

Abstract
The myofibroblast is a specialized fibroblast that expresses α-smooth muscle actin (α-SMA) and participates in wound contraction and fibrosis. The fibroblast to myofibroblast transition depends on chemical and mechanical signals. A fibroblast senses the changes in the environment (extracellular matrix (ECM)) and transduces these changes to the cytoskeleton and the nucleus, resulting in activation or inhibition of α-SMA transcription in a process called mechanosensing. A stiff matrix greatly facilitates the transition from fibroblast to myofibroblast, and although the aging heart is much stiffer than the young one, the aging fibroblast has difficulties in transitioning into the contractile phenotype. This suggests that the events occurring downstream of the matrix, such as activation or changes in expression levels of various proteins participating in mechanotransduction can negatively alter the ability of the aging fibroblast to become a myofibroblast. In this review, we will discuss in detail the changes in ECM, receptors (integrin or non-integrin), focal adhesions, cytoskeleton, and transcription factors involved in mechanosensing that occur with aging.
Keywords: Cardiac fibroblast, myofibroblast, mechanosensing, aging
1. Introduction
The myofibroblast, a specialized fibroblast that expresses the contractile protein α-smooth muscle actin (α-SMA), appears in the myocardium upon injury. In fact, in the uninjured young heart, there are very few myofibroblasts present (mostly around arterioles) (Shinde et al., 2017). Myofibroblasts appear in the heart after injury and play a distinct role depending on the type of injury; they are indispensable for proper scar formation and contraction after myocardial infarction (Hinz, 2006) but in other types of cardiac injuries (pressure overload) they participate in adverse remodeling (Moore-Morris et al., 2014). The transition from the fibroblast into myofibroblast depends on chemical and mechanical signals (Czubryt, 2019), but the distinction in vivo between the contribution of these two classes of signals is challenging. The major chemical activator is TGF-β, and it is released from its latent form upon demand (Buscemi et al., 2011; Klingberg et al., 2014). Mechanical signals often come from changes in protein composition of extracellular matrix (ECM) (see Table 1 for abbreviations) that translates to either increased tension and/or altered ECM composition. Changes in matrix very often are accompanied by an altered expression of different components, or activation of various receptors that connect cells with the ECM, such as integrins or transient receptor potential (TRP) channels. These mechanical signals translate into rearrangements and activation of various proteins that form focal adhesions (FA), a functional bridge between ECM and cytoskeleton. The cytoskeleton then undergoes a transformation, and a key functional protein, actin, becomes polymerized (F-actin). Polymerized actin then facilitates Myocardin-Related Transcription Factor A (MRTF-A) translocation to the nucleus where it can pair with another transcription factor, Serum Response Factor (SRF). Each factor binds to its cognate site on the α-SMA promoter, leading to its transcriptional activation.
Table 1.
Abbreviations.
