

Abstract
Objective:
Apolipoprotein A-I binding protein (AIBP) is an effective and selective regulator of lipid rafts modulating many metabolic pathways originating from the rafts, including inflammation. The mechanism of action was suggested to involve stimulation by AIBP of cholesterol efflux, depleting rafts of cholesterol, which is essential for lipid raft integrity. Here we describe a different mechanism contributing to the regulation of lipid rafts by AIBP.
Approach and Results:
We demonstrate that modulation of rafts by AIBP may not exclusively depend on the rate of cholesterol efflux or presence of the key regulator of the efflux, ATP binding cassette transporter A-I (ABCA1). AIBP interacted with phosphatidylinositol 3-phosphate [PI(3)P], which was associated with increased abundance and activation of Cdc42 and re-arrangement of the actin cytoskeleton. Cytoskeleton re-arrangement was accompanied with reduction of the abundance of lipid rafts, without significant changes in the lipid composition of the rafts. The interaction of AIBP with PI(3)P was blocked by AIBP substrate, NADPH, and both NADPH and silencing of Cdc42 interfered with the ability of AIBP to regulate lipid rafts and cholesterol efflux.
Conclusions:
Our findings indicate that an underlying mechanism of regulation of lipid rafts by AIBP involves PIP-dependent rearrangement of the cytoskeleton.
Keywords: Lipid rafts; apolipoprotein A-I binding protein; cytoskeleton; phosphatidylinositol phosphates, Cdc42.
Subject terms: Cell Biology/Structural Biology, Lipids and Cholesterol, Mechanisms
Graphical Abstract
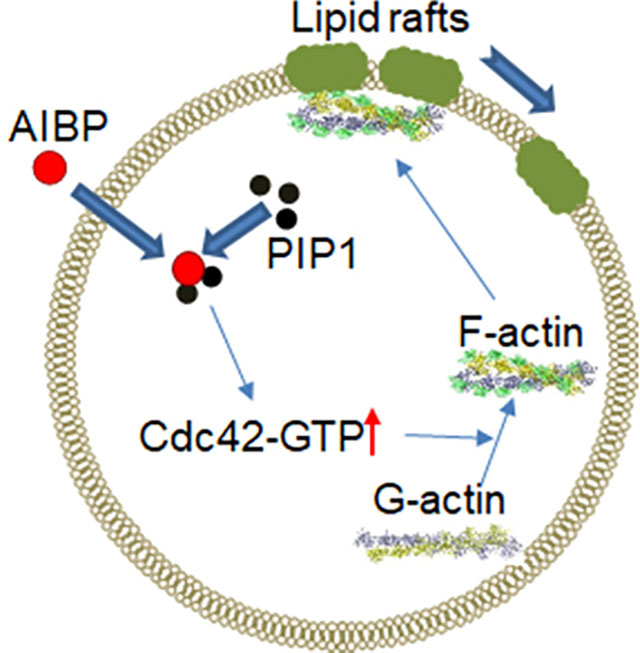
Introduction
Apolipoprotein A-I Binding Protein (AIBP) is a ubiquitously expressed protein: its expression and secretion was characterized in humans, mice and zebrafish, and homologous genes were found in organisms of various taxa1. Physiologic functions of AIBP, both intracellular and systemic, are unclear, but interest in this protein was ignited because of its ability to mitigate inflammation in a wide range of pathological conditions. Intrathecal injections of AIBP in mice were able to reduce neuroinflammation, effectively reversing established tactile allodynia2. Systemic overexpression of AIBP reduced inflammation and development of atherosclerosis in murine models of this disease3, 4. Inhalation of AIBP reduced acute lung injury in mice5. Further, AIBP was able to regulate angiogenesis6, 7 and haematopoiesis8, had an anti-cancer effects9 and reduced HIV replication in T cells and macrophages10. The mechanistic basis for such a broad range of beneficial activities is believed to be the capacity of AIBP to bind to apolipoprotein A-I (apoA-I), lipid-free or as a constituent of high density lipoprotein (HDL), stimulate cholesterol efflux and modify lipid rafts in target cells1. Lipid rafts are dynamic “ordered” domains in the generally “liquid-disordered” plasma membrane; they host many inflammatory receptors and serve as a platform for assembly and signalling of these receptors11. Properties and abundance of lipid rafts are important elements of the regulation of activity of many inflammatory receptors and, consequently, of inflammation in general12. Furthermore, lipid rafts are intimately involved in regulation of endo- and exocytosis, apoptosis, cell cycle and cell mobility. Modification or breakdown of lipid rafts by AIBP may therefore be a key contributor to the anti-inflammatory and other beneficial properties of AIBP.
According to the current hypothesis, AIBP binds to a raft-associated receptor, most likely TLR42. After binding to the rafts, AIBP binds apoA-I and/or HDL, brings them to the rafts and facilitates their interaction with ABCA1 prompting rapid and effective efflux of cholesterol from lipid rafts to apoA-I and/or HDL1. Cholesterol is an essential structural component of lipid rafts and cholesterol depletion changes functional properties and eventually results in disintegration of rafts1, 13. Increased binding of apoA-I to ABCA1 also leads to stabilization of ABCA113, the latter is capable of regulation of lipid rafts not only through potentiating cholesterol efflux, but also through changes in cytoskeleton14, 15. In this study we propose a different mechanism of regulation of lipid rafts by AIBP that does not rely on interaction with apoA-I and does not involve ABCA1 and cholesterol efflux, but instead involves interaction with phosphoinositides and rearrangement of the cytoskeleton.
Materials and Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Cells
THP-1 human monocyte cells (ATCC, Manassas, VA) were grown in RPMI1640 + Glutamax (Life Technologies) containing 10% heat inactivated foetal bovine serum (FBS; GE Healthcare) and 100 U/mL penicillin-streptomycin (Pen-Strep; Life Technologies). THP-1 cells were differentiated into macrophages by incubation with 100 ng/mL phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) for 72 hours.
Human umbilical vein endothelial cells (HUVECs; ATCC, Manassas, VA) were grown in Medium 199 (Sigma-Aldrich) containing 10% FBS, 20 mM HEPES (Life-Technologies), 100 U/mL Pen-Strep, 300 U/mL Heparin (Sigma-Aldrich) and 70 μg/mL endothelial cell growth supplement (ECGS; Bio-Rad). Prior to seeding HUVECs, cell-culture plates were coated with 0.2% gelatine.
HeLa cells (ATCC, Manassas, VA) were grown in DMEM (Life Technologies) containing 10% FBS, 100 U/mL Pen-Strep, 150 μg/ml G-418 (Gibco) and 200 μg/ml hygromycin B (Life Technologies). HeLa cells stably expressing GFP-tagged ABCA1 or ABCG1 were a kind gift of Dr. Alan Remaley and were described previously16.
Human skin fibroblasts, normal and from donor with Tangier disease, were a kind gift from Dr. Alan Remaley. Fibroblasts were grown in DMEM containing 10% FBS and 100 U/mL Pen-Strep.
SH-SY5Y human neuroblastoma cells (ATCC, Manassas, VA) were grown in Dulbecco’s modified Eagle’s/F12 medium (DMEM/F12; Life Technologies) supplemented with 10% FBS. Prior to seeding SH-SY5Y cells, cell-culture plates were with collagen-coated. SH-SY5Y cells were differentiated for five days with complete media supplemented with 10 μM retinoic acid (Sigma-Aldrich), followed by a secondary differentiation for two days with serum-free DMEM/F12 medium with 5 ng/ml brain derived neurotrophic factor (BDNF) (Abcam). Differentiated cells were subsequently maintained in serum-free BDNF-containing media.
BV-2 murine microglial cells (ATCC, Manassas, VA) were grown in high-glucose DMEM (Gibco) containing 5% FBS and 100 U/mL Pen-Strep. All cells were grown in a humidified 37°C, 5% CO2 incubator.
Treatments
Recombinant His-tagged AIBP was from Sino Biological or custom ordered from Selvita, Inc. AIBP was added to cells in serum-free medium at concentration 0.4 μg/ml unless a different concentration is indicated. For cholesterol efflux experiments, AIBP was co-incubated with cholesterol acceptors for 2–24 h. When cells were treated with lipopolysaccharide (LPS; Sigma-Aldrich), it was added at the final concentration of 1 μg/ml for 6 hours prior to adding AIBP and was included into incubation with AIBP, unless indicated otherwise. For cholesterol efflux experiments, LPS was added during serum free as well as cholesterol efflux incubations. To eliminate lipid rafts, cells were treated with 5 mM methyl-β-cyclodextrin (MβCD; Sigma-Aldrich) for 15 minutes prior to AIBP treatment or incubation with acceptors for cholesterol efflux experiments. To determine if NADPH blocks AIBP’s ability to increase cholesterol efflux, indicated concentrations of NADPH were co-incubated with cholesterol acceptors and cells in the presence or absence of AIBP. To test the effect of wortmannin, it was added in cell culture medium with 5% FBS at a final concentration of 200 nM 30 min prior to incubating cells with LPS (15 min, 100 ng/ml).
