

Abstract
Pharmacoresistance in epilepsy is a major challenge to successful clinical therapy. Glucocorticoid receptor (GR) dysregulation can affect the underlying disease pathogenesis. We recently reported that local drug biotransformation at the blood-brain barrier is upregulated by GR, which controls drug-metabolizing enzymes (e.g., cytochrome P450s, CYPs) and efflux drug transporters (MDR1) in human epileptic brain endothelial cells (EPI-ECs). Here we establish that this mechanism is influenced upstream by GR and its association with heat shock proteins/co-chaperones (Hsps) during maturation, which differentially affect human epileptic (EPI) tissue and brain endothelial cells. Overexpressed GR, Hsp90, Hsp70, Hsp40 were found in EPI vs. NON-EPI brain regions. Elevated neurovascular GR expression and co-localization with Hsps was evident in the EPI regions with cortical dysplasia, predominantly in the brain micro-capillaries and neurons. A corresponding increase in ATPase activity (*p < 0.05) was found in the EPI regions. The GR-Hsp90/Hsp70 binding patterns indicated a faster chaperone-promoted maturation of GR, leading to its overactivation in both the tissue and EPI-ECs derived from EPI/focal regions and GR silencing in EPI-ECs slowed such GR-Hsp interactions. Significantly accelerated GR nuclear translocation was determined in EPI-ECs following treatment with GR modulators/ligands dexamethasone, rifampicin or phenytoin. Our findings reveal that overexpressed GR co-localizes with Hsps in the neurovasculature of EPI brain, increased GR maturation by Hsps accelerates EPI GR machinery and further that this change in EPI and NON-EPI GR-Hsp interaction alters with the age of seizure-onset in epileptic patients, together affecting the pathophysiology and drug regulation in the epileptic brain endothelium.
Keywords: glucocorticoid receptor, heat shock proteins, drug resistance, blood-brain barrier, nuclear translocation
Introduction
Pathophysiological brain alterations during disease progression trigger several critical molecular determinant factors that affect drug therapies and cures. Of these, the glucocorticoid receptor (GR) is a critical target that belongs to the superfamily of steroid receptor proteins and functions as a ligand-dependent transcription factor regulating multiple metabolic, immune and behavioral functions [1–4]. However, the role of GR in epilepsy, particularly in the context of drug regulatory properties at the blood-brain barrier (BBB), remains unclear, and may be a mechanism contributing to pharmacoresistance in a significant proportion of epileptic patients [5,6]. Further, the association of GR to heat shock proteins (Hsps), ubiquitous molecular chaperones which play important roles in functions from cellular stressors to receptor trafficking, could be a vital element in the GR regulation of drugs in focal epilepsies. Thus, this association warrants further investigation.
We recently showed that (a) GR is overexpressed in human epileptic BBB endothelial cells (EPI-EC) compared with control human brain endothelial cells (HBMEC) [7]; (b) GR upregulation in the neuro-vasculature is observed in pharmacoresistant temporal lobe epilepsy, particularly in regions with reactive gliosis [7]; and (c) modulation of GR in EPI-EC at the BBB affects drug pharmacokinetics, demonstrated by improved oxcarbazepine bioavailability and increased phenytoin permeability across the BBB [8].
In the resting stage, GR is a ligand-dependent transcription factor residing in the cytosol. Upon pathophysiological trigger, ligand binding induces a conformational change in the receptor, exposing a nuclear localization signal and exchanging interaction partners, after which the receptor translocates into the nucleus. Heat shock protein (Hsps) Hsp70 and Hsp90-based chaperone machinery functions with co-chaperones to guarantee GR folding, maturation, nuclear accumulation, and enhancement or repression of transcription of target genes by binding to specific regions of their DNA via its DNA binding domains [3,9]. Given the essential role of Hsp90 and other chaperones in GR translocation, and in the possible dysfunction in translocation of GR, their association to several disease states or conditions, including aging, depression and post-traumatic stress disorders has been previously investigated [10,11,3,12]. However, knowledge of the underlying molecular cascade of GR in epilepsy is still emerging and is poorly understood [13], despite evidence that these chaperones could be effective potential drug targets in the pathophysiology of stress-related psychiatric disorders [14,12]. It is therefore imperative to better understand the role of the brain GR regulatory mechanisms that involve association of GR with chaperone/co-chaperone complexes in focal epilepsies. This is especially true in EPI-ECs subsequent to drug exposure, as in the recent past the EPI-ECs have been found to be metabolically active and engage in local drug biotransformation [15–17]. Studying the interaction of GR with modulators [such as dexamethasone (DEX, steroid), rifampicin (RIF, antibiotics), phenytoin (PHT, anti-seizure medication)] in EPI-ECs opens new avenues to better understand the regulatory effect of GR activators which influence GR-nuclear translocation and possibly other downstream events. Reports suggest that treatment of brain endothelial cells with DEX, a synthetic glucocorticoid, influences the differentiation of tight-junction proteins altering microvessel permeability [18,19]. This suggests that DEX treatment of EPI-ECs is an intriguing process and needs to be further understood to regulate GR function.
The objective of this study was to further determine the molecular mechanisms of GR regulation during pathophysiological conditions at the human BBB endothelial cells of intractable epilepsy by investigating : 1) The expression of GR, the role of constitutive (Hsp90 and Hsp40) and inducible Hsps (Hsp70) and adapter chaperone, Hop-1 (Hsp90/70 organizer protein) in epileptic (EPI) vs non-epileptic (NON-EPI) regions of the same epileptic brain; 2) The localization pattern of GR and molecular chaperones/Hsps in the neurovasculature; 3) The nature of GR-Hsp90/Hsp70 interactions, its relation with seizure onset age and the importance of ATPase function of the chaperones in GR maturation in EPI and NON-EPI regions of the brain tissue and its corresponding ECs; 4) The consequence of GR silencing in EPI-ECs on GR-Hsp interactions, and 5) Endothelial GR nuclear translocation in epilepsy following induction by GR ligands to better understand the pathophysiology and its effect on drug regulation (or therapy).
Materials and Methods
Human subjects
Brain specimens from subjects (n = 21) with pharmacoresistant epilepsy were obtained following focal surgical resections, according to the principles outlined in the Declaration of Helsinki and the Cleveland Clinic Institutional Review Board–approved protocol (IRB 07–322). Brain tissues from EPI (epileptic) and NON-EPI (non-epileptic) regions were resected after prior non-invasive (scalp video-EEG monitoring, magnetic resonance imaging, positron emission tomography) and invasive (stereo-electro encephalography) evaluations. A small portion of the NON-EPI area from the excision in each subject was considered as relative/ internal control. Experimental outline is provided in Supplemental Fig. 1. Patient information such as age, gender, race, AEDs, experimental use of the specimens, seizure frequency, epilepsy duration, seizure onset age, resected tissue region and pathology is summarized in Table 1.
Table 1.
Demographic details
| ID | Age (years)/Race/Gender | AEDs | Exp. use | Seizure Freq. (per week) | Seizure onset (years) | Duration of epilepsy (years) | Resected tissue regions | Pathology |
|---|---|---|---|---|---|---|---|---|
| 1 | 8/CAU/M | VPA; DZ | IHC | 1 | 4.5 | 3.5 | Left frontal lobe | Decreased subcortical/deep white matter; cortical architectural distortion; ulegyria |
| 2 | 30/CAU/F | LCM; LOR; TPM; OXC | IHC | 1 | 9 | 21 | Left frontal lobe | Chronic perivascular 1ymphocyticinflammationi; Marked gliosis |
| 5 | 26/CAU/F | LTG; LEV; LCM | WB, IP, IHC | 7 ± 1 | 13 | 13 | Right parieto-occipital lobe | Perivascular chronic inflammation: Perivascular atrophy; Subpial Gliosis |
| 7 | 17/CAU/F | LEV, ZNS | IHC | 7 ± 2 | 9 | 5 | Left temporal lobe | Mild FCD; Focal subpial calcification and gliosis |
| 9 | 23/CAU/M | PHT; CLB; LTG | IP, IHC | 1 | 5 | 18 | Eight frontal lobe | Perivascular white matter atrophy: Gliosis |
| 11 | 34/CAU/F | LEV; LCM | WB, IP | n/a | 31 | 3 | Focal gliosis | |
| 13 | 43/CAU/M | LTG; LOR | IP | 105 ± 2 | 8 | 35 | Left parietal lobe | Focal cortical atrophy and meningeal fibrosis: gliosis |
| 16 | 16/CAU/M | OXC; LTG | WB, IP, HPLC | 7 ± 3 | 2 | 14 | Right temporal lobe | Subpial gliosis, focal vascular sclerosis, mild perivascular chronic inflammation, focal neuron loss and gliosis |
| 18 | 28/CAU/M | LCM; TPM | WB, IP | 1 ± 1 | 20 | 8 | Frontal temporal lobe | Subpial gliosis |
| 23 | 22/CAU/M | OXC; LTG | IHC, HPLC | 1 | 14 | 8 | Right frontal lobe | FCD; Focal perivascular chronic and meningeal inflammation; focal contusional damage |
| 26 | 27/CAU/M | LTG; ZNS | WB, IP, IHC | 1 | 16 | 11 | Left frontal lobe | Subpial gliosis micro-calcification, focal changes consistent with contusional damage/infarct |
| 25 | 64/CAU/F | OXC; TPM, LEV | WB, IP, IHC, HPLC | 1 | 37 | 27 | Left frontal lobe | Meningeal fibrosis; Perivascular and meningeal chronic inflammation; gliosis |
| 29 | 47/CAU/F | ZNS | WB, IP | 1 | 44 | 2 | Left lateral lobe | FCD |
| 30 | 23/CAU/F | LTG LEV, LCM | IP, WB | 1 | 2.5 | 20.5 | Lateral temporal lobe, Hippocampus | HS. diffuse mild subpial gliosis |
| 33 | 37/CAU/M | LCM; LTG | WB, IP | 1 | 22 | 15 | Left temporal lobe | FCD; Perivascular white matter atrophy; Gliosis |
| 35 | 66/CAU/M | LEV; CLB; OXC | WB, IP | 7 ± 1 | 1.5 | 64 | Right frontal lobe | Gliosis; Perivascular chronic inflammation |
| 36 | 44/CAU/M | ESL; CLB | WB, IP | 1 | 8 | 37 | Right temporal lobe | Focal gliosis and perivascular atrophy |
| 43 | 33/CAU/M | LEV; OXC; ZNS | WB, IP | 4 ± 1 | 1 day old | 33 | Left parietal lobe | FCD |
| 44 | 14/CAU/M | ZNS; LTG | WB, IP | 3 ± 1 | 4 | 10 | Right frontal lobe | FCD |
| 61 | L0/CAU/M | CLB; IBM; LTG | IHC | 1 | 2 | 8 | Right frontal lobe | Rare perivascular chronic lymphocytic inflammation |
| 62 | 28/CAU/F | OXC; CLB; LEV | IHC, HPLC | 1 ± 1 | 6 | 22 | Left mesial frontal lobe | FCD; Mild focal perivascular chronic inflammation: Focal perivascular white matter atrophy and subpial gliosis |
Abbreviations: AEDs: antiepileptic drags: VPA, valproic acid: DZ, Diazepam; LCM lacosamide; LEV, levetiracetam; LOR, lorazepam; LTG, lamotrigine: OXC, oxcarbazepine; PHT. phenytoin; CLB, clobazam; ESL, edicarbazepime acetate; VGB, vigabatrin; ZNS, zonisamide; TPM. topiramate; CLZ, clonazepam; FBM, felbamate; FCD, focal cortical dysplasia; HS, hippocampal sclerosis; CAU, Caucasian; M, male. F, female; Exp. Use, experimental use; WB. western blot; IHC, mimmunohistochemistry: IP. immunoprecipitation; HPLC. high-performance liquid chromatography.
Cell culture
Endothelial cells and hepatocytes.
We used primary endothelial cells derived from brain specimens resected from patients with drug-resistant epilepsy (human epileptic endothelial cells [EPI-ECs], n = 10), as described earlier [20,21], obtained from both EPI and compared with relatively non-epileptic (NON-EPI-EC). Briefly, surgical specimens were incubated in collagenase type II (2 mg/ml; Worthington Biochemical Corp., Lakewood, NJ, USA) at 37 °C for 20 min to dissociate the ECs. The collagenase was then washed off with medium (1.5 g/100 ml, MCDB 105 supplemented with EC growth supplement 15 mg/100 ml, heparin 800 U/100 ml, 10% fetal bovine serum, and penicillin/streptomycin 1%). Cells were stained positive for von Willebrand factor and negative for glial fibrillary acidic protein (GFAP). EPI-ECs were initially expanded in 75 cm2 flasks pre-coated with fibronectin, 3 μg /cm2 [8,7]. Control human brain microvascular cerebral ECs (HBMECs) were purchased from Cell Systems (Kirkland, WA, USA; catalog number: ACBRI 376). The HBMECs (n = 3 in triplicate) were obtained from different batches to confirm the results obtained from control human brain ECs. According to the information provided by the company, the HBMECs were dissociated from normal human brain cortex tissue (obtained from healthy donors), isolated with a Beckman elutriation system, and characterized by staining for von Willebrand factor. Other specific details are available on the company website (Cell Systems, http://www.cell-systems.com/cert/22/137/Products/Products/ACBRI-Site-S). Human primary hepatocytes (n = 3, in triplicate) were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA; catalog number CRL-11233), and the suggested media components were used in the following cell culture experiments [21].