| Abbreviation | Full name |
|---|---|
| ART | Ak strain Transforming, Protein Kinase B |
| Arp2/3 | Actin Related Proteins 2/3 |
| Cdc42 | Cell Division Control Protein 42 homolog |
| ECM | Extracellular Matrix |
| F-actin | Filamentous Actin |
| FA | Focal Adhesions |
| FAK | Focal Adhesion Kinase |
| FERM | Protei 4.1-Ezrin-Radixin-Moesin |
| FN | Fibronectin |
| FN-EDA | Fibronectin with Extra Domain A |
| G-actin | Monomeric Actin |
| GSK3 | Glycogen Synthase Kinase-3 |
| GTPases | Guanosine Triphosphatases |
| JNK | c-Jun N-terminal Kinase |
| LATS1/2 | Large Tumor Suppressor Kinase 1/2 |
| MAP kinase | Mitogen Activated Protein Kinase |
| MDia | Mammalian Diaphanous |
| MRTF-A | Myocardin-Related Transcription Factor A |
| MST1/2 | Mammalian Sterile 20-like Kinases 1/2 |
| mTOR | Mammalian Target of Rapamycin |
| NFAT4 | Nuclear Factor of Activated T-cells 4 |
| N-WASP | Neural Wiskott-Aldrich Syndrome Protein |
| PDGFRα | Platelet Derived Growth Factor Receptor α |
| PI3K | Phosphatidylinositol-3-kinase |
| Rac1 | Ras-related C3 botulinum toxin 1 |
| Rho GTPase | Ras homolog GTPase |
| SMAD 3, 4, 7 | (Small) Mothers against decapentaplegic homolog 3, 4, 7 |
| α-SMA | α-Smooth Muscle Actin |
| Sos | Son of sevenless |
| SRF | Serum Response Factor |
| Taz | Transcriptional co-Activator with a PDZ motif |
| TGF-β | Transforming Growth Factor-β |
| TRP | Transient Receptor Potential |
| TRPV4 | Transient Receptor Potential Vanilloid 4 |
| Yap | Yes-Associated Protein |
| ZBP-1 | Zip-code-Binding Protein 1 |
In the past, we examined the role of TGF-β in myofibroblast transdifferentiation in aged hearts (Trial and Cieslik, 2018). We have found defects in the canonical TGF-β Smad-dependent pathway (Cieslik et al., 2011b). Specifically, we found that the fibroblasts derived from the aging hearts had reduced responsiveness to TGF-β, which translated to a weaker α-SMA expression in vitro and reduced myofibroblast numbers after myocardial infarction in vivo, as noted by others (Bujak et al., 2008). This defect may cause reduced tensile strength of the scar, altered heart geometry and ultimately leads to increased post-infarction remodeling. In this review, we concentrated our efforts on understanding how mechanical signaling and mechanosensing are altered in the aged cardiac fibroblasts. The idea for this review originated from our observation that even without chemical signaling (TGF-β), in vitro α-SMA expression in the fibroblasts isolated from the