Cholesterol efflux
Cholesterol efflux was measured as described previously17. Briefly, cells were labelled by incubation in serum-containing medium supplemented with [3H]-cholesterol (75 kBq/ml, American Radiolabeled Chemicals) for 72 hours. Cells were washed and incubated for 6 hours in serum-free media. For 2 and 4 hour efflux experiments, human apoA-I (kind gift from CSL Behring) was added to the final concentration of 30 μg/ml unless stated otherwise. For 24 hour and time-course cholesterol efflux experiments, apoA-I, HDL or 5A mimetic peptide were added to the final concentration of 10 μg/ml, 20 μg/ml or 20 μg/ml respectively. Specific cholesterol efflux was calculated as a proportion of radioactivity moved from cells to medium; non-specific efflux (i.e. the efflux to the medium without acceptor) was subtracted. When release of cholesterol with extracellular vesicles (EV) and nascent lipoproteins was investigated, cells were activated and labelled as described above, and incubated for 24 h in serum-free medium in the presence or absence of 0.4 μg/ml AIBP in absence of an acceptor. After incubation cell culture medium was centrifuged for 5 min at 200 × g to remove cell debris and then for 90 min at 60,000 × g using TLA-120.2 rotor to pellet EVs. The radioactivity remaining in the supernatant after centrifugation was designated as “nascent lipoprotein” fraction.
Western blotting
Cultured cells were lysed in RIPA buffer containing protease/phosphatase inhibitor cocktails (Roche) prior to SDS-PAGE. Lysates or lipid raft fractions were separated by SDS-PAGE, transferred to PVDF or nitrocellulose membrane and following a block in 4% skim milk probed with antibodies listed below. Images were taken and quantitated using GBox (Syngene).
Confocal microscopy
Cells were grown in μ-slide 8 well chamber slides (ibidi). The abundance of lipid rafts was assessed using Vybrant lipid raft labelling kit (Life Technologies) according to manufacturer’s protocol. Briefly, cells were incubated with AlexaFluor 488-conjugated CT-B (Life Technologies) for 10 min, washed and cross-linked by incubation with anti-CTB antibody for 10 min at 4°C, then fixed with 4% paraformaldehyde for 10 minutes at room temperature.
F-actin was visualized with LifeAct-TagGFP2 Protein (ibidi) according to manufacturer’s protocol. Briefly, cells were fixed with 4% paraformaldehyde for 10 minutes at room temperature, washed with PBS, permeabilised with 0.1% Triton X-100 for 5 minutes and then washed again with PBS. Cells were incubated with LifeAct-TagGFP2 Protein 1 hour and imaged.
Membrane fluidity was measured with di-4-ANEPPDHQ (Life Technologies) as described18. Briefly, cells were washed and incubated with di-4-ANEPPDHQ for 15 minutes. Cells were then washed with PBS and imaged on confocal by exciting di-4-ANEPPDHQ at 488nm. Two-channel image acquisition was performed at 595±25 nm (ordered phase) and at 700 (Long Pass) nm (disordered phase). Focussing on the cellular membrane, results are then expressed as a ratio of ordered: disordered phase.
Images were captured using a Nikon A1r+ confocal microscope equipped with 60x Water Immersion objective (Nikon 60x or 40x Plan Apo VC, WI NA 1.2) and were analysed using Fiji software.
In the experiments detecting intracellular AIBP colocalization with early endosomes and testing the effects of PI3K inhibition, cells were fixed with 4% buffered PFA (10 min), permeabilised by 1% Triton X-100 for 20 min before staining with anti-AIBP (1:250 v/v) or anti-EEA1 (1:250 v/v, Abcam) for 2 h, followed by a 2 h incubation with AlexaFluor-labelled secondary antibodies (1:750 v/v).
In the experiments detecting colocalization of internalized AIBP, cells were stained with anti-His, anti-EEA1, Anti-LAMP1 (Abcam), ER-Tracker™ Blue-White DPX (ThermoFisher), MitoTracker™ Deep Red FM (ThermoFisher), Alexa Fluor™ 635 Phalloidin (ThermoFisher). Images were captured with a Leica SP8 confocal microscope with LIGHTING super-resolution, and individual z-sections were analysed for colocalizations using Imaris software.
Protein expression silencing
THP-1 cells were silenced using siRNAscram (Origene) or siRNACdc42 (Origene) using INTERFERin (Polyplus Transfections) according to manufacturer’s protocol with minor modifications. Briefly, cells were passaged to 0.5 × 106 cells/ml a day before transfection. Cells were then seeded in antibiotic-free RPMI-1640 containing 10% FBS and co-incubated with INTERFERin/siRNA complex for 6 hours. Complete RPMI-1640 was added to cells and left to incubate for 24 hours. Cells were then differentiated by adding PMA to a 100 ng/ml final concentration.
Cdc42 activity
Cdc42 activation pull down assay (Cytoskeleton) was performed according to the manufacturer’s protocol with minor modifications. Briefly, THP-1 cells were seeded at 40% confluency and treated as indicated. Cells were then washed with PBS and activated with 100 ng/ml bradykinin (Sigma-Aldrich) in serum-free RPMI for 5 minutes at 37°C. Cells were washed with PBS, lysed and the supernatant was snap frozen. 400 μg of total protein was incubated with Sepharose beads with affinity to activated Cdc42. Beads were washed, incubated with Laemmli buffer and boiled for 2 minutes then separated with spin columns. Eluates were finally subjected to SDS-PAGE probing for Cdc42.
Lipid raft isolation
Lipid raft isolation analysis was performed as described previously19. Briefly, hypo-osmotic shock and ultracentrifugation was performed on treated cells to obtain the membrane fraction. The fraction was then mixed with iodixanol to a 23% final concentration and subjected to two rounds of gradient ultracentrifugation. Sixteen fractions were collected and analysed by immunoblotting for ABCA1 and flotillin 1, or by MS (lipidomics, see below). In some experiment cellular cholesterol was trace-labeled using protocol described for cholesterol efflux and the radioactivity in fractions were analysed on a liquid scintillation counter.
Lipidomics
Lipidomic analysis was performed as described previously20. Lipid raft fractions were combined and sonicated on ice; lipids were extracted using a, single phase CHCl3:CH4OH method. The analysis was performed by liquid chromatography electrospray ionization-tandem mass spectrometry (LC ESI-MS/MS) using an Agilent 1290 liquid chromatography system (Agilent Technologies), Agilent 6490 Triple Quadrapole Mass Spectrometer and Analyst 1.5 and MultiQuant data systems (AB SCIEX). Lipid concentrations were calculated by relating the peak area of each species to the peak area of the corresponding internal standard.
Apoptosis
Apoptosis was assessed as reported previously20 using Cell Death Detection ELISA kit determining cytoplasmic histone-associated DNA fragments (TUNEL assay). Positive control (a DNA-histone complex) was supplied by the manufacturer, negative control was untreated THP-1 cells.
Interaction with PIP
Interaction between AIBP and PIP was assessed using PIP strips (Echelon Biosciences) according to manufacturer’s protocol. Briefly, PIP strip was blocked with blocking buffer (TBS containing 0.1% Tween-20 and 3% fatty acid free BSA (Sigma-Aldrich)) followed by an overnight incubation with 0.5 μg/ml AIBP at 4°C. Membrane was washed and probed for AIBP. To determine if NADPH blocks AIBP binding to PIP, AIBP was pre-incubated with 1.44 mM β-NADPH (Sigma-Aldrich) for 10 minutes at room temperature prior to incubation with PIP strip.
Differential scanning fluorimetry
AIBP or lysozyme (Sigma) at a final concentration of 50 μg/ml in PBS (without Ca++ or Mg++) was mixed with SYPRO® Orange Protein Gel Stain (Sigma), with or without addition of NADPH (Cayman, final concentration 10 mM,). Melting temperature was determined using Rotor-Gene Q machine from Qiagen and calculated using Rotor-Gene Q series software.
Statistical Analysis
Data is shown as mean ± SD unless otherwise stated. GraphPad Prism 8 and RStudio software were used to examine the data for statistical significance between groups. ANOVA was used to assess the presence of statistically significant differences between the means of three or more independent groups, in all cases data followed a normal distribution. Post-hoc Bonferroni correction was performed for multiple group comparisons. Otherwise, Student’s t-test was used when data followed a normal distribution, alternatively Mann-Whitney test on ranks was used. Experiments were conducted at a minimum of triplicates and repeated 3–4 times. A p-value of < 0.05 was considered statistically significant.
