

Abstract
We combined mRNA display technology with lipid-nanodisc based selections and identified high-affinity ligands targeting the integral membrane sensor domain of the histidine kinase AgrC as potent inhibitors of Staphylococcus aureus quorum sensing-modulated virulence. Our study highlights the potential of this integrated approach for identifying functional modulators of integral membrane proteins.
Graphical Abstract
By integrating lipid nanodisc and the RaPID technologies, we identified novel peptidic quorum quenchers targeting S. aureus histidine kinase AgrC.
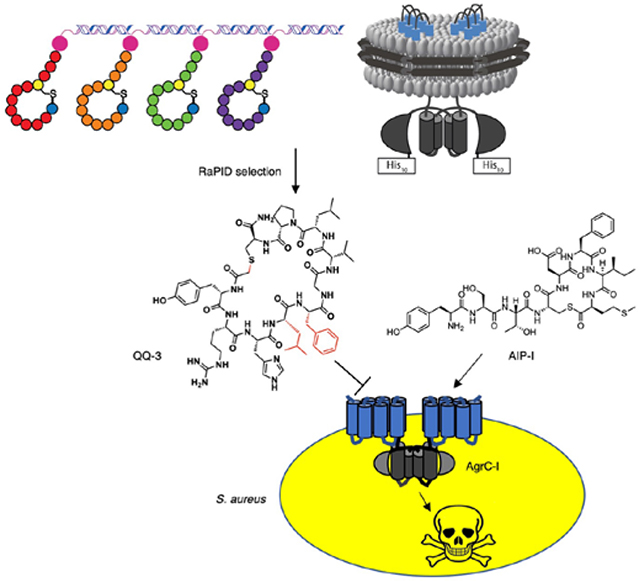
Infections caused by the Gram-positive pathogen Staphylococcus aureus (S. aureus) pose an ever growing threat to public health due by the spread of antibiotic resistance.1 To tackle this crisis, an anti-virulence strategy has been proposed where therapeutic agents are administered to interfere with production of virulence factors rather than directly killing the bacterium.2 This approach is considered advantageous due to its low selection pressure for resistance build-up and minimal perturbation of the host microbiota. In S. aureus, virulence gene expression is under the control of the accessory gene regulator (agr) quorum sensing system, and accordingly there has been great interest in developing inhibitors of the agr response (also termed quorum quenchers).3 While the agr signaling circuit offers multiple potential nodes for inhibitor design, the receptor histidine kinase, AgrC, is perhaps the most promising target due to its accessibility on the cell surface. The membrane-embedded sensor domain of AgrC binds the quorum signal, a thiolactone containing autoinducing peptide (AIP), leading to activation of a two-component signaling cascade to turn on Staphylococcal virulence. Four different agr subgroups have been identified in S. aureus, each producing their own AIP and AgrC. Among them, AgrC of a specific subgroup is activated by its cognate AIP while the heterologous AIPs are generally inhibitory, a phenomenon referred to as agr interference.4 There have been extensive efforts made towards harnessing the cross-inhibitory activities of native AIPs for the development of global quorum quencher agents.5–9 While exploring the chemical space around the native AIP scaffold has been a productive approach, there remains a need for novel molecular scaffolds distinct from AIPs that still target the AgrC sensor domain.3 Identification of such quorum quenchers would not only provide new solutions to inhibition of S. aureus virulence, but likely also yield insights into the molecular signatures required for AgrC engagement. Despite this potential, such efforts have been constrained by the lack of defined biochemical systems compatible with high-throughput approaches for ligand discovery. Herein, we report the design and implementation of such a screening platform for AgrC, leading to the discovery of a suite of novel macrocyclic peptide scaffolds as highly potent agr quorum quenchers.
We elected to use the Random non-standard Peptide Integrated Discovery (RaPID) system to select for AgrC binders from a highly diverse library (>1012) of ribosomally synthesized macrocyclic peptides.10, 11 This screening platform has been widely employed to discover ligands for soluble protein targets.12–14 By contrast, only a handful of examples of using the RaPID system on integral membrane proteins have been reported, and in all cases the membrane protein targets were solubilized in detergent micelles.15–17 Such an approach is incompatible with AgrC since it requires a lipid bilayer environment to adopt a functional homodimeric state.18 The domain architecture of AgrC presents an additional challenge for screening; in addition to the polytopic membrane sensor domain, the protein contains a large cytoplasmic dimerization and histidine kinase module. Thus, any screen would have to discriminate between ligands that bind to the AgrC sensor (desired) and those that bind elsewhere on the protein or to the lipids themselves. To address these various issues, we devised a two-step selection process involving full-length and truncated versions of AgrC proteins each reconstituted into lipid nanodiscs compatible with the RaPID system (Figure 1 and S1).18, 19
Fig 1.