BBB in vitro setup and phenytoin permeability
The BBB in vitro setup is established by using the previously used flow based in vitro / DIV modules [8], which were purchased from FiberCell Systems Inc. (New Market, Maryland; catalog C2025). In the Fiber Cell Polysulfide Plus cartridge, each module contains 20 hollow-fiber capillaries embedded inside a clear plastic chamber, which is attached to a reservoir for media circulation and connected to a pulsatile pump. EPI-ECs and NON EPI-ECs (n = 3, in triplicate) and HBMECs (4 × 106/device) were seeded in the luminal side in different devices. Astrocytes (3 × 106/device) were co-cultured in the abluminal side of the DIV. Once the BBB has been formed (evaluated by transendothelial electrical resistance measurement, TEER data not shown), the permeability of boluses (1.0 mL each) of the radioactive tracers [14C] phenytoin (PerkinElmer, Boston, Massachusetts; catalog NEC-246) was injected upstream into the lumen as described previously [8]. The permeability across the cerebrovascular bed was calculated by graphical integration of drug concentration in the corresponding lumen and ECSs over 10 min (n = 3, in triplicate per condition). Phenytoin permeability values were obtained by integrating the area under the ECSs and the lumen according to an equation described earlier [22,8].
Protein isolation, subcellular fractionation, and Western blotting
EPI-ECs used for the present study were passaged no more than two times after isolation [7]. Total proteins were extracted from epileptic brain tissue (EPI-EC) and NON-EPI-EC regions and also HBMECs, and hepatocytes as described earlier [7]. Subcellular nuclear fractions from EPI-ECs and HBMECs were fractionated using a Subcellular Protein Fractionation Kit (Pierce Biotechnology, Thermo Scientific, Rockford, IL, USA; catalog numbers 78833/78835) according to the manufacturer’s guidelines.
Western blot analysis:
Proteins from EPI-ECs and HBMECs were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride membranes (PVDF, EMD Millipore Corp., Billerica, MA, USA). The membranes were probed overnight at 4 °C with the primary antibody (GR, Santa Cruz Biotechnology, Dallas, TX) and the appropriate secondary antibody (Supplemental Table 1). Proteins from EPI and NON-EPI brain tissues (n = 8) were similarly separated with SDS-PAGE and transferred onto PVDF membranes. To quantify the relative expression of GR and various Hsps and co-chaperones the membranes were probed to identify target proteins such as Hsp90, Hsp70, Hop-1 (Hsp90/70 organizing protein-1 or adapter protein) and Hsp40. The membranes were later probed with Hsp90, Hsp70, GR, Hop-1, Hsp40 and respective secondary antibodies (listed in Supplemental Table 1). PVDF membranes were incubated for 30 min at 50 °C in stripping buffer and later normalized with β-actin or proliferating cell nuclear antigen (PCNA) (for nuclear fractions) where used as loading controls [7]. Protein expression was quantified by Image-J software (National Institutes of Health, Bethesda, MD).
Immunoprecipitation and Western blotting
We performed the immunoprecipitation (IP) assays [23,24] by homogenizing the resected human epileptic brain tissue specimen (both EPI and NON-EPI), isolated brain endothelial cells (EPI-ECs, NON-EPI-ECs), HBMECs and hepatocytes, which were lysed in RIPA Buffer (Sigma, St. Louis, MO) with appropriate protease inhibitors (Sigma). Total protein was estimated using the Bradford Assay (Bio-Rad Protein Assay kit, Hercules, CA). Protein concentration for all samples was then diluted to 1 μg/μl with IP buffer (RIPA in 0.25 % NP-40 and Protease Inhibitor). Also, 500 μl of protein from each sample were precleared with 30 μl of Protein A/G agarose beads (Thermo Scientific, Waltham, MA) by rotating overnight at 4 °C.
The following day, clear proteins were collected by centrifuging at 14000 rpm for 1 h at 4 °C. Also, 100 μl from each sample was saved separately as inputs. Samples were incubated in a rotator with anti-GR (Cell Signaling Technology, Danvers, MA) at 4 °C for 6 h. 30 μl of the Protein A/G agarose beads were added to each sample and continued rotating at 4 °C overnight. Beads were spin down by centrifuging for 1 min at 2000 rpm and washed three times with IP buffer. After final wash the buffer was carefully removed and the beads were resuspended with 2X loading buffer (Laemmli). Proteins, along with the beads, were boiled and separated by SDS-PAGE and then transferred to PVDF membrane (EMD Millipore Corporation Billerica, MA, USA). Membranes were incubated with GR antibody (Santa Cruz Biotechnology, Dallas TX) to confirm IPs. Membranes were then stripped and re-probed with anti-Hsp90 or anti-Hsp70 (Origene Antibodies, Rockville MD), anti-Hsp40 (R&D Systems, Minneapolis, MN) and anti-Hop-1 antibodies (Santa Cruz Biotechnology, Dallas TX) to determine the co-immunoprecipitation (co-IP) pattern. All protein bands were quantified by ImageJ software (National Institutes of Health, Bethesda, MD, USA.). The percentage (%) binding of GR with each of the co-chaperones (Hsp90, Hsp70, Hsp40, and Hop-1) was graphically plotted. Similarly, western blot analysis was performed and quantified for the other target proteins, CYP3A4 and P-gp/MDR1, and normalized with β-actin in the EPI and NON-EPI brain tissue (all antibodies details are listed in Supplemental Table 1).
Small interfering RNA (siRNA) gene silencing of GR in EPI-ECs
Small interfering RNA (siRNA) gene silencing of GR was performed in EPI-ECs. Gene-specific human NR3C1 GR (catalog: E-003424–000020) siRNA oligonucleotides were purchased from GE Dharmacon, Inc. (Lafayette, CO, USA) and used at a final concentration of 1 μmol/L for EPI-EC [8,7]. GR silencing (human NR3C1) was performed using Accell SMART pool siRNAs containing four siRNAs designed for the GR gene: (5′-GGAGCAAAUAUAAUUGGUA-3′, 5′-GCAUGUACGACCAAUGUAA-3′, 5′-GGGUGGA GUUUCGUAAUUU-3′, and 5′-CUAACAUGAUUUGU GUCUA-3′). The effects of GR siRNA on EPI-EC gene expression were confirmed by western blotting (n = 3 subjects, in triplicate per condition). Then, immunoprecipitation analysis in EPI-ECs focal regions, with and without GR siRNA was conducted to further confirm the interaction of GR and Hsps (Hsp90, Hsp70, Hsp40) (as described above).
Histology and immunohistochemistry
We used brain tissue specimens from patients who had undergone surgical resection for intractable epilepsy from EPI (n = 6) and compared them with the respective NON-EPI region. Gross anatomical evaluation was made by cresyl violet histological staining, which was performed on brain slices (n = 5 per specimen) for cytoarchitectural analysis to identify dyslamination, ectopic neurons, and vascular malformations [16]. Immunohistochemical staining was performed on contiguous sections (10 μm; n = 5 per specimen, in triplicate) obtained from resected brain tissues (Table 1).
Diaminobenzidine (DAB) staining:
Brain sections were incubated at 4 °C overnight with primary GR, Hsp90 and Hsp70 antibodies. The detailed method has been described previously [21,16]. After washes, sections were incubated for 1 h at 25 °C with a biotinylated anti-mouse IgG or anti-rabbit IgG, based on the species (primary and secondary antibodies used are listed in Supplemental Table 1) followed by 1 h with the avidin/biotin complex (Elite Vectastain ABC kit, cat. PK-6102; Vector Labs, Burlingame, CA, USA), visualization with DAB (peroxidase substrate kit, SK-4100; Vector Labs), dehydration, and mounting.
Immunofluorescence staining:
We also determined expression and localization pattern of GR and specific chaperone proteins (Hsp90 and Hsp70) in the GR maturation cascade [25] by immunofluorescence staining on contiguous brain slices. These were similarly 10 μm thick slices, stained/co-stained for GR, Hsp90, Hsp70. Astrocytic (GFAP) and neuronal (NeuN, neuronal nuclei) markers were also used [7,16] to confirm the precise cellular localization of the staining. The concentrations and sources of all primary and secondary antibodies used are listed in Supplemental Table 1. In brief, after blocking, the sections were incubated in the first primary antibody at 4 °C overnight, followed by 2 h re-incubation at 25 °C in the corresponding secondary antibody. Sections were then incubated in the second primary antibody at 4 °C overnight, followed by 2 h incubation at 25 °C in the second secondary antibody to achieve understanding of the localization of two target proteins. Blocking for autofluorescence with Sudan B preceded mounting with VECTASHIELD® Mounting Medium with DAPI (Vector Laboratories, cat. H-1200). Sections were analyzed by fluorescence microscopy and the acquired images were processed using ImageJ software.
Quantifications of the immunostaining (n = 20/subject) were performed using ImageJ software (National Institutes of Health) as previously described [26]. Background was removed using the brightness and contrast controls and the Rolling Ball Radius function. Images were then converted to 8-bit from color and threshold was maintained by using the Threshold function. Resulting highlights after adjustment were then measured for average relative intensity using the Measure function. The analysis of variance analysis (ANOVA) was used (Origin Pro 9.0 Software) to identify significant differences in expression between the EPI and NON-EPI brain tissue regions.
Drug treatment and immunocytochemical evaluation
To assess the effects of drug exposure (dexamethasone, DEX; rifampicin, RIF, and phenytoin, PHT) as GR ligand or modulators, HBMECs and EPI-ECs were cultured in 6-well plates or chambered slides. The HBMEC and EPI-ECs were treated with DEX, 10 μM; RIF 10 μM and PHT, 50 μM, or not treated for 24 h [27–30]. Subcellular nuclear fractions from EPI-ECs and HBMECs were fractionated using a Subcellular Protein Fractionation Kit (Pierce Biotechnology, Thermo Scientific, Rockford, IL, USA; catalog numbers 78833/78835) used earlier [7] according to the manufacturer’s guidelines. Later GR nuclear expression was evaluated by western blot, with and without drug exposure. Polyvinylidene fluoride membranes were incubated for 30 min at 50 °C in stripping buffer and later normalized with proliferating cell nuclear antigen (PCNA) (for nuclear fractions) as loading controls [7]. Protein expression was quantified by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Primary brain endothelial cells isolated (n = 4, in triplicates) from the EPI-ECs and NON EPI-EC regions were grown on fibronectin-coated chamber slides at a seeding density of 30 × 103 cells/well. Each chamber was separately designated as in the treated or untreated group. GR ligand, DEX 10 μM was treated for 24 h on both sets of endothelial cells (EPI and NON EPI-ECs). The EPI-ECs and NON-EPI ECs were subsequently washed, fixed with 2 % formalin, washed again with 1x PBS and then processed by immunofluorescence staining. Primary antibodies (GR and Hsp90) and respective secondary antibodies were used to follow the co-localization pattern in the EPI-EC vs NON EPI-ECs. The primary and secondary antibodies used, concentration, and sources are listed in Supplemental Table 1.
The immunocytochemistry procedure was followed as described earlier [31]. Cells were visualized using the fluorescence microscope and compared within the EPI and compared to NON EPI-EC (n = 3 subjects/in triplicates). Images were subsequently processed using ImageJ (National Institutes of Health, Bethesda, MD, USA) and Q-Capture-Pro™ software (QImaging, Inc., Surrey, BC, Canada). All images were converted to an 8-bit format; brightness/contrast were adjusted using the same parameters for all fluorophores. Threshold signals were set and segmented via the watershed feature of ImageJ.
The number of cells per square millimeter intensity was determined in each individual field. Values were collected and expressed as mean ± standard error of the mean (SEM). The nuclear intensity of each stain was determined using the corresponding DAPI. The DAPI images were rendered in black and white using the Make Binary function, and the intensity of each nucleus was measured by mapping to the original image and using the Analyze Particles function (size limit 0.005 in2-infinity). For cytoplasmic staining, Hsp images were rendered in black and white by the Make Binary function to determine the cell shape. The Image Calculator function was then used to remove the binary DAPI/nuclear image(s) from the black and white Hsp image(s), and the resulting image used to measure cytoplasmic staining by mapping and the Analyze Particles function (size limit 0.005 in2-infinity).
ATPase assay
The ATPase activity from the tissue lysates of EPI and NON-EPI (n = 5, triplicate) and cell lysates (EPI-ECs, NON EPI-EC, HBMEC/control ECs and HEPATO) was measured by detecting the free inorganic phosphate (Pi) using a Pi-per-Phosphate Assay kit (Molecular Probe, catalog numbers P22061). The assay measures an increase in fluorescence absorption of an Amplex Red reagent that is proportional to the amount of Pi in the samples [32,33]. Briefly, a standard curve was obtained by using different known concentration of Pi. The standards and samples were reacted with Amplex Red reagent for 60 min at 37 °C in the dark. The Amplex Red reagent reaction product was detected fluorometrically using a fluorescence microplate reader (BioTek, Synergy HT, USA), with a setting for excitation in the 530–560 nm and emission detection at 590 nm. The levels are represented as μmol of Pi per μg of protein in each specimen.