aged hearts was reduced when compared with the ones derived from the young hearts, suggesting impaired mechanosensing (Cieslik et al., 2011b). Most studies related to the topic are based on observations from various cardiac injury models in young rodents. However, it became apparent that the multiple signaling and structural changes that we observe in the aging heart or fibroblasts derived from the old heart contribute to the alternative mechanosensing that translates into the reduced fibroblast-to-myofibroblast transition (Figure 1). This review attempts to depict the role of these effectors and to provide insight into their potential interactions.
Figure 1.
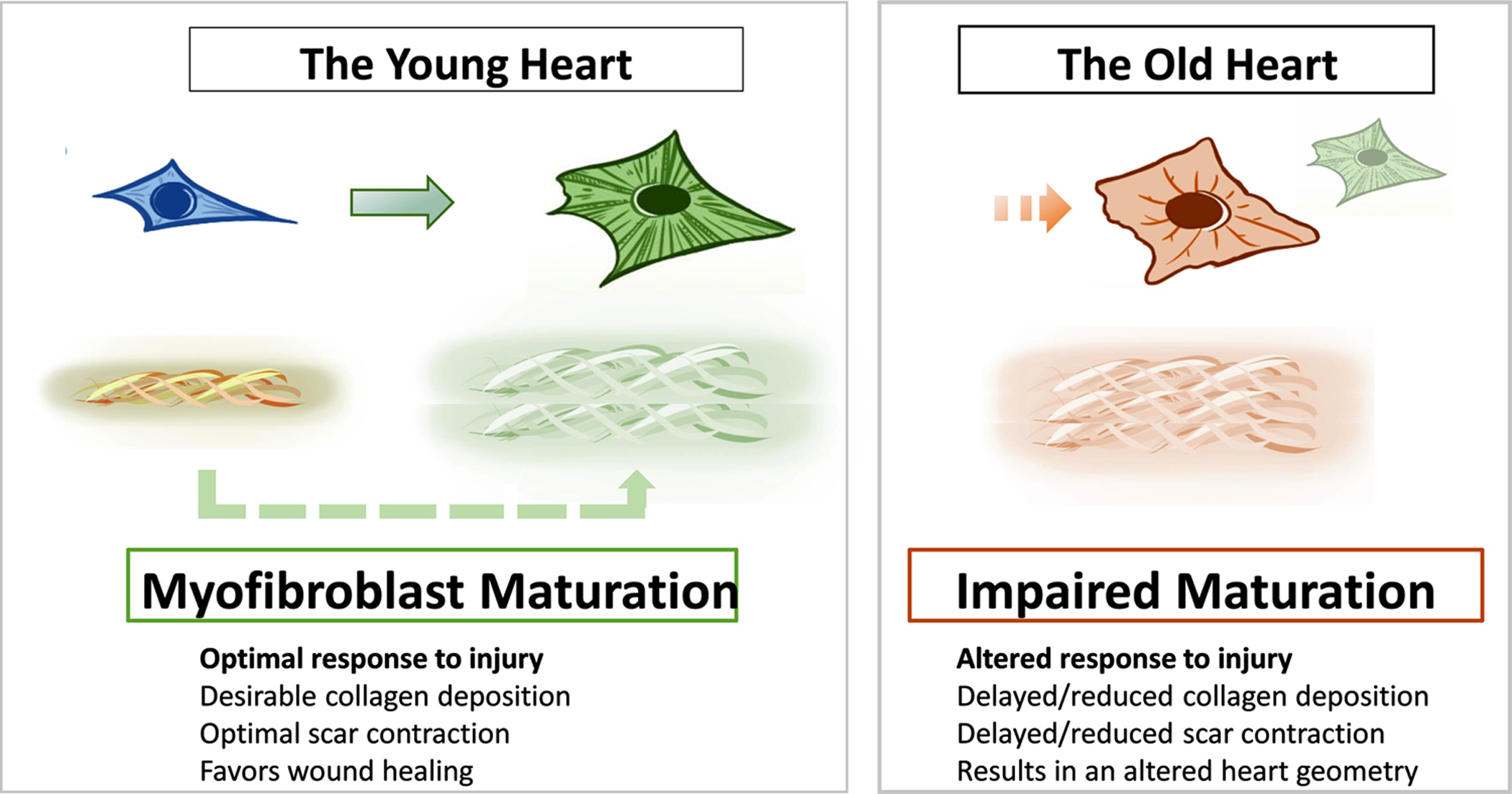
Schematic representation of fibroblast-myofibroblast maturation in the young and old heart.
2. The cardiac fibroblast and its role in cardiovascular diseases
The primary fibroblast function in the healthy and diseased heart is synthesis, deposition, and crosslinking of the matrix. In disease, this is characterized by excessive ECM deposition and crosslinking, leading to fibrosis. Fibrosis manifests with nearly all forms of heart disease, and its progression is clinically linked to heart failure. Cardiac fibrosis can develop in aging, aortic stenosis, hypertension, cardiomyopathies, and coronary heart disease without infarction (Hinderer and Schenke-Layland, 2019; Kong et al., 2014) or as a result of a poorly formed scar after myocardial infarction in the old (Bujak et al., 2008) or diabetic heart (Thakker et al., 2006); however, the prevalence for these diseases increases with aging.
There are no diagnostic assays or tests that will directly evaluate fibroblast function in vivo. The only possibility to have an insight into fibroblast “health” is to detect and quantify fibrosis. This can be achieved via invasive and non-invasive methods. For the direct method, the collagen level evaluation in the endomyocardial biopsy is the only method currently available (Hinderer and Schenke-Layland, 2019). The non-invasive imaging methods include magnetic resonance imaging, integrated backscatter echocardiography, single-photon emission tomography, and positron emission tomography. Finally, there is also a possibility to assess the levels of circulating carboxy-terminal pro-peptide of collagen type I that reflects on the level of newly synthesized collagen, but this method does not identify which organ is undergoing fibrotic changes (Hinderer and Schenke-Layland, 2019).
The pathological activation of fibroblasts that contribute to fibrosis can be curbed using several different approaches, such as angiotensin-converting enzyme blockers, angiotensin receptor 1 antagonist, beta-blockers, endothelin antagonists, statins, cyclic GMP-specific phosphodiesterase type 5A inhibitor, and loop diuretics (Fang et al., 2017). Most of these available treatments directly affect collagen synthesis, but statins work by quenching inflammation. It has been shown that inflammation can promote fibrosis (Trial et al., 2017).
As the heart ages, fibroblasts start to undergo phenotypic changes. They become activated, which leads to matrix deposition and crosslinking and they start to secrete various cytokines, switching to a pro-inflammatory phenotype (Cieslik et al., 2015). It is not known if the entire fibroblast population expands with aging or if any sub-population is preferentially activated. Fibroblast heterogeneity has been extensively studied in various injury mouse models in young animals (Tallquist and Molkentin, 2017), but not much has been reported in regards to aged animals. Nevertheless, it has been demonstrated that the aging heart displays increased interstitial and adventitial fibrosis (Alex et al., 2018), suggesting that at least two different populations are activated; PDGFRα+ (Moore-Morris et al., 2014) and Gli+ fibroblasts (Kramann et al., 2015). The former is associated with interstitial and the latter with adventitial fibrosis. Our group previously reported an increased number of activated fibroblasts in the aged myocardium (Cieslik et al., 2013b); however, we did not identify the sub-population. Interestingly, the number of α-SMA+ cells is not increased with aging (in the uninjured heart) (Alex et al., 2018), although after myocardial infarction, their number is reduced when compared with the young heart (Bujak et al., 2008) suggesting an impaired myofibroblast differentiation program. Recent studies have also demonstrated that cardiac and dermal fibroblasts from old male and/or female mouse hearts have a higher transcriptional heterogeneity (Ali et al., 2014; Farbehi et al., 2019; Mahmoudi et al., 2019; Vidal et al., 2019). The reason for this heterogeneity remains unclear since it could be due to mixed origins of resident fibroblasts in aged hearts, or to the accumulation of different events leading to transcriptional (and epigenetic) drift between distinct fibroblast populations. Yet, the consequences are obvious, since it means that both ECM synthesis and applied mechanosensing may differ from one fibroblast population to another, with young-like versus aged islets of cells and secreted matrix, leading to an unsynchronized response to mechanical stress.
3. Extracellular matrix
The ECM forms a mechanical scaffold necessary to maintain the geometry of organs. The ECM also plays a significant role in signal transduction regulating cellular function. The activated fibroblast/myofibroblast is the primary ECM protein producer (Hortells et al., 2019). Because all cardiac cells can influence each other via secreted factors (Frangogiannis, 2019) or through coupling (MacCannell et al., 2007), the changes in ECM directly or indirectly can affect all cells residing in the interstitium and may result in changes in heart function, including heart failure.
The major differences in ECM in the aging heart (when compared with the young heart) are the expansion of the interstitial and perivascular ECM repository, increased collagen cross-linking, or deposition of different isoforms of fibronectin (FN) (Cieslik et al., 2017; Gazoti Debessa et al., 2001).
The main components of the cardiac ECM collagen network are fibrillar collagen I and III (Weber et al., 1988), that are best characterized in various pathologies, aging included. Since there is a noticeable discrepancy between mRNA and protein levels of collagens in the old heart, we will discuss here only changes related to protein levels. Data from the human and mouse heart demonstrate increased levels of collagen type I (Cieslik et al., 2011a; Gazoti Debessa et al., 2001). These data are also supported by in vitro observations wherein aged fibroblasts synthesize twice as much procollagen type I as the young ones (Cieslik et al., 2013b), consistent with reports showing that the total collagen content in the old male and female heart doubles when compared with the young heart (Eghbali et al., 1989; Lin et al., 2008). Data from human hearts also point to the altered ratio between collagen I and III, with increased collagen I and reduced collagen III deposition (Mendes et al., 2012). The altered collagen I/III ratio may impair left ventricle function via increased stiffness as shown in both sexes (Wei et al., 1999). Cross-linking of collagen can occur either through an enzymatic process (via lysyl oxidase) or via a non-enzymatic reaction (between glucose and lysine or hydroxylysine residues leading to the formation of advanced glycation end products). Both enzymatic and non-enzymatic cross-linking are augmented in the aging myocardium (Asif et al., 2000; Thomas et al., 1992), leading to the increased stiffness of the matrix.
The other collagens (non-fibrillar) or ECM matrix proteins also change with aging and can affect directly or indirectly the fibroblast phenotype and their transition into myofibroblasts. As we have described above, the matrix stiffness is increased in aging (Wei et al., 1999). The presence of elastin very often compensates for collagen-related stiffness; however, it has been demonstrated that with age, its expression is reduced in human valves (McDonald et al., 2002), which may indicate similar changes in the ventricles. Interestingly, matrix stiffness by itself can increase α-SMA transcriptional activation in both males and females (Herum et al., 2017). However, fibroblasts derived from aged hearts (Cieslik et al., 2011b) or fibroblasts in injured old hearts (Cieslik et al., 2013a) have a reduced ability to become α-SMA-expressing myofibroblasts, suggesting additional or even multiple defects that contribute to this abnormality.
Aging also affects other important components that play various roles in the matrix. Biglycan and decorin, for example, have been shown to control myofibroblast transdifferentiation via downregulation of focal adhesions (Melchior-Becker et al., 2011) and expression of α-SMA (Melchior-Becker et al., 2011; Nakatani et al., 2008). Interestingly, the expression of these proteins is increased in the aged pig heart (Stephens et al., 2008). It has also been reported that certain glycoproteins such as periostin, tenascin-C, and thrombospondin-2 are upregulated in the aged heart or fibroblasts derived from the old heart (Li et al., 2014; Sato and Shimada, 2001; Swinnen et al., 2009). None of these proteins directly affects α-SMA transcriptional regulation; however, periostin may have an indirect effect because it affects ECM homeostasis by interacting with collagen I, FN, tenascin-C, and heparin (Norris et al., 2007).
Finally, FN and its variants play a role in myofibroblast differentiation. FN serves as a “bridge” between cells and other ECM proteins by binding to cell membrane receptors and ECM proteins (collagens, fibrillin, tenascin-C, heparin) (Chung and Erickson, 1997; Kadler et al., 2008; Sabatier et al., 2009). After stress or injury, there are several splice variants of FN that are transcribed with extra domain A (FN-EDA) or extra domain B, but specifically, the presence of FN-EDA has been attributed to an increased fibroblast to myofibroblast transition (Kohan et al., 2010). Interestingly, in the aging uninjured heart, there is FN-EDA (Cieslik et al., 2017), but myofibroblasts were not detected in the interstitium of the male or female heart (Alex et al., 2018). Increased stretching of FN fibrils can lead to the partial unfolding of the secondary structure and change of binding site availability (Antia et al., 2008), which may explain why in the aging heart, even in the presence of FN-EDA, there are very few myofibroblasts.
4. Mechanosensing at the membrane
4.1. Integrin-mediated mechanosensing
Mechanosensing from the ECM to the cytoplasm of the fibroblasts is directed through transmembrane adhesion molecules, many of which are integrins (Giancotti and Ruoslahti, 1999). Integrins are heterodimers, with α and β chains (Figure 2). The class of integrins that are able to bind to the ECM are mainly β1 coupled to a variety of α chains, which have the specificity to allow binding to various ECM components such as collagen, fibronectin, or laminin. Integrins and other adhesion molecules are essential mediators of fibroblast functions such as migration and contractility (Figure 2), which are diminished in aged cells (Cieslik et al., 2011b). It is, therefore, likely that reduced mechanosensing by these cells could be responsible for the compromised functions. Although the expression levels of various α and β integrin chains have been reported to change in aged animals (reviewed in (Meschiari et al., 2017)), the effect of these changes on the functions of the cells cannot be determined because of the complex nature of the α-β pairing (availability of each chain for assembly) and their subsequent activation to allow binding to the ECM.
Figure 2: Schematic representation of the Focal Adhesion (FA) core complex.
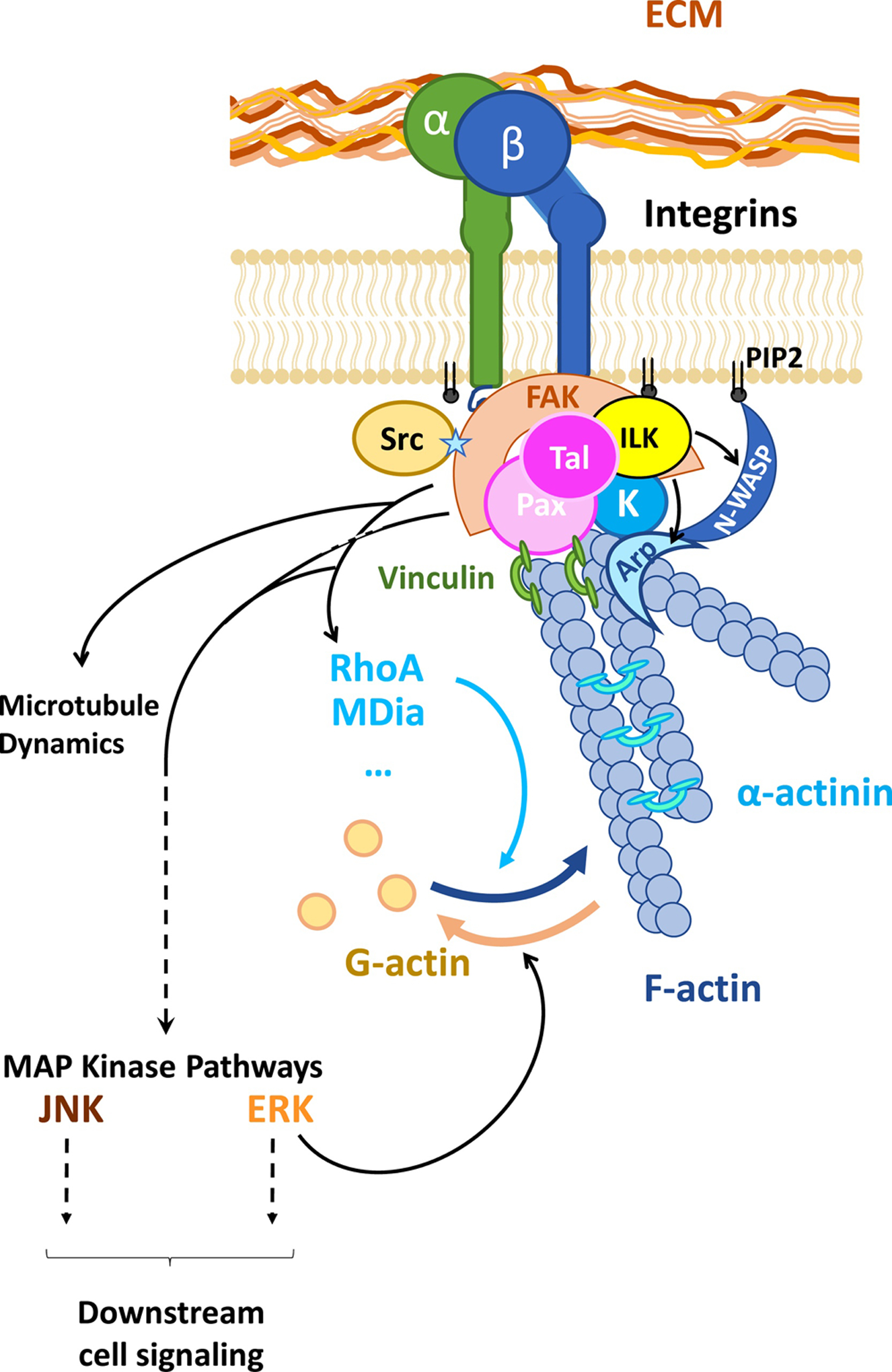
The anchorage of integrin subunits to the ECM promotes the recruitment of talin (Tal), paxillin (Pax), kindlin-2 (K), which facilitates the docking and catalytic activity of the integrin-linked kinase (ILK) and focal adhesion kinase (FAK). FAK ensures the recruitment and the binding of Src, and the enrichment of the FA with phosphoinositide (PIP2). Downstream signaling of FAK, integrin-linked kinase, and Src includes RhoGTPase that can trigger actin polymerization to generate fibers (F-actin) or MAP Kinase pathways. Enzymatic activity at the FA core also favors the recruitment of actin-binding proteins such as: vinculin that binds to Pax and reinforces the tight connection between the FA complex and the cytoskeleton, α-actinin that helps the stabilization of parallel F-actin, Arp2.3 (Arp) or N-WASP that favor branching of F-actin.
Integrins signal to the cell bidirectionally, called “outside-in” or “inside-out”, depending on whether the originating signal comes from the external matrix or from focal adhesion molecules bound to the integrin cytoplasmic tail within the cytoplasm. The signaling may also be simultaneous. Signaling and increased binding to ligands is accomplished by a conformational change in the heterodimer, including the change from a bent to an open configuration (reviewed in (Liu et al., 2015b)). Whereas the integrins of most hematopoietic cells are in the “resting” configuration to prevent their adhesiveness while in the blood, fibroblasts have some of their integrins in the active state because of their requirement for anchorage in a tissue (Norris et al., 2007). No comparison of the integrins in the active versus resting states has been done in young versus old cardiac fibroblasts.
Outside-in signaling is accomplished by integration of mechanical and biochemical forces. Integrins respond to properties of the ECM and the tissue in which it resides: force, rigidity, and the spatial arrangement of the ECM (Kechagia et al., 2019). Changes in the heart occur with aging, so that there is an increase in stiffness (rigidity) and the appearance of new matrix proteins such as FN-EDA that may affect the ECM spatial arrangement (Cieslik et al., 2017). The added stiffness may also be a result of matrix cross-linking (Meschiari et al., 2017). These may result in forces that change the conformation of the integrin to its fully active state, thus increasing its affinity for matrix. Further, the changes can have a “memory”, in that they can persist after the force has been removed (Kechagia et al., 2019). Inside-out signaling may derive from cellular responses to cytokines and chemokines (Gahmberg et al., 2019), some of which are markedly increased in the aging heart (Cieslik et al., 2011a; Cieslik et al., 2015), or from other stimuli. The fibroblast then accumulates molecules of the focal adhesion at the site of the integrin cytoplasmic tail, and the integrin assumes its activated conformation.
4.2. Non-integrin mechanosensing
Cardiac fibroblasts also express TRP channels, which are members of a superfamily of molecules that sense an extraordinary range of environmental changes, including pH, osmolarity, temperature, and chemical (reviewed in (Venkatachalam and Montell, 2007)). One of the most characterized for its induction by mechanical signals is TRP vanilloid 4 (TRPV4). It is a calcium channel that can be activated when a force is applied to β1 integrins; this force is transduced by a mechanical strain in the cytoskeletal backbone of the focal adhesions associated with the integrins (Matthews et al., 2010). In addition to this indirect mechanism for mechanosensing, direct activation of a calcium influx can be induced by shear stress, stretch, and many other stimuli (Michalick and Kuebler, 2020; Zhan and Li, 2018). TRPV4 activation has been linked to fibrosis in many tissues (Zhan and Li, 2018). This may be because differentiation of fibroblasts into myofibroblasts is dependent on non-canonical TGF-β signaling in association with TRPV4, which responds to increased ECM stiffness (Adapala et al., 2013). The latter study of cardiac fibroblasts reported that mechanical stiffness transduced via TRPV4 resulted in signaling through focal adhesions, including Rho-GTPases (Adapala et al., 2013). In lung fibroblasts, signaling through TRPV4 facilitates actomyosin remodeling, thereby causing nuclear translocation of the Myocardin-Related Transcription Factor-A (MRTF-A) to enhance α-SMA transcription (Rahaman et al., 2014). It has been suggested that this pathway might be operative in the cardiac ventricle (Inoue et al., 2019). Although little is known about changes in TRPV4 expression in aging fibroblasts, a noted increase of TRPV4 expression in aged cardiomyocytes is associated with hypercontractility and a decline of cardiac function (Jones et al., 2019). Responsiveness to the known increase in matrix stiffness with aging may be the link between TRPV4 as a mechanosensor and fibrosis.
5. Focal adhesions
Focal adhesions (FAs) represent multi-unit structures that are organized around active integrins at the plasma membrane to bridge the ECM to the inner cytoskeleton (Burridge, 2017) (Figure 2). By transducing ECM mechanical tension into biochemical cell signaling and mechanical inner force, FAs thus play a key role in cell motility, proliferation, adhesion and migration. The size of FAs can predict cell migration capacity in both mouse embryonic and human male skin fibroblasts during wound healing (Kim and Wirtz, 2013). Due to the multiple functions of FAs, the integrin-actin protein binding complex (the so-called integrin “adhesome”) includes a wide variety of kinases, actin-binding proteins and adapters, for more than 180 dynamic and context-actionable protein-protein interactions (Horton et al., 2016; Horton et al., 2015; Zaidel-Bar and Geiger, 2010). Therefore, its composition and role in the aging cardiac fibroblast is both complex and multi-layered. Critical FA core proteins are shown in Figure 2.
5.1. Talin and vinculin
Talin is an actin-binding protein that is indispensable and sufficient for integrin engagement in FAs (Chinthalapudi et al., 2018). It interacts with the integrin β-chain via its “head” FERM domain (Protein 4.1- Ezrin- Radixin- Moesin), while its “tail” domain binds to a polymerized actin filament (F-actin) and to vinculin, an actin-binding protein that interacts with F-actin and also with α-actinin, another actin-binding protein promoting F-actin parallel cross-linkage (Figure 2) (Klapholz and Brown, 2017). Main adaptors of the integrin adhesome are kindlins and paxillin (Figure 2). Neither possesses enzymatic activity by itself, but both rather promote the recruitment of kinases and phosphatases, which locally initiates intracellular signaling in response to mechanical forces. Vinculin levels are increased in the left ventricle in aging female rats and male rhesus monkeys, both at the inter-cardiomyocyte connection site (intercalated disk) and at the lateral border of these cells, which may thus include fibroblasts as well (Kaushik et al., 2015). Fruit flies overexpressing vinculin exhibited an increased lifespan and a delayed age-associated diastolic dysfunction (Kaushik et al., 2015). Therefore, the increase in vinculin in the aging mammal heart probably represents an adaptive response to tension at the FA site and downstream gene expression, but its beneficial effect may be eventually overwhelmed due to accumulated changes along the mechanosensing axis.
5.2. Kindlins and paxillin
Kindlins represent a family of conserved FERM-domain proteins that are recently described as central integrin regulators. The three family members have a tissue-restricted distribution, while holding specific binding partners: while kindlin-1 is more ubiquitous, kindlin-3 is restricted to hematopoietic cells (podosomes) (Rognoni et al., 2016). No disease-causing mutation is reported for kindlin-2, but a knock-out is lethal in the mouse due to cardiovascular defects (Dowling et al., 2008a; Montanez et al., 2008). Kindlin-2 is the most predominant in cardiac cells, including in fibroblasts, which may indicate a key role in myocardial tissue integrity during both development and post-natal maintenance (Dowling et al., 2008a; Dowling et al., 2008b; Zhan et al., 2014). Conditional knock-out in the adult mouse leads to dilated cardiomyopathy characterized by invading and massive fibrosis (Zhang et al., 2016), while shRNA against kindlin-2 reduces fibroblast mechanical strength and myofibroblast differentiation (He et al., 2011). According to biochemical studies, