

Abstract
Objective:
Currently, there are no approved drugs for abdominal aortic aneurysm (AAA) treatment, likely due to limited understanding of the primary molecular mechanisms underlying AAA development and progression. BAF60a, a unique subunit of the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex, is a novel regulator of metabolic homeostasis, yet little is known about its function in the vasculature and pathogenesis of AAA. In this study, we sought to investigate the role and underlying mechanisms of vascular smooth muscle cell (VSMC)-specific BAF60a in AAA formation.
Approach and Results:
BAF60a is upregulated in human and experimental murine AAA lesions. In vivo studies revealed that VSMC-specific knockout of BAF60a protected mice from both AngII- and elastase-induced AAA formation with significant suppression of vascular inflammation, monocyte infiltration, and elastin fragmentation. Through RNA-sequencing and pathway analysis, we found that the expression of inflammatory response genes in cultured human aortic smooth muscle cells (HASMCs) was significantly downregulated by siRNA mediated BAF60a knockdown while upregulated upon adenovirus mediated BAF60a overexpression. BAF60a regulates VSMC inflammation by recruiting BRG1, a catalytic subunit of the SWI/SNF complex, to the promoter region of NF-κB target genes. Furthermore, loss of BAF60a in VSMCs prevented the upregulation of the proteolytic enzyme cysteine protease cathepsin S, thus ameliorating extracellular matrix (ECM) degradation within the vascular wall in AAA.
Conclusions:
Our study demonstrated that BAF60a is required to recruit the SWI/SNF complex in order to facilitate the epigenetic regulation of VSMC inflammation, which may serve as a potential therapeutic target in preventing and treating AAA.
Keywords: abdominal aortic aneurysm, inflammation, SWI/SNF, BAF60a, cathepsin S
Subject Codes: vascular biology, aneurysm, inflammation, gene expression and regulation
Graphical Abstract
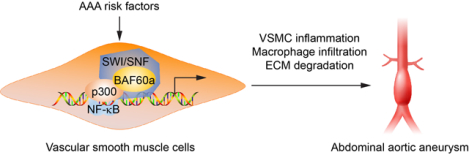
Introduction
Abdominal aortic aneurysm (AAA) affects more than 5% of men1 and 1.3% of women over 65 years old2 and is associated with a mortality rate of 50%−80% upon rupture. AAA is a focal dilation of the abdominal aorta to a diameter of ≥ 30mm in human and is mostly asymptomatic.3 Although endovascular and open surgical repair can spare patients from fatal or severe outcomes, no medical therapy has been conclusively shown to prevent or treat AAA, partially due to the incomplete understanding of the molecular mechanisms of AAA development and progression.
Recent evidence has highlighted the importance of innate and adaptive immunity in the initiation and expansion of AAA.4 As a hallmark of AAA, the intense transmural inflammatory response involves different kinds of cells constituting the vascular wall, including endothelial cells,5 VSMCs6 and resident immune cells, along with a major influx of exogenous leukocytes to the media and adventitia.3, 4 VSMC inflammatory response promotes vascular disease progression through enhanced secretion of pro-inflammatory cytokines, adhesion molecules, as well as proteolytic enzymes.7 Nevertheless, the molecular biology aspects linking VSMC inflammation and its role in AAA development remain largely elusive.
As one of the four major families of ATP-dependent chromatin remodelers, SWI/SNF complexes have been suggested to play an essential role in the pathogenesis of many human diseases by controlling physiological relevant gene expression through alterations of chromatin accessibility.8, 9 Assembled from up to 15 components, the SWI/SNF complexes contain one of the two catalytic subunits, BRG1 or BRM, and other subunits collectively referred to as BAFs (BRG1/BRM associated factors).10 BAF60a, encoded by the SMARCD1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily d, member 1) gene, is one of the three mutually exclusive 60-kDa subunits and functions by recruiting a number of transcription factors.11–13 In vivo studies indicated that BAF60a plays an important part in regulating hepatic fatty acid oxidation,12 restricting embryonic stem cell pluripotency networks,14 integrating circadian signals,15 and controlling systemic metabolic homeostasis.16 A recent study has demonstrated that BAF60a deficiency in hepatocytes protected mice from diet-induced atherosclerosis,16 which was the first study to link BAF60a with cardiovascular disease. Data from several microarray-based genome-wide human gene expression profiles showed an upregulation of BAF60a in AAAs17 and intracranial aneurysms,18 indicating a potential role of BAF60a in altering vascular homeostasis.
In the current study, we identified a causative role of BAF60a-dependent chromatin remodeling in AAA development through regulation of VSMC inflammation and the proteolytic enzyme cathepsin S expression. Disrupting this pathway by VSMC-specific loss of BAF60a prevented AAA formation in two different mouse models. Our findings here expand our knowledge of the epigenetic regulation of AAA and position BAF60a as an attractive potential therapeutic target for AAA prevention and treatment.
Materials and Methods
The authors declare that all supporting data are available within the article and its Data Supplement.
Human Patients’ Aortic Samples
Human aortic tissues from AAA patients or heart transplant donors were obtained from the Cardiovascular Health Improvement Project (CHIP) core of the Cardiovascular Center at the University of Michigan, with the Institutional Review Board approval from the Human Research Protection Program and Institutional Review Boards of the University of Michigan Medical School.
Mice AAA models
The BAF60af/f mice in the C57BL/6J background were described previously,16 and were crossbred with SMMHC-CreERT2 mice19 to generate the SMMHC-CreERT2-BAF60af/f mice. Tamoxifen (80 mg/kg/day) was given to 6–8 weeks old male SMMHC-CreERT2-BAF60af/f mice and BAF60af/f mice for consecutive 5 days by intraperitoneal injection to induce VSMC-specific BAF60a knockout (BAF60aSMKO). The AngII-induced murine AAA model was performed as previously described,20, 21 with a release rate of AngII (Bachem, #H-1706, Vista, CA) at 1,500 ng/kg/min (or an equivalent volume of saline solution, as sham control) for 28 days. In the elastase-induced AAA model, 30μL elastase (MilliporeSigma, #E1250, 44 units/mL) or heat-inactivated elastase (as sham control) was applied to the adventitial layer of the infrarenal aorta as described previously.22, 23 The maximal external diameters of suprarenal or infrarenal abdominal aortas were assessed in situ using a digital caliper (Fisher Scientific, #14-648-17) in the AngII- and elastase-induced AAA models, respectively. AAA was defined as a dilation of the abdominal aortic diameter >50% larger than its adjacent region.24 All animal procedures were approved by the Institutional Animal Care & Use Committee (IACUC) at the University of Michigan.
Immunostaining and Immunohistochemistry
Human and murine aortic samples were fixed with 10% buffered formalin in PBS (Fisher Healthcare, #245685) for 24h and embedded in paraffin. Histology was made in serial sections (5 μm thick, 200 μm apart) of mouse suprarenal abdominal aortas (AngII-induced AAA) and infrarenal abdominal aortas (elastase-induced AAA). Sections were applied for Verhoeff Van Gieson’s staining or immunofluorescence staining. At least 6 sections were analyzed for elastin degradation grade25 and 4 sections were used to measure the macrophage content in the vascular wall.
Flow Cytometry Analysis of Murine Aorta
Mouse abdominal aortas were harvested, minced, and digested in enzymatic digestion solution containing 450 U/mL collagenase type I (Gibco, #17100–017), 125 U/mL collagenase type XI (Sigma-Aldrich, #C7657), 60 U/mL hyaluronidase type I-s (Sigma-Aldrich, #H3506) and 60 U/mL DNase-I (Roche, #10104159001) at 37°C for 60 minutes. Single cells were released using a 70μm cell strainer and stained with the indicated antibodies, followed by flow cytometry analysis on a MoFlo Astrios cell sorter (Beckman Coulter, Brea, CA). Antibodies used for flow cytometry are listed in the major resource table. Dot plots were generated using the FlowJo Software.
RNAscope In Situ Hybridization
RNAscope® 2.0 HD Brown Chromogenic Reagent Kit was used for RNA in situ hybridization assay according to the manufacturer’s instructions (ACD, Hayward, CA).26 Briefly, abdominal aortas were fixed and processed into 8 μm cross-sections before being subsequently treated with a peroxidase blocker, target retrieval solution, and protease plus reagent. Probes targeting mouse Ctss (ACD #579891) and DapB (ACD #310043, serves as negative control) were hybridized on the sections, followed by signal amplification and detection.
Isolation of Mouse Aortic Smooth Muscle Cells
Mouse aortic smooth muscle cells (MASMCs) were isolated from 8-week-old male BAF60af/f and BAF60aSMKO mice as described previously.27 To ensure BAF60a KO in vitro, MASMCs isolated from BAF60af/f and BAF60aSMKO mice were infected with adenovirus expressing GFP (Ad-CMV-GFP) or Cre recombinase (Ad-CMV-iCre, Vector Biolabs, #1045N) respectively before experiments. MASMCs at passage 5–7 were used for co-Immunoprecipitation and ChIP assay experiments.