HPLC-UV analysis on epileptic tissue and serum
Extraction procedure:
Brain tissues from both EPI and NON-EPI regions were homogenized in methanol/water (60/40 v/v; 10 mg tissue/100 μl) as previously described [34]. Then, 100 μl of tissue or serum sample was taken from the respective subject and 0.2 M perchloric acid added, vortexed for 30 sec, and then samples were centrifuged at 4,900 g for 10 min.
Standard and chromatographic conditions:
Standard OXC was purchased from Sigma Aldrich (OXC, catalog O3764). OXC levels in the EPI and NON-EPI brain tissue and serum samples (n = 4 subjects) were measured by using a reversed-phase high-performance liquid chromatography-ultraviolet detection (RPHPLC- UV; Agilent 1200 Series) and a Zorbax Eclipse Plus C18 column (4.6 × 150 mm, 3.5 μm) as described earlier [8].
Stability:
OXC is stable in serum/plasma when stored at −20 °C for at least 4 weeks. The extracted drug samples were stored at room temperature (25 °C) for 24 h prior to analysis.
Statistical analysis
As stated above, all data are expressed as mean ± standard error of the mean. The Wilcoxon signed rank test and post t-test was used to compare means of obtained data sets as required. Other data were analyzed by one-way or two-way analysis of variance (ANOVA) followed by a Bonferroni test for multiple comparison. A p-value of < 0.05 was considered statistically significant. The linear regression (Pearson’s coefficient, r) is applied to determine the associations between GR-Hsp interaction and clinical characteristics (e.g. seizure-onset age). Origin 9.0 software (OriginLab Corp., Northampton, MA, USA) was used for statistical analyses.
Results
Extensive cellular disorganization coincides with increased GR-Hsp co-localization at the neurovascular interface of the epileptic brain
We analyzed patient cohorts and evaluated the pre-characterized epileptic regions by histopathological (post-resection) staining as performed previously (17). This showed extensive cellular disorganization with heterotopic and dysmorphic neurons in the cortex and hippocampal regions (Fig. 1a). Balloon cells in the tissue slices were identified across multiple specimens, confirming the pathology of focal cortical dysplasia type IIb. We further identified increased GR expression pattern (*p < 0.001, Fig. 1b, e) across the immunostained brain tissue slices. The upregulated trend was localized on the brain micro-capillaries and neurons in the EPI regions relative to the NON-EPI. Among the Hsps, we identified Hsp90 (Fig. 1c) and Hsp70 (Fig. 1d) to be highly immunoreactivity at the neurovascular regions of the EPI brain. The increased Hsp90 (*p < 0.05) and Hsp70 (*p < 0.05) expression in EPI vs. NON-EPI regions indicated significant differences in value within the analyzed specimens. Immunofluorescence staining (Fig. 2) supported the diaminobenzene staining pattern (Fig. 1) on these EPI tissue sections and additionally confirmed the localization pattern and expression of GR, Hsp90 and Hsp70 (Hsp90, Fig. 2a; Hsp70, Fig. 2b) in the brain microvessels, astrocytes and neurons. In EPI tissue, we found Hsp90 staining (Fig. 2a, d) to be prominent in the microvessels and extensive in the neurons and astrocytes as well, and GR-Hsp90 co-localization within these areas was observed to a greater extent (Fig. 2a). In contrast, Hsp70 expression was limited across the astrocytes (n = 6) but was profound in the microvessels and neurons in EPI relative to NON-EPI (Fig. 2b) specimens. However, this GR-Hsps co-localization pattern to a limited extent was also seen in NON-EPI regions (Fig. 2a), suggesting that a common mechanism of GR maturation operates in both EPI and NON-EPI regions of the brain.
Fig. 1.
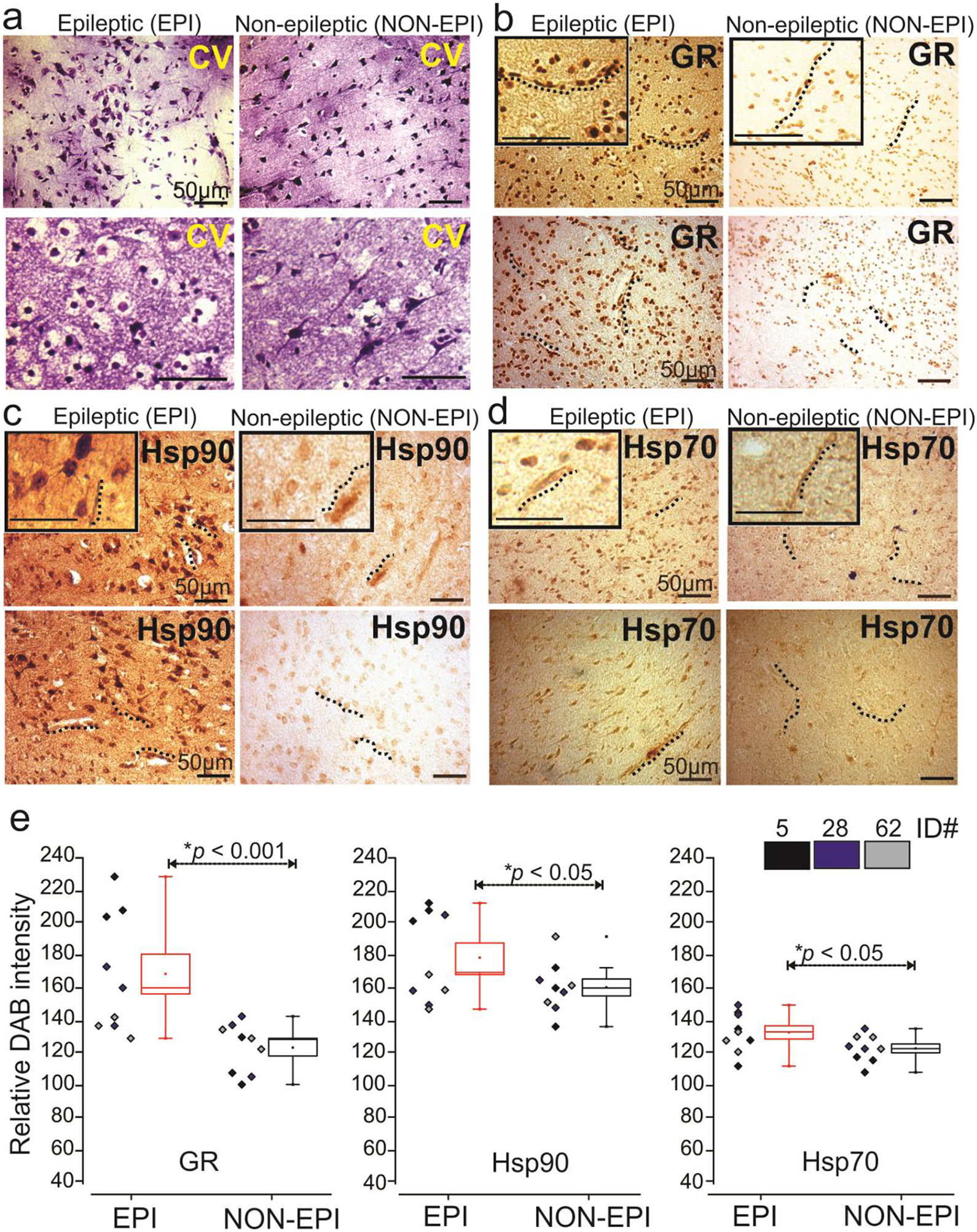
Histological characterization of epileptic (EPI) and non-epileptic (NON-EPI) brain tissue and GR-Hsp localization in focal epilepsies. (a) Cresyl violet staining showing dysmorphic region and gross disorganization in cellular pattern in mostly EPI compared to NON-EPI regions. The dysmorphic and balloon cells were present more extensively in EPI brain regions compared to NON-EPI. (b-d) Diaminobenzidine (DAB) immunohistochemical staining of GR, Hsp90 and Hsp70 exhibited positive staining, predominantly in the micro-capillaries and across neurons in the lesioned brain areas. Sporadically, Hsp90-positive staining was also noticed in the astrocytes in these EPI regions, which was negligible in NON-EPI regions. The magnified images are provided as inserts. (e) Results are expressed as mean ± S.E.M by one-way ANOVA (n = 3 subjects, ID # 5, 28, 62/ triplicates) depicts significant increase in GR (*p < 0.001), Hsp90 (*p < 0.05) and Hsp70 (*p < 0.05) expression in EPI compared to NON-EPI brain tissue regions.
Fig. 2.
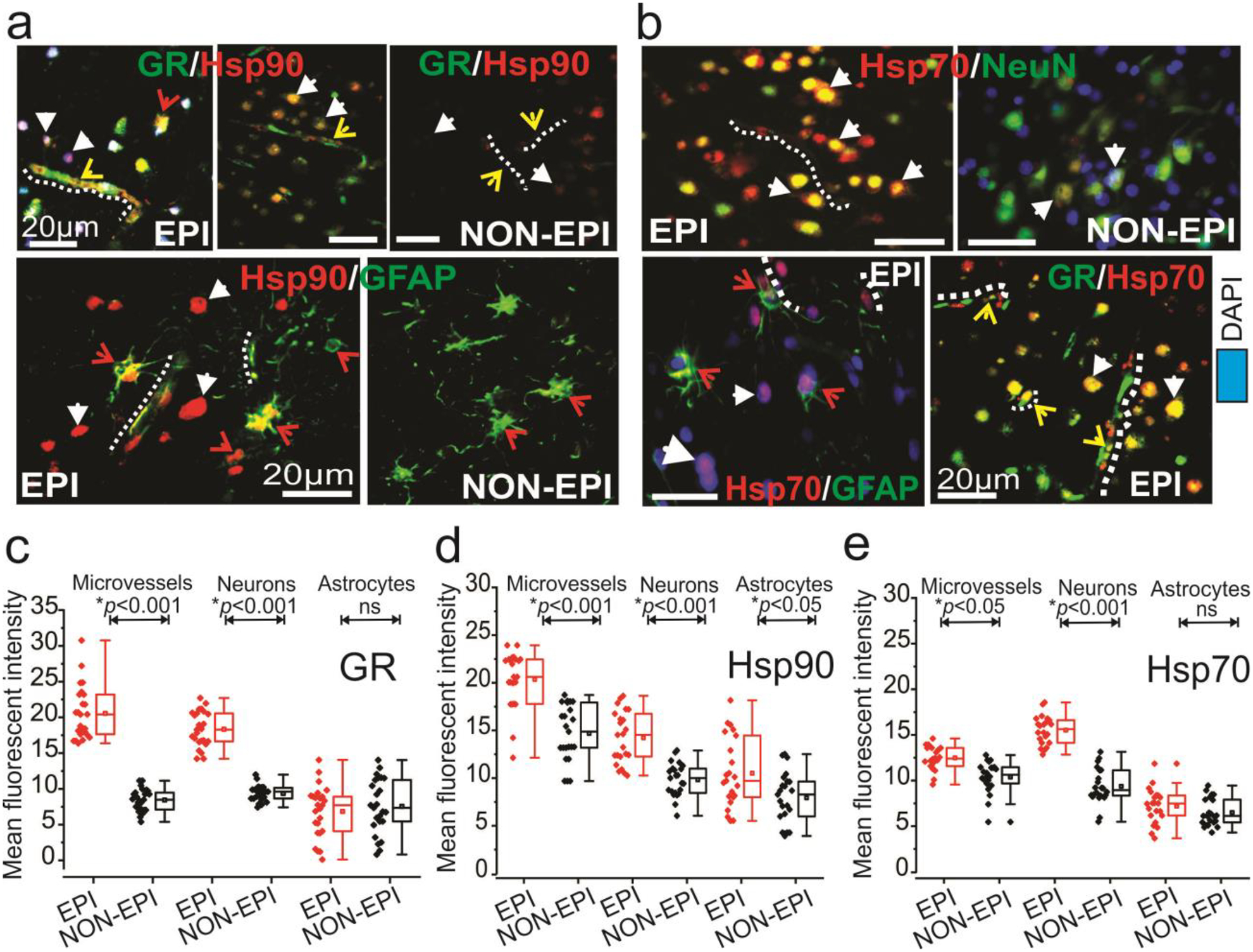
Neurovascular GR-Hsp overexpression and co-localization at the epileptic brain regions. (a) Immunohistochemistry results indicate co-localization and increased expression of GR and Hsp90 in the micro-capillaries and across neurons in EPI brain, as shown by the representative images. Hsp90 staining across the astroglial cells was also evident. However, GR and Hsp70 staining (b) was mostly restricted to micro-capillaries and neurons. The EPI regions showed increased staining for GR and Hsp70 compared to NON-EPI regions. (c, d, e) Quantification of immunohistochemistry in EPI compared to NON-EPI indicates increased GR and Hsp90 expression in the microvessels (*p < 0.001) and neurons (*p < 0.001) and Hsp90 staining was also found in the astrocytes (*p < 0.05). However, Hsp70 expression was significantly elevated in the microvessels (*p < 0.001) and neurons (*p < 0.001) in EPI compared to NON-EPI tissues. Results are expressed as mean ± SEM, *p < 0.05, ***p<0.001 ANOVA, analysis of variance was used to compare data sets. Note: The standard markers for neuronal nuclei (NeuN) and glial fibrillary actin protein (GFAP) were used. The white-dotted lines indicate micro-capillaries; arrowheads are neurons, and astrocytes are highlighted with red arrows.