Results
AIBP and cellular cholesterol metabolism
In this study, we investigated the effects of AIBP on cholesterol metabolism and lipid rafts. Consistent with the previous report13, incubation of differentiated THP-1 macrophages with a mixture of AIBP with apoA-I or HDL over 24 h promoted cholesterol efflux (Fig. 1 A). The rate of specific cholesterol efflux to apoA-I or HDL was modestly, but statistically significantly higher in the presence of AIBP compared to the efflux to the same acceptor in the absence of AIBP; AIBP alone did not support cholesterol efflux. Time-course of cholesterol efflux is shown in Fig. 1 B revealing that the stimulation of cholesterol efflux by AIBP is not apparent at early time-points and requires at least 24 h exposure to AIBP to manifest. Cells in these experiments were pre-treated with LPS to mimic an inflammatory state, AIBP did not stimulate cholesterol efflux in cells not pre-treated with LPS (Fig. 1 C). We also tested the effects of AIBP on cholesterol efflux in two other cell types, human umbilical cord vascular endothelial cells (HUVEC) and human neuroblastoma cells SH-SY5Y; AIBP stimulated cholesterol efflux from both these cell types (Fig. 1 D). These findings confirm the previous reports on stimulation of cholesterol efflux by AIBP5, 6, 8, 13 and collectively suggest that the modulation of cholesterol efflux by AIBP most likely results from the effects on cellular mechanisms, rather than from changes in the properties of cholesterol acceptor.
Figure 1. The effect of AIBP on cholesterol efflux and apoptosis.
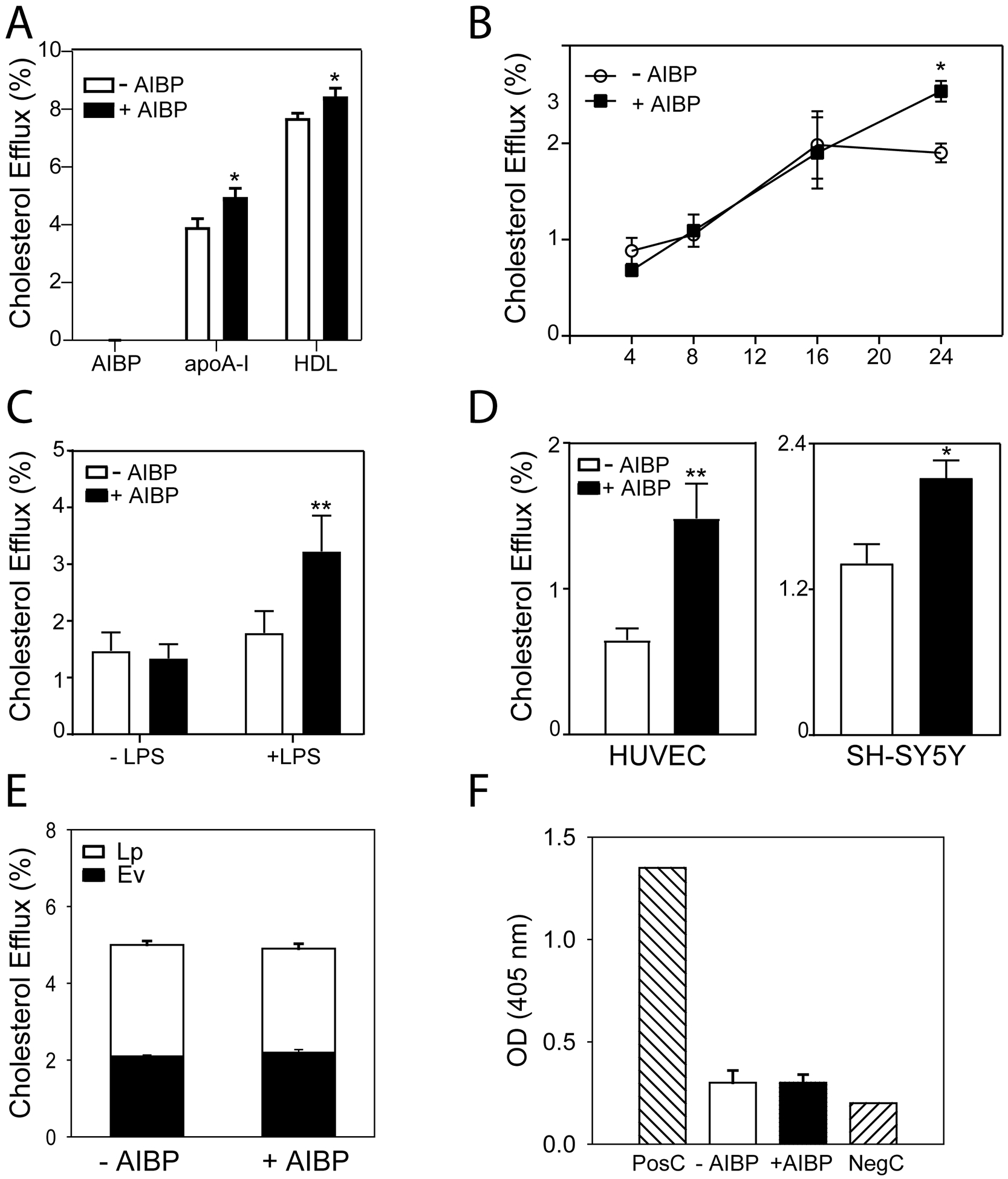
A – Specific cholesterol efflux from THP-1 macrophages over 24 h to AIBP alone (0.4 μg/ml) or to apoA-I (10 μg/ml) or HDL (20 μg/ml) in the presence or absence of AIBP (0.4 μg/ml). Mean ± SD are shown; *p<0.05 versus no AIBP, n=4. B – Time course of specific cholesterol efflux from THP-1 macrophages to apoA-I (10 μg/ml) in the presence or absence of AIBP (0.4 μg/ml). Mean ± SD are shown; *p<0.05 versus no AIBP, n=4. C – The effect of LPS (1 μg/ml, 6 h) on specific cholesterol efflux from THP-1 macrophages to apoA-I (10 μg/ml) in the presence or absence of AIBP (0.4 μg/ml) over 24 h. Mean ± SD are shown; **p<0.01 versus no AIBP, n=4. D – Specific cholesterol efflux from HUVEC (2 h) or SH-SY5Y human neural cells (24 h) to apoA-I alone or a mixture of AIBP (0.4 μg/ml) with apoA-I (30 μg/ml for HUVEC and 10 μg/ml for other cells). Mean ± SD are shown; *p<0.05, **p<0.01; n=4. E – Total cholesterol efflux from THP-1 macrophages over 24 h in the presence or absence of AIBP (0.4 μg/ml). After efflux incubation, extracellular vesicles (EV) were isolated from cell culture media by ultracentrifugation, the reminder of the radioactivity in the medium was designated as “Lipoprotein” fraction (Lp). Mean ± SD are shown; n=4. F – Apoptosis in THP-1 macrophages after incubation over 24 h with AIBP (0.4 μg/ml). PosC – positive control, NegC – negative control. Mean ± SD are shown; n=4.
In addition to cholesterol efflux, macrophages may release cholesterol with extracellular vesicles (EV) and nascent apoE-containing lipoproteins. We examined the effects of AIBP on these pathways by incubating THP-1 cells for 24 h with or without AIBP in the absence of any cholesterol acceptors. After the incubation, EV were precipitated by ultracentrifugation, the remainder of the labelled cholesterol released from cells was designated as “nascent lipoprotein” fraction. Cholesterol export with neither EV nor nascent lipoproteins was affected by AIBP (Fig. 1 E). Further, we tested if AIBP at the same conditions can cause apoptosis in THP-1 cells; no apoptosis in the presence or absence of AIBP was detected (Fig. 1 F).
It was previously suggested that one consequence of enhanced cholesterol efflux due to action of AIBP is reduction in the abundance of lipid rafts2, 3, 6. Indeed, incubation of THP-1 macrophages with AIBP/apoA-I complex at conditions when AIBP stimulated cholesterol efflux to apoA-I led to a significant reduction in the abundance of lipid rafts (Fig. 2 A,B). Unexpectedly, however, AIBP alone also reduced the abundance of lipid rafts (Fig. 2 A,B) despite lack of cholesterol efflux in the absence of apoA-I or HDL (Fig. 1 A). Furthermore, the effect of AIBP and AIBP/apoA-I on lipid rafts was evident after 4 h exposure (Fig. 2 C) when no effect of AIBP on the efflux was detected (Fig. 1 B). ApoA-I in the absence of AIBP also reduced the abundance of lipid rafts after 4 h, but the effect of apoA-I alone was not seen after 24 h (when cholesterol efflux presumably is saturated) (Fig. 2 B,C). These findings are inconsistent with a hypothesis that reduction of the abundance of lipid rafts by AIBP is a consequence of stimulation of cholesterol efflux. Rather, they argue that AIBP may have an intrinsic capacity to modify lipid rafts and elevation of cholesterol efflux may be a consequence of modification of lipid rafts. This suggestion was supported by experiments where rafts were eliminated by treatment of cells with MβCD prior to treatment with AIBP. We used the previously described conditions where MβCD breaks down the rafts, but does not affect cellular cholesterol content21, and under these conditions elimination of rafts prevented the effect of AIBP on cholesterol efflux (Fig. 2 D). Thus, it appears that AIBP does not require apoA-I or cholesterol efflux to rapidly exert its effect on lipid rafts.
Figure 2. The effect of AIBP on the abundance of lipid rafts.
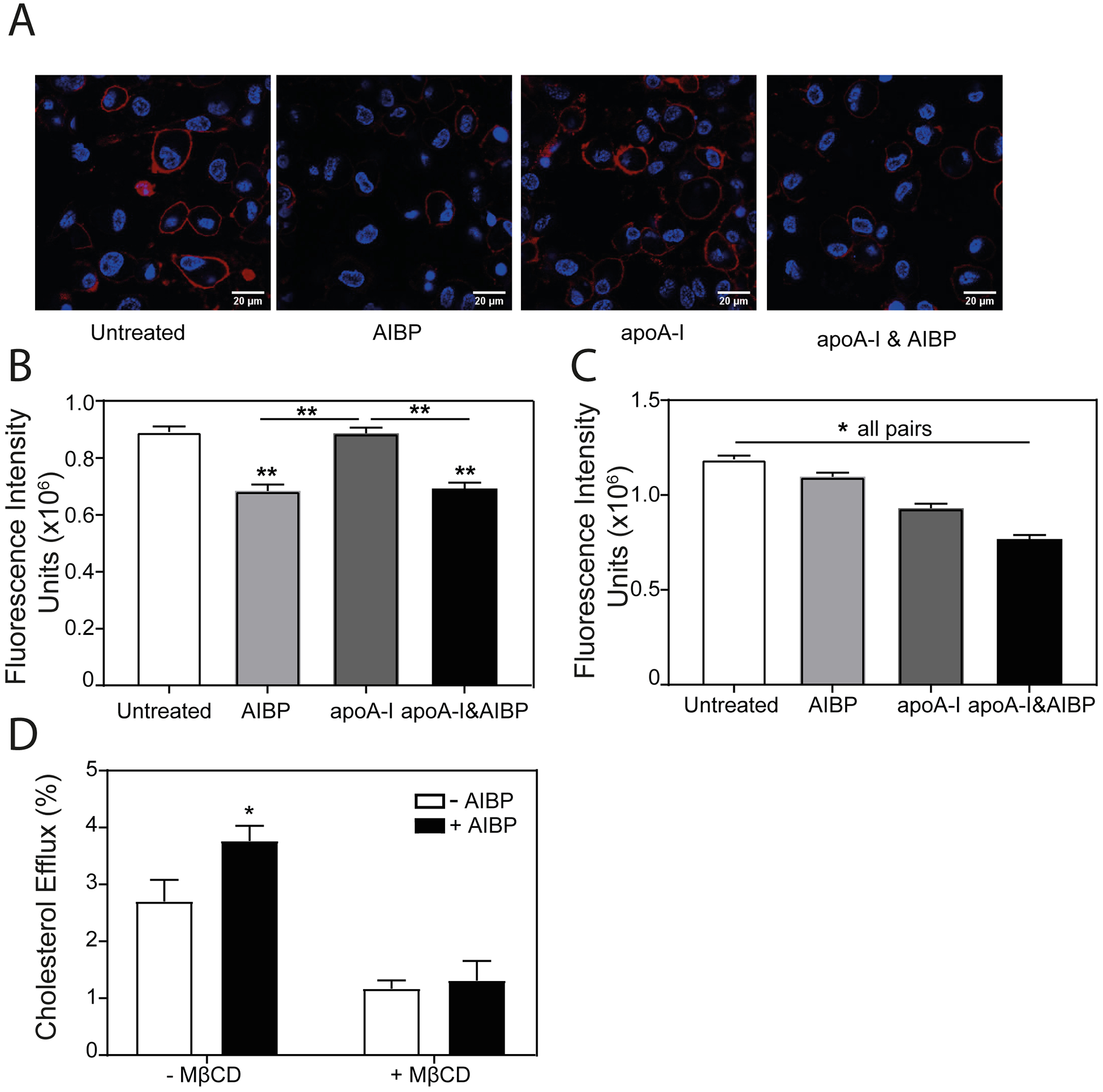
A – The effect of AIBP (0.4 μg/ml), apoA-I (10 μg/ml) or their mixture (24 h incubation) on the abundance of lipid rafts in THP-1 cells (cholera toxin subunit B (CTB) staining, confocal microscopy, red - CTB, blue - DAPI). Scale bar – 20 μm. B – Quantitation of the effect of AIBP, apoA-I or their mixture (24 h incubation) on the abundance of lipid rafts in THP-1 cells by confocal microscopy (CTB staining, mean fluorescence intensity). Mean ± SEM are shown, **p<0.01, n=6. C – Quantitation of the effect of AIBP, apoA-I or their mixture (4 h incubation) on the abundance of lipid rafts in THP-1 cells by confocal microscopy (CTB staining, mean fluorescence intensity). Mean ± SEM are shown, **p<0.01, n=6. D – Specific cholesterol efflux from THP-1 cells over 24 h to apoA-I (10 μg/ml) in the presence or absence of AIBP (0.4 μg/ml) with or without pre-treatment with MβCD (5 mM, 15 min). Mean ± SD are shown; *p<0.05, n=4.
ABCA1 is not involved in AIBP-mediated modulation of lipid rafts
ABCA1 is a key element of cellular cholesterol efflux machinery and a key regulator of lipid rafts, exerting its raft-regulatory capacity both through and independently of cholesterol efflux. To further investigate the involvement of ABCA1 in the regulation of lipid rafts by AIBP, we investigated its effect on the abundance of lipid rafts in skin fibroblasts from a patient with Tangier disease (familial ABCA1 deficiency), cells completely lacking ABCA122, and compared them with normal skin fibroblasts. When normal and Tangier fibroblasts were incubated with AIBP in the absence of apoA-I or HDL for 2 h or 24 h, in both instances AIBP caused a significant reduction in the abundance of lipid rafts (Fig. 3 A,B). We next tested the effect of AIBP on HeLa cells, a human cell line not expressing ABC transporters, and compared it with the effects on the HeLa cells stably transfected with human ABCA1 (HeLa/ABCA1)23, 24. Again, incubation with AIBP for 2 or 24 h caused a reduction in the lipid rafts abundance (Fig. 3 C,D) in ABCA1-deficient HeLa cells. Conversely, AIBP had no effect on the abundance of lipid rafts in HeLa/ABCA1 cells (Fig. 3 D). It is important to recognize that ABCA1 in HeLa/ABCA1 cells is expressed under CMV promoter resulting in very high, possibly saturating, levels of ABCA1 - a likely explanation of a discrepancy between the effects of AIBP on HeLa/ABCA1 cells and skin fibroblasts expressing physiological levels of ABCA1. As expected, no specific cholesterol efflux from HeLa cells to apoA-I was detected in the presence or absence of AIBP (Fig. 3 E). There was considerable cholesterol efflux to apoA-I from HeLa/ABCA1 cells, however the efflux was not affected by AIBP (Fig. 3 E). There was minimal cholesterol efflux to apoA-I from HeLa cells stably transfected with ABCG1 (HeLa/ABCG1), and AIBP also didn’t affect this efflux (Fig. 3 E). There was significant cholesterol efflux from HeLa-ABCG1 to HDL and AIBP reduced this efflux (Fig. 3 E), possibly by shifting HDL binding to lipid rafts. Thus, the effect of AIBP on the abundance of lipid rafts was not dependent on the presence of ABCA1 and was not determined by the rate of cholesterol efflux making it unlikely that ABCA1 is mechanistically involved in the action of AIBP.
Figure 3. ABCA1 is not involved in the action of AIBP.
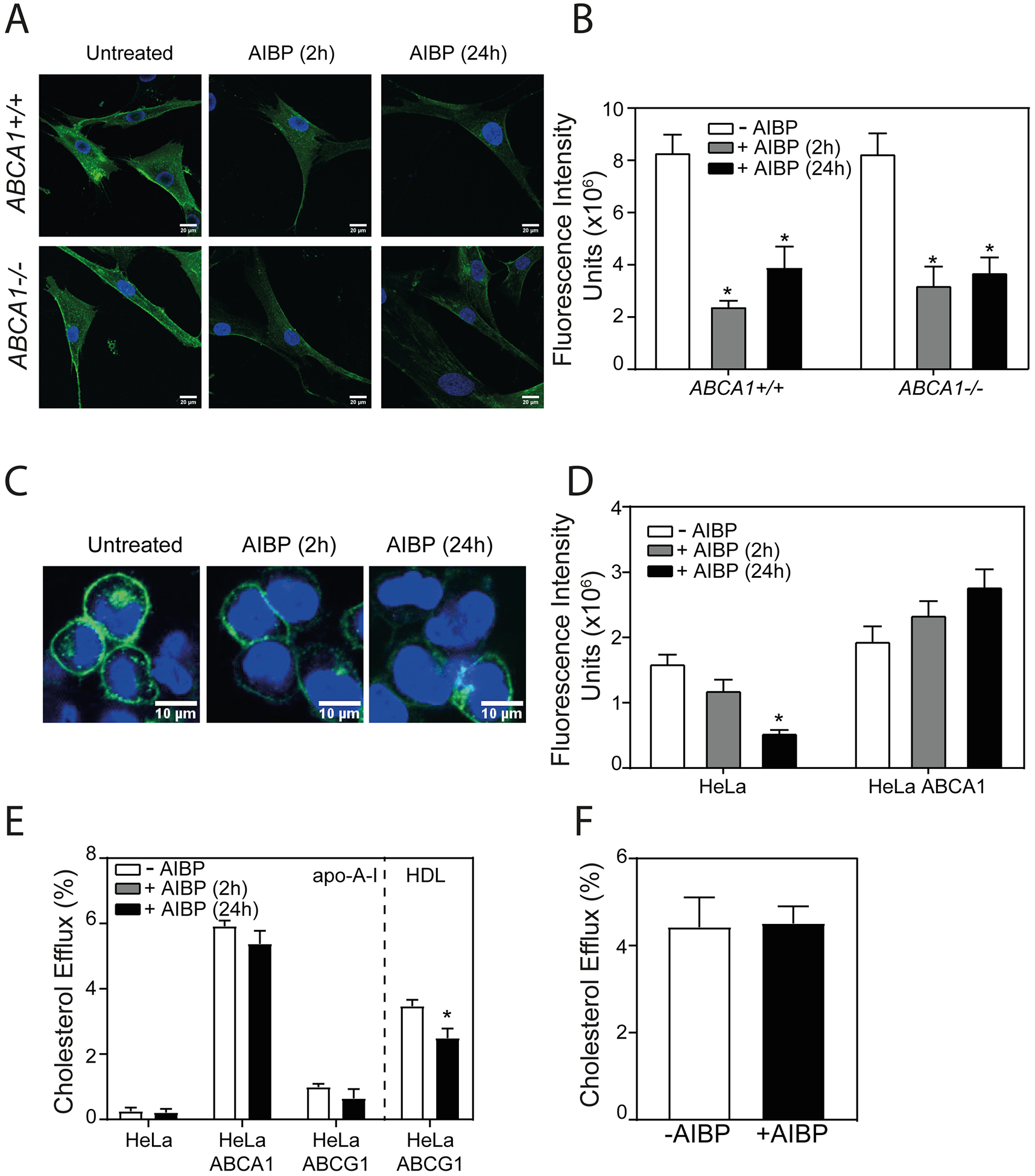
A – The effect of AIBP (0.4 μg/ml) (2 h and 24 h incubation) on the abundance of lipid rafts in human skin fibroblasts from normal subject (ABCA1+/+) and from subject with Tangier disease (ABCA1−/−) (cholera toxin subunit B (CTB) staining, confocal microscopy). Scale bar – 20 μm. B – Quantitation of the effect of AIBP (2 h and 24 h incubation) on the abundance of lipid rafts in human skin fibroblasts from normal subject (ABCA1+/+) and from subject with Tangier disease (ABCA1−/−) by confocal microscopy (CTB staining, mean fluorescence intensity). Mean ± SEM are shown, *p<0.05 versus no AIBP, n=6. C – The effect of AIBP (0.4 μg/ml) (2 and 24 h incubation) on the abundance of lipid rafts in HeLa cells (cholera toxin subunit B (CTB) staining, confocal microscopy). Scale bar – 10 μm. D – Quantitation of the effect of AIBP (2 and 24 h incubation) on the abundance of lipid rafts in HeLa and HeLa/ABCA1 cells by confocal microscopy (CTB staining, mean fluorescence intensity). Mean ± SEM are shown, *p<0.05 versus no AIBP, n=6. E – Specific cholesterol efflux from HeLa, HeLa/ABCA1 and HeLa/ABCG1 cells to apoA-I or HDL (10 μg/ml, in the presence or absence of AIBP (0.4 μg/ml), 24 h incubation. Mean ± SD are shown; *p<0.05 versus no AIBP, n=4. F – Specific cholesterol efflux from THP-1 cells to 5A peptide (20 μg/ml, 24 h) in the presence or absence of AIBP (0.4 μg/ml) Mean ± SD are shown; n=4.
Finally, we tested the effect of AIBP on cholesterol efflux to apoA-I mimetic peptide 5A, a peptide supporting ABCA1-dependent cholesterol efflux25. AIBP failed to stimulate cholesterol efflux to 5A (Fig. 3 F). This finding suggests that the presence of an acceptor capable of supporting ABCA1-dependent cholesterol efflux is not sufficient for the cholesterol efflux stimulating activity of AIBP. It should be noted, however, that primary structure of 5A has no homology with that of apoA-I and 5A most likely does not bind to AIBP, which may be an alternative explanation for the lack of the effect of AIBP.
AIBP modifies lipid rafts
To investigate how AIBP modifies lipid rafts, we assessed the effect of AIBP on fluidity of the plasma membrane using staining of cells with di-4-ANEPPDHQ26. Treatment of THP-1 cells with AIBP for 2 h or 24 h as well as treatment with MβCD shifted the emission spectrum from emission wavelength band peaking at 560 nm (liquid ordered state (Lo)) to that peaking at 620 nm (Liquid disordered state (Ld)) indicating increased fluidity of the plasma membrane, consistent with reduced abundance of lipid rafts (Supplemental Fig. I, A).
Next, we tested the effects of AIBP on the properties of isolated lipid rafts. THP-1 cells were labelled with [3H]cholesterol tracer and lipid rafts were isolated using a two-step continuous gradient centrifugation (see Materials and Methods). Supplemental Fig. I, B–C presents the analysis of rafts in untreated cells versus cells treated with AIBP showing distribution of [3H]cholesterol (Supplemental Fig. I, B) and of a lipid raft marker, flotillin-1, (Supplemental Fig. I, C) after final centrifugation. Based on distribution of [3H]cholesterol and raft markers in untreated cells, we designated fractions 2–9 as lipid raft-enriched fractions (Supplemental Fig. I, B). When compared to untreated cells, treatment with AIBP resulted in a dramatic reduction in the abundance of lipid rafts on plasma membrane as evidenced by distribution of [3H]cholesterol and flotillin-1; the total amount of [3H]cholesterol in the lipid raft fractions was almost halved compared to control (1.4 × 104 dpm versus 2.6 × 104 dpm).
To investigate if lipid composition of the rafts is affected by AIBP, we conducted a lipidomics analysis of isolated lipid raft fractions. The full lipidomics analysis is presented in the Supplemental Table 1 and the relative abundance of the most relevant lipid species is shown in Supplemental Fig. II, A. There was little change in relative abundance of major lipid species after treatment of cells with AIBP, apoA-I or AIBP/apoA-I (Supplemental Fig. II, A); absolute abundancies of major lipid species in the isolated rafts followed the effect on lipid raft abundance (Supplemental Table 1). This finding is consistent with a suggestion that AIBP is primarily affecting the abundance of lipid rafts rather than their lipid composition. There was no reduction of relative abundance of cholesterol (Supplemental Fig. II, A) or redistribution of cholesterol to the non-raft fractions (Supplemental Fig. II, B), as would be expected if ABCA1 and cholesterol efflux were the driving force of changes in lipid raft abundance. Interestingly, lipid rafts of THP-1 cells contained an unusually high proportion of phosphatidylserine (PS, Supplemental Fig. II A)), which might be indicative of apoptosis27, however no apoptosis was detected in THP-1 cells under conditional used in this study (Fig. 1 F). Most likely, high PS concentration is a reflection of a redistribution of PS to the cell surface characteristic for monocyte differentiation and initiation of phagocytosis28.
Mechanism of the effect of AIBP on lipid rafts
Two mechanisms could be potentially responsible for modulation of the lipid raft abundance: raft depletion of cholesterol and/or sphingomyelin or rearrangement of the cytoskeleton. The findings presented above argue against the involvement of the former mechanism, at least under the conditions of our experiments, leaving the latter as a likely alternative. To investigate this mechanism, we used confocal microscopy to assess the effect of AIBP on actin polymerization. The abundance of F-actin in THP-1 cells increased when cells were treated with AIBP (Fig. 4 A,B). The increase of F-actin was most notable in cell filopodia and higher magnification revealed the presence of microfilament-like structures in cells treated with AIBP, but not in untreated cells (Fig. 4 A, right sub-panels).
Figure 4. Role of cytoskeletal rearrangement and Cdc42 activity in the effect of AIBP on lipid rafts.
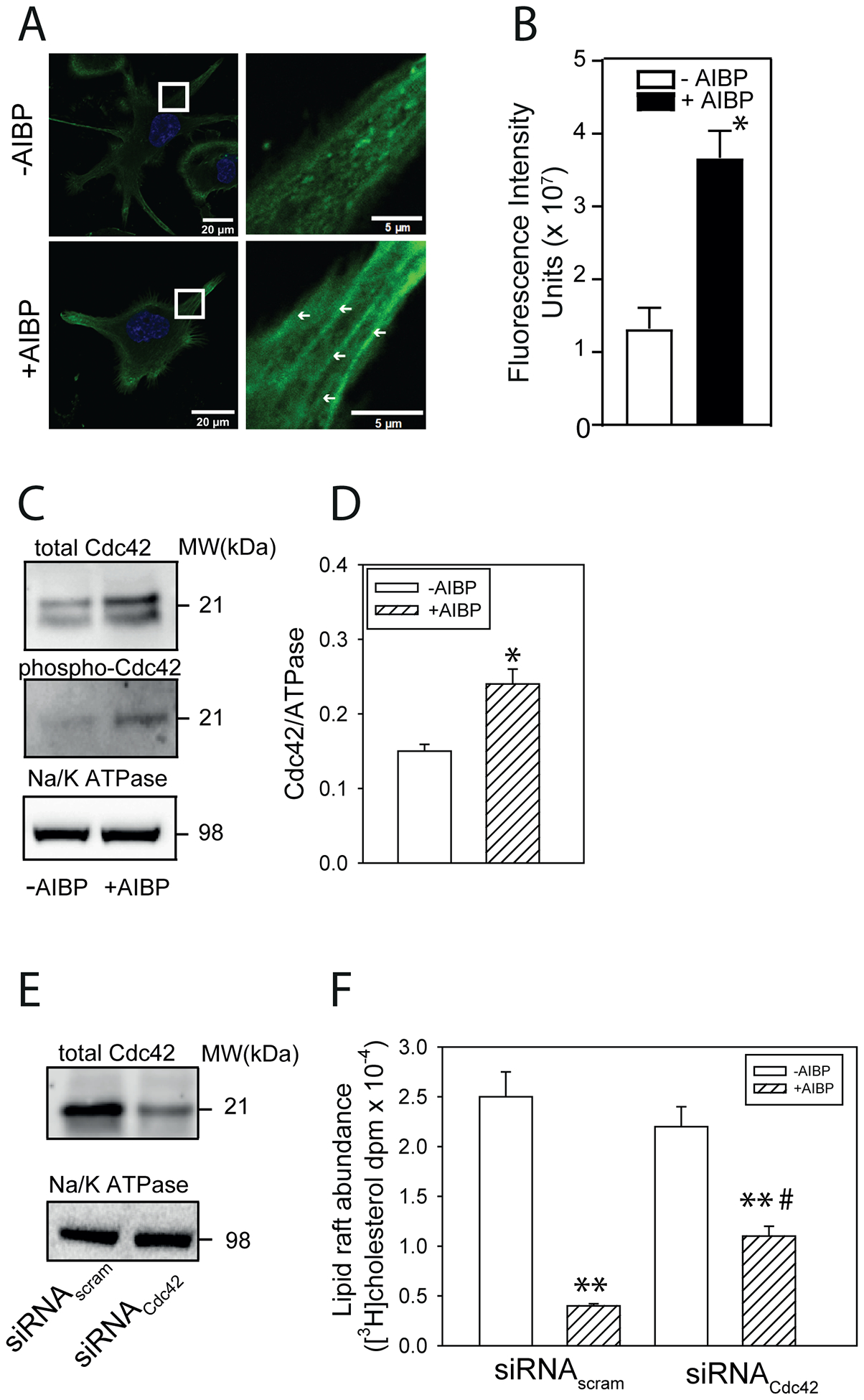
A – The effect of AIBP (0.4 μg/ml, 2 h) on the abundance of F-actin in THP-1 cells. Panels on the right show higher magnification of the designated area from the left panels, arrows point to the microfilament-like F-actin structures (LifeAct staining, confocal microscopy). Scale bars left panels – 20 μm, right panels - 5 μm. B – Quantitation of the effect of AIBP (0.4 μg/ml, 2 h) on the abundance of F-actin by confocal microscopy (LifeAct staining, mean fluorescence intensity). Mean ± SEM are shown, *p<0.05, n=6. C - The effect of AIBP (0.4 μg/ml, 4 h) on the abundance of total CdC42 and phosphorylated Cdc42 in THP-1 cells (Western blot). D – Quantitation of the effect of AIBP on the abundance of total CdC42 in THP-1 cells (densitometry of Western blot). *p<0.05, n=5. E - The abundance of total CdC42 after treatment of THP-1 cells with siRNACdc42 (Western blot). F – The abundance of [3H]cholesterol in lipid raft fractions after isolation of rafts by sequential gradient centrifugation from THP-1 cells untreated or treated with AIBP (0.4 μg/ml, 4 h) with or without Cdc42 silencing. Mean ± SEM are shown, **p<0.01 versus no AIBP, #p<0.05 versus siRNAscram; n=3.
A key element connecting rearrangement of cytoskeleton and lipid metabolism is small GTPase Cdc4229; activation (phosphorylation) of Cdc42 leads to actin polymerization30 and disruption of lipid rafts in macrophages19. We tested the effect of AIBP on Cdc42 abundance and activity and found that the abundance of both total and phosphorylated Cdc42 increased after treatment with AIBP (Fig. 4 C,D). Silencing of Cdc42 with siRNA reduced the abundance of Cdc42 by approximately 80% (Fig. 4 E) and partially reversed AIBP-mediated reduction of the abundance of rafts (Fig. 4 F).
To further elucidate the mechanism of activation of Cdc42 by AIBP, we investigated the interaction of AIBP with phosphatidylinositol phosphates (PIPs), known regulators of Cdc42 activity and actin assembly31. While we detected no binding of AIBP to an established regulator of actin polymerisation, PI(4,5)P2, there was a strong binding of AIBP to another essential component of actin polymerization machinery, PI(3)P, and, to a lesser degree, to PI(4)P (Fig. 5 A).
Figure 5. The role of PIPs in the effect of AIBP on lipid rafts.

A – Binding of AIBP to PIP strips. B – The effect of NADPH (1.44 mM) on binding of AIBP to PIP strips. C - The effect of NADPH on binding of anti-PIP2 antibody to PIP strips. D – The effect of wortmannin (200 nM, 30 min pre-treatment) on co-localization of AIBP and early endosomes in BV2 microglia stimulated with LPS (100 ng/ml, 15 min). E –Quantitation of effect of wortmannin on co-localization of AIBP and early endosomes. Manders’ tM1 coefficient, Mean ± SD are shown; ***p<0.001, n=8. F - The effect of NADPH (10 mM) on melting temperature of AIBP or lysozyme (50 μg/ml) determined using Rotor-Gene Q machine G – Dose-dependence of the effect of NADPH on cholesterol efflux from THP-1 macrophages to apoA-I (10 μg/ml, 24 h) in the presence or absence of AIBP (0.4 μg/ml). Mean ± SD are shown versus no AIBP; ***p<0.001, n=4. H – PIP1 content in lipid rafts isolated from THP-1 cells untreated or treated with AIBP (0.4 μg/ml), apoA-I (10 μg/ml) or their mixture (24 h incubation). Mean ± SD are shown; *p<0.05, n=3.
Although presence of PI(3)P on plasma membrane has been recently demonstrated32, 33, most of the published data point to a predominant localization of PI(3)P in early endosomes34. To test the interaction of AIBP with PI(3)P in a cellular system, we studied co-localization of endogenous AIBP with early endosomes in BV2 microglial cells in which endocytosis was stimulated with LPS. We found considerable co-localization of AIBP with the marker of early endosomes EEA1, which was significantly reduced after treatment of cells with wortmannin, a broad spectrum PI3 kinase inhibitor that reduces the PI(3)P abundance (Fig. 5 D,E). These findings point to a possibility that AIBP interacts with PI(3)P in early endosomes.
AIBP crystal structure demonstrates the existence of a binding pocket for nucleotide ligands, and the protein was suggested to have an NAD(P)H-hydrate epimerase or an ADP-ribosyl transferase activity35, 36. Therefore, we hypothesized that NADPH could impair binding of AIBP to PI(3)P either directly or by inflicting changes to the AIBP tertiary structure. Addition of NADPH changed AIBP melting temperature (Tm) without affecting Tm of lysozyme, a protein that does not interact with NADPH, suggesting that NADPH affects the tertiary structure of AIBP (Fig. 5 F). Excess of NADPH completely blocked binding of AIBP to PI(3)P and PI(4)P (Fig. 5 A,B). As an additional control and to ensure that NADPH does not interfere with the antibody binding, PIP2 strip was preincubated with NADPH prior to adding the anti-PIP2 antibody, NADPH did not block this binding (Fig. 5 C). Furthermore, NADPH at minimal tested concentration of 0.24 mM also completely blocked the effect of AIBP on cholesterol efflux from macrophages (Fig. 5 G). These results suggest that either NADPH and PI(3)P compete for the same binding site in AIBP, or there is an allosteric effect of NADPH inhibiting the PI(3)P binding. Interestingly, lipidomics analysis showed that treatment of cells with AIBP led to a decrease in the relative abundance of total phosphatidylinositol monophosphates (PIP1) in lipid rafts (Fig. 5 H) pointing to a possibility that AIBP may trigger PIP1 internalization.
To reconcile the results showing cytoskeletal effects of recombinant AIBP added to the cell culture media and the predominantly intracellular localization of PI(3)P, we investigated if extracellularly added AIBP is internalized. BV-2 microglia cells were incubated with 0.2 μg/ml of His-tagged AIBP for 30 min, followed by a 15 min incubation with 100 ng/ml LPS, fixed and stained with an anti-His antibody and organelle markers. We found that AIBP rapidly internalizes, with majority of internalized AIBP co-localizing with early endosomes (Fig. 6 A, F), mitochondria (Fig. 6, B,F) and F-actin (Fig. 6 C,F); there was also some co-localization with endoplasmic reticulum (Fig. 6, D,F) and lysosomes (Fig. 6 E,F). Collectively, our findings along with the results shown in Fig. 5G indicate that externally added AIBP is likely internalizes and exerts its action inside the cell.
Figure 6. Intracellular localization of internalized AIBP.

BV-2 microglia cells were incubated with 0.2 ug/ml AIBP for 30 min, followed by a 15 min incubation with 100 ng/ml LPS, fixed and stained with an anti-His antibody (green) and organelle markers of early endosomes (A, yellow), mitochondria (B, red), F-actin (C, red), lysosomes (D, yellow) and endoplasmic reticulum & DAPI (E, blue). F – colocalization ratio with different organelles, Mean ± SD are shown, n=7.
In conclusion, findings of our study suggest that binding of AIBP to PI(3)P and activation of Cdc42 followed by actin polymerisation may mechanistically contribute to the reduction of lipid raft abundance by AIBP.
Discussion
Changes in the abundance and/or functional properties of lipid rafts are key elements of pathogenesis of many diseases. This attracted interest to the experimental approaches regulating lipid rafts. Cyclodextrins, non-specific cholesterol acceptors, have been successfully used on a number of occasions37–39, but lack of selectivity and inherent problem of disposal of cholesterol removed from cells may limit their long-term applicability. Recently, another “raft-reducing” approach has been described involving AIBP. AIBP offered a more selective effect, reducing the abundance of rafts in “pathological” (e.g. inflamed) cells to the normal level, but not below2, 3, 10. AIBP stimulated cholesterol efflux and it was assumed that the mechanism is fundamentally similar to that of cyclodextrins, but included a feedback loop: the receptor for AIBP was suggested to be TLR4 located in pathological lipid rafts, or inflammarafts40, and disintegration of inflammarafts would lead to re-localization of the TLR4 from rafts reducing binding of AIBP1.
Current understanding of the mechanism of cholesterol efflux involves 2-step process: apoA-I-dependent stabilization of ABCA1 leading to an appearance of cholesterol in the “activated lipid domains”, followed by ABCA1-independent solubilisation of this domain by another molecule of apoA-I with formation of nascent HDL41. In this context, it was reasonable to assume that conversion of lipid rafts into activated lipid domains and/or redistribution of cholesterol from the former to the latter, would be breaking down the lipid rafts even before cholesterol efflux occurs. Indeed, overexpression of ABCA1 was associated with disruption of lipid rafts independently of cholesterol efflux42. The second mechanism of regulation of lipid rafts is through changes in the cytoskeleton and we have previously demonstrated that disruption of rafts through reorganization of the cytoskeleton leads to increased cholesterol efflux20. Furthermore, there is an established reciprocal connection between regulation of lipid rafts, lipid metabolism and rearrangements of the cytoskeleton through the action of phosphoinositides and small GTPases, primarily, PI(4,5)P2 and Cdc4243. Thus, although AIBP both stimulates cholesterol efflux and disrupts lipid raft, the mechanisms may not be limited to boosting extraction of cholesterol from lipid rafts to an acceptor.
In this study, we demonstrated that AIBP added to human macrophages in the absence of apoA-I, HDL, serum or any other cholesterol acceptor modified lipid rafts without triggering cholesterol efflux. This effect was demonstrated using confocal microscopy in situ and by studying isolated lipid rafts, and it was further confirmed by lipidomics analysis of isolated lipid rafts. The effects of AIBP on lipid rafts were apparent within 4 h, while the effects on cholesterol efflux required 24 h to appear and stimulation of cholesterol efflux was modest. Early findings with HUVEC cells also suggested that AIBP can modify lipid rafts without apoA-I or HDL6. Lack of requirement for apoA-I or HDL is also supported by findings that AIBP effectively reduced the abundance of rafts in alveolar macrophages where surfactant was used as cholesterol acceptor5. It was suggested that AIBP can stabilize ABCA14, 13 and ABCA1 can disrupt lipid rafts by mechanism independent of cholesterol efflux15. However, in our studies AIBP effectively reduced lipid rafts in ABCA1-deficient cells indicating that mechanisms unrelated to ABCA1 may be involved.
In this context, we hypothesized that another point of interaction between AIBP and lipid rafts, the lipid metabolism – cytoskeleton axis, may mechanistically contribute to regulation of lipid rafts by AIBP. Indeed, we found that a key element of the mechanism responsible for the effect of AIBP on lipid rafts is interaction of AIBP with PI(3)P, activation of small GTPase Cdc42 and rearrangement of cytoskeleton. AIBP likely binds PI(3)P in a complex with PI(4,5)P2, as presence of both PIPs seems to be required for efficient activation of actin polymerisation31. PI(3)P is considered a predominantly endosomal lipid, however, recently its formation and presence on the plasma membrane has been documented32, 33. Alternatively, initial AIBP binding to TLR4 may be followed by internalization of an AIBP/TLR4 complex toward early endosomes, as our data suggest, where interaction of AIBP with PI(3)P may occur. It is yet to be examined whether or how AIBP can be transported from the luminal to the cytoplasmic surface of an endosome, or if an alternative mechanism of AIBP internalisation may exist. If internalization occurs, AIBP seems to be involved in recruitment and activation by PI(3)P/PI(4,5)P2 of an actin polymerisation complex, SNX9/Cdc42.GTP/N-WASP/WIP/Arp2/344. In the actin polymerisation complex, activation of Cdc42 has been shown to be a major factor connecting restructuring of cytoskeleton with lipid metabolism29 and in our study it was found to be another key element causally linking AIBP with reduction of the lipid raft abundance. More studies are required to provide a detailed understanding of all elements of this pathway.
In conclusion, our findings suggest a novel mechanism of modulation of lipid rafts by AIBP involving binding of AIBP to PI(3)P, activation of Cdc42 and modification of cytoskeleton. The scope of this study is limited to mechanistic investigation and does not address possible implications of our findings to the physiological and pathological regulation of lipid rafts, physiological consequences of AIBP deficiency or administration and potential utility as a therapeutic approach. These aspects require further studies.
Supplementary Material
Highlights.
Apolipoprotein A-I binding protein (AIBP) is an effective and selective regulator of lipid rafts mitigating inflammation
AIBP stimulates cholesterol efflux, but this is unrelated to the effects on lipid rafts
The mechanism of lipid raft regulation involves interaction of AIBP with phosphatidylinositol 3-phosphate, activation of Cdc42 and re-arrangement of the cytoskeleton
Acknowledgements
The authors acknowledge the assistance of Monash Micro Imaging Facility. We are grateful to Ms Anh Hoang, Ms Natalie Mellett and Ms Kajsa Broman for excellent technical assistance and to Dr Chad Johnson for his help in quantitative analysis of confocal images.
Sources of Funding
This study was supported by grants from the National Institutes of Health: HL131473 (to DS), NS102432, HL135737, HL136275, NS104769 (to YIM) and supported in part by the Victorian Government’s OIS Program (DS).
Abbreviations:
- ABCA1
ATP binding cassette transporter type A1
- ABCG1
ATP binding cassette transporter type G1
- apoA-I
apolipoprotein A-I
- AIBP
apolipoprotein A-I binding protein
- CTB
cholera toxin, subunit B
- HDL
high density lipoprotein
- HUVEC
human umbilical cord endothelial cells
- LPS
popolysaccharide
- MβCD
methyl-β-cyclodextrin
- PIP
phosphatidylinositol phosphate
Footnotes
Disclosures
DS, MB and YIM and are inventors listed in patents and patent applications related to the topic of this article. YIM is scientific co-founder of Raft Pharmaceuticals LLC. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. Other authors have no competing interests to declare.
References
- 1.Fang L, Miller YI. Regulation of lipid rafts, angiogenesis and inflammation by AIBP. Curr Opin Lipidol. 2019;30:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woller SA, Choi S-H, An EJ, Low H, Schneider DA, Ramachandran R, Kim J, Bae YS, Sviridov D, Corr M, Yaksh TL, Miller YI. Inhibition of Neuroinflammation by AIBP: Spinal Effects upon Facilitated Pain States. Cell Reports. 2018;23:2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schneider DA, Choi S-H, Agatisa-Boyle C, Zhu L, Kim J, Pattison J, Sears DD, Gordts PLSM, Fang L, Miller YI. AIBP protects against metabolic abnormalities and atherosclerosis. Journal of Lipid Research. 2018;59:854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang M, Zhao GJ, Yao F, Xia XD, Gong D, Zhao ZW, Chen LY, Zheng XL, Tang XE, Tang CK. AIBP reduces atherosclerosis by promoting reverse cholesterol transport and ameliorating inflammation in apoE(−/−) mice. Atherosclerosis. 2018;273:122–130. [DOI] [PubMed] [Google Scholar]
- 5.Choi S-H, Wallace AM, Schneider DA, Burg E, Kim J, Alekseeva E, Ubags NDJ, Cool CD, Fang L, Suratt BT, Miller YI. AIBP augments cholesterol efflux from alveolar macrophages to surfactant and reduces acute lung inflammation. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang L, Choi S-H, Baek JS, Liu C, Almazan F, Ulrich F, Wiesner P, Taleb A, Deer E, Pattison J, Torres-Vazquez J, Li AC, Miller YI. Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature. 2013;498:118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mao R, Meng S, Gu Q, Araujo-Gutierrez R, Kumar S, Yan Q, Almazan F, Youker KA, Fu Y, Pownall HJ, Cooke JP, Miller YI, Fang L. AIBP Limits Angiogenesis Through gamma-Secretase-Mediated Upregulation of Notch Signaling. Circ Res. 2017;120:1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu Q, Yang X, Lv J, et al. AIBP-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate. Science. 2019;363:1085–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang T, Wang Q, Wang Y, Wang J, Su Y, Wang F, Wang G. AIBP and APOA-I synergistically inhibit intestinal tumor growth and metastasis by promoting cholesterol efflux. J Transl Med. 2019;17:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dubrovsky L, Ward A, Choi S-H, Pushkarsky T, Brichacek B, Vanpouille C, Adzhubei AA, Mukhamedova N, Sviridov D, Margolis L, Jones RB, Miller YI, Bukrinsky M. Inhibition of HIV Replication by Apolipoprotein A-I Binding Protein Targeting the Lipid Rafts. mBio. 2020;11:e02956–02919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol. 2010;11:688–699. [DOI] [PubMed] [Google Scholar]
- 12.Sorci-Thomas MG, Thomas MJ. Microdomains, Inflammation, and Atherosclerosis. Circ Res. 2016;118:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M, Li L, Xie W, et al. Apolipoprotein A-1 binding protein promotes macrophage cholesterol efflux by facilitating apolipoprotein A-1 binding to ABCA1 and preventing ABCA1 degradation. Atherosclerosis. 2016;248:149–159. [DOI] [PubMed] [Google Scholar]
- 14.Nofer J-R, Remaley AT, Feuerborn R, Wolinnska I, Engel T, von Eckardstein A, Assmann G. Apolipoprotein A-I activates Cdc42 signaling through the ABCA1 transporter. J Lipid Res. 2006;47:794–803. [DOI] [PubMed] [Google Scholar]
- 15.Tsukamoto K, Hirano K, Tsujii K, Ikegami C, Zhongyan Z, Nishida Y, Ohama T, Matsuura F, Yamashita S, Matsuzawa Y. ATP-Binding Cassette Transporter-1 Induces Rearrangement of Actin Cytoskeletons Possibly through Cdc42/N-WASP. Biochem Biophys Res Commun. 2001;287:757–765. [DOI] [PubMed] [Google Scholar]
- 16.Mukhamedova N, Escher G, D’Souza W, Tchoua U, Grant A, Krozowski Z, Bukrinsky M, Sviridov D. Enhancing apolipoprotein A-I-dependent cholesterol efflux elevates cholesterol export from macrophages in vivo. J Lipid Res. 2008;49:2312–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Low H, Hoang A, Sviridov D. Cholesterol Efflux Assay. J Vis Exp. 2012:e3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Owen DM, Rentero C, Magenau A, Abu-Siniyeh A, Gaus K. Quantitative imaging of membrane lipid order in cells and organisms. Nat Protoc. 2011;7:24–35. [DOI] [PubMed] [Google Scholar]
- 19.Mukhamedova N, Hoang A, Dragoljevic D, et al. Exosomes containing HIV protein Nef reorganize lipid rafts potentiating inflammatory response in bystander cells. PLoS Pathog. 2019;15:e1007907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Low H, Mukhamedova N, Cui Huanhuan L, et al. Cytomegalovirus Restructures Lipid Rafts via a US28/CDC42-Mediated Pathway, Enhancing Cholesterol Efflux from Host Cells. Cell Reports. 2016;16:186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta. 2007;1768:1311–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Remaley AT, Schumacher UK, Stonik JA, Farsi BD, Nazih H, Brewer HB, Jr. Decreased reverse cholesterol transport from Tangier disease fibroblasts. Acceptor specificity and effect of brefeldin on lipid efflux. Arterioscler Thromb Vasc Biol. 1997;17:1813–1821. [DOI] [PubMed] [Google Scholar]
- 23.Mukhamedova N, Fu Y, Bukrinsky M, Remaley AT, Sviridov D. The Role of Different Regions of ATP-Binding Cassette Transporter A1 in Cholesterol Efflux. Biochemistry. 2007;46:9388–9398. [DOI] [PubMed] [Google Scholar]
- 24.Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney AM, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie J, Santamarina-Fojo S, Brewer HB, Jr. Cellular Localization and Trafficking of the Human ABCA1 Transporter. J Biol Chem. 2001;276:27584–27590. [DOI] [PubMed] [Google Scholar]
- 25.Sethi AA, Stonik JA, Thomas F, Demosky SJ, Amar M, Neufeld E, Brewer HB, Davidson WS, D’Souza W, Sviridov D, Remaley AT. Asymmetry in the Lipid Affinity of Bihelical Amphipathic Peptides: A STRUCTURAL DETERMINANT FOR THE SPECIFICITY OF ABCA1-DEPENDENT CHOLESTEROL EFFLUX BY PEPTIDES. J Biol Chem. 2008;283:32273–32282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owen DM, Rentero C, Magenau A, Abu-Siniyeh A, Gaus K. Quantitative imaging of membrane lipid order in cells and organisms. Nat Protoc. 2011;7:24–35. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Chen X, Gueydan C, Han J. Plasma membrane changes during programmed cell deaths. Cell Research. 2018;28:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Callahan MK, Halleck MS, Krahling S, Henderson AJ, Williamson P, Schlegel RA. Phosphatidylserine expression and phagocytosis of apoptotic thymocytes during differentiation of monocytic cells. J Leukoc Biol. 2003;74:846–856. [DOI] [PubMed] [Google Scholar]
- 29.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. [DOI] [PubMed] [Google Scholar]
- 30.Chadda R, Howes MT, Plowman SJ, Hancock JF, Parton RG, Mayor S. Cholesterol-sensitive Cdc42 activation regulates actin polymerization for endocytosis via the GEEC pathway. Traffic. 2007;8:702–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daste F, Walrant A, Holst MR, Gadsby JR, Mason J, Lee JE, Brook D, Mettlen M, Larsson E, Lee SF, Lundmark R, Gallop JL. Control of actin polymerization via the coincidence of phosphoinositides and high membrane curvature. J Cell Biol. 2017;216:3745–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin HW, Hayashi M, Christoforidis S, Lacas-Gervais S, Hoepfner S, Wenk MR, Modregger J, Uttenweiler-Joseph S, Wilm M, Nystuen A, Frankel WN, Solimena M, De Camilli P, Zerial M. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J Cell Biol. 2005;170:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kale SD, Gu B, Capelluto DG, Dou D, Feldman E, Rumore A, Arredondo FD, Hanlon R, Fudal I, Rouxel T, Lawrence CB, Shan W, Tyler BM. External lipid PI3P mediates entry of eukaryotic pathogen effectors into plant and animal host cells. Cell. 2010;142:284–295. [DOI] [PubMed] [Google Scholar]
- 34.Marat AL, Haucke V. Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. EMBO J. 2016;35:561–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marbaix AY, Noël G, Detroux AM, Vertommen D, Van Schaftingen E, Linster CL. Extremely Conserved ATP- or ADP-dependent Enzymatic System for Nicotinamide Nucleotide Repair. J Biol Chem. 2011;286:41246–41252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shumilin Igor A, Cymborowski M, Chertihin O, Jha Kula N, Herr John C, Lesley Scott A, Joachimiak A, Minor W. Identification of Unknown Protein Function Using Metabolite Cocktail Screening. Structure. 2012;20:1715–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimmer S, Grebe A, Bakke SS, et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med. 2016;8:333ra350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merscher-Gomez S, Guzman J, Pedigo CE, et al. Cyclodextrin Protects Podocytes in Diabetic Kidney Disease. Diabetes. 2013;62:3817–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prior M, Lehmann S, Sy M-S, Molloy B, McMahon HEM. Cyclodextrins Inhibit Replication of Scrapie Prion Protein in Cell Culture. J Virol. 2007;81:11195–11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller YI, Navia-Pelaez JM, Corr M, Yaksh TL. Lipid rafts in glial cells: role in neuroinflammation and pain processing. J Lipid Res. 2020;61:655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips MC. Molecular Mechanisms of Cellular Cholesterol Efflux. J Biol Chem. 2014;289:24020–24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Landry YD, Denis M, Nandi S, Bell S, Vaughan AM, Zha X. ATP-binding Cassette Transporter A1 Expression Disrupts Raft Membrane Microdomains through Its ATPase-related Functions. J Biol Chem. 2006;281:36091–36101. [DOI] [PubMed] [Google Scholar]
- 43.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. [DOI] [PubMed] [Google Scholar]
- 44.Wu CY, Lin MW, Wu DC, Huang YB, Huang HT, Chen CL. The role of phosphoinositide-regulated actin reorganization in chemotaxis and cell migration. Br J Pharmacol. 2014;171:5541–5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.