RNA-sequencing (RNA-seq)
Total RNA was extracted from HASMCs transfected with siControl or siBAF60a for 72h (n=3/group) or HASMCs infected with AdGFP or AdBAF60a12 (n=3/group) and submitted to the DNA sequencing core of the University of Michigan. Briefly, the RNA library was prepared with TruSeq RNA Library Prep Kit v2 (Illumina) and 50bp non-stranded single-end sequencing was performed on a HiSeq 4000 platform (Illumina), with an average of 43 million reads for each sample. RNAseq read mapping was performed as described previously.28 Gene expression quantification was performed using Salmon v0.14.0,29 and differential expression analysis was performed with DeSeq2 package in R.30 Gene Set Enrichment Analysis (GSEA)31 was performed to interpret gene expression profiles of siControl/siBAF60a or AdGFP/AdBAF60a treated HASMCs. Genes were mapped to the HALLMARK gene set in the Molecular Signatures Database (MSigDB) for pathway analysis.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed according to the protocol from SimpleChIP Enzymatic Chromatin IP Kit (CST, #9003). In brief, cells were treated with 2mM disuccinimidyl glutarate (DSG, ThermoFisher, #20593) at room temperature for 45 min for cross-link before 1% paraformaldehyde incubation for 10 min. Chromatin was immunoprecipitated with indicated antibodies at 4°C overnight with rotation. DNA was captured, eluted, cross-link reversed, and purified followed with real-time PCR amplification. ChIP assay primer sequences are listed in the Online Supplemental Table VIII.
Chromatin immunoprecipitation followed by sequencing (ChIP-seq)
Samples containing 30 ng of DNA were immunoprecipitated with antibodies against BRG1, H3K9Ac and H3K27Ac, together with their respective inputs, were sent to the DNA sequencing core at the University of Michigan for quality control, library preparation (NEBNext® Ultra™ II FS DNA Library Prep Kit for Illumina) and sequencing on an Illumina Novaseq S1 flowcell with 100bp pair-end reads. An average of 40 million reads was generated for each sample. ChIP-seq reads were aligned to the human genome (GRCh19) using bowtie2 version 2.3.5. Peaks were called by MACS232 software. Samples were normalized to adjust for sequencing depth. The peaks were visualized in Integrative Genomics Viewer (IGV).33
Raw data of RNA-seq and ChIP-seq have been deposited in the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) under the accession code: GSE153449.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA) or R (for RNA-seq and ChIP-seq). Data are presented as mean ± standard error of the mean (SEM). If data were normally distributed and with similar variances, Student’s t test (parametric) was used to compare the difference between two groups, and one-way ANOVA followed by Tukey’s post hoc analysis or two-way ANOVA followed by Holm-Sidak post hoc analysis were performed for comparison among three or more groups. For data that did not pass the normality test, Mann-Whitney test (nonparametric), Chi-squared analysis (distribution) or Mantel-Cox test (survival percentage) were used to compare differences between the two groups. Results with p < 0.05 were considered as statistically significant. All results are representative of at least 3 independent experiments.
Results
BAF60a is Highly Expressed in Human and Murine AAA Lesions
We first compared the expression of BAF60a in human AAA samples and normal abdominal aortic tissues (Table I in the online-only Data Supplement) by quantitative real-time PCR (qPCR) and Western blot, and the results showed significantly increased BAF60a mRNA and protein abundance in aneurysmal lesions (Figure 1A; Figure I-A in the online-only Data Supplement). The upregulated BAF60a expression in human AAA was also confirmed by immunofluorescence staining (Figure 1B). We also assessed BAF60a levels in two distinct murine AAA models. In the AngII model20 using adeno-associated virus (AAV) carrying a gain-of-function mutation of the mouse proprotein convertase subtilisin/kexin type 9 (AAV-Pcsk9.D377Y, hereafter AAV-Pcsk9) (Figure I-B in the online-only Data Supplement), both the mRNA and the protein levels of BAF60a exhibit a significant increase in mouse AAAs (Figure 1C and 1D). We then monitored BAF60a abundance during the pathogenic process of elastase-induced AAA by immunofluorescence staining (Figure I-C in the online-only Data Supplement). Similarly, increased expression of BAF60a was observed at 7 and 14 days after elastase exposure (Figure 1E). Altogether, these data indicate a potential pathogenic role of BAF60a in AAA development.
Figure 1. BAF60a is upregulated in human and murine AAA lesions.
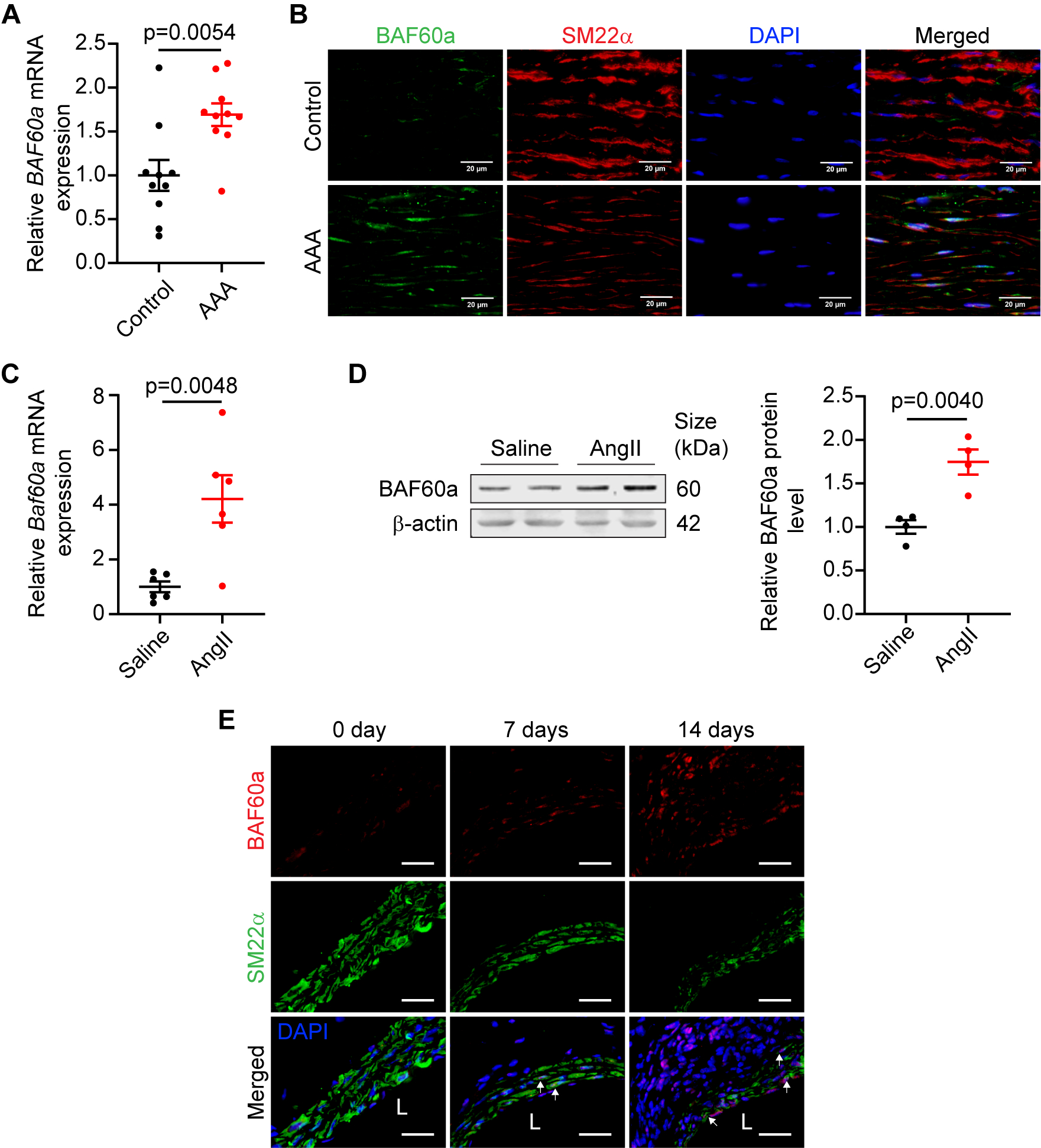
A, BAF60a mRNA expression was determined by qPCR in human AAA lesions (n=10) and control abdominal aortas (n=10). B, Representative immunofluorescence staining of BAF60a (green) in human AAA samples (n=3) and control abdominal aortas (n=3). SM22α, red; DAPI blue. Scale bar=20 μm. For the images, the upper side is the adventitial side and lower side is the lumen side. C-D, Relative BAF60a mRNA and protein levels were determined by qPCR (C, n=6 for each group) and Western blotting (D, n=4 for each group) in abdominal aortas of C57BL/6J mice infused with saline or AngII for 28 days. E, Representative immunofluorescence staining of BAF60a (red) in the infrarenal abdominal aortas of C57BL/6J mice at 0, 7, or 14 days after elastase exposure (n=3 for each group). SM22α, green; DAPI, blue. Scale bar=100 μm; L, lumen. Data are presented as mean±SEM. Student’s t-test for A, C and D.
BAF60a Deficiency in VSMCs Attenuates AngII- and Elastase-Induced Murine AAA Formation
VSMCs play a crucial role in maintaining vascular homeostasis, and their dysfunction is a hallmark of AAA.34 To determine the function of BAF60a in the pathogenesis of AAA, we generated VSMC-specific BAF60a knockout mice (BAF60aSMKO) by crossbreeding BAF60a floxed mice (BAF60af/f) with SMMHC-CreERT2 mice followed with tamoxifen induction (Figure II-A in the online-only Data Supplement). BAF60a knockout efficiency was verified by qPCR and Western blot (Figure II-B and II-C in the online-only Data Supplement), with some remaining signal likely from adventitia and endothelium. Next, 8 to 10 weeks old male BAF60af/f and BAF60aSMKO mice were subjected to AngII-induced AAA formation (Figure III-A in the online-only Data Supplement) or sham treatment. No significant differences were found in survival rate, systolic blood pressure, body weight, serum total cholesterol and total triglyceride levels between AngII infused BAF60af/f and BAF60aSMKO mice (Figure III-B through III-F and Table II in the online-only Data Supplement). In sham control groups, no mice developed AAA (Figure III-G through III-I in the online-only Data Supplement). In the presence of AngII, a 57.14% AAA incidence was observed in BAF60af/f mice, while only 8.33% of BAF60aSMKO mice developed aneurysms in their suprarenal aortas (Figure 2A and 2B). Furthermore, AngII-infused BAF60aSMKO mice showed a significant decrease in maximal outer diameters compared with BAF60af/f mice (BAF60af/f 1.41 ± 0.09mm vs. BAF60aSMKO 1.05 ± 0.05mm, p=0.0001, Figure 2C).
Figure 2. VSMC-specific BAF60a knockout attenuates AngII-induced AAA in mice.
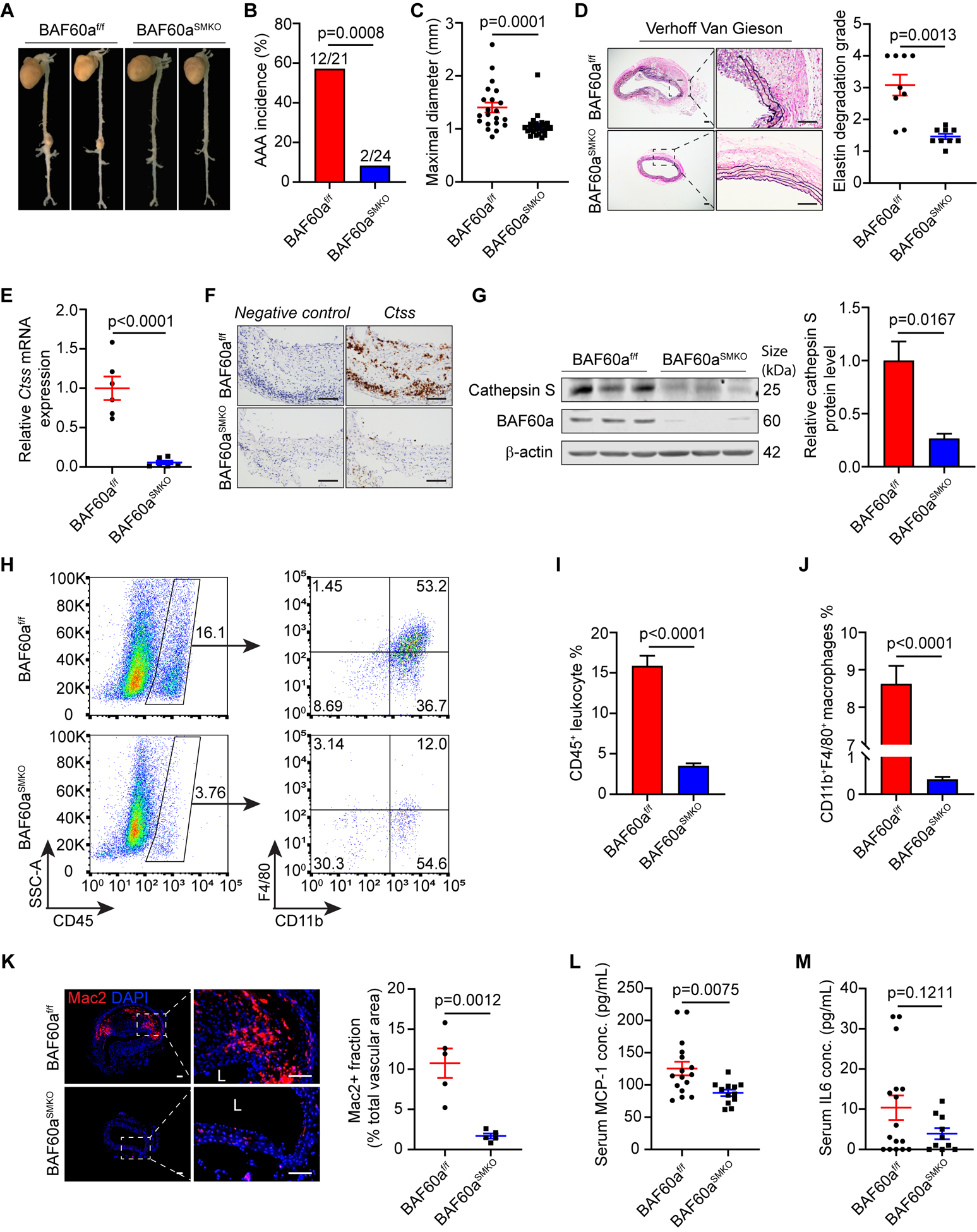
In the AngII-induced AAA model, 8 to 10 weeks old male BAF60af/f (n=24) and BAF60aSMKO (n=25) mice were given AAV-Pcsk9 (i.p.) and Western diet to generate hypercholesterolemia followed by AngII (1,500 ng/kg/min) infusion for 28 days. A, Representative morphology of aortas. B, Incidence of AAA; rupture events in the thoracic aorta were excluded from the AAA incidence. C, Quantification of maximal external diameters of suprarenal abdominal aortas. D, Representative images of Verhoff Van Gieson staining in suprarenal abdominal aortic sections and quantification analysis of elastin fragmentation (n=9 for each group). Scale bar=100 μm. E, Cathepsin S mRNA expression was determined by qPCR in the aortic RNA extracts (n=6 for each group). F, Representative images of RNAscope in situ hybridization assay to detect Cathepsin S mRNA in aortic sections (n=3 for each group). Scale bar=100 μm. G, Cathepsin S protein level was determined by Western blot in abdominal aortic protein extracts (n=3 for each group). H-J, Single cells were isolated from abdominal aortas of BAF60af/f (n=3) and BAF60aSMKO (n=3) mice after 28 days of AngII infusion and subjected to flow cytometry analysis. H, Representative dot plots showing the gating strategy to obtain the macrophage population (CD11b+F4/80+) from gated CD45+ leukocytes. I, Quantitative analysis of the percentage of CD45+ leukocytes. J, Quantitative analysis of the percentage of CD11b+F4/80+ macrophages. K, Representative images of Mac2 staining (red) and quantification analysis of Mac2+ fraction in abdominal aortas of BAF60af/f (n=5) and BAF60aSMKO (n=5) mice. Scale bar=100 μm; L, lumen. L-M, Serum concentrations of MCP-1 (BAF60af/f, n=16; BAF60aSMKO, n=12) (L) and IL-6 (BAF60af/f, n=16; BAF60aSMKO, n=10) (M) were determined by ELISA. Data are presented as mean ± SEM. Chi-squared test for B, Mann-Whitney test for C, Student’s t-test for D, E, G, and I-M.
To further demonstrate the role of VSMC-specific BAF60a deletion in attenuating AAA formation, the elastase-induced aneurysm model22, 23 was performed on 10 to 12 weeks old male BAF60af/f and BAF60aSMKO mice (Figure IV-A in the online-only Data Supplement). Fourteen days after elastase exposure, systolic blood pressure, body weight, serum total cholesterol, and serum triglyceride levels were comparable between the two groups (Figure IV-B through IV-E and Table III in the online-only Data Supplement). No mice developed AAA after exposure to the heat-inactivated elastase (Figure V-A through V-C in the online-only Data Supplement). However, and consistent with the AngII model, BAF60a depletion in VSMCs significantly reduced elastase-induced AAA incidence (BAF60af/f 72.22% vs. BAF60aSMKO 6.67%, p=0.0002, Figure V-D and V-E in the online-only Data Supplement) and maximal infrarenal aortic diameter (BAF60af/f 0.83 ± 0.04mm vs. BAF60aSMKO 0.58 ± 0.02mm, p<0.0001, Figure V-F in the online-only Data Supplement).
BAF60a Deficiency in VSMCs Reduces ECM Degradation in AAA Lesion
Elastin fragmentation is well recognized to play a pivotal role in aortic wall dilation and can be detected through Verhoff Van Gieson staining.25 No elastin breakage was identified in either suprarenal or infrarenal aortic sections from mice subjected to control treatment in each model (Figure VI-A and VI-B in the online-only Data Supplement). Notably, BAF60aSMKO mice were protected from severe ECM degradation induced by AngII infusion (Figure 2D; Figure VII-A in the online-only Data Supplement) or elastase exposure (Figure VII-B and VII-C in the online-only Data Supplement), when compared to the BAF60afl/fl mice, indicating a reduced level of protease production or activity.
We then checked the expression of several proteolytic cysteine cathepsins reported to contribute to the pathogenesis of AAA35 in the medial layer of aortas harvested from the AngII-induced AAA model. Notably, the mRNA level of cathepsin S, encoded by the CTSS gene, was significantly downregulated in aortas from BAF60aSMKO mice compared with BAF60af/f mice (Figure 2E). This finding was confirmed by RNAscope assay, a novel RNA in situ hybridization technology using specific probes to measure gene expression within tissues.26 Abundant brown staining representing Ctss RNA was detected in the aortic sections of BAF60af/f mice, mainly located in the media and adventitia, while the equivalent aortic area from BAF60aSMKO mice demonstrated faint staining with a reduction in both the amount and density of the punctate signals (Figure 2F). Furthermore, deletion of BAF60a in VSMCs largely inhibited the aneurysm-associated increase35 in cathepsin S protein expression as indicated by Western blot (Figure 2G).
Next, we investigated the effect of BAF60a depletion in VSMCs on matrix metalloproteinases (MMPs) expression, specifically MMP2 and MMP9, two well-recognized members of MMP family reported to contribute to the ECM degradation during AAA formation.36, 37 qPCR showed that the mRNA abundance of MMP9, not MMP2, was significantly decreased in the aortic media of AngII-infused BAF60aSMKO mice (Figure VII-D in the online-only Data Supplement). Immunofluorescence staining further confirmed the reduction of MMP9 in the tunica media of the abdominal aortas of AngII and elastase treated BAF60aSMKO mice (Figure VII-E and VII-F in the online-only Data Supplement). Interestingly, reduced signals for both MMP9 and MMP2 were noted in the adventitia of the abdominal aortas of BAF60aSMKO mice. Given that macrophages are a major source of MMPs, the discrepancy between MMP2 qPCR and immunofluorescence staining may be due to suppressed recruitment of immune cells into the aortic wall of BAF60aSMKO mice.
BAF60a Deficiency in VSMCs Suppresses inflammatory response in AAA Lesion
To explore whether the protective effect of VSMC-specific BAF60a knockout in AAA development is related to reduced immune cell recruitment, flow cytometry analysis was performed on single cells isolated from abdominal aortas.38 There was no significant difference in the proportions of CD45+ leukocytes and CD11b+/F4/80+ macrophages between BAF60af/f and BAF60aSMKO mice subjected to control treatment (Figure VIII-A through VIII-F in the online-only Data Supplement). However, upon exposure to AngII or elastase, significantly decreased numbers of CD45+ leukocytes and CD11b+/F4/80+ macrophages were noticed in BAF60aSMKO aortas (Figure 2H through 2J; Figure IX-A through IX-C in the online-only Data Supplement). Further immunofluorescence staining of Mac2 confirmed the attenuated macrophage accumulation in the aortic wall of BAF60aSMKO mice compared with BAF60af/f mice (Figure 2K; Figure IX-D in the online-only Data Supplement). Systemic inflammatory response was then determined by ELISA measuring the concentration of circulating monocyte chemoattractant protein-1 (MCP-1/CCL2) and interleukin-6 (IL-6), two inflammatory cytokines that were reported as elevated in human AAA disease.6, 39 In the AngII model, the serum MCP-1 concentration in BAF60aSMKO mice was significantly lower compared with that in BAF60af/f mice (Figure 2L); the serum IL-6 level was also decreased in BAF60aSMKO mice, although without statistical significance due to wide variation (Figure 2M). In elastase-induced AAA model, however, circulating MCP-1 and IL-6 concentration were not significantly changed in the BAF60aSMKO mice (Figure IX-E and IX-F in the online-only Data Supplement), likely as a result of the highly localized nature of the stimulus in this model.
BAF60a Regulates Vascular Smooth Muscle Cell Inflammation
To further explore the molecular mechanisms underlying the function of BAF60a in AAA development, small interfering RNA (siRNA) was used to knockdown BAF60a (siBAF60a) in cultured human aortic smooth muscle cells (HASMCs) (Figure 3A), and RNA sequencing was performed to uncover the transcriptomic changes relative to HASMCs transfected with control siRNA (siControl). Gene Set Enrichment Analysis (GSEA)31 was employed to search for overrepresented pathways across the Molecular Signatures Database (MSigDB) and the top ten up-regulated and down-regulated HALLMARK pathways are listed (Figure X-A and Table IV in the online-only Data Supplement). Notably, inflammatory response and TNFα signaling via NF-κB were highly enriched as the top two down-regulated pathways in HASMCs transfected with siBAF60a, indicating suppressed inflammation upon BAF60a knockdown in VSMCs (Figure 3B). Decreased expression of several pro-inflammatory cytokines regulated by NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) (Figure X-B and Table V-a in the online-only Data Supplement) was validated through qPCR (Figure 3C). Silencing of BAF60a in HASMCs decreased TNFα (tumor necrosis factor alpha)-induced MCP-1 secretion to the culture medium, as determined by ELISA (Figure 3D). Consistently, macrophage migration assay25 also indicated a decreased level of cytokine secretion, as demonstrated by significantly reduced recruitment of RAW264.7 cells (a mouse macrophage cell line) towards the conditioned medium of BAF60a-deficient HASMCs (Figure 3E).
Figure 3. BAF60a regulates vascular smooth muscle cell inflammation.
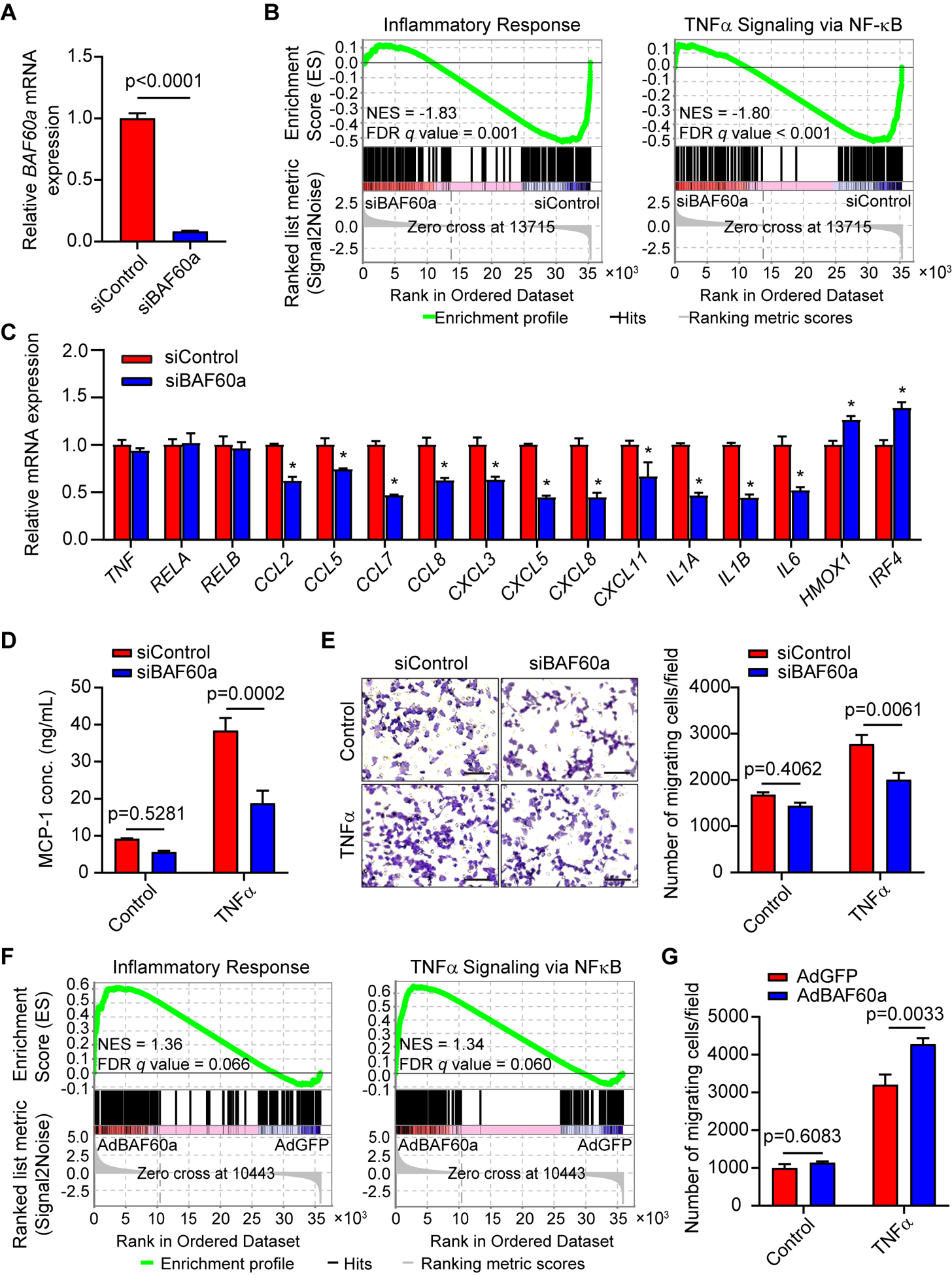
A-B, HASMCs were transfected with siControl or siBAF60a (30 nM). After 72 hours, total RNA was extracted for RNA-seq (n=3 for each group). A, BAF60a mRNA expression was determined by qPCR. B, Negative enrichment in the pathways for Inflammatory Response (left) and the TNFα Signaling via NF-κB (right) is shown in GSEA plots (siBAF60a vs. siControl). C, qPCR Validation of representative NF-κB target genes identified by RNA-seq in 3 independent sets of samples treated as in A. n=3 for each group. D, MCP-1 concentration was measured by ELISA in the cell culture medium of HASMCs transfected with siControl or siBAF60a for 48 hours followed by treatment with or without TNFα (20 ng/ml, 24h). n=3. E, Representative images (magnified field, left) and quantitative analysis (right) of the macrophages migrated through the transwell toward the conditioned media from HASMCs transfected with siControl or siBAF60a, with or without TNFα stimulation (20 ng/ml, treated for 4 hours before changing to fresh medium and incubated for another 20 hours). n=3. Scale bar=100 μm. F, HASMCs were infected with AdGFP or AdBAF60a (20 MOI). After 48 hours, total RNA was extracted for RNA-seq (n=3 for each group). Positive enrichment in the Inflammatory Response (left) and the TNFα Signaling via NF-κB (right) pathways is shown in GSEA plots (AdBAF60a vs. AdGFP). G, quantitative analysis of the macrophages migrated through the transwell toward the conditioned media from HASMCs infected with AdGFP or AdBAF60a, with or without TNFα stimulation (20 ng/ml, treated for 4 hours before changing to fresh medium and incubate for another 20 hours). n=3. Data are presented as mean ± SEM. *p< 0.05. Student’s t-test for A, one-way ANOVA followed by Tukey’s post hoc analysis for C, and two-way ANOVA followed by Holm-Sidak post hoc analysis for D-E and G.
As a complementary approach, BAF60a was overexpressed in HASMCs (Figure X-C in the online-only Data Supplement) followed by RNA sequencing. GSEA uncovered that the inflammatory response and TNFα signaling via NF-κB pathways are highly enriched in the BAF60a-overexpressing HASMCs (Figure 3F and Table VI in the online-only Data Supplement). Moreover, BAF60a overexpression upregulated the mRNA levels of several NF-κB target genes encoding pro-inflammatory cytokines (Figure X-D and Table V-b in the online-only Data Supplement) and enhanced macrophage migration (Figure 3G). Taken together, these data indicate that BAF60a induces VSMC inflammatory response.
BAF60a Regulates Cathepsin S Expression in VSMCs
RNA-seq in HASMCs also indicated an alteration in the mRNA levels of various cathepsins upon BAF60a knockdown and overexpression (Table VII in the online-only Data Supplement). Notably, cathepsin S expression was significantly down-regulated (log2 fold change = −1.080, adjusted p value = 2.5E-21) in BAF60a deficient HASMCs, while up-regulated in BAF60a-overexpressing HASMCs (log2 fold change = 1.430, adjusted p value = 5.1E-4), which was later confirmed by qPCR (Figure 4A and Figure 4B), respectively. Furthermore, TNFα stimulation upregulated cathepsin S protein level in cultured HASMCs, while knockdown of BAF60a significantly reduced cathepsin S protein expression (Figure 4C). Consistently, silencing of BAF60a decreased the concentration of cathepsin S secreted into the culture media of HASMCs either in the presence or absence of TNFα, as measured by ELISA (Figure 4D).
Figure 4. BAF60a regulates cathepsin S expression in VSMCs.
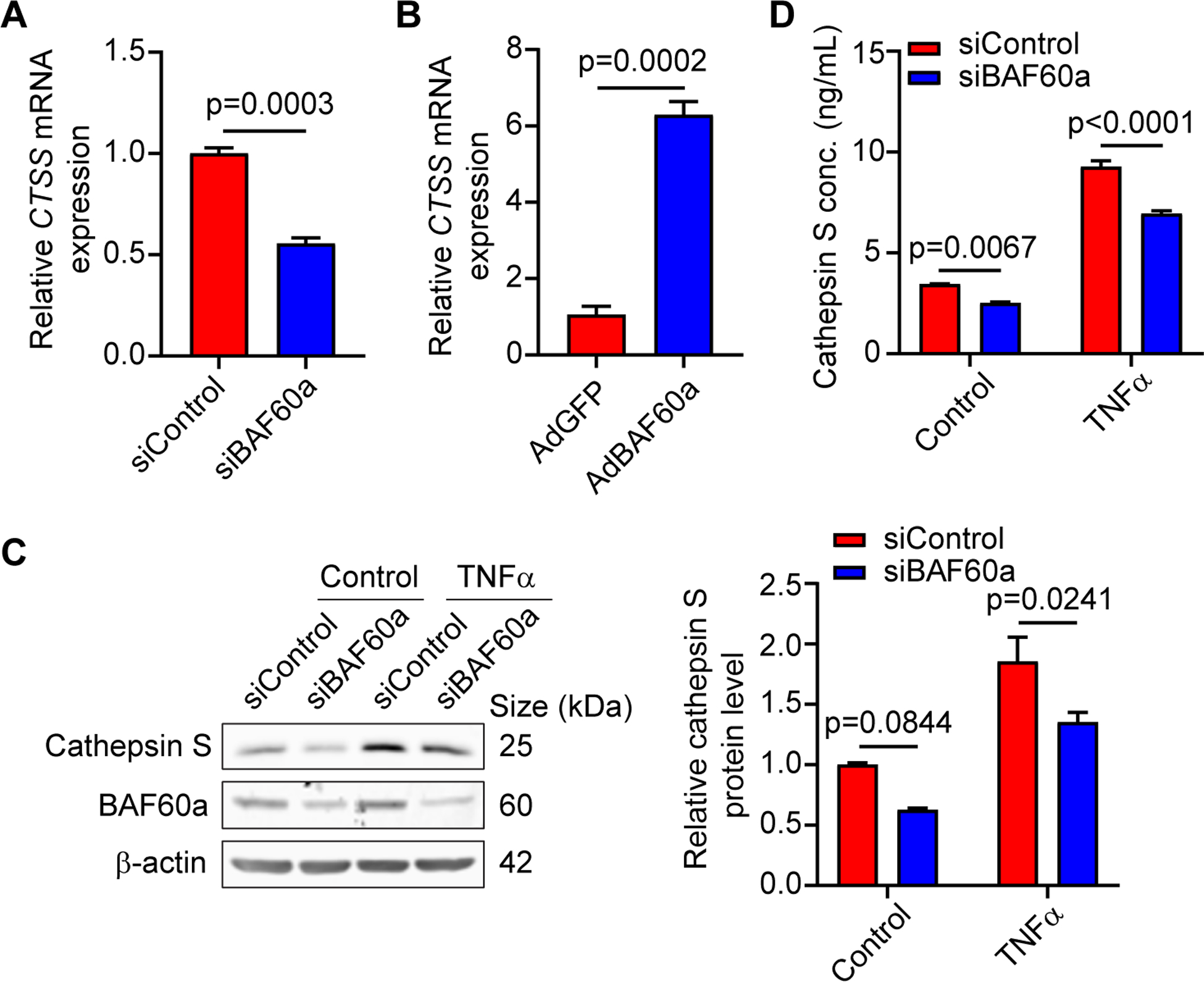
A, HASMCs were transfected with siControl or siBAF60a (30 nM). After 72 hours, CTSS mRNA expression was determined by qPCR. n=3. B, HASMCs were infected with AdGFP or AdBAF60a (30 MOI). After 48 hours, CTSS mRNA expression was determined by qPCR. n=3. C-D, HASMCs were transfected with siControl or siBAF60a (30 nM). After 48 hours, cells were treated with or without TNFα (20 ng/ml) for 24h. C, Cathepsin S protein level was determined by Western blot. n=3. D, Cathepsin S concentration in the culture medium was measured by ELISA. n=3. Data are presented as mean ± SEM. Student’s t-test for A-B, two-way-ANOVA followed by Holm-Sidak post hoc analysis for C-D.
BAF60a is Required for SWI/SNF Recruitment to the Promoters of NF-κB Target Genes
As the NF-κB family of transcription factors plays a central role in inflammatory gene expression during immune response, regulation of the NF-κB pathway is crucial to maintain cellular homeostasis upon environmental stimuli. Consistent with BAF60a’s role in regulating transcription at the chromatin level, its deficiency in cultured HASMCs did not alter the rapid nuclear translocation of NF-κB subunit p65 (RelA) after TNFα stimulation (Figure XI-A and XI-B in the online-only Data Supplement). To address whether BAF60a mediates SWI/SNF-dependent chromatin remodeling to facilitate NF-κB target gene transcription,40 chromatin immunoprecipitation followed by sequencing (ChIP-seq) was performed for BRG1 in HASMCs transfected with either siBAF60a or siControl. In this study, 8699 genome-wide binding sites for BRG1 were discovered (Figure 5A). Compared with the siControl group, BAF60a knockdown in HASMCs resulted in significantly decreased BRG1 genomic binding (Figure 5A; Figure XII-A in the online-only Data Supplement), without altering the protein abundance of BRG1 or other core SWI/SNF subunits (Figure XII-B in the online-only Data Supplement). Next, we plotted the binding of BRG1 within a ±3kb region surrounding the transcription start site (TSS) of NF-κB target genes, according to a recently published database (http://www.bu.edu/nf-kb/gene-resources/target-genes/). Similarly, knockdown of BAF60a in HASMCs resulted in a significantly decreased enrichment of BRG1 around the TSS of NF-κB target genes (Figure 5B). Through ChIP-seq, we also examined changes in the histone modification markers H3K9Ac and H3K27Ac, two chromatin signatures reported to be enriched at active promoters.41, 42 A decreased binding of H3K9Ac was identified around the TSS of NF-κB target genes, in concert with the impaired activation of the NF-κB pathway (Figure 5C). Additionally, we performed ChIP assay in primary aortic smooth muscle cells27 isolated from BAF60af/f and BAF60aSMKO mice, and reduced binding of BRG1 was identified in the promoters of several NF-κB target genes (Figure 5D; Figure XII-C in the online-only Data Supplement).
Figure 5. BAF60a is required for SWI/SNF recruitment to the promoters of NF-κB target genes.
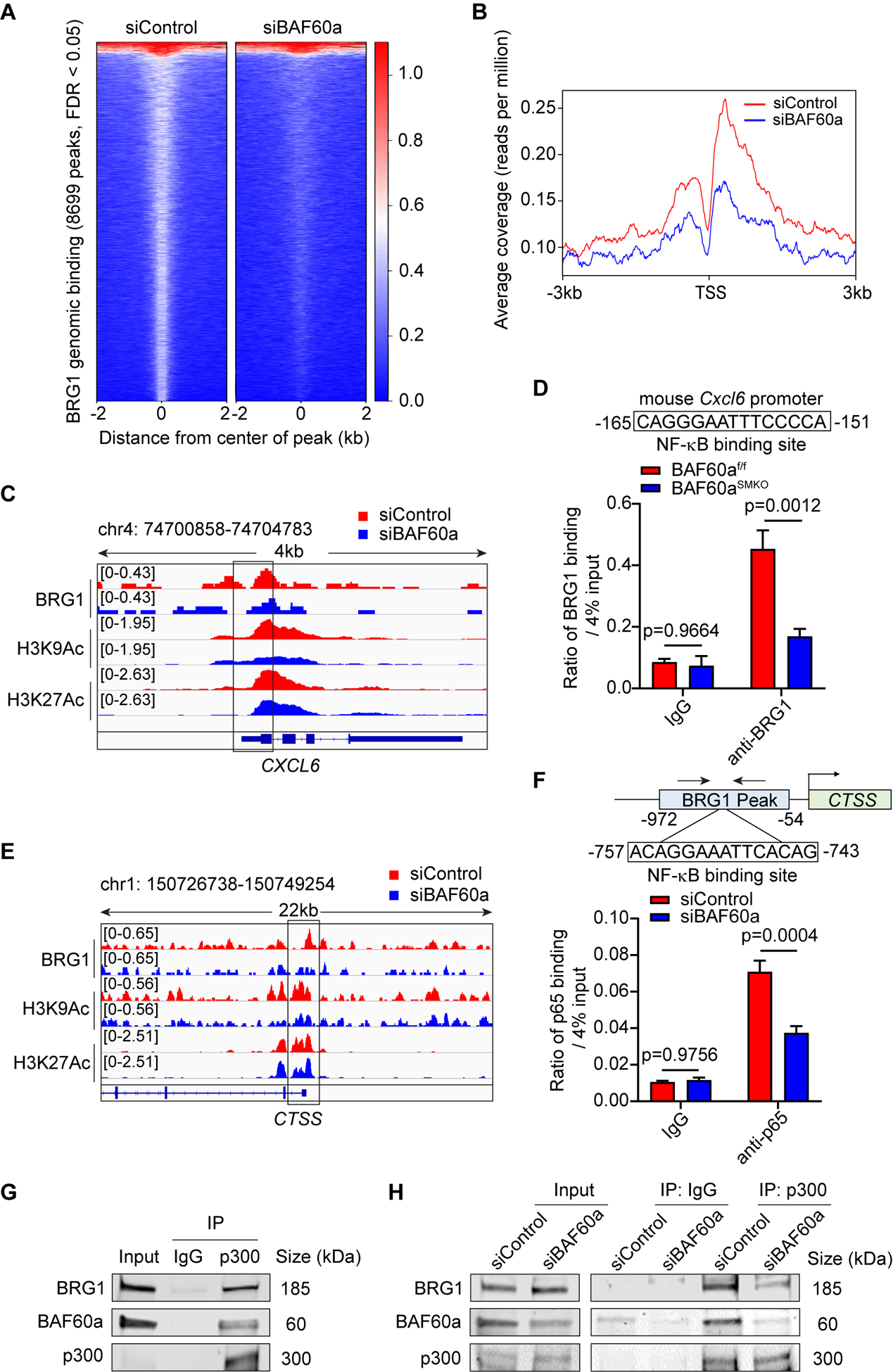
A-C and E, HASMCs were transfected with siControl or siBAF60a (30 nM). After 72 hours, ChIP-seq was performed for BRG1, H3K9Ac and H3K27Ac. A, BRG1 genomic binding on target sites within a ±2kb range centered on each peak is presented by a heatmap. B, Histogram of ChIP-seq reads of BRG1 ±3kb surrounding the TSS of NF-κB target genes. C, Normalized ChIP-seq reads of BRG1, H3K9Ac and H3K27Ac in the CXCL6 gene promoter and coding region are shown in IGV. D, BRG1 binding on the NF-κB binding site located in the mouse Cxcl6 promoter was detected by ChIP assay in primary aortic smooth muscle cells isolated from abdominal aortas of BAF60af/f and BAF60aSMKO mice. n=3. E, Normalized ChIP-seq reads of BRG1, H3K9Ac and H3K27Ac around the TSS of the CTSS gene shown in IGV. F, p65 binding on the NF-κB binding site located in the CTSS promoter was detected by ChIP assay in HASMCs transfected with 30 nM siControl or siBAF60a for 72h followed by 1h treatment with TNFα (20 ng/ml). n=3. G, Physical interaction between p300, BRG1, and BAF60a in cultured HASMCs was detected by Co-IP. IgG was used as a negative control. n=3. H, HASMCs were transfected with siControl or siBAF60a (30 nM). After 72 hours, physical interaction between p300 and BRG1 was detected by Co-IP. IgG was used as a negative control. n=3. Data are presented as mean ± SEM. Two-way-ANOVA followed by Holm-Sidak post hoc analysis for D and F.
To further explore the mechanism of BAF60a-dependent regulation of cathepsin S expression, we analyzed the BRG1 ChIP-seq data and identified a significantly reduced signal at the promoter region of the CTSS gene upon knockdown of BAF60a in HASMCs (Figure 5E). Similarly, silencing of BAF60a resulted in a suppressed enrichment of the activation histone marker H3K9Ac within the CTSS promoter (Figure 5E), indicating impaired transcription. Interestingly, an NF-κB binding site was found to reside within the BRG1 peak by MatInspector43 in the human CTSS promoter, and a reduced binding of BRG1 to this NF-κB site was confirmed in HASMCs transfected with siBAF60a (Figure XII-D in the online-only Data Supplement). In parallel, attenuated recruitment of BRG1 to the corresponding NF-κB binding site in the mouse Ctss promoter was validated by ChIP assay in primary aortic smooth muscle cells isolated from BAF60aSMKO mice compared with BAF60af/f mice (Figure XII-E in the online-only Data Supplement). Further investigation uncovered that p65 accumulation to the CTSS promoter induced by TNFα was significantly dampened upon silencing of BAF60a in VSMCs (Figure 5F). These data define CTSS as a novel direct target of p65 regulated by BAF60a-dependent recruitment of the SWI/SNF complex to the promoter.
As one of the best-characterized co-factors of NF-κB, p300, a member of the histone acetyltransferase (HAT) family, is associated with increased transcription of inflammatory response genes.44 Besides, previous reports have demonstrated a physical interaction between the SWI/SNF complex and p300, suggesting a synergistic role of these chromatin remodelers in modulating transcription factor accessibility.45, 46 Consequently, we performed co-immunoprecipitation (co-IP) and found that p300 interacts with both BAF60a and BRG1 in cultured HASMCs (Figure 5G). Direct interaction between BAF60a and p300 was further confirmed by using a promiscuous biotin ligase-BirA*-fused to the BAF60a protein,47 BAF60a-BirA*, stably expressed in A7r5 cells (a rat aortic smooth muscle cell line). We found that p300 interacting with BAF60a-BirA* was labeled with biotin and immunoprecipitated by streptavidin (Figure XIII-A and XIII-B in the online-only Data Supplement). Furthermore, knockdown of BAF60a in HASMCs significantly reduced the interaction between p300 and BRG1 (Figure 5H), which may serve as an explanation to the reduced histone acetylation. In addition, the attenuated interaction between p300 and BRG1 was further identified in primary aortic smooth muscle cells isolated from BAF60aSMKO mice compared with BAF60af/f mice (Figure XIII-C in the online-only Data Supplement). Collectively, these data indicate that BAF60a is required for SWI/SNF recruitment in mediating NF-κB target gene expression in VSMC inflammation.
In summary, our data provide evidence that loss of BAF60a in VSMCs impaired NF-κB transcriptional activity and reduced cysteine protease cathepsin S expression, thus contributing to protect from AAA formation by simultaneously inhibiting ECM degradation and reducing the overall inflammatory response.
Discussion
Although the SWI/SNF chromatin remodeling complex has been demonstrated to be an important regulator of many chromatin accessibility checkpoints for maintaining cellular homeostasis in various human diseases,8, 9 its role in the biology and pathophysiology of the vasculature remains largely unknown. The mutually exclusive incorporation of BAF60 family members (BAF60a, b and c) into distinct SWI/SNF complexes suggests different biological functions. Thus, BAF60a regulates fatty acid oxidation and cholesterol homeostasis, while BAF60c is involved in glucose metabolism in liver and muscle.48 BAF60a is the only 60-kDa subunit involved in all three mammalian SWI/SNF assemblies including canonical BAF (cBAF), polybromo-associated BAF (PBAF) and non-canonical BAF (ncBAF)49. However, its role in vascular biology remains unexplored. Our results here show that loss of BAF60a in VSMCs protected mice from AngII- and elastase-induced AAA formation by suppressing vascular inflammation and ECM degradation, which sheds new light on the molecular mechanisms of this disease. In this study, BAF60a was found to be required for recruiting the catalytic subunit BRG1 of the SWI/SNF complex to the promoter of NF-κB target genes, underscoring a critical role of BAF60a in regulating VSMC inflammatory response. Furthermore, we uncovered that NF-κB regulates the expression of cathepsin S in a BAF60a-dependent fashion, thus providing the first evidence that NF-κB and BAF60a regulate concurrently the VSMC inflammatory response and the ECM remodeling, two hallmark features of AAA.
The transcription factors of the NF-κB family regulate a large fraction of the inflammatory transcriptome. According to the kinetics of transcriptional activation, NF-κB target genes can be divided into two classes: primary response genes with chromatin landscape poised for immediate activation, such as TNF; and secondary response genes with delayed transcription, such as most of the cytokines and chemokines, with promoters that are frequently found to contain a positioned nucleosome overlapping the TATA box.50 In this scenario, chromatin remodelers, such as the SWI/SNF complex which alters DNA/histone contacts, are essential to increase the accessibility for transcription. BRG1, the catalytic subunit of the SWI/SNF complex, contains a DNA dependent ATPase domain and was shown to cooperate with other co-factors such as histone acetyltransferase (HAT) to configure the chromatin and facilitate NF-κB target gene expression.40 In this study, by performing ChIP-seq, we identified BAF60a as a bridge able to recruit BRG1 in the SWI/SNF complex to the transcriptional target genes of NF-κB. Silencing of BAF60a in HASMCs resulted in a noticeable decrease in BRG1 genome-wide occupancy, especially within a 6-kb region around the transcriptional start site of NF-κB-dependent secondary response genes, without altering the expression of primary response genes. As one of the best-characterized co-factors of NF-κB, p300 functions as an acetyltransferase to enhance transcriptional activity through acetylation of p65 and histones residing in the promoters.41 Acetylated p65 presents stabilized binding to DNA and suppresses interaction with IkBα, leading to enhanced NF-κB transcription activation.44 Interestingly, in this study, we identified a physical interaction between BAF60a and p300. Based on our observations, we propose a novel model for the BAF60a-dependent regulation of NF-κB transcription of inflammatory genes (mainly secondary response genes) where BAF60a plays a central role in linking NF-κB transcriptional co-factors with the SWI/SNF chromatin remodeling complex (Figure 6).
Figure 6. Schematic model of BAF60a regulation of VSMC inflammation and ECM degradation in AAA development.
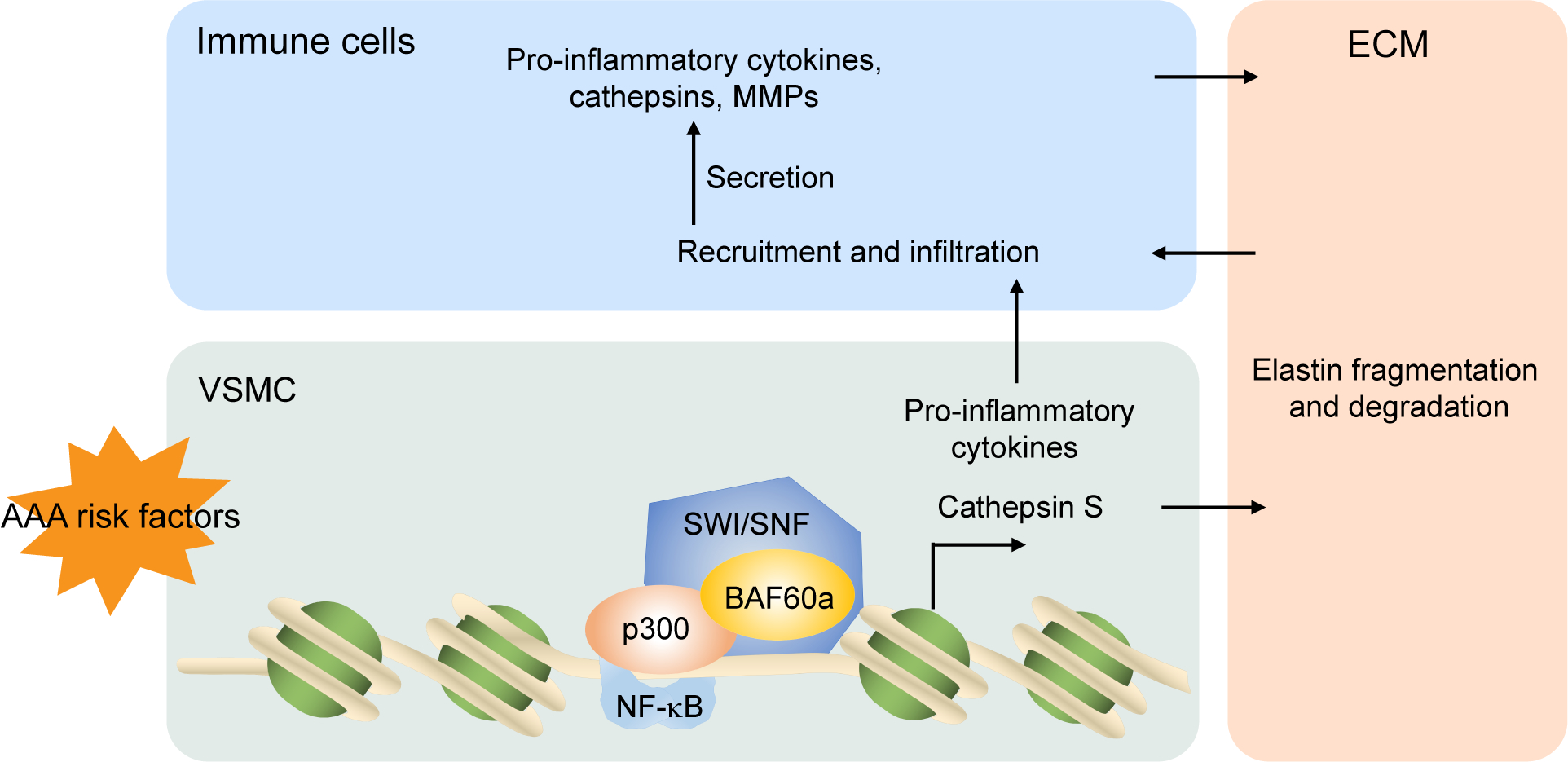
VSMC, vascular smooth muscle cells; ECM, extracellular matrix; MMPs, matrix metalloproteinases.
In summary, through the use of VSMC-specific BAF60a-deficient mice in AngII- and elastase-induced murine aneurysm models, our study demonstrates for the first time that BAF60a regulates inflammatory signaling and cysteine protease cathepsin S expression in VSMCs, crucial in AAA development. Mechanistically, BAF60a regulates inflammatory gene expression in VSMCs by recruiting the SWI/SNF chromatin remodeling complex to the promoter of NF-κB target genes and by interacting with the NF-κB transcriptional co-factor p300. Of relevance, it is remarkable that targeting of individual genes or pathways have failed to translate into clinically relevant drugs so far. A coordinated, genome-wide chromatin remodeling, such as that uncovered here for BAF60a, in response to the stimuli from AAA risk factors, could help explain the paucity of favorable results for targeted treatments in AAA. As local and tissue-specific delivery strategies continue to develop, inhibition of VSMC BAF60a may eventually contribute to the pharmacological treatment of AAA and other inflammatory diseases of the vascular wall.
Supplementary Material
Highlights.
BAF60a is upregulated in human and experimental murine abdominal aortic aneurysm (AAA) lesions.
BAF60a plays a critical role in regulating vascular smooth muscle cell (VSMC) inflammation and extracellular matrix degradation.
VSMC-specific knockout of BAF60a attenuates both AngII- and elastase-induced murine AAA formation.
Acknowledgments
We thank the Cardiovascular Health Improvement Project (CHIP) at the University of Michigan Cardiovascular Center to provide patients’ AAA and healthy control abdominal aorta samples. We thank Dr. Zhuoxian Meng from the Zhejiang University for critical discussions.
Sources of Funding
This work was partially supported by National Institutes of Health grants HL138139 (J. Zhang), HL068878 and HL134569 (Y.E. Chen), HL122664 (L. Chang), HL145176 (Y. Fan), HL150233 (O. Rom) and American Heart Association grants 20P0ST35110064 (G. Zhao).
Nonstandard Abbreviations and Acronyms
- AAA
abdominal aortic aneurysm
- AngII
angiotensin
- ChIP
chromatin immunoprecipitation
- Co-IP
co-immunoprecipitation
- CTSS
cathepsin S
- ECM
extracellular matrix
- HASMC
human aortic smooth muscle cell
- MASMC
mouse aortic smooth muscle cell
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PCSK9
proprotein convertase subtilisin/kexin type 9
- siRNA
small interfering RNA
- SWI/SNF complex
SWItch/Sucrose Non-Fermentable complex
- TNFα
tumor necrosis factor alpha
- VSMC
vascular smooth muscle cell
- MMP
matrix metalloproteinase
Footnotes
Disclosures
None
References
- 1.Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8:92–102 [DOI] [PubMed] [Google Scholar]
- 2.Moll FL, Powell JT, Fraedrich G, Verzini F, Haulon S, Waltham M, van Herwaarden JA, Holt PJ, van Keulen JW, Rantner B, Schlosser FJ, Setacci F, Ricco JB, European Society for Vascular S. Management of abdominal aortic aneurysms clinical practice guidelines of the european society for vascular surgery. Eur J Vasc Endovasc Surg. 2011;41 Suppl 1:S1–S58 [DOI] [PubMed] [Google Scholar]
- 3.Raffort J, Lareyre F, Clement M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017;14:457–471 [DOI] [PubMed] [Google Scholar]
- 4.Li H, Bai S, Ao Q, Wang X, Tian X, Li X, Tong H, Hou W, Fan J. Modulation of immune-inflammatory responses in abdominal aortic aneurysm: Emerging molecular targets. J Immunol Res. 2018;2018:7213760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siasos G, Mourouzis K, Oikonomou E, Tsalamandris S, Tsigkou V, Vlasis K, Vavuranakis M, Zografos T, Dimitropoulos S, Papaioannou TG, Kalampogias A, Stefanadis C, Papavassiliou AG, Tousoulis D. The role of endothelial dysfunction in aortic aneurysms. Curr Pharm Des. 2015;21:4016–4034 [DOI] [PubMed] [Google Scholar]
- 6.Chen HZ, Wang F, Gao P, al. e. Age-associated sirtuin 1 reduction in vascular smooth muscle links vascular senescence and inflammation to abdominal aortic aneurysm. Circ Res. 2016;119:1076–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ackers-Johnson M, Talasila A, Sage AP, Long X, Bot I, Morrell NW, Bennett MR, Miano JM, Sinha S. Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arterioscler Thromb Vasc Biol. 2015;35:817–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang W, Agrawal DK, Boosani CS. Cellspecific histone modifications in atherosclerosis (review). Mol Med Rep. 2018;18:1215–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian swi/snf complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schick S, Rendeiro AF, Runggatscher K, Ringler A, Boidol B, Hinkel M, Majek P, Vulliard L, Penz T, Parapatics K, Schmidl C, Menche J, Boehmelt G, Petronczki M, Muller AC, Bock C, Kubicek S. Systematic characterization of baf mutations provides insights into intracomplex synthetic lethalities in human cancers. Nat Genet. 2019;51:1399–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito T, Yamauchi M, Nishina M, Yamamichi N, Mizutani T, Ui M, Murakami M, Iba H. Identification of swi.Snf complex subunit baf60a as a determinant of the transactivation potential of fos/jun dimers. J Biol Chem. 2001;276:2852–2857 [DOI] [PubMed] [Google Scholar]
- 12.Li S, Liu C, Li N, Hao T, Han T, Hill DE, Vidal M, Lin JD. Genome-wide coactivation analysis of pgc-1alpha identifies baf60a as a regulator of hepatic lipid metabolism. Cell Metab. 2008;8:105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh J, Sohn DH, Ko M, Chung H, Jeon SH, Seong RH. Baf60a interacts with p53 to recruit the swi/snf complex. J Biol Chem. 2008;283:11924–11934 [DOI] [PubMed] [Google Scholar]
- 14.Alajem A, Biran A, Harikumar A, Sailaja BS, Aaronson Y, Livyatan I, Nissim-Rafinia M, Sommer AG, Mostoslavsky G, Gerbasi VR, Golden DE, Datta A, Sze SK, Meshorer E. Differential association of chromatin proteins identifies baf60a/smarcd1 as a regulator of embryonic stem cell differentiation. Cell Rep. 2015;10:2019–2031 [DOI] [PubMed] [Google Scholar]
- 15.Tao W, Chen S, Shi G, Guo J, Xu Y, Liu C. Switch/sucrose nonfermentable (swi/snf) complex subunit baf60a integrates hepatic circadian clock and energy metabolism. Hepatology. 2011;54:1410–1420 [DOI] [PubMed] [Google Scholar]
- 16.Meng ZX, Wang L, Chang L, Sun J, Bao J, Li Y, Chen YE, Lin JD. A diet-sensitive baf60a-mediated pathway links hepatic bile acid metabolism to cholesterol absorption and atherosclerosis. Cell Rep. 2015;13:1658–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lenk GM, Tromp G, Weinsheimer S, Gatalica Z, Berguer R, Kuivaniemi H. Whole genome expression profiling reveals a significant role for immune function in human abdominal aortic aneurysms. BMC Genomics. 2007;8:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakaoka H, Tajima A, Yoneyama T, Hosomichi K, Kasuya H, Mizutani T, Inoue I. Gene expression profiling reveals distinct molecular signatures associated with the rupture of intracranial aneurysm. Stroke. 2014;45:2239–2245 [DOI] [PubMed] [Google Scholar]
- 19.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12-g13-larg-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68 [DOI] [PubMed] [Google Scholar]
- 20.Lu H, Howatt DA, Balakrishnan A, Graham MJ, Mullick AE, Daugherty A. Hypercholesterolemia induced by a pcsk9 gain-of-function mutation augments angiotensin ii-induced abdominal aortic aneurysms in c57bl/6 mice-brief report. Arterioscler Thromb Vasc Biol. 2016;36:1753–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu H, Sun J, Liang W, Chang Z, Rom O, Zhao Y, Zhao G, Xiong W, Huilun W, Zhu T, Guo Y, Chang L, Garcia-Barrio Minerva T, Zhang J, Chen YE, Fan Y. Cyclodextrin prevents abdominal aortic aneurysm via activation of vascular smooth muscle cell tfeb. Circulation. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhamidipati CM, Mehta GS, Lu G, Moehle CW, Barbery C, DiMusto PD, Laser A, Kron IL, Upchurch GR Jr., Ailawadi G. Development of a novel murine model of aortic aneurysms using peri-adventitial elastase. Surgery. 2012;152:238–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He L, Fu Y, Deng J, al e. Deficiency of fam3d (family with sequence similarity 3, member d), a novel chemokine, attenuates neutrophil recruitment and ameliorates abdominal aortic aneurysm development. Arterioscler Thromb Vasc Biol. 2018;38:1616–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang C, van der Voort D, Shi H, Zhang R, Qing Y, Hiraoka S, Takemoto M, Yokote K, Moxon JV, Norman P, Rittié L, Kuivaniemi H, Atkins GB, Gerson SL, Shi G-P, Golledge J, Dong N, Perbal B, Prosdocimo DA, Lin Z. Matricellular protein ccn3 mitigates abdominal aortic aneurysm. The Journal of Clinical Investigation. 2016;126:1282–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao G, Fu Y, Cai Z, Yu F, Gong Z, Dai R, Hu Y, Zeng L, Xu Q, Kong W. Unspliced xbp1 confers vsmc homeostasis and prevents aortic aneurysm formation via foxo4 interaction. Circ Res. 2017;121:1331–1345 [DOI] [PubMed] [Google Scholar]
- 26.Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y. Rnascope: A novel in situ rna analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 2012;14:22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamblin M, Chang L, Chen YE. Isolation and culture of vascular smooth muscle cells. Manual of Research Techniques in Cardiovascular Medicine. 2014:125–130 [Google Scholar]
- 28.Lu H, Sun J, Liang W, Zhang J, Rom O, Garcia-Barrio MT, Li S, Villacorta L, Schopfer FJ, Freeman BA, Chen YE, Fan Y. Novel gene regulatory networks identified in response to nitro-conjugated linoleic acid in human endothelial cells. Physiol Genomics. 2019;51:224–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods. 2017;14:417–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014;15:550–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of chip-seq (macs). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petsophonsakul P, Furmanik M, Forsythe R, Dweck M, Schurink Geert W, Natour E, Reutelingsperger C, Jacobs M, Mees B, Schurgers L. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2019;39:1351–1368 [DOI] [PubMed] [Google Scholar]
- 35.Qin Y, Cao X, Guo J, Zhang Y, Pan L, Zhang H, Li H, Tang C, Du J, Shi GP. Deficiency of cathepsin s attenuates angiotensin ii-induced abdominal aortic aneurysm formation in apolipoprotein e-deficient mice. Cardiovasc Res. 2012;96:401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadoglou NP, Liapis CD. Matrix metalloproteinases: Contribution to pathogenesis, diagnosis, surveillance and treatment of abdominal aortic aneurysms. Curr Med Res Opin. 2004;20:419–432 [DOI] [PubMed] [Google Scholar]
- 37.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. The Journal of Clinical Investigation. 2002;110:625–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gjurich BN, Taghavie-Moghadam PL, Galkina EV. Flow cytometric analysis of immune cells within murine aorta. Methods Mol Biol. 2015;1339:161–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Golledge AL, Walker P, Norman PE, Golledge J. A systematic review of studies examining inflammation associated cytokines in human abdominal aortic aneurysm samples. Dis Markers. 2009;26:181–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramirez-Carrozzi VR, Nazarian AA, Li CC, Gore SL, Sridharan R, Imbalzano AN, Smale ST. Selective and antagonistic functions of swi/snf and mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006;20:282–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heintzman ND, Stuart RK, Hon G, al e. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318 [DOI] [PubMed] [Google Scholar]
- 42.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. Matinspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942 [DOI] [PubMed] [Google Scholar]
- 44.Calao M, Burny A, Quivy V, Dekoninck A, Van Lint C. A pervasive role of histone acetyltransferases and deacetylases in an nf-kappab-signaling code. Trends Biochem Sci. 2008;33:339–349 [DOI] [PubMed] [Google Scholar]
- 45.Huang ZQ, Li J, Sachs LM, Cole PA, Wong J. A role for cofactor-cofactor and cofactor-histone interactions in targeting p300, swi/snf and mediator for transcription. EMBO J. 2003;22:2146–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, Haswell JR, Park PJ, Roberts CW. The swi/snf chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun. 2017;8:14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 2012;196:801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang RR, Pan R, Zhang W, Fu J, Lin JD, Meng ZX. The swi/snf chromatin-remodeling factors baf60a, b, and c in nutrient signaling and metabolic control. Protein Cell. 2018;9:207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michel BC, D’Avino AR, Cassel SH, Mashtalir N, McKenzie ZM, McBride MJ, Valencia AM, Zhou Q, Bocker M, Soares LMM, Pan J, Remillard DI, Lareau CA, Zullow HJ, Fortoul N, Gray NS, Bradner JE, Chan HM, Kadoch C. A non-canonical swi/snf complex is a synthetic lethal target in cancers driven by baf complex perturbation. Nature Cell Biology. 2018;20:1410–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saccani S, Pantano S, Natoli G. Two waves of nuclear factor kappab recruitment to target promoters. J Exp Med. 2001;193:1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.