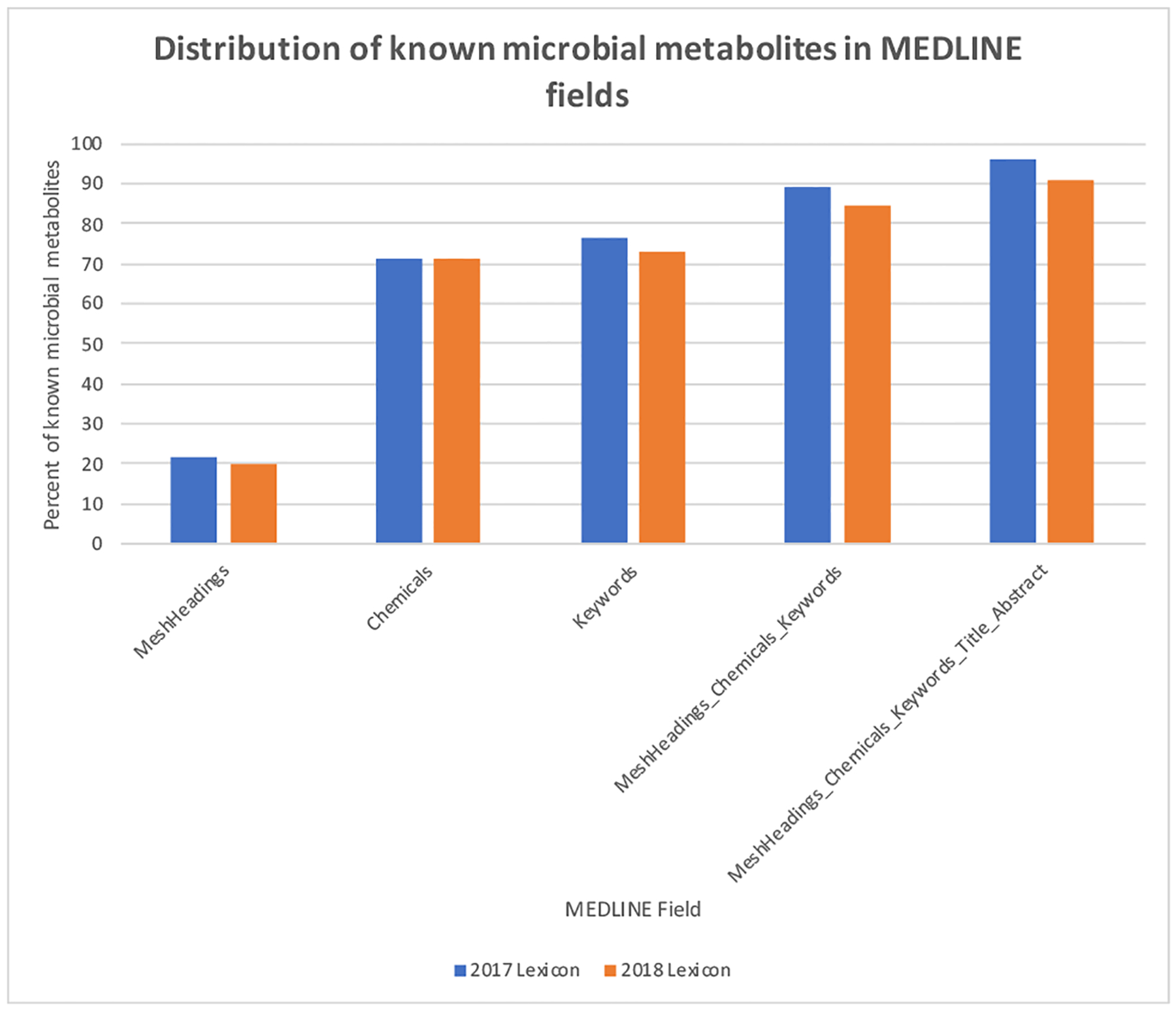
Distribution of 172 known microbial metabolites in different fields of MEDLINE records (MeshHeadings, Chemicals, Keywords, Title, Abstract).
Table 1 shows the performance of the dictionary-based NER (baseline approach) from the combined five fields of MEDLINE records. A total of 11,846 unique metabolites were extracted. The recall is high (0.959) and the precision is extremely low as evaluated with the 172 known microbial metabolites. These results demonstrates that (1) MEDLINE records is a comprehensive resource for microbial metabolite extraction (high recall); and (2) microbial metabolites constitute only a small portion of all extracted metabolites (low precision), therefore further prioritization of microbial metabolites among all extracted human metabolites is necessary. Also shown in the table, though the 2018 version of HMDB-based lexicon contains significantly more metabolites than the 2017 version, extraction based on it had both lower coverage and precision. One reason if the low precision is due to the mapping errors in HMDB lexicon. For example, in the mapping “HMDB0000958- trans-aconitic acid - acid”, the general term ‘acid’ is mapped to the specific term ‘trans-aconitic acid’. The term ‘acid’ appeared in MEDLINE 1317752 times. When it was extracted from MEDLINE, it was incorrectly mapped to the concept “trans-aconitic acid”. While both versions of HMDB contain these mapping errors, the version of HMDB may have more errors due to its much large size of mappings (11,131,600 versus 365,632 mappings). The lower coverage of 2018 version HMDB as compared to 2017 version HMDB may due to the fact that many concepts in 2018 version HMDB may not appear in MEDLINE. Therefore, subsequent CoMN construction and prioritization were based on metabolites extracted using the 2017 version lexicon.
Table 1.
Performance of the dictionary-based metabolite extraction from the combined fields of MEDLINE records.
| Lexicon | Precision | Recall | FI |
|---|---|---|---|
| 2017 HMDB Lexicon | |||
| (42,003 concepts, 65,632 terms) | 0.014 | 0.959 | 0.027 |
| 2018 HMDB Lexicon | |||
| (114,100 concepts, 1,131,600 terms) | 0.012 | 0.909 | 0.024 |
4.2. CoMNRank effectively prioritized microbial metabolites
We further prioritized extracted metabolites in order to enrich true microbial metabolites among top. When a known microbial metabolite (butyric acid or trimethylamine n-oxide) was used as the seed, top ranked metabolites were highly enriched with known microbial metabolites (Figure 2).
Fig. 2.
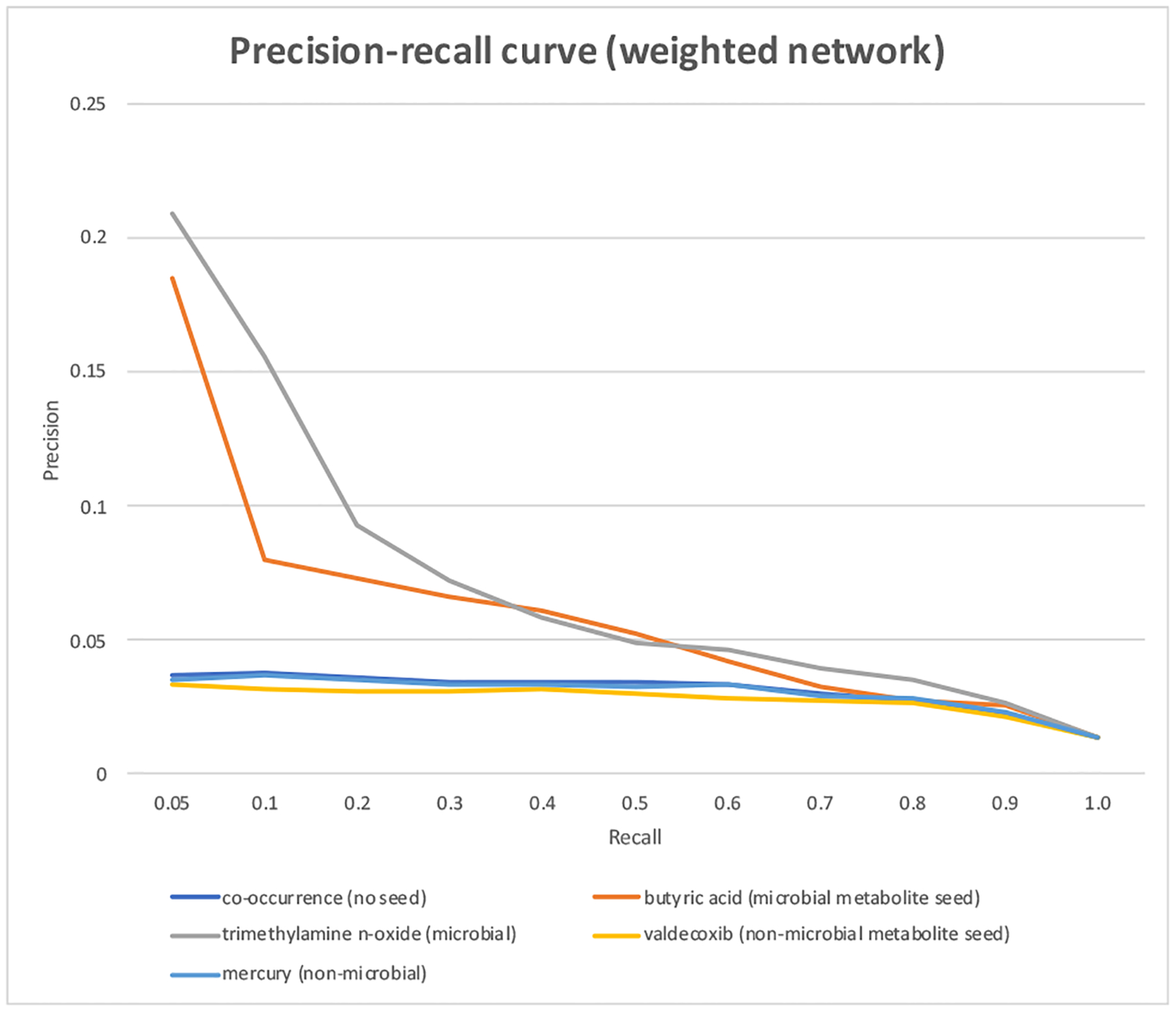
Precision-recall curves for CoMNRank from weighted CoMN with microbial metabolite seeds, non-microbial metabolite sees or no seed.
For example, when the seed “butyric acid” was used, the top ranked metabolites (at recall of 0.05) has a precision of 0.185, representing a 12.2-fold enrichment as compared to the overall precision of 0.014 (at recall of 1.0). When the microbial metabolite seed “trimethylamine n-oxide” was used, the top ranked metabolites (at recall of 0.05) has a precision of 0.209, which represents a 13.9-fold enrichment as compared to the overall precision of 0.014. On the other hand, when a non-microbial metabolite seed (mercury or valdecoxib) or no seed (pure frequency-based ranking) was used, the top ranked metabolites are not enriched with known microbial metabolites (Figure 2). These results support our hypotheses that (1) if an article contains a known microbial metabolite (e.g., butyric acid, trimethylamine n-oxide), then the article is likely related to microbial metabolism study (“Implicit Text Classification”); (2) other metabolites extracted from microbial metabolism-related articles are likely microbial metabolites (“Implicit Microbial Metabolite Classification”).
4.3. The weighted CoMN has better overall performance
We investigated how the edge weights of CoMN affect the overall performance. When the unweighted CoMN was used, the performance was not as good as that for the weighted CoMN (Figure 3). For example, when the seed “butyric acid” was used for the unweighted CoMN, the top ranked metabolites (at recall of 0.05) has a precision of 0.125, which represents a 7.9-fold enrichment as compared to the overall precision of 0.014. When the seed “trimethylamine n-oxide” was used, the top ranked metabolites (at recall of 0.05) has a precision of 0.132, which represents a 8.4-fold enrichment as compared to the overall precision of 0.014. These results support our hypothesis that if a metabolite co-occurs with known microbial metabolites many times (captured by the edge weights), then the metabolite is more likely a microbial metabolite than those co-occurring with known microbial metabolites few times.
Fig. 3.
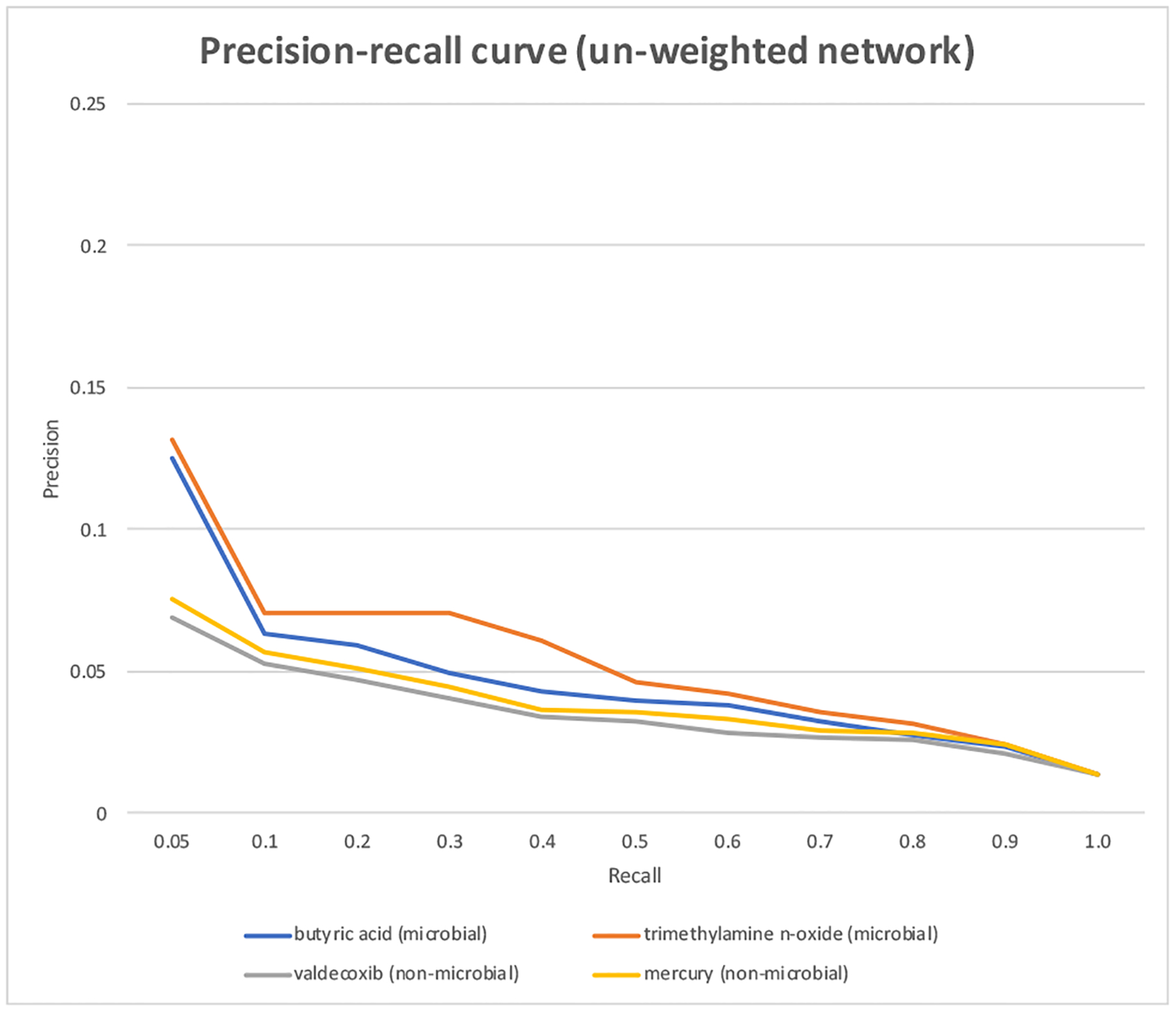
Precision-recall curves of CoMNRank from un-weighted CoMN, when a microbial or non-microbial seed was used.
4.4. The frequency of seeds affects the overall performance
Here we investigated how the frequencies of seeds in the entire MEDLINE collection (well-known/studied vs. less known/studied microbial metabolites) affected the performance of CoMNRank. We used 12 microbial metabolite seeds with different frequency and 12 non-microbial metabolite seeds with matching frequency (Table 2). Butyric acid is one of the most abundant short chain fatty acids (SCFAs) and the primary end-products of fermentation of non-digestible dietary fiber by the gut microbiota (Rooks et al., 2016). Trimethylamine n-oxide (TMAO) is a gut microbial metabolite of red meat and high fat diet (Tang et al., 2018; Brown et al., 2018).
Table 2.
Microbial vs non-microbial seeds and their MEDLINE frequencies.
| Microbial Seed | Non-microbial Seed | ||
|---|---|---|---|
| Metabolite | Frequency | Metabolite | Frequency |
| acetic acid | 135678 | valdecoxib | 135362 |
| propionic acid | 27784 | ampicillin | 27919 |
| butyric acid | 24036 | substance p | 24190 |
| trimethylamine n-oxide | 1793 | hydroxyzine | 1793 |
| taurodeoxycholic acid | 700 | 6- acetylmorphine | 699 |
| p-cresol sulfate | 311 | ligustilide | 311 |
| n-acetylputrescine | 102 | pisatin | 102 |
| glycoursodeoxycholic acid | 86 | 13-cis-retinal | 86 |
| indole | 6 | citalopram n-oxide | 6 |
| chenodeoxycholic acid 3-sulfate | 3 | pterolactam | 3 |
| n2-succinyl-l-omithine | 1 | momordicilin | 1 |
As shown in Figure 4, MEDLINE frequency of the input seed affected the overall performance. When rare microbial metabolite seeds (e.g., n2-succinyl-l-ornithine, chenodeoxycholic acid 3-sulfate, indole) were used, the performance is not as good as that for frequent seeds. For example, when n2-succinyl-l-ornithine (appeared once in the MEDLINE collection) was used as the input seed, the top ranked metabolites (at recall of 0.05) has a precision of 0.086, a 5.1-fold enrichment as compared to the overall precision of 0.014. On the other hand, when p-cresol sulfate (appeared in 311 times in MEDLINE) was used, the top ranked metabolites (at recall of 0.05) has a precision of 0.278, an 18.8-fold enrichment over the overall precision of 0.014. However, the performance is not proportional to seed frequency. For example, acetic acid appeared in 135,678 MEDLINE articles. When acetic acid was used as the seed, the precision at recall of 0.05 is 0.143, which is lower than that for less frequent seeds such as butyric acid, propionic acid, TMAO or p-cresol. One explanation is that when acetic acid, a very general term, is mentioned in MEDLINE, it often does not specifically refer to microbiota-derived metabolite, therefore the articles it appears are often not microbial metabolism-related. CoMNRank using the frequency-matched non-microbial metabolite seeds in general was not able to enrich true positives among top. (Figure 5).
Fig. 4.
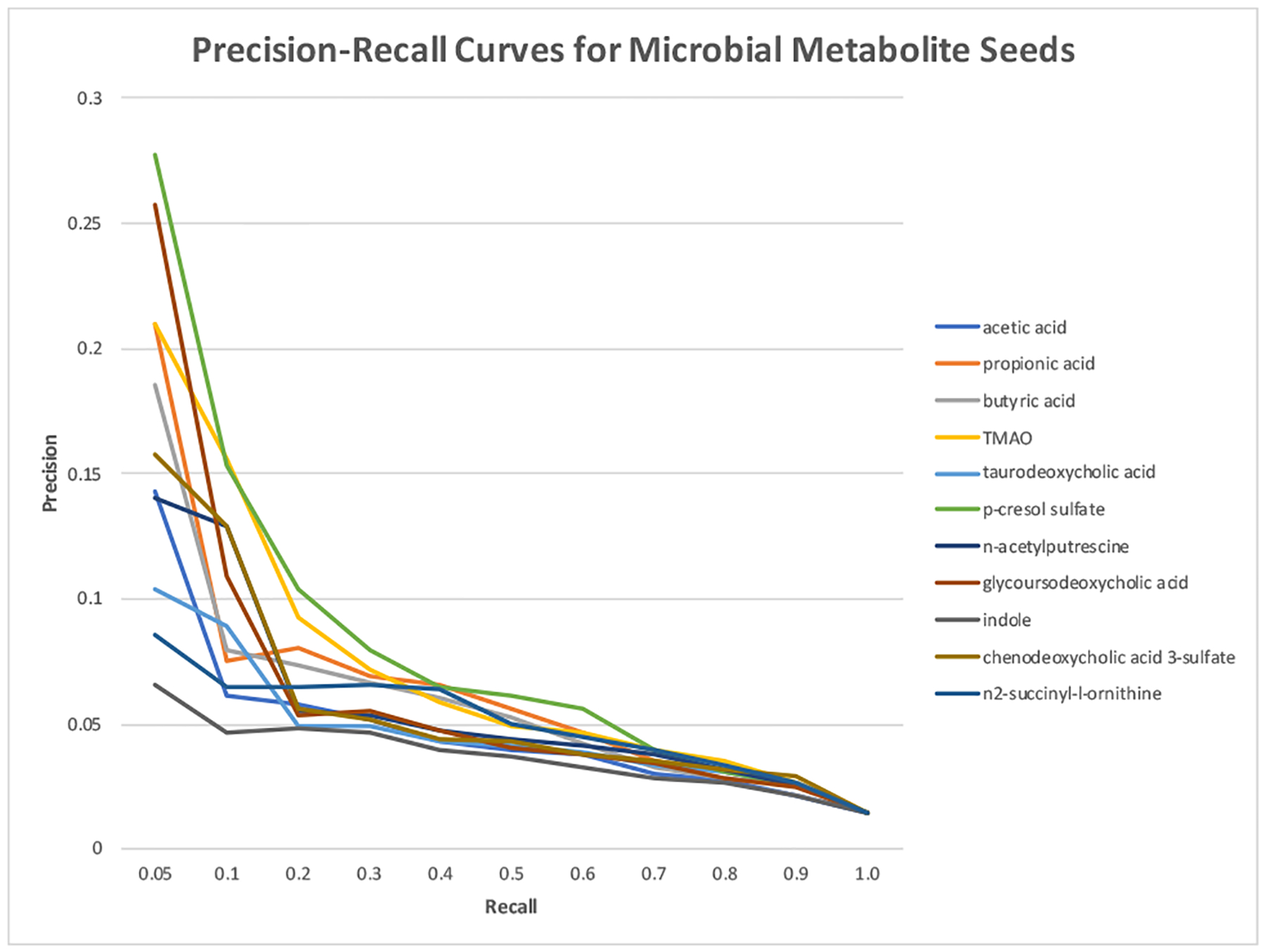
Precision-recall curves of CoMNRank using microbial metabolite seeds with different frequencies.
Fig. 5.
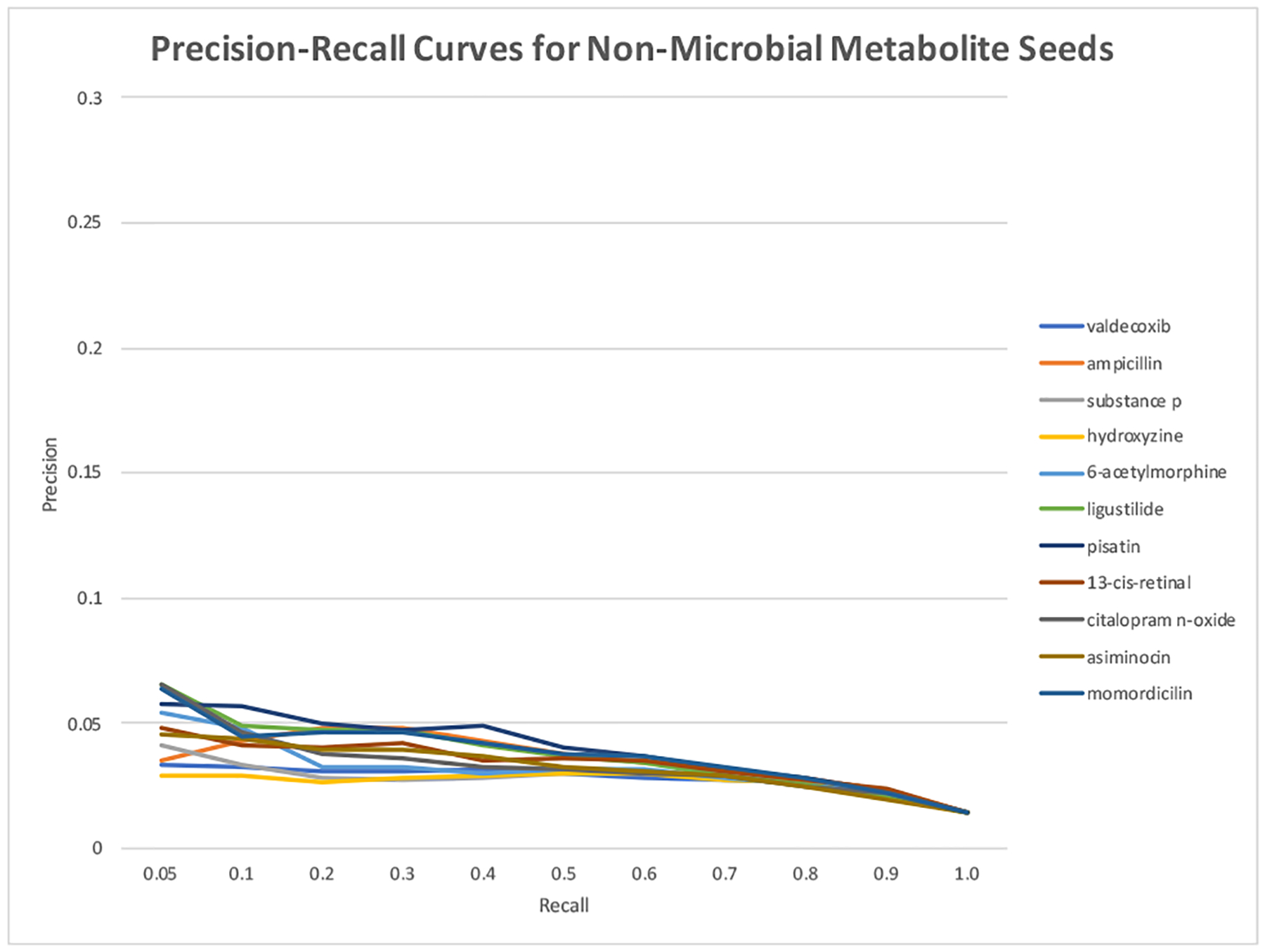
Precision-recall curves of CoMNRank using non-microbial metabolite seeds with different frequencies. Y-axis has the same scale with that in Figure 4.
4.5. Combined seeds further improved performance
We investigated how combining different types of microbial metabolite seeds affected the overall performance. Butyric acid and propionic acid are both short chain fatty acids (SCFAs) produced by the gut microbiota in fermentation of non-digestible dietary fiber (Rooks et al., 2016). Trimethylamine n-oxide (TMAO) is a gut microbial metabolite of red meat and high fat diet (Tang et al., 2018; Brown et al., 2018). P-Cresol is an end-product of protein breakdown and microbial metabolites produced from tyrosine (Verbeke et al., 2015). SCFAs often co-occur together in MEDLINE articles, for example in the sentence “… as well as between treated CD patients and healthy adults, regarding acetic acid, propionic acid, butyric acid, and total SCFAs.” (PMID 22542995). When two similar seeds butyric acid and propionic acid were used together as seeds, the overall performance (precision of 0.184 at recall of 0.05) did not improve as compared to that when they were used individually (precision of 0.185 for butyric acid and 0.209 for propionic acid) (Figure 6).
Fig. 6.
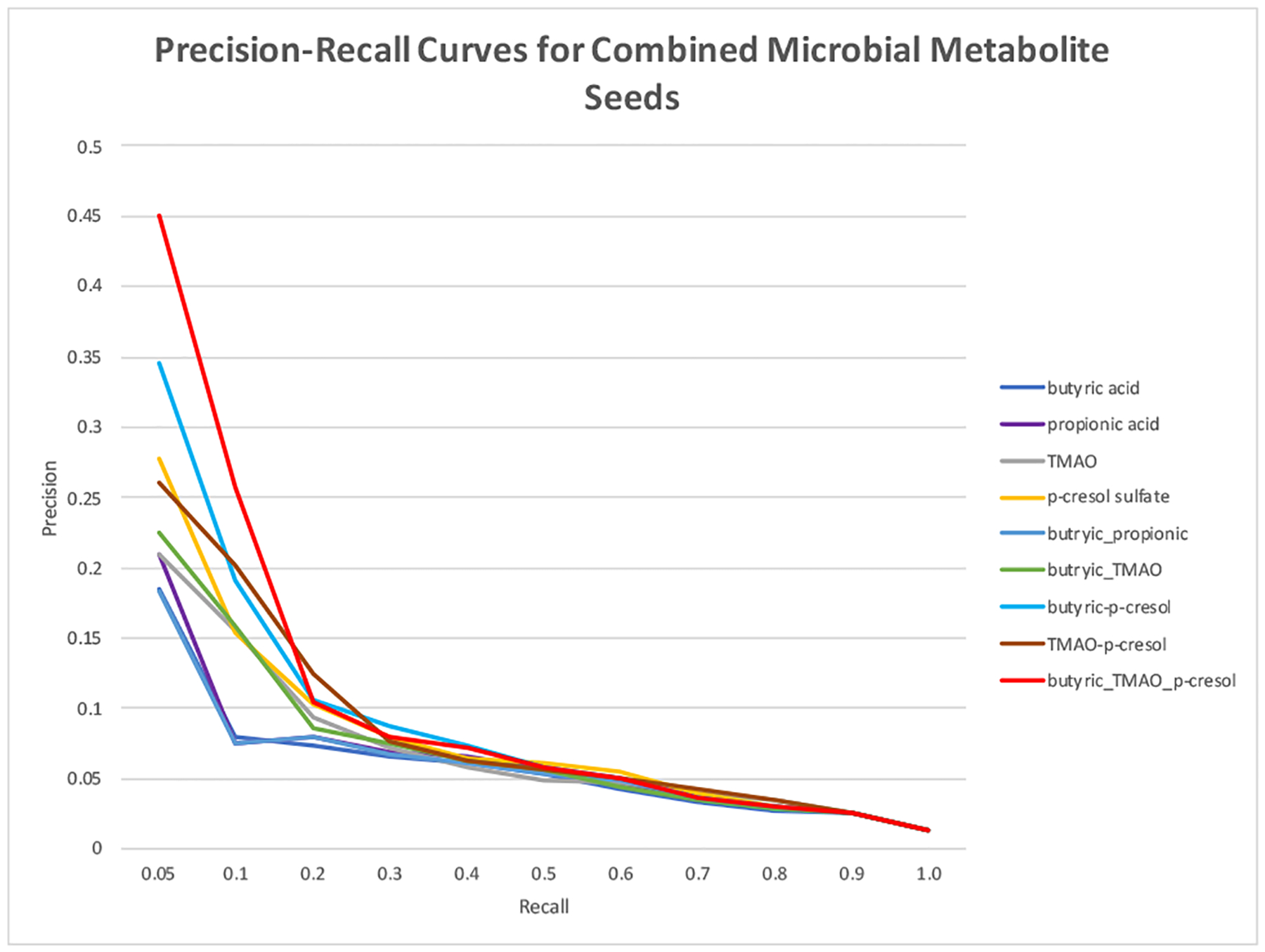
Precision-recall curves of CoMNRank for combined seeds. Weighted CoMN was used. Evaluation dataset: 172 known microbial metabolites.
On the other hand, when different types of seeds were combined, the performance of CoMNRank improved (Figure 6). Combining butyric acid and TMAO, the precision (at recall of 0.05) is 0.224, which is higher than that for butyric acid (0.185) and TMAO (0.209) alone. By combining butyric acid and p-cresol, the precision (at recall of 0.05) is 0.346, which is higher than that for butyric acid (0.185) and p-cresol (0.278) alone. Combining all three together, the precision (at recall of 0.05) is 0.45, which is higher than that for each individual seed (0.185 for butyric acid, 0.209 for TMAO and 0.278 for p-cresol). When three SCFAs (the same type of metabolites) were used as the seeds, the precision at recall of 0.05 is 0.170 (data not shown), which is significantly lower than that when three different types of seeds (butyric acid, TMAO and p-cresol) were used. These results indicate that if a metabolite co-occur with two or more other types of microbial metabolites, it is highly likely a microbial metabolite.
4.6. Manual curation and error analysis
Since many microbial metabolites are not captured by HMDB, evaluation using 172 known microbial metabolites from HMDB under-estimated the true precision of CoMNRank. Therefore we used it only to compare the performance of different configurations of CoMNRank (metabolite dictionary choice, different fields of MEDLINE records, weighted vs unweighted CoMNs, and different seeds). These evaluations showed that CoMN effecvtively enriched true positives among top ranked metabolites. We then manually curated top 100 metabolites output from CoMN with configuration of combined seeds (butyric acid, TMAO and p-cresol), weighted CoMN, 2017 version of HMDB lexicon, and five fields of MEDLINE records. We calculated the true precision of these top 100 metabolites. Note that recall was difficult to calculate since we don’t know how many metabolites are produced by human microbiota. Given that CoMN has effectively enriched true positives among top and that manually curating all extracted 11,864 metabolites is extremely labor intensive, we only curated top 100 metabolites. Results in Table 1 indicate that MEDLINE records are indeed a comprehensive resource for known microbial metabolites (recall of 0.959).
A total of 67 out of the top 100 metabolites are true positives, including 19 from HMDB and additional 48 microbial metabolites that are reported in MEDLINE but not captured in HMDB. The true precision of top 100 metabolites is 0.67, which is significantly higher than the estimated precision of 0.19 evaluated using 172 microbial metabolites from HMDB. This manual curation demonstrates that (1) the ranking algorithm indeed significantly enriched true positives among top (67% are true positives); (2) the majority (as much as 48%) of microbial metabolites have not been captured by HMDB; and (3) the 28 million MEDLINE records are indeed a rich resource for microbial metabolite extraction.
We performed error analysis of five highly ranked non-microbial metabolites (trans-aconitic acid, sodium, alpha-amyrin acetate, oxygen, omeprazole). The three non-microbial metabolites (Trans-aconitic acid, alpha-amyrin acetate, omeprazole) ranked highly due to synonym mapping errors from HMDB. Based on HMDB, trans-aconitic acid (HMDB00958) has 13 synonyms, among which is an incorrect synonym mapping “acid!trans-aconitic acid”. The term “acid” frequently appears in MEDLINE and does not refer to “trans-aconitic acid”. During the metabolite extraction and mapping process, the common term “acid” was extracted and incorrectly mapped to the concept “trans-aconitic acid”. Since the term “acid” universally appeared in MEDLINE articles and often co-occurs with many known microbial metabolites, its mapped term “trans-aconitic acid” was ranked highly when microbial metabolite seeds were used. The same mapping errors occurred for “alpha-amyrin acetate” (“alpha→alpha-amyrin acetate”) and “omeprazole” (“result→meprazole”). Both terms “alpha” and “result” frequently appeared in MEDLINE records. The other two top ranked non-microbial metabolites (sodium and oxygen) ranked highly not due to mapping errors but to their high frequencies in MEDLINE.
5. Discussion
We developed an integrated approach CoMNRank by combining named entity recognition, network construction and network-based prioritization to extract and prioritize microbial metabolites from over 28 million MEDLINE records. A few points warrant further discussion and future investigation.
This study was motivated by our previous studies showing that the list of microbial metabolites from HMDB had limited coverage and that an accurate and comprehensive catalog of microbial metabolites that are also present in human body is the key to data-driven studies of how human microbial metabolism affects human health. While the main goal of this study is to demonstrate the novel algorithm CoMNRank in prioritizing microbial metabolites by leveraging their occurrences in biomedical literature, our next step is to demonstrate how the newly extracted microbial metabolites facilitate data-driven analysis of microbial metabolism related to human diseases. For this purpose, the extracted metabolites need further manual curation. CoMNRank prioritized true positives among top, which will greatly facilitate the manual curation process.
In this study, we used the fields of Title, Abstract, MeshHeadings, Keywords, and Chemicals of MEDLINE records and showed that these fields captured as much as 95.9% of the 172 known microbial metabolites from HMDB. While important findings are often captured by these fields, it is likely that some microbial metabolites are listed in full-text fields, including embedded tables or even supplementary data. In our future works, we will investigate if using full-text archive from PubMed Central (PMC., 2018) can identify additional microbial metabolites.
During the error analysis, we found that many non-microbial metabolites ranked highly because of the synonym mapping errors from HMDB. Given the large number of synonym→concept mappings (365,632) in HMDB, manual curation of all mappings is not feasible. Discarding common terms is also not a good option since some true microbial metabolites (e.g., hydrogen, urea, and acetic acid) frequently appear in MEDLINE. One possible solution is to only manually curate mappings containing frequent terms. These common synonyms, when incorrectly mapped, will have big impact on the overall performance given their frequent appearances in MEDLINE. Incorrect mappings containing rare terms will not have big effect on overall performance since these terms will not be ranked highly from the weighted CoMN. In our previous study, we curated five Million UMLS Metathesaurus Terms based on their appearances in all MEDLINE articles (Xu et al., 2010). In our future studies, we will first find the frequencies of all metabolite terms (names and synonyms) in the entire MEDLINE collection and then manually curate mappings containing top frequent terms.
The main goal of this study was to identify and prioritize microbial metabolites that are included in HMDB but not classified as microbial origin, therefore we performed dictionary-based NER using a lexicon constructed from HMDB. During the experiment, we noticed that some microbial metabolites were clearly stated in biomedical literature, but the names were not included in HMDB. For example, the sentence “… trans-resveratrol and resveratrol-derived microbial metabolites (dihydroresveratrol and lunularin) were also identified” (PMID 26156396) contains two microbial metabolites dihydroresveratrol and lunularin. None of these two metabolites are included in HMDB, therefore they were missed from our dictionary-based extraction. For our future studies, we will complement the dictionary-based NER with de-novo NER techniques, including the pattern-based iterative learning approaches that we previously developed for both NER (Xu et al., 2008, 2009) and relationship extraction (Xu et al., 2009, 2013, 2014), to further increase the recall of microbial metabolite extraction from biomedical literature.
Microbial metabolites in this study are defined as metabolites produced by microbes, by not necessarily exclusively so. The goal of this study, motivated by our previous data-driven studies in understanding how microbial metabolism contributed to human health and diseases, is to automatically prioritize microbial metabolites among all human metabolites. While some microbial metabolites are exclusively produced by microbes, many others are produced by both human hosts and microbes. It will be an interesting classification question to further categorize the extracted microbial metabolites into microbial only or dual (produced by both microbes and human hosts.
Our future directions also include metabolite-bacteria and microbial metabolite-disease relationship extractions. For microbial metabolite relationship extraction, a comprehensive and accurate lexicon of microbial metabolites is necessary. In this study, we manually curated top 100 ranked metabolites and demonstrated that top ranked metabolites have a precision of 0.68. In the future, manual curation of more top-ranked metabolites (e.g., top 1000 metabolites) will be necessary in order to build a more comprehensive lexicon of microbial metabolites.
6. Conclusions
We developed an integrated approach CoMNRank to extract human microbial metabolites from MEDLINE records. We demonstrated that CoMNRank effectively enriched microbial metabolites among top. Manual curation of top ranked metabolites showed as much as 48% true microbial metabolites are reported in biomedical literature but have not been captured by existing databases. In summary, our study sets the foundation for future microbial metabolite relationship extraction and will facilitate data-driven studies of how gut microbial metabolites interact with host genetics in different human diseases.
Highlights.
Gut microbiota is important in human health.
Biomedical literature is a rich knowledge resource of microbial metabolites.
We developed a novel algorithm CoMNRank to extract and prioritize microbial metabolites from biomedical articles.
A comprehensive list of microbial metabolites is important for developing data-driven approaches in understanding microbial metabolism in human health.
A comprehensive list of microbial metabolites is important for future microbial metabolite-related information extraction tasks from free-text documents.
Funding
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under the NIH Director’s New Innovator Award (DP2HD084068, Xu), NIH National Institute of Aging (AG057557-01, Xu; AG061388-01, Xu; AG062272-01, Xu), American Cancer Society Research Scholar Grant (RSG-16-049-01 - MPC, Xu), NIH Clinical and Translational Science Collaborative of Cleveland (1UL1TR002548-01, Konstan).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Availability of data and material
Data and results are available at nlp.case.edu/public/data/CoMNRank
References
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI (2007) The human microbiome project. Nature, 449(7164), 804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, … Nelson KE (2006) Metagenomic analysis of the human distal gut microbiome. Science, 312(5778), 1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I, Blaser MJ (2012) The human microbiome: at the interface of health and disease. Nature Reviews Genetics, 13(4), 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer F, Bäckhed F (2013) The gut microbiota—masters of host development and physiology. Nature Reviews Microbiology, 11(4), 227. [DOI] [PubMed] [Google Scholar]
- Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R (2018). Current understanding of the human microbiome. Nature medicine, 24(4), 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremaroli V, Bäckhed F (2012) Functional interactions between the gut microbiota and host metabolism. Nature, 489(7415), 242–249. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S (2012) Host-gut microbiota metabolic interactions. Science, 336(6086), 1262–1267. [DOI] [PubMed] [Google Scholar]
- Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, … Marsland BJ (2014) Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nature medicine, 20(2), 159. [DOI] [PubMed] [Google Scholar]
- Koppel N, Rekdal VM, Balskus EP (2017). Chemical transformation of xenobiotics by the human gut microbiota. Science, 356(6344), eaag2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WW, Li DY, Hazen SL (2018). Dietary metabolism, the gut microbiome, and heart failure. Nature Reviews Cardiology, 1. doi: 10.1038/s41569-018-0108-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Hazen SL (2018). Microbial modulation of cardiovascular disease. Nature Reviews Microbiology, 16(3), 171.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfora EE, Meex RC, Venema K, Blaak EE (2019). Gut microbial metabolites in obesity, NAFLD and T2DM. Nature Reviews Endocrinology, 1. doi: 10.1038/s41574-019-0156-z [DOI] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, … Patterson PH (2013). Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell, 155(7), 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PA (2015). The tantalizing links between gut microbes and the brain. Nature News, 526(7573), 312. [DOI] [PubMed] [Google Scholar]
- Valles-Colomer M, Falony G, Darzi Y, Tigchelaar EF, Wang J, Tito RY, … Claes S (2019). The neuroactive potential of the human gut microbiota in quality of life and depression. Nature microbiology, 1. doi: 10.1038/s41564-018-0337-x [DOI] [PubMed] [Google Scholar]
- Louis P, Hold GL, Flint HJ (2014). The gut microbiota, bacterial metabolites and colorectal cancer. Nature Reviews Microbiology, 12(10), 661. [DOI] [PubMed] [Google Scholar]
- Schwabe RF, Jobin C (2013). The microbiome and cancer. Nature Reviews Cancer, 13(11), 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-y M, … Garrett WS (2013). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science, 341(6145), 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooks MG, Garrett WS (2016). Gut microbiota, metabolites and host immunity. Nature Reviews Immunology, 16(6), 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke KA, Boobis AR, Chiodini A, Edwards CA, Franck A, Kleerebezem M, … Tuohy KM (2015). Towards microbial fermentation metabolites as markers for health benefits of prebiotics. Nutrition research reviews, 28(1), 42–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, … Krishnamurthy B (2017). Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nature immunology, 18(5), 552. [DOI] [PubMed] [Google Scholar]
- Jangi S, Gandhi R, Cox LM, Li N, Von Glehn F, Yan R, … Cook S (2016). Alterations of the human gut microbiome in multiple sclerosis. Nature communications, 7, 12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase S, Haghikia A, Wilck N, Müller DN, Linker RA (2018). Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology, 154(2), 230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Wang Q, Li L. (2015) Genome-wide systems analysis reveals strong link between colorectal cancer and trimethylamine N-oxide (TMAO), a gut microbial metabolite of dietary meat and fat. BMC Genomics, 16(Suppl 7):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li L, Xu R (2018) A systems biology approach to predict and characterize human gut microbial metabolites in colorectal cancer, Scientific reports, 8(1), 6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Wang Q. (2016) Towards understanding brain-gut-microbiome connections in Alzheimer’s disease. BMC Systems Biology, 10:63 DOI: 10.1186/s12918-016-0307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, McCormick TS, Ward NL, Cooper KD, Conic R, Xu R (2017). Combining mechanism-based prediction with patient-based profiling for psoriasis metabolomics biomarker discovery In AMIA Annual Symposium Proceedings (Vol. 2017, p. 1734). American Medical Informatics Association. [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Xu R (2017). MetabolitePredict: A de novo human metabolomics prediction system and its applications in rheumatoid arthritis. Journal of biomedical informatics, 71, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Xu R (2019) Data-driven multiple-level analysis of gut-microbiome-immune-joint interactions in rheumatoid arthritis, BMC Genomics, 20:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Ulrich CM, Neuhouser ML, Malysheva O, Bailey LB, Xiao L, … Miller JW (2014). Plasma choline metabolites and colorectal cancer risk in the Women’s Health Initiative Observational Study. Cancer research, 74(24), 7442–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt NM, Romano KA, Darst BF, Engelman CD, Johnson SC, Carlsson CM, … Rey FE (2018). The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimer’s research & therapy, 10(1), 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, … Bouatra S (2013). HMDB 3.0-the human metabolome database in 2013. Nucleic Acids Research, 41(D1), D801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz M, Stein A, Uncini T (2012). 5-HTP efficacy and contraindications. Neuropsychiatric disease and treatment, 8, 323. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Pincemail J (1995). Free radicals and antioxidants in human diseases In Analysis of free radicals in biological systems (pp. 83–98). Birkhäuser Basel. [Google Scholar]
- Xu R, Li L, Wang Q (2013). Towards building a disease-phenotype relationship knowledge base: large scale extraction of disease-manifestation relationship from literature. Bioinformatics, 29(17), 2186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Wang Q (2013). Large-scale extraction of accurate drug-disease treatment pairs from biomedical literature for drug repurposing. BMC bioinformatics, 14(1), 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Li L, Wang Q (2014). dRiskKB: a large-scale disease-disease risk relationship knowledge base constructed from biomedical text. BMC bioinformatics, 15(1), 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Xu R (2018). Immunotherapy-related adverse events (irAEs): extraction from FDA drug labels and comparative analysis. JAMIA open, 2(1), 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badal VD, Wright D, Katsis Y, Kim HC, Swafford AD, Knight R, Hsu CN (2019). Challenges in the construction of knowledge bases for human microbiome-disease associations. Microbiome, 7(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Zhang L, Zeng P, Huang C, Li J, Geng B, … Cui Q (2016). An analysis of human microbe–disease associations. Briefings in bioinformatics, 18(1), 85–97. [DOI] [PubMed] [Google Scholar]
- Janssens Y, Nielandt J, Bronselaer A, Debunne N, Verbeke F, Wynendaele E, … De Spiegeleer B (2018). Disbiome database: linking the microbiome to disease. BMC microbiology, 18(1), 50–55. MEDLINE: https://www.nlm.nih.gov/databases/download/pubmed_medline.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Xu R (2015) Phenome-driven Disease Genetics Prediction Towards Drug Discovery. Bioinformatics 2015 31 (12): i276–i283 doi: 10.1093/bioinformatics/btv245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Xu R (2017). Context-sensitive network based disease genetics prediction and its implications in drug discovery. Bioinformatics 2017; btw737 DOI: 10.1093/bioinformatics/btw737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Wang Q (2015) PhenoPredict: a disease phenome-wide drug repositioning approach towards schizophrenia drug discovery. Journal of Biomedical Informatics, 56, 348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Wang Q. (2016) A genomics-based systems approach towards drug repositioning for rheumatoid arthritis. BMC genomics, 17(7), 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Xu R (2018) Disease comorbidity-guided drug repositioning: a case study in schizophrenia. The 2018 Annual American Medical Informatics Association Symposium, Nov 3–7, San Francisco CA. [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Chen Y, Xu R (2018). A Drug-Side Effect Context-Sensitive Network approach for Drug Target Prediction. Bioinformatics. bty906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning CD, Raghavan P, Schutze H (2008). Introduction to information retrieval (Vol. 1, p. 6). Cambridge: Cambridge university press. [Google Scholar]
- Davis J, Goadrich M (2006) The relationship between Precision-Recall and ROC curves. In Proceedings of the 23rd international conference on Machine learning, (pp. 233–240). [Google Scholar]
- Xu R, Musen M, Shah N, (2010) A Comprehensive Analysis of Five Million UMLS Metathesaurus Terms Using Eighteen Million MEDLINE Citations. The Annual American Medical Informatics Association Symposium 2010 pp. 907–911. PubMed Central: https://www.ncbi.nlm.nih.gov/pmc/. [PMC free article] [PubMed] [Google Scholar]
- Xu R, Supekar K, Morgan A, Das A, Garber A (2008). Unsupervised method for automatic construction of a disease dictionary from a large free text collection In AMIA annual symposium proceedings (Vol. 2008, p. 820). American Medical Informatics Association. [PMC free article] [PubMed] [Google Scholar]
- Xu R, Morgan A, Das AK, Garber A (2009, June). Investigation of unsupervised pattern learning techniques for bootstrap construction of a medical treatment lexicon In Proceedings of the workshop on current trends in biomedical natural language processing (pp. 63–70). Association for Computational Linguistics. [Google Scholar]
- Xu R, Das AK, Garber AM (2009). Unsupervised method for extracting machine understandable medical knowledge from a large free text collection In AMIA annual symposium proceedings (Vol. 2009, p. 709). American Medical Informatics Association. [PMC free article] [PubMed] [Google Scholar]
- Xu R, Li L, Wang Q (2013). Towards building a disease-phenotype knowledge base: extracting disease-manifestation relationship from literature. Bioinformatics, 29(17), 2186–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Li L, Wang Q, (2014) dRiskKB: a large-scale disease-disease risk (causal) relationship knowledge base constructed from biomedical text. BMC Bioinformatics 2014, 15:105. [DOI] [PMC free article] [PubMed] [Google Scholar]