Schematic of the RaPID system employed for selection of macrocyclic peptide binders to the integral membrane sensor domain of AgrC-I. Notably, the strategy integrates negative and positive selections (steps 5 & 6) in the selection cycle.
Four different libraries were screened, initiated with either an N-chloroacetylated D-tyrosine (ClAcD-Tyr) or an N-chloroacetylated L-tyrosine (ClAcL-Tyr) residue at the AUG start codon through flexizymeassisted genetic code reprogramming.20 The initiator amino acid was followed by a random sequence encoded by either (NNK)4–9 or (NNK)10–15 codons (where N represents any of the four RNA bases and K represents either G or U) and a cysteine residue required for macrocyclization via a non-hydrolyzable thioether bond (Figure S2). Use of the RaPID display format allowed for identification of selected peptide binders using next generation sequencing. In a typical selection cycle, we first subjected the peptide library to a nanodisc-embedded truncated group-I AgrC with only the last transmembrane helix present (1TM-AgrC-I), which is incapable of binding AIPs (Figure S1b). In this negative selection step, non-specific binders to either the nanodisc or the intracellular histidine kinase domain of AgrC were removed. In the ensuing positive selection step, peptides that bound to the transmembrane sensor domain of full-length AgrC were enriched from the remaining library. The cDNAs encoding for the enriched peptide pool were amplified and used for subsequent rounds of RaPID selection. After multiple cycles, sufficient enrichment of the cDNA sequences encoding for AgrC-I binders was observed (Figure S3). Deep sequencing of each selected pool revealed the sequences with highest abundance, from which we selected four cyclic peptides for further study, designated as quorum quenching peptides 1 through 4 (QQ-1 to QQ-4) (Figure 2a, S4 and S5). Notably, these QQ peptides are structurally distinct from the native AIPs. In addition to having a stable thioether linkage in place of the labile thiolactone, the selected peptides bear no immediate sequence similarity and are much larger macrocycles compared with the native AgrC ligands (e.g. 14 amino acids for QQ-4, vs. 5 amino acids for the AIPs). Principal component analysis on the molecular descriptors of the QQ and AIP peptides using ClustVis also supports the idea that the selected peptides reside in different chemical space from the native AIPs (Figure S6, Table S1).21
Figure 2. Validation and characterization of the quorum-quenching (QQ) peptide hits.

a) The chemical structures of the top macrocyclic peptide hits (QQ-1 to QQ-4) from the RaPID selection. b) Inhibition dose-response curves for the QQ peptides (QQ-1 to QQ-4). Exponential phase cultures of a group-I S. aureus reporter strain (RN9222) were incubated with varying concentrations of the QQ peptides in the presence of 100 nM AIP-I agonist. β-lactamase activity was measured and plotted as % maximal activation versus QQ peptide concentration. Error bars represent SEM (n = 3). c) Summary of the structure-activity relationship analysis of the QQ-3 peptide. Red: >50 fold increase in IC50 upon sidechain removal; orange: 10–50 fold increase in IC50; olive: <10 fold increase in IC50. Note that in addressing the role of the thioether linkage, both the linear peptide and the methylene substituted variant (QQ-5) were made.
Next, the biological activities of the QQ peptides were explored using synthetic versions of the macrocycles (Figure 2a, S7). First, we used an in vitro autokinase assay in which either AIPs or the QQ peptides were incubated with three subgroups of AgrC (AgrC-I/II/III) reconstituted into nanodiscs and the autophosphorylation of the receptor was monitored by autoradiography.18, 19 As expected, the cognate AIP strongly activated AgrC histidine kinase activity (Figure S8). The QQ peptides, by contrast, minimally altered the basal kinase activity of AgrC-I and AgrC-II. Interestingly, partial activation on AgrC-III was observed for all four QQ peptides. Given that the QQ peptides were selected as tight binders to the AgrC-I sensor domain, we hypothesized that they may act as competitive inhibitors of the cognate AgrC-AIP interaction. To test this, we investigated the impact of the QQ peptides on agr induction in S. aureus cells of different subgroups using a previously established β-lactamase reporter assay.22 All four QQ peptides displayed inhibitory activity on agr induction in the three agr systems tested, with the IC50 values against group-I cells being the lowest (Figure 2b, S9 and Table 1). This is not entirely unexpected since the QQ peptides were selected based on their binding affinity to AgrC-I. Among the selected peptides, QQ-3 inhibits group-I agr signaling most effectively, with its IC50 value (~1 nM) comparable to the most potent AIP-derived AgrC-I inhibitors reported to date.8, 23 Therefore, we focused our subsequent analyses on QQ-3.
Table 1.
The inhibitory activity of the QQ peptides and AIPs on three agr subgroups of S. aureus using a β-lactamase reporter cell assay
| QQ peptides/AIPs | IC50 (95% CI) [nM] |
||
|---|---|---|---|
| Group-I | Group-II | Group-III | |
| QQ-1 | 4.2 (3.2 – 5.7) | 331.6 (199.1 – 552.4) | 5.6 (4.3 – 7.3) * |
| QQ-2 | 1.8 (1.1 – 2.9) | 1144 (192.2 – 6815.0) | 524.7 (417.9 – 658.8) * |
| QQ-3 | 1.1 (0.9 – 1.4) | 135.4 (111.7 – 164.2) | 175.6 (92.9 – 331.8) * |
| QQ-4 | 106.5 (53.0 – 213.7) | 998.3 (660.0 – 1510.0) | 47.6 (31.2 – 72.7) * |
| AIP-I | – | 25 (14 – 45)⋄ | 3 (2 – 5)⋄ |
| AIP-II | 40 (12 –140)⋄ | – | 1 (0.7 – 2.6)⋄ |
| AIP-III | 70 (30 –150)⋄ | 6 (5 – 6.5)⋄ | – |
| tr-AIP-II | 260 (95 – 695)⋄ | 230 (190 – 270)⋄ | 4 (3 – 5)⋄ |
Against 500 nM AIP-III
IC50 values have been previously reported in ref 6 and cited for comparison
Examination of the QQ peptides revealed a consensus sequence motif “LFG” preceded by a polar residue (Figure S10a). We postulated that this motif is essential for the competitive inhibition of AgrC by the QQ peptides. To test this, we performed an alanine scan on QQ-3 and tested the inhibitory activity of each analog using the β-lactamase reporter cell assay (Figure 2c, Figure S7, S10b, S10c and Table S2). Substitution of residues Tyr1, His3, Leu4, Phe5 and Gly6, which map to one hemisphere of the macrocycle, was found to have the biggest impact on activity. For example, replacement of Leu4 and Phe5, part of the aforementioned motif, both decrease the inhibitory potency of QQ-3 by at least 50-fold. This result is interesting in light of previous structure-activity relationship studies performed on AIPs that have revealed receptor binding primarily resides in two adjacent bulky hydrophobic residues within the AIP thiolactone macrocycle.24 Furthermore, a similar structural motif is also found in the 16-membered macrocycle of Solonamide B, a cyclodepsipeptide previously reported to competitively inhibit AgrC (Figure S11), with the order of the Leu and Phe reversed in Solonamide B and the former residue being in the D-configuration.25 Notably, replacement of Gly6 in QQ-3 with D-Ala only modestly (2-fold) compromised its inhibitory activity, whereas the corresponding L-Ala substitution had a more profound effect (12-fold, Figure 2c, Figure S7, S10b, S10c and Table S2). We interpret this to mean that the presence of the glycine within the “LFG” motif allows access to unique dihedral angles around the αC, presumably permitting access to an optimal backbone conformation for AgrC-I binding. Similar to what is known for AIPs, the macrocyclic constraint in the QQ peptides was also found to be critical for agr inhibition (Figure 2c, Figure S7, S10d and Table S2).22 For example, the linear version of QQ-3 was ~800-fold less active than the corresponding cyclic version of the peptide.
We also replaced the sulfur atom in QQ-3 with a methylene group, generating the des-thio analog, QQ-5 (Figure 3a, S7). Both QQ-3 and QQ-5 peptides showed improved chemical stability and in vitro serum stability compared to a native AIP, AIP-II (Figure S12a and S12b). Moreover, the QQ-3 and QQ-5 peptides were found to have similar affinity for AgrC-I nanodiscs as measured by fluorescence anisotropy (Figure 3b) and comparable potency in inhibiting group-I agr quorum sensing in the β-lactamase reporter cell assay (Figure 3c). To validate that QQ-3 and QQ-5 peptides target the sensor domain of AgrC in S. aureus, we utilized an R238A mutant of the receptor, which possesses constitutive histidine kinase activity independent of AIP binding to the sensor domain.26 We compared the extent of inhibition on AIP biosynthesis by either QQ-3 or QQ-5 between isogenic S. aureus strains carrying either wild type agr genes (WT) or an R238A mutation in agrC. As expected, QQ-3 and QQ-5 significantly reduced the amount of AIP secreted from the WT S. aureus strain (Figure 3d, Figure S13). By contrast, we observed no reduction in AIP production when QQ-3 and QQ-5 were administered to the AgrC-R238A containing mutant strain, a finding that is fully consistent with a mechanism of action in WT cells involving binding to the AgrC sensor domain.
Fig 3. Characterization of selected QQ peptides as lead compounds to inhibit S. aureus virulence.

a) The chemical structure of QQ-5 peptide. b) Binding affinity measurement between QQ-3/QQ-5 peptide and AgrC-I. Competitive displacement of FAM-AIP-I from AgrC-I nanodiscs was monitored by steady-state anisotropy change (ΔSSA). One representative titration of three is shown. Error bars represent SEM (n = 10, technical). c) Inhibition dose-response curves for QQ-3, QQ-5 and AIP-II on group-I S. aureus reporter strain RN9222. Error bars represent SEM (n = 3). d) Inhibition of S. aureus AIP biosynthesis by QQ-3 and QQ-5. 500 nM QQ-3 (left panel), QQ-5 peptide (right panel) or DMSO control was added to S. aureus carrying either wild-type agr or bearing a constitutive R238A mutation in the agrC gene when the culture reached an OD600 ~0.4 and was defined as t = 0h. Then AIP secreted into the medium was quantified in reference to an internal standard using LC-MS for t = 0h and 6h. e) S. aureus hemolysis assay. AIP-II or QQ peptides were separately mixed with group-I S. aureus RN12132 cells with intact agr and spotted on a blood agar plate. Inhibition of hemolysis was monitored after 24 h incubation at 37°C. Note, the higher concentration of peptide required here for inhibition compared to the reporter cell assays is likely due to diffusion of peptides on the blood agar plate.
Finally, we compared the anti-virulence activity of QQ-3 and QQ-5 with AIP-II on S. aureus cells with intact agr. We confirmed that QQ-3 and QQ-5 inhibited the agr response more potently than AIP-II in a luciferase-based reporter cell assay (Figure S14). Then, we examined the effect of QQ-3 and QQ-5 on S. aureus induced hemolysis, which is mediated by upregulated expression of hemolysins upon agr activation. Gratifyingly, both QQ-3 and QQ-5 effectively inhibited hemolysis, with QQ-3 exhibiting superior activity to AIP-II (Figure 3e).
In this study, we demonstrated the first successful application of the RaPID system to select ligands that target the orthosteric site of a membrane protein reconstituted in lipid nanodiscs. This endeavor led to the discovery of novel peptidic scaffolds that potently inhibit S. aureus quorum sensing via competitive binding to the sensor histidine kinase AgrC. Characterization of these QQ peptide ligands provided insights on their mode of interaction with AgrC, highlighting a hydrophobic dyad in the macrocycle as being a key pharmacophore. Collectively, our studies identified QQ-3 as a promising new peptide scaffold for developing group-I agr quorum quenchers. We envision that our AgrC screening platform can be adapted to identify macrocycles that potently inhibit all four agr subgroups by targeting AgrC of different subgroups in a staggered fashion in the RaPID selection cycles. Moreover, the integration of the RaPID system with the nanodisc technology should be broadly applicable to other functionally important membrane protein targets such as GPCRs and ion channels.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grant AI042783 to T.W.M., and Japan Agency for Medical Research and Development (AMED), Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research) under JP19am0101090 to H.S.. M.M.W. is an International Research Fellow of the Japanese Society for the Promotion of Science (P16810). We thank current members of the Novick, Suga and Muir laboratories for discussions and comments. Dr. Geeta Ram from Novick lab kindly provided us the group-I S. aureus RN12312 cells.
Footnotes
Conflicts of interest
There is no conflict of interest to declare.
This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication.
Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available.
You can find more information about Accepted Manuscripts in the Information for Authors.
Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains.
References
- 1.Ansari S, Jha RK, Mishra SK, Tiwari BR and Asaad AM, Infect Drug Resist, 2019, 12, 1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dickey SW, Cheung GYC and Otto M, Nat Rev Drug Discov, 2017, 16, 457–471. [DOI] [PubMed] [Google Scholar]
- 3.Wang B and Muir TW, Cell Chem Biol, 2016, 23, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ji G, Beavis R and Novick RP, Science, 1997, 276, 2027–2030. [DOI] [PubMed] [Google Scholar]
- 5.Lyon GJ, Mayville P, Muir TW and Novick RP, Proc Natl Acad Sci U S A, 2000, 97, 13330–13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyon GJ, Wright JS, Muir TW and Novick RP, Biochemistry, 2002, 41, 10095–10104. [DOI] [PubMed] [Google Scholar]
- 7.Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW and Blackwell HE, J Am Chem Soc, 2013, 135, 7869–7882. [DOI] [PubMed] [Google Scholar]
- 8.Tal-Gan Y, Ivancic M, Cornilescu G, Yang T and Blackwell HE, Angew Chem Int Ed Engl, 2016, 55, 8913–8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vasquez JK and Blackwell HE, ACS Infect Dis, 2019, 5, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hipolito CJ and Suga H, Curr Opin Chem Biol, 2012, 16, 196–203. [DOI] [PubMed] [Google Scholar]
- 11.Passioura T and Suga H, Chem Commun (Camb), 2017, 53, 1931–1940. [DOI] [PubMed] [Google Scholar]
- 12.Sakai K, Passioura T, Sato H, Ito K, Furuhashi H, Umitsu M, Takagi J, Kato Y, Mukai H, Warashina S, Zouda M, Watanabe Y, Yano S, Shibata M, Suga H and Matsumoto K, Nat Chem Biol, 2019, 15, 598–606. [DOI] [PubMed] [Google Scholar]
- 13.Nawatha M, Rogers JM, Bonn SM, Livneh I, Lemma B, Mali SM, Vamisetti GB, Sun H, Bercovich B, Huang Y, Ciechanover A, Fushman D, Suga H and Brik A, Nat Chem, 2019, 11, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleming SR, Himes PM, Ghodge SV, Goto Y, Suga H and Bowers AA, J Am Chem Soc, 2020, 142, 5024–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hipolito CJ, Tanaka Y, Katoh T, Nureki O and Suga H, Molecules, 2013, 18, 10514–10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kodan A, Yamaguchi T, Nakatsu T, Sakiyama K, Hipolito CJ, Fujioka A, Hirokane R, Ikeguchi K, Watanabe B, Hiratake J, Kimura Y, Suga H, Ueda K and Kato H, Proc Natl Acad Sci U S A, 2014, 111, 4049–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Passioura T, Watashi K, Fukano K, Shimura S, Saso W, Morishita R, Ogasawara Y, Tanaka Y, Mizokami M, Sureau C, Suga H and Wakita T, Cell Chem Biol, 2018, 25, 906–915 e905. [DOI] [PubMed] [Google Scholar]
- 18.Wang B, Zhao A, Novick RP and Muir TW, Mol Cell, 2014, 53, 929–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang B, Zhao A, Xie Q, Olinares PD, Chait BT, Novick RP and Muir TW, Cell Chem Biol, 2017, 24, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goto Y, Katoh T and Suga H, Nat Protoc, 2011, 6, 779–790. [DOI] [PubMed] [Google Scholar]
- 21.Metsalu T and Vilo J, Nucleic Acids Res, 2015, 43, W566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick RP and Muir TW, Proc Natl Acad Sci U S A, 1999, 96, 1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson JG, Wang B, Debelouchina GT, Novick RP and Muir TW, Chembiochem, 2015, 16, 1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright JS 3rd, Lyon GJ, George EA, Muir TW and Novick RP, Proc Natl Acad Sci U S A, 2004, 101, 16168–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen A, Mansson M, Bojer MS, Gram L, Larsen TO, Novick RP, Frees D, Frokiaer H and Ingmer H, PLoS One, 2014, 9, e84992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie Q, Zhao A, Jeffrey PD, Kim MK, Bassler BL, Stone HA, Novick RP and Muir TW, Cell Chem Biol, 2019, 26, 548–558.e544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.