GR and Hsp are overexpressed in the epileptic regions of the brain
As depicted in Fig. 3, we found significantly increased expression of GR (*p = 0.01) and Hsp40 (*p = 0.03), and elevated concomitant expression of Hsp90, Hsp70 and Hop-1(Hsp90/70 organizing protein-1 or adapter protein) in EPI vs. NON-EPI tissue regions (Fig. 3a, b). Together, our data indicate that pathological alteration in the epileptic regions show increased GR expression and elevated overall expression of chaperones; Hsp90, Hsp70, Hsp40 and Hop-1. The effect of increased expression of GR and Hsps on GR-Hsp90/Hsp70 co-localization at the neurovascular regions may result in higher cytosolic GR maturation in the epileptic brain.
Fig. 3.
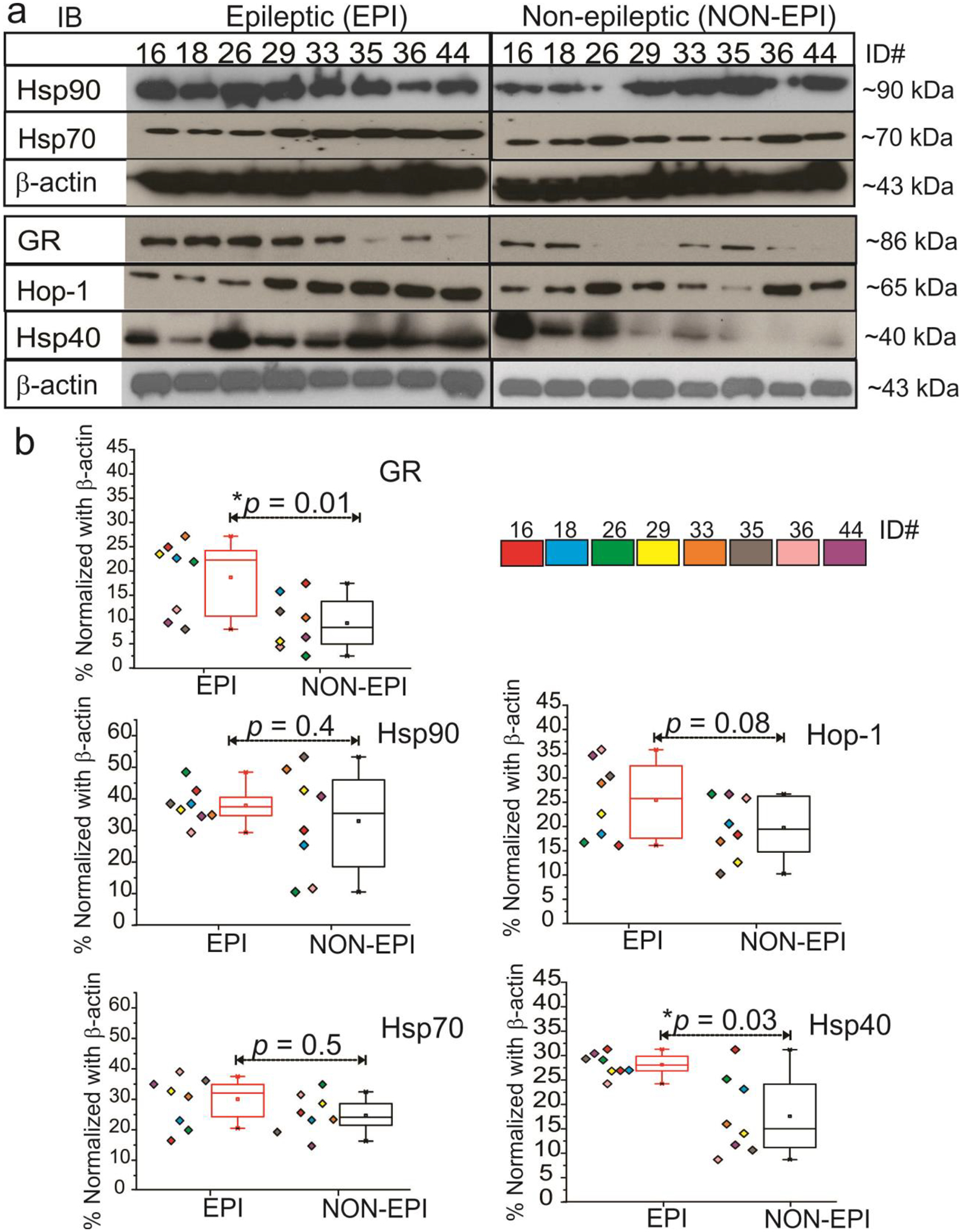
GR-Hsp and Hop expression pattern in EPI and NON-EPI brain tissue regions (a, b). Western blot or immunoblot (IB) showed a significant increase in GR (**p < 0.01) expression and Hsp40 (*p < 0.03) in EPI vs. NON-EPI; however, Hsp90, Hsp70, and Hop-1 expression showed non-significant alterations (n = 8 subjects, color-coded, #ID, 16,18,26,29,33,35,36,44; EPI and NON-EPI) although a trend toward an increased level is evident among the subjects, as shown in the quantification (b). Results are expressed as mean ± SEM; *p < 0.05. ANOVA, analysis of variance.
GR-Hsp associations with increased ATPase activity accelerates GR maturation machinery in the epileptic brain regions. Association with seizure-onset age
Since GR maturation involves chaperoning by an active Hsp90 chaperone complex [35,36] we investigated the binding dynamics of GR with chaperones Hsp90 and Hsp70. We found that the GR-Hsp90/Hsp70 interactions were variable across EPI and NON-EPI brain regions (Fig. 4a, b). However, the overall binding pattern suggested greater levels of Hsp90 and Hsp70 associated with GR in NON-EPI, while lesser amounts of the chaperones were bound to GR in the EPI samples. Since higher Hsp90/Hsp70 bound to its client proteins (GR in this case) is indicative of an immature GR undergoing maturation [37,38] while a GR both unbound to ligand and with lower Hsp90/Hsps70 retained on client proteins suggest an already matured GR [37,38] inferred that GR maturation is faster in EPI vs. NON-EPI regions. To further support this finding, we estimated the ATPase activity (monitored by the release of phosphate using ATPase activity) in tissue supernatants from EPI and NON-EPI (n = 5) regions. We found significantly higher ATPase activity (Fig. 4c) in the EPI (*p < 0.05) vs. NON-EPI regions. Since higher ATPase dependence of chaperones indicates acceleration in its chaperoning activity [32,39], an enhanced overall ATPase accelerated the GR-Hsp90/70 maturation machinery in the epileptic brain resulting in faster GR maturation. This is evident from the low GR-Hsp90/70 association events (Fig. 4a, b), despite GR and Hsp90/70 expressions being predominant in the EPI regions (Fig. 3). Within this patient cohort, we also found a correlation with age of seizure onset among the subjects and their respective GR-Hsp90 (r = 0.60) and GR-Hsp70 (r = 0.56) binding percentages in EPI and NON EPI brain tissues regions (Fig. 4d). The difference in GR-Hsp90/70 interactions in the EPI and NON-EPI regions was lower when the onset in seizure occurred at a very young age and this difference was higher when the seizure onset happened at higher age. This may also suggest that seizure onsets occurring at higher ages with increased GR-Hsp interactions maybe be more prone to pharmacoresistance.
Fig. 4.
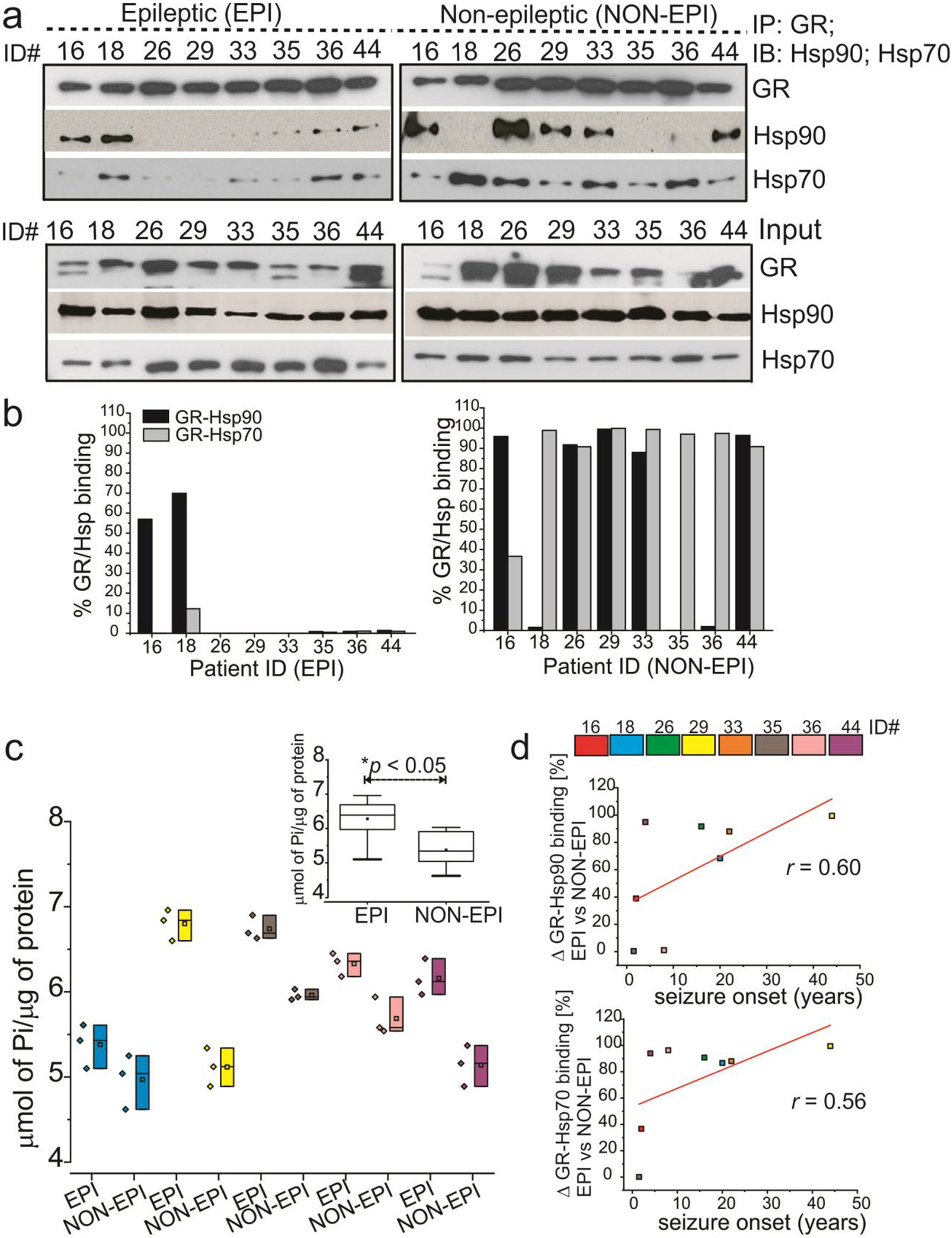
Immunoprecipitations (IPs) depicting GR-Hsp90/Hsp70 interactions on brain tissue. (a) The interaction of GR-Hsp90 and GR-Hsp70 is variable across epileptic (EPI) and non-epileptic (NON-EPI) brain regions (n = 8 subjects, color-coded, #ID, 16, 18, 26, 29, 33, 35, 36, 44; evaluated), but the overall low GR-Hsp90/Hsp70 binding pattern in the EPI suggests faster GR maturation. The lanes marked as Input indicate that 1 ⁄5th of total extract was loaded and analyzed by immunoblot (IB) for GR and Hsp90/Hsp70. (b) GR-Hsp90/Hsp70 binding percentages have been plotted, depicting the difference in GR-Hsp90/Hsp70 interactions in the EPI vs NON-EPI regions. (c) Enhanced focal ATPase activity and accelerated GR-Hsp maturation machinery in EPI compared to NON-EPI region is evident. Individually inorganic phosphate levels of each subject (n = 5) in EPI vs. NON-EPI tissue are plotted. Analysis of the data in the insert (c) showed the average values plotted post doc t test. (d) Difference in GR-Hsp90 (r = 0.60) and GR-Hsp70 (r = 0.56) binding percentages, depicting difference in these interactions (Δ) in EPI and NON-EPI brain tissue (n = 8 subjects) were directly correlated with individual seizure onset age (years) among the small specimen cohort analyzed. Results are expressed as mean ± SEM by ANOVA, *p < 0.05.
Likewise, we also determined GR interaction with the Hsp90-Hsp70 complex involving co-chaperones, Hop-1 and Hsp40 in EPI-ECs. As depicted in Fig. 5, we again found lower GR-Hsp interactions in the GR maturation complex of EPI-ECs relative to NON-EPI-ECs, suggesting elevated GR maturation machinery in the endothelial cells of the EPI regions. ATPase activity also showed increased ATPase levels in EPI-ECs (Fig. 5c). The NON-EPI-ECs, control HBMECs and hepatocytes displayed higher GR-Hsp associations, indicating slower GR maturation. Also consistent with the earlier finding in brain tissues (Fig. 4d), we found a correlation (r = 0.96) with the age of seizure-onset and percentage change in GR-Hsps/co-chaperones Hsp90, Hsp70, Hop-1 and Hsp40 interactions in EPI and NON EPI-ECs (Fig. 5d). Together, these data reiterate the findings that the epileptic brain tissue and epileptic brain endothelial cells show enhanced GR maturation as evident from high ATPase levels and low GR-Hsp interactions during GR maturation cascades that could also be associated with higher seizure-onset age in these subjects.
Fig. 5.
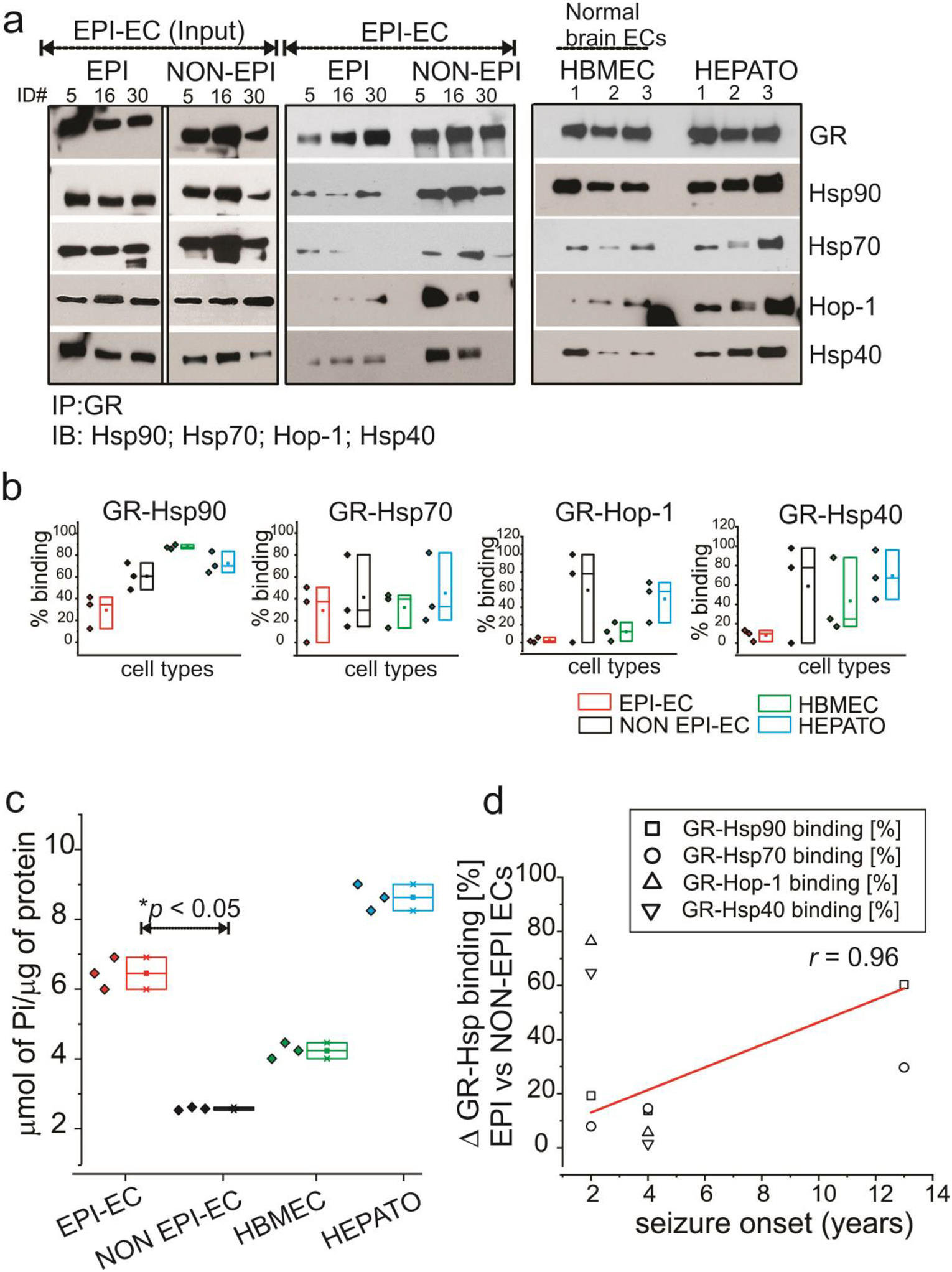
Brain endothelial cell GR-Hsp interactions. (a) The interaction of GR in human brain endothelial cells with the binding partners Hsp90, Hsp70, Hsp40 and Hop is more evident in NON EPI-ECs compared to EPI-ECs (n = 3, #ID, 5, 6, 30). The lanes marked as Input indicate that 1 ⁄5th of total EPI-EC extract loaded. Control ECs (HBMEC, n = 3) and hepatocytes (HEPATO, n = 3) display comparable GR-Hsp binding profiles similar to NON EPI-ECs. The low binding pattern of GR-Hsps in EPI-ECs relative to NON EPI-ECs is evident and suggests faster GR maturation in the EPI-ECs. (b) The quantitation of the GR-Hsp90/Hsp70/Hop-1/Hsp40 interactions depicts a distinct pattern between individual chaperone proteins with GR. (c) The increased maturation of GR is supported by significant increases (*p < 0.05) in ATPase activity of EPI-ECs vs. NON EPI-ECs. The limited ATPase activity in NON EPI-EC is more comparable to HBMEC. (d) Difference in GR-Hsp90, GR-Hsp70, GR-Hop-1 and GR-Hsp40 binding percentages, depicting difference in these interactions (Δ) in EPI and NON-EPI-ECs (n = 3 subjects) were directly correlated (r = 0.96) with individual seizure onset age (years) among the small specimens cohort analyzed. Results are expressed as mean ± SEM by ANOVA, *p < 0.05.
GR modulation alters GR-Hsp interactions in focal EPI-ECs
GR silencing on EPI-ECs was successfully performed (Fig. 6a), as demonstrated by significantly reduced GR protein levels by siRNA (*p < 0.05), compared with non-siRNA of focal EPI-ECs. We found increased GR-Hsp interaction in the complex, with increased levels of Hsp90, Hsp70 and Hsp40 binding (Fig. 6b), suggesting a possible slowdown in the GR maturation process when GR expression was reduced by siRNA in EPI-ECs compared to its corresponding non-siRNA EPI-ECs. These data confirm that GR maturation can be regulated by silencing GR in EPI-ECs.
Fig. 6.
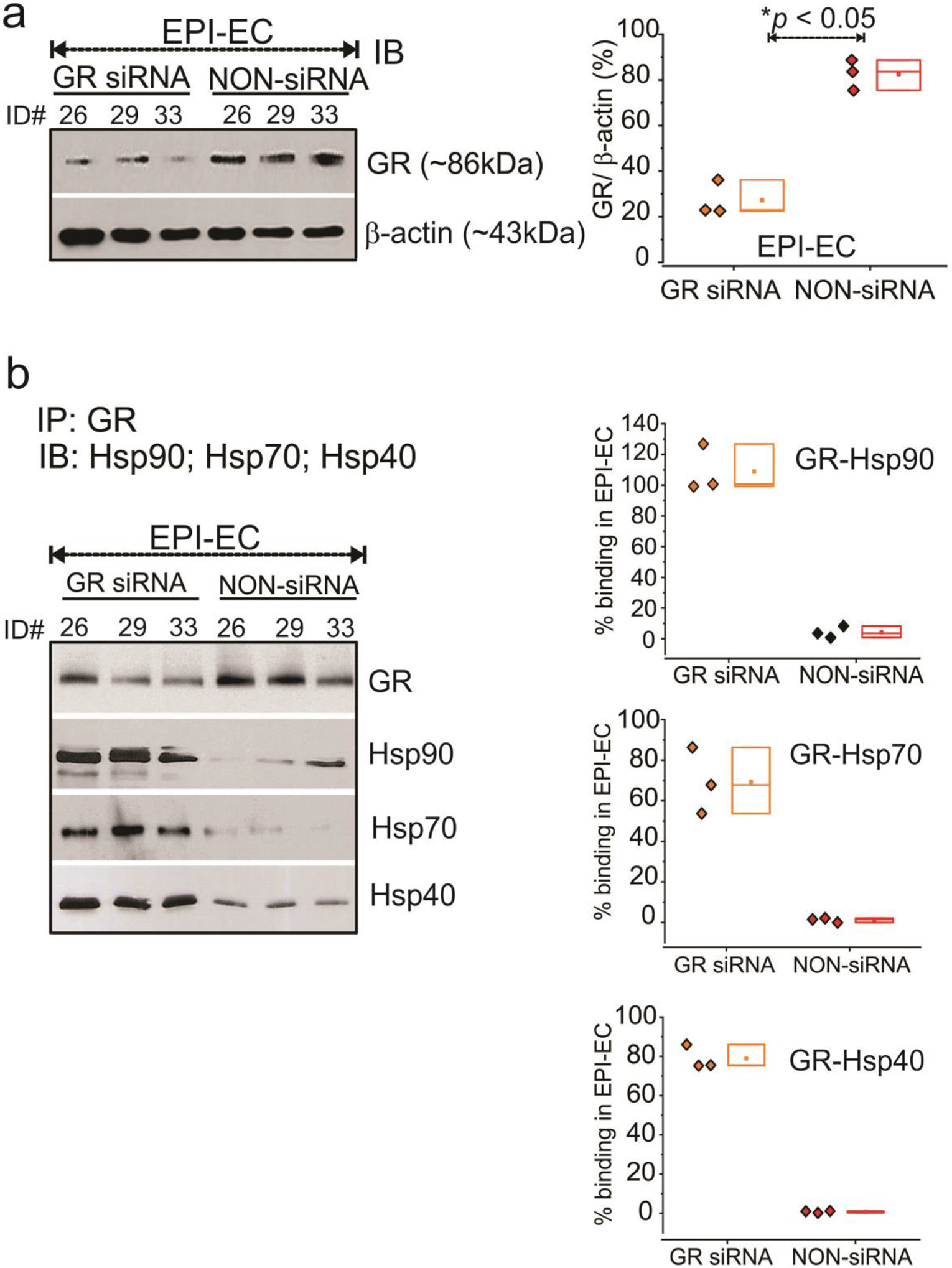
GR silencing in human EPI-ECs alters GR-Hsp interaction. (a) GR silencing of genes in EPI-ECs was evaluated by western blot and compared with non-siRNA EPI-ECs counterparts (n = 3 subjects #ID, 26, 29, 33). β-actin was used as a loading control and normalization. (b) The interaction of GR and Hsps were compared within the GR-silenced and non-silenced EPI-ECs by immunoprecipitation analysis and binding percentages were quantified. GR-siRNA on EPI-ECs showed higher trend of GR-Hsp90, GR-Hsp70 and GR-Hsp40 binding compared to non-siRNA EPI-ECs, suggesting that lowering GR levels can regulate GR-Hsp interactions and downregulates the GR maturation process in EPI-ECs. The results are expressed as mean ± SEM by ANOVA, *p < 0.05.
GR nuclear translocation is significantly increased with GR modulators in epileptic brain endothelial cells. Implications for disease pathology and drug resistance
GR nuclear translocation is a vital event regulating transcription of genes involved in conferring possible drug resistance in epilepsy. We therefore first studied GR nuclear expression in EPI-ECs and control ECs (HBMEC). This showed increased nuclear GR levels at steady-state in EPI-ECs (Fig. 7a). Further treatment with GR modulators and ligands like dexamethasone (DEX), rifampicin (RIF) or phenytoin (PHT) significantly increased nuclear GR in EPI-ECs relative to HBMEC. Likewise, we also determined increased nuclear translocation of GR with DEX treatment in focal EPI-ECs analyzed by immunocytochemistry (Fig. 7b). However, Hsp90 expression remained unaltered with DEX exposure under similar conditions. Quantifying the cytoplasmic and nuclear fluorescent staining (Fig. 7c) further confirmed overall increased GR levels in the nucleus relative to the cytoplasm in EPI-ECs and NON-EPI-ECs, and showed a more significant increase in GR levels (*p < 0.01) in the nucleus subsequent to DEX exposure in EPI-ECs compared to the cytoplasm or when compared to nuclear GR of EPI-ECs in the steady state without DEX treatment. These data suggest that disease pathology in the epileptic brain and the presence of GR ligands such as with steroidal pretreatment (e.g. DEX) may synergize to cause greater GR translocation to the nucleus in focal EPI-ECs, which subsequently contributes to pharmacoresistance.
Fig. 7.
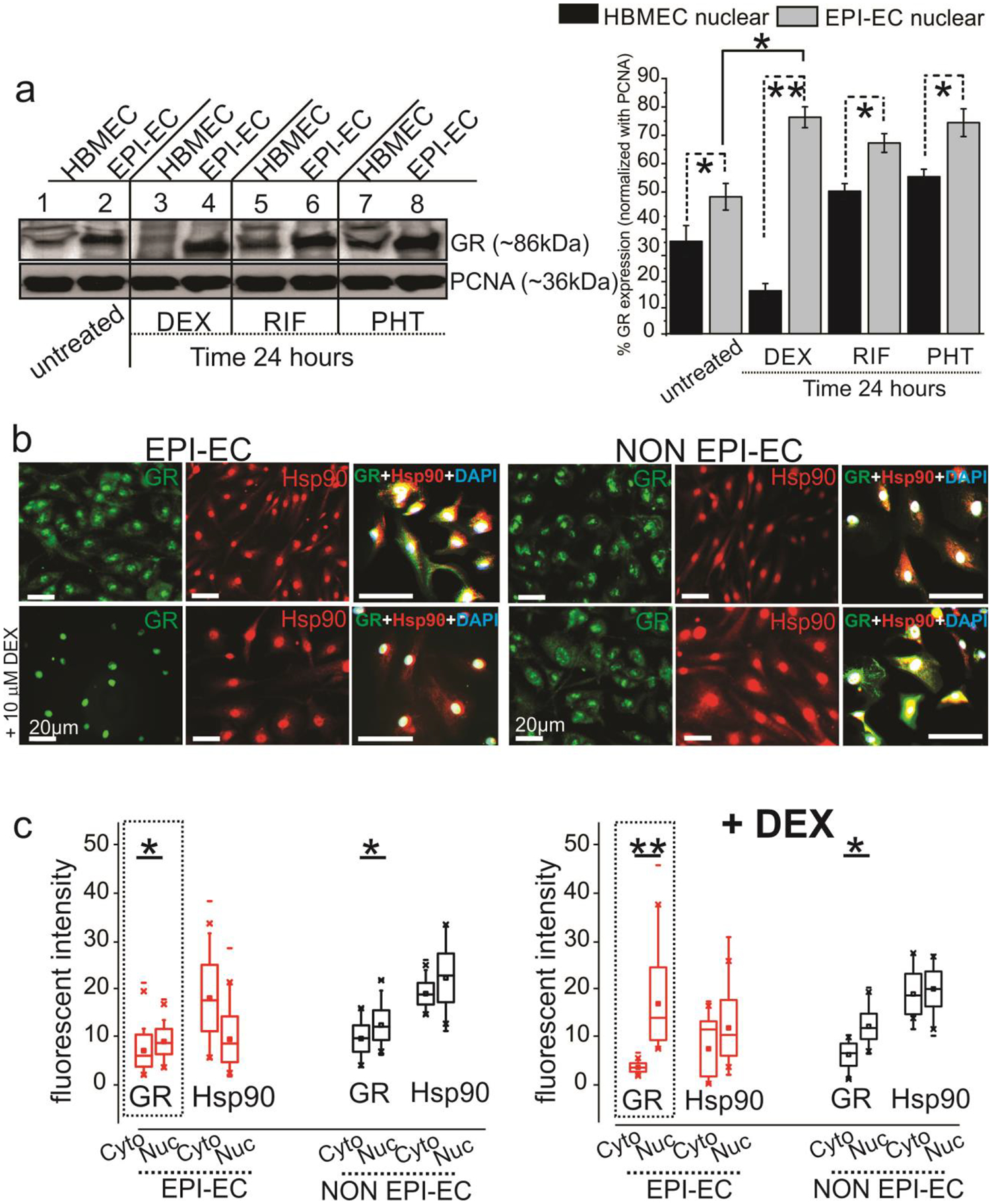
GR levels in brain endothelial cells are affected by disease state and drug exposure and increased GR nuclear translocation occurs with dexamethasone (DEX) treatment in EPI-ECs. (a) A significant increase in GR nuclear expression in EPI-ECs compared to control HBMEC is observed in steady state. The GR levels were followed, with and without GR ligand (DEX, RIF, and PHT) treatment. DEX/RIF/PHT treatment showed elevated GR levels in EPI-EC nuclear fractions, suggesting that nuclear translocation was induced both in disease state (EPI-EC) and by GR ligands. Proliferating cell nuclear antigen (PCNA) was used as loading control for nuclear fractions. (b) Representative immunofluorescent images of increased nuclear translocation of GR with DEX treatment is seen more in focal EPI-ECs compared to NON EPI-ECs, while Hsp90 pattern did not change significantly after treatment. The merged images of GR, Hsp90 and DAPI are indicative of the cytoplasmic or nuclear GR-Hsp90 localization pattern, pre- and post-DEX treatment. DAPI is used as a nuclear counterstain. (c) Analysis of data (n = 3 subjects/in triplicates) by paired Wilcoxon test indicated a significant effect of DEX in intracellular GR translocation (cytoplasm to nucleus, **p < 0.01) and is more pronounced in EPI-ECs and relative to NON EPI-ECs (cytoplasm vs nucleus, *p < 0.05) or EPI-EC without treatment. Results are expressed as mean ± SEM by ANOVA and paired Wilcoxon-test, asterisks indicate *p < 0.05; **p < 0.01.
Pharmacoresistance attenuates phenytoin penetration across epileptic BBB and diminishes GR-CYP-MDR-dependent oxcarbazepine (OXC) bioavailability in focal epilepsies
A decreased phenytoin (PHT) permeability across EPI BBB in vitro was found when compared with NON-EPI BBB (Fig. 8a) or control/HBMEC BBB. The 14C phenytoin showed a 1.53-fold increase in permeability levels (1.03 × 10−7 cm/s) in NON-EPI DIV-BBB (*p < 0.05) compared with EPI-EC DIV-BBB (6.7 × 10−8 cm/s). The permeability pattern of phenytoin in NON EPI-EC DIV-BBB was comparable to that of HBMEC DIV-BBB (6.8 × 10−7 cm/s). In addition, OXC penetration was absent or non-detectable (Fig. 8b) along with significantly increased GR, P-gp/MDR1 and CYP expression in EPI brain tissue vs NON-EPI (Fig. 8c) evaluated in the same individuals (n = 4 subjects, ex vivo). Likewise OXC levels were consistently high in the serum of the same individuals (Fig. 8b). Together, this suggests that reduced AED penetration and bioavailability in the focal EPI regions correlates to a GRCYP-MDR1 overexpression pattern. A model describing our present findings is depicted in Fig. 9, the implications of which are discussed in the following section.
Fig. 8.
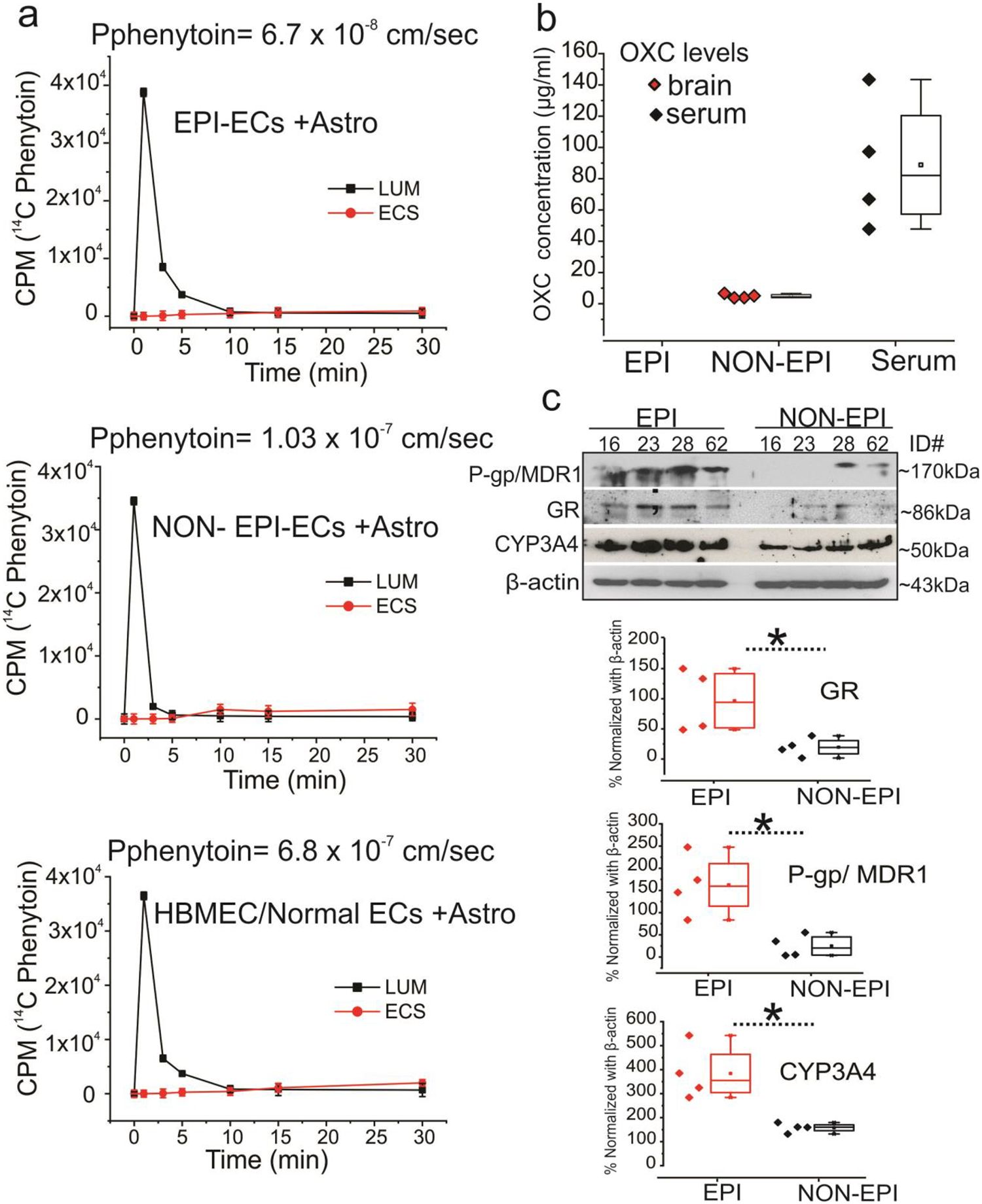
Decreased phenytoin permeability across in vitro BBB in EPI compared with NON-EPI BBB and no oxcarbazepine (OXC) penetration in EPI brain tissue is linked to increased GR, P-gp/MDR1 and CYP levels ex vivo. (a) The 14C phenytoin showed a 1.53-fold increase in permeability levels (1.03 ×10−7 cm/s) in NON-EPI in vitro BBB (*p < 0.05) compared with EPI-EC in vitro BBB (6.7 × 10−8 cm/s). The permeability pattern of phenytoin in NON EPI-EC in vitro BBB was comparable to that of HBMEC in vitro BBB (6.8 × 10−7 cm/s). (b) OXC is measured in the NON-EPI regions by HPLC-UV, as well as in the serum (n = 4 subjects, #ID, 16, 23, 28, 62) however, was non-detectable in EPI regions (ex vivo) of the same individual. (c) In addition, the western blot analysis shows a significant increase in GR, P-gp/MDR1 and CYP expression levels in the EPI vs. NON-EPI regions in the same brain tissue specimens. Data are mean ± SEM by ANOVA, *p < 0.05.
Fig. 9.
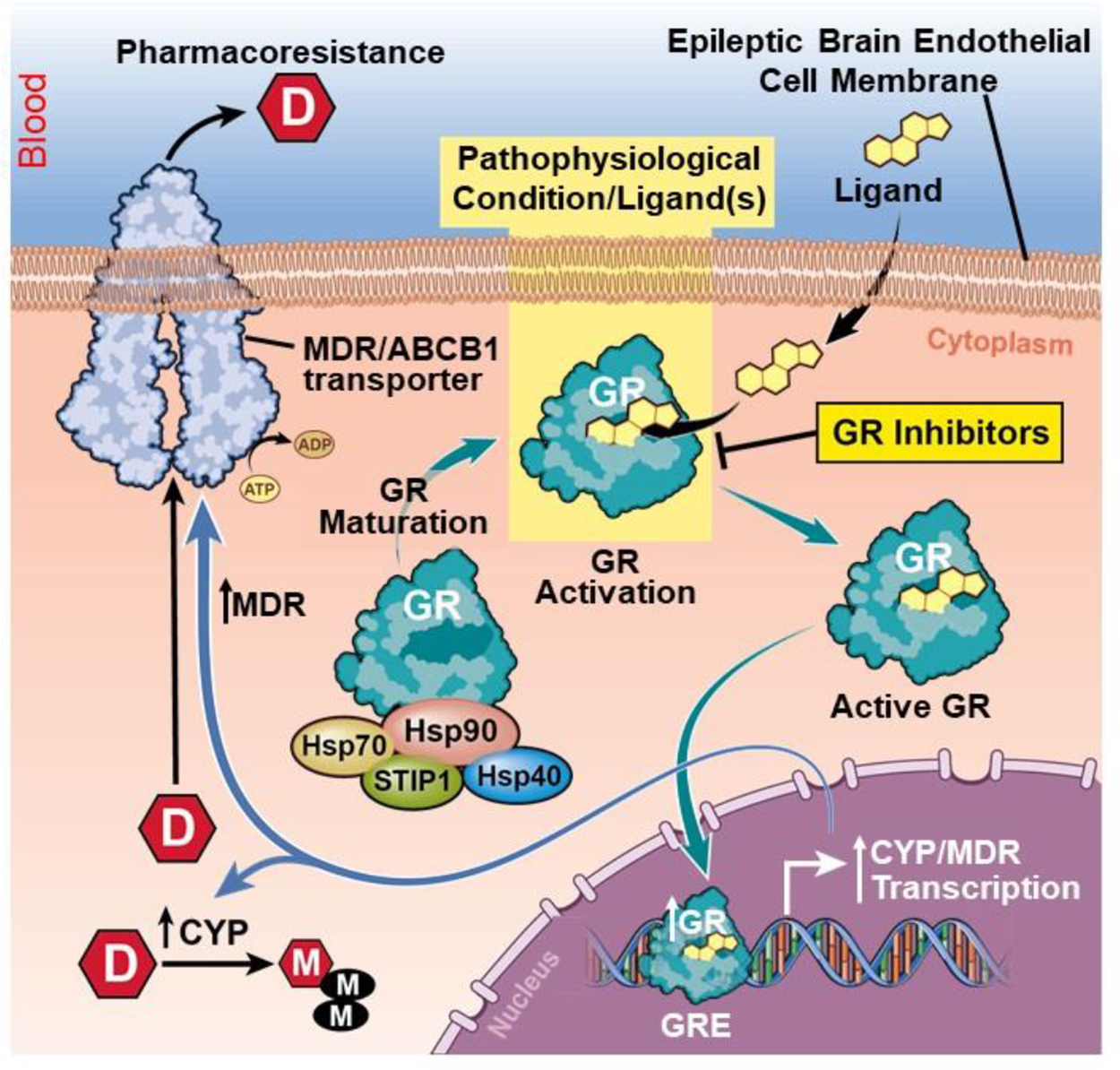
Schematic diagram depicting pharmacoresistance initiated by GR-Hsp binding, enabling GR maturation, reliant on GR activation by specific ligands leading to its nuclear translocation, CYP-MDR transcription and subsequent drug regulation in epileptic brain endothelial cells. GR activation by ligands or pathophysiological conditions initiate GR nuclear translocation. This is followed by multiple regulatory steps, including CYP-MDR transcription and upregulated CYP or MDR protein levels in the human epileptic brain endothelial cells, which causes a corresponding increase in drug biotransformation [8,15,7] and contributes to drug resistance in focal epilepsies.
Discussion
Since it is widely understood that the coordinated action of chaperones enhances GR stability, function and regulation [25], studying GR-Hsp/chaperone/co-chaperone interactions within different regions of the same epileptic brain is a novel attempt to determine mechanisms that contribute to the disease pathology in epilepsy. Our current study reveals for the first time how the upstream molecular cascade in GR protein maturation/activation is distinctly enhanced in specific EPI vs NON-EPI regions of the human brain, by virtue of higher ATPase activity of chaperone proteins involved in the post-translational processing of GR. The significance of GR modulation on drug biotransformation at the BBB vasculature is insightful for clinical considerations of drug bioavailability in pharmacoresistant epilepsy (Fig. 9) [8] considering that human epileptic brain endothelial cells are the most metabolically potent cell type, after hepatocytes [7,16,19]. In the current study, we found that: (1) the overexpressed neurovascular GR co-localizes with Hsps extensively in the EPI brain regions; (2) increased ATPase activity coupled with faster GR maturation accelerates the GR machinery in EPI and EPI-ECs faster than respective NON-EPI or NON EPI-ECs; and, (3) the disease state, coupled with GR-ligand binding, affects nuclear translocation such that brain endothelial nuclear GR expression is greatly enhanced in the EPI-ECs.
In the EPI tissue regions with dysmorphic neuronal organization, we found a profound increase in GR expression in the microvessels, neurons and was scattered sparsely across the astrocytes compared to NON-EPI regions. While GR expression was noticed inside the nucleus, as well as the cytoplasm, in the EPI brain tissue regions [7], GR-Hsp90 and Hsp70 show differential staining patterns within EPI and NON-EPI areas, and GR upregulation is focused more in the EPI areas. This is conceivable with the fact that Hsp90 and Hsp70 are key members of the chaperone complex that is associated with cytoplasmic GR regulation and proper ligand binding, as well as for initiating the maturation process [1,35]. We also found a significant increases in GR levels (*p < 0.01) in the EPI vs. NON-EPI regions among these individuals, which is again consistent with recent findings [26].
In the current study, the localization pattern confirmed that the Hsp90 and Hsp70 immunostaining is overexpressed in the micro-capillaries, neurons and astrocytes, and co-localization with neurovascular GR is persistent across the EPI regions of the brain. The NON-EPI portions showed varied levels of Hsp90 and Hsp70, which was confirmed by western blotting. Increased Hsp expression in the key hippocampal subfield, such as CA3, CA4, CA1, and the hilus, has been reported in the past in mesial temporal lobe epilepsy patients [13]. The studies also showed that Hsp90 and Hsp70 immunoreactivity values decreased in these patients, with complete remission after surgery, suggesting an association between these molecular players and the phenomena of epilepsy versus seizure-freedom [13]. Similar to our results, in animal models of epilepsy/seizure increased Hsp70 expression during the acute [40,41] and chronic phases [42] has been reported. Interestingly, the neuronal overexpression of inducible Hsp70 and Hsp90 by GR has also been shown to have a neuroprotective effect in epilepsy (due to physiological trigger or intracellular calcium influx during seizure). This needs to be better understood, since higher Hsp90/70 in EPI regions, as shown by our current study, promotes faster GR maturation which may contribute to pharmacoresistance. Since the neuroprotective effect of Hsp has also been identified in ischemic models [43], it remains to be explicitly determined whether some form of neuroprotection may also coexist along with the phenomena of pharmaoresistance in epilepsy, or whether these processes are exclusively linked to the activity or association of GR with specific Hsps.
Given that GR requires Hsps for ligand binding and the first accessory proteins found to interact with GR in the cytoplasm are Hsp90 and Hsp70, we found an interesting pattern of GR interaction with these co-chaperones in the EPI vs NON-EPI region. In this study, the GR-Hsp machinery seems to accelerate more quickly, as suggested by elevated ATPase levels (*p < 0.05) in the EPI vs NON-EPI regions. Thus, enhanced GR maturation through rapid interactions of the GR-Hsp complex at any given time will increase the proportion of readily available GR to be activated by ligands (e.g., steroids) and translocate to the nucleus and activate transcription of genes (including CYPs/MDRs), which subsequently causes pharmacoresistance (Fig. 8). The quicker kinetics of GR maturation/activation is likely one of the reasons why we found low GR-Hsp90 or Hsp70 interactions in EPI regions or in the corresponding EPI-ECs (Figs. 4 and 5). This maturation process of GR was however decelerated by silencing GR in the EPI-ECs (Fig 6), thereby suggesting that downregulation of GR in EPI-ECs slows GR maturation by altering GR-Hsp interactions. It is also well-known that Hsp machinery is associated with GR maturation [35,44]. However, in case of human epileptic endothelial cells it would be interesting to explore in future which specific heat shock protein has a particularly pronounced influence on the maturation of GR, or to prove that a collective chaperoning mechanism operates whereby the heat shock proteins act in a concerted fashion to mature GR in the cytosol. Moreover, it has been found that when small molecule inhibitors of hsp90, like radicicol are used to inhibit the chaperone, hsp90 binds back or traps to its client protein [45]. This is similar to what we found in case of GR siRNA in EPI-ECs (Fig. 6), where GR-Hsps interaction increases, which is indicative of a slower process.
Our current study also evaluated the EPI-ECs, which have proven to be metabolically active for drugs at the BBB. When compared, the EPI-ECs isolated from both the EPI and NON-EPI regions, revealed a pattern of GR-Hsp90/Hsp70 binding that was strikingly different within the two regions. Similar to the EPI brain tissue, the EPI-ECs also had increased ATPase levels compared to NON EPI-ECs (*p < 0.05), suggesting an increased ATPase activity in these EPI-ECs that is somewhat comparable to hepatocytes. Evidence also suggests that other factors could affect GR-Hsp90 associations, such as the circadian GR signaling in the hippocampus vs. hypothalamus, considering the negative feedback regulation of the hypothalamus-pituitary adrenal axis, which has deleterious consequences as reported in depression and has been connected to age-related memory loss [10,46]. Further, the seizure onset-age of individual patients seems a relevant factor potentially modulating the GR-Hsps/co-chaperone interactions in epileptic brain regions. However, this would require further studies to test the implications with regard to GR-Hsp90 interactions in epilepsy.
Nuclear GR expression indicates several downstream molecular effects at the cellular level in the brain endothelium, and is found to be upregulated in EPI-ECs compared to HBMECs (control ECs) at a steady state. Subsequent to GR ligand or GR modulators exposure such as DEX, PHT or RIF on EPI-ECs, the nuclear GR levels are further elevated in EPI-ECs vs. HBMEC. We also observed that DEX exposure assisted further nuclear GR translocation which was elevated in EPI-ECs compared to NON EPI-ECs; however, the Hsp90 distribution pattern remained unaltered post-DEX treatment, which might suggest that GR maturation by Hsps and nuclear translocation of GR are two separate events and Hsp90 expression in the latter event is unaffected by DEX treatment.
Our studies identified a correlation between an increase in GR-CYP-MDR1 expression and 1) low phenytoin permeability across the in vitro BBB in EPI compared to NON-EPI or control HBMECs, and 2) non-detectable oxcarbazepine penetration in EPI compared to NON-EPI brain regions or serum levels (ex vivo) as observed previously [17,26] in epileptic tissue. This reduced AED tissue bioavailability and pharmacoresistance may be due to impaired BBB endothelial cells and local CYP-induced drug metabolism. The overall summary of the GR-regulatory mechanism in epileptic human brain endothelial cells (Fig. 8), based on the current and past findings, supports the hypothesis of endothelial GR regulation and its contribution to pharmacoresistance in epilepsy [47]. Recent publications [11,8] suggest that GR in human EPI-ECs modulates expression of CYP3A4, CYP2C9, PXR and MDR1 at the BBB and affects AED bioavailability to the brain. Together, the GR-Hsp regulatory mechanism observed in this study may be pivotal in enhancing CYP-MDR activity, thereby limiting the access of drugs to the target epileptic brain region and contributing to pharmacoresistance.
Future directions in epileptic therapies.
Current evidence suggests that pharmacological inhibition of Hsp90 prevents glutamate transporter GLT-1 degradation by disruption of Hsp90β (not Hsp70) and GLT-1 interaction, and as a therapy target for the treatment of epilepsy and excitotoxicity [48]. Modulation of epileptic endothelial cell GR is also found to improve drug penetration across the brain vasculature [8], therefore both GR and Hsp90 could be pertinent molecular clients for drug regulation in epilepsy therapy. A similarity we find in our studies in epileptic brain regions to that of many different forms of cancer is the upregulation of chaperone Hsp90 and GR [32,49]. Upregulated GR has been shown to cause drug resistance in cancer [50]. Since these proteins are critical druggable targets in cancer, such therapies could also be tried, and the inhibitors of Hsp90 or GR may have the potential to be repurposed for epilepsy treatments.
Supplementary Material
{kind=link}
Acknowledgements:
This work is supported in part by the National Institute of Neurological Disorders and Stroke/National Institutes of Health grants R01NS095825 and R01NS078307 (to CG), and National Heart, Lung, and Blood Institute/National Institutes of Health grant R56HL139564 (to AG). We are thankful for the support of the Cleveland Clinic Center of Research Excellence in Epilepsy and Comorbidities grant award and Lerner Research Institute Recognition award 2019.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest I.N. serves on the Speaker’ bureau and as a member of ad hoc advisory board for Eisai, Inc. None of the other authors has any potential conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Desmet SJ, De Bosscher K (2017) Glucocorticoid receptors: finding the middle ground. J Clin Invest 127 (4):1136–1145. doi: 10.1172/JCI88886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jenkins BD, Pullen CB, Darimont BD (2001) Novel glucocorticoid receptor coactivator effector mechanisms. Trends Endocrinol Metab 12 (3):122–126 [DOI] [PubMed] [Google Scholar]
- 3.Kadmiel M, Cidlowski JA (2013) Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci 34 (9):518–530. doi: 10.1016/j.tips.2013.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams S, Ghosh C (2019) Neurovascular glucocorticoid receptors and glucocorticoids: implications in health, neurological disorders and drug therapy. Drug Discov Today. doi: 10.1016/j.drudis.2019.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Granata T, Marchi N, Carlton E, Ghosh C, Gonzalez-Martinez J, Alexopoulos AV, Janigro D (2009) Management of the patient with medically refractory epilepsy. Expert Rev Neurother 9 (12):1791–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwan P, Schachter SC, Brodie MJ (2011) Drug-resistant epilepsy. N Engl J Med 365 (10):919–926. doi: 10.1056/NEJMra1004418 [DOI] [PubMed] [Google Scholar]
- 7.Ghosh C, Hossain M, Solanki J, Najm IM, Marchi N, Janigro D (2017) Overexpression of pregnane X and glucocorticoid receptors and the regulation of cytochrome P450 in human epileptic brain endothelial cells. Epilepsia 58 (4):576–585. doi: 10.1111/epi.13703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh C, Hossain M, Mishra S, Khan S, Gonzalez-Martinez J, Marchi N, Janigro D, Bingaman W, Najm I (2018) Modulation of glucocorticoid receptor in human epileptic endothelial cells impacts drug biotransformation in an in vitro blood-brain barrier model. Epilepsia 59 (11):2049–2060. doi: 10.1111/epi.14567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, De Bosscher K (2013) How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol 380 (1–2):41–54. doi: 10.1016/j.mce.2012.12.014 [DOI] [PubMed] [Google Scholar]
- 10.Furay AR, Murphy EK, Mattson MP, Guo Z, Herman JP (2006) Region-specific regulation of glucocorticoid receptor/HSP90 expression and interaction in brain. J Neurochem 98 (4):1176–1184. doi: 10.1111/j.1471-4159.2006.03953.x [DOI] [PubMed] [Google Scholar]
- 11.Garabedian MJ, Harris CA, Jeanneteau F (2017) Glucocorticoid receptor action in metabolic and neuronal function. F1000Res 6:1208. doi: 10.12688/f1000research.11375.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saaltink DJ, Vreugdenhil E (2014) Stress, glucocorticoid receptors, and adult neurogenesis: a balance between excitation and inhibition? Cell Mol Life Sci 71 (13):2499–2515. doi: 10.1007/s00018-014-1568-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kandratavicius L, Hallak JE, Carlotti CG Jr., Assirati JA Jr., Leite JP (2014) Hippocampal expression of heat shock proteins in mesial temporal lobe epilepsy with psychiatric comorbidities and their relation to seizure outcome. Epilepsia 55 (11):1834–1843. doi: 10.1111/epi.12787 [DOI] [PubMed] [Google Scholar]
- 14.Lesuis SL, Weggen S, Baches S, Lucassen PJ, Krugers HJ (2018) Targeting glucocorticoid receptors prevents the effects of early life stress on amyloid pathology and cognitive performance in APP/PS1 mice. Transl Psychiatry 8 (1):53. doi: 10.1038/s41398-018-0101-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh C, Hossain M, Solanki J, Dadas A, Marchi N, Janigro D (2016) Pathophysiological implications of neurovascular P450 in brain disorders. Drug Discov Today. doi: 10.1016/j.drudis.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh C, Marchi N, Desai NK, Puvenna V, Hossain M, Gonzalez-Martinez J, Alexopoulos AV, Janigro D (2011) Cellular localization and functional significance of CYP3A4 in the human epileptic brain. Epilepsia 52 (3):562–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh C, Marchi N, Hossain M, Rasmussen P, Alexopoulos AV, Gonzalez-Martinez J, Yang H, Janigro D (2012) A pro-convulsive carbamazepine metabolite: quinolinic acid in drug resistant epileptic human brain. Neurobiol Dis 46 (3):692–700. doi:S0969–9961(12)00081–2 [pii]; 10.1016/j.nbd.2012.03.010 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hue CD, Cho FS, Cao S, Dale Bass CR, Meaney DF, Morrison B 3rd (2015) Dexamethasone potentiates in vitro blood-brain barrier recovery after primary blast injury by glucocorticoid receptor-mediated upregulation of ZO-1 tight junction protein. J Cereb Blood Flow Metab 35 (7):1191–1198. doi: 10.1038/jcbfm.2015.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salvador E, Shityakov S, Forster C (2014) Glucocorticoids and endothelial cell barrier function. Cell Tissue Res 355 (3):597–605. doi: 10.1007/s00441-013-1762-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dombrowski SM, Desai SY, Marroni M, Cucullo L, Goodrich K, Bingaman W, Mayberg MR, Bengez L, Janigro D (2001) Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia 42 (12):1501–1506 [DOI] [PubMed] [Google Scholar]
- 21.Ghosh C, Gonzalez-Martinez J, Hossain M, Cucullo L, Fazio V, Janigro D, Marchi N (2010) Pattern of P450 expression at the human blood-brain barrier: roles of epileptic condition and laminar flow. Epilepsia 51 (8):1408–1417. doi: 10.1111/j.1528-1167.2009.02428.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cucullo L, Hossain M, Rapp E, Manders T, Marchi N, Janigro D (2007) Development of a humanized in vitro blood-brain barrier model to screen for brain penetration of antiepileptic drugs. Epilepsia 48 (3):505–516. doi: 10.1111/j.1528-1167.2006.00960.x [DOI] [PubMed] [Google Scholar]
- 23.Golemis EA, Tew KD, Dadke D (2002) Protein interaction-targeted drug discovery: evaluating critical issues. Biotechniques 32 (3):636–638, 640,, 642 passim. doi: 10.2144/02323dd01 [DOI] [PubMed] [Google Scholar]
- 24.Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, Gavin BJ, Kley N, Kaelin WG Jr., Iliopoulos O (1998) The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell 1 (7):959–968 [DOI] [PubMed] [Google Scholar]
- 25.Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA (2014) Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 157 (7):1685–1697. doi: 10.1016/j.cell.2014.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams S, Hossain M, Ferguson L, Busch RM, Marchi N, Gonzalez-Martinez J, Perucca E, Najm IM, Ghosh C (2019) Neurovascular Drug Biotransformation Machinery in Focal Human Epilepsies: Brain CYP3A4 Correlates with Seizure Frequency and Antiepileptic Drug Therapy. Mol Neurobiol. doi: 10.1007/s12035-019-01673-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alms D, Fedrowitz M, Romermann K, Noack A, Loscher W (2014) Marked differences in the effect of antiepileptic and cytostatic drugs on the functionality of P-glycoprotein in human and rat brain capillary endothelial cell lines. Pharm Res 31 (6):1588–1604. doi: 10.1007/s11095-013-1264-4 [DOI] [PubMed] [Google Scholar]
- 28.Ott M, Fricker G, Bauer B (2009) Pregnane X receptor (PXR) regulates P-glycoprotein at the blood-brain barrier: functional similarities between pig and human PXR. J Pharmacol Exp Ther 329 (1):141–149. doi: 10.1124/jpet.108.149690 [DOI] [PubMed] [Google Scholar]
- 29.Pascussi JM, Robert A, Nguyen M, Walrant-Debray O, Garabedian M, Martin P, Pineau T, Saric J, Navarro F, Maurel P, Vilarem MJ (2005) Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J Clin Invest 115 (1):177–186. doi: 10.1172/JCI21867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider N, Goncalves Fda C, Pinto FO, Lopez PL, Araujo AB, Pfaffenseller B, Passos EP, Cirne-Lima EO, Meurer L, Lamers ML, Paz AH (2015) Dexamethasone and azathioprine promote cytoskeletal changes and affect mesenchymal stem cell migratory behavior. PLoS One 10 (3):e0120538. doi: 10.1371/journal.pone.0120538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams S, Hossain M, Mishra S, Gonzalez-Martinez J, Najm I, Ghosh C (2018) Expression and Functional Relevance of Death-Associated Protein Kinase in Human Drug-Resistant Epileptic Brain: Focusing on the Neurovascular Interface. Mol Neurobiol. doi: 10.1007/s12035-018-1415-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ (2003) A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425 (6956):407–410. doi: 10.1038/nature01913 [DOI] [PubMed] [Google Scholar]
- 33.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP (1997) A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem 253 (2):162–168. doi: 10.1006/abio.1997.2391 [DOI] [PubMed] [Google Scholar]
- 34.Marchi N, Betto G, Fazio V, Fan Q, Ghosh C, Machado A, Janigro D (2009) Blood-brain barrier damage and brain penetration of antiepileptic drugs: role of serum proteins and brain edema. Epilepsia 50 (4):664–677. doi: 10.1111/j.1528-1167.2008.01989.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grad I, Picard D (2007) The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol 275 (1–2):2–12. doi: 10.1016/j.mce.2007.05.018 [DOI] [PubMed] [Google Scholar]
- 36.Hawle P, Siepmann M, Harst A, Siderius M, Reusch HP, Obermann WM (2006) The middle domain of Hsp90 acts as a discriminator between different types of client proteins. Mol Cell Biol 26 (22):8385–8395. doi: 10.1128/MCB.02188-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghosh A, Chawla-Sarkar M, Stuehr DJ (2011) Hsp90 interacts with inducible NO synthase client protein in its heme-free state and then drives heme insertion by an ATP-dependent process. FASEB J 25 (6):2049–2060. doi: 10.1096/fj.10-180554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghosh A, Dai Y, Biswas P, Stuehr DJ (2019) Myoglobin maturation is driven by the hsp90 chaperone machinery and by soluble guanylyl cyclase. FASEB J:fj201802793RR. doi: 10.1096/fj.201802793RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russo-Abrahao T, Lacerda-Abreu MA, Gomes T, Cosentino-Gomes D, Carvalho-de-Araujo AD, Rodrigues MF, Oliveira ACL, Rumjanek FD, Monteiro RQ, Meyer-Fernandes JR (2018) Characterization of inorganic phosphate transport in the triple-negative breast cancer cell line, MDA-MB-231. PLoS One 13 (2):e0191270. doi: 10.1371/journal.pone.0191270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim YJ, Kim JY, Ko AR, Kang TC (2013) Reduction in heat shock protein 90 correlates to neuronal vulnerability in the rat piriform cortex following status epilepticus. Neuroscience 255:265–277. doi: 10.1016/j.neuroscience.2013.09.050 [DOI] [PubMed] [Google Scholar]
- 41.Lively S, Brown IR (2008) Extracellular matrix protein SC1/hevin in the hippocampus following pilocarpine-induced status epilepticus. J Neurochem 107 (5):1335–1346. doi: 10.1111/j.1471-4159.2008.05696.x [DOI] [PubMed] [Google Scholar]
- 42.Kharlamov EA, Lepsveridze E, Meparishvili M, Solomonia RO, Lu B, Miller ER, Kelly KM, Mtchedlishvili Z (2011) Alterations of GABA(A) and glutamate receptor subunits and heat shock protein in rat hippocampus following traumatic brain injury and in posttraumatic epilepsy. Epilepsy Res 95 (1–2):20–34. doi: 10.1016/j.eplepsyres.2011.02.008 [DOI] [PubMed] [Google Scholar]
- 43.Yenari MA, Fink SL, Sun GH, Chang LK, Patel MK, Kunis DM, Onley D, Ho DY, Sapolsky RM, Steinberg GK (1998) Gene therapy with HSP72 is neuroprotective in rat models of stroke and epilepsy. Ann Neurol 44 (4):584–591. doi: 10.1002/ana.410440403 [DOI] [PubMed] [Google Scholar]
- 44.Pratt WB (1993) The role of heat shock proteins in regulating the function, folding, and trafficking of the glucocorticoid receptor. J Biol Chem 268 (29):21455–21458 [PubMed] [Google Scholar]
- 45.Ghosh A, Stuehr DJ (2012) Soluble guanylyl cyclase requires heat shock protein 90 for heme insertion during maturation of the NO-active enzyme. Proc Natl Acad Sci U S A 109 (32):12998–13003. doi: 10.1073/pnas.1205854109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Q, Van Heerikhuize J, Aronica E, Kawata M, Seress L, Joels M, Swaab DF, Lucassen PJ (2013) Glucocorticoid receptor protein expression in human hippocampus; stability with age. Neurobiol Aging 34 (6):1662–1673. doi: 10.1016/j.neurobiolaging.2012.11.019 [DOI] [PubMed] [Google Scholar]
- 47.Ghosh C, Hossain M, Spriggs A, Ghosh A, Grant GA, Marchi N, Perucca E, Janigro D (2015) Sertraline-induced potentiation of the CYP3A4-dependent neurotoxicity of carbamazepine: An in vitro study. Epilepsia 56 (3):439–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sha L, Wang X, Li J, Shi X, Wu L, Shen Y, Xu Q (2017) Pharmacologic inhibition of Hsp90 to prevent GLT-1 degradation as an effective therapy for epilepsy. J Exp Med 214 (2):547–563. doi: 10.1084/jem.20160667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whitesell L, Lindquist SL (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5 (10):761–772. doi: 10.1038/nrc1716 [DOI] [PubMed] [Google Scholar]
- 50.Lewis-Tuffin LJ, Cidlowski JA (2006) The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci 1069:1–9. doi: 10.1196/annals.1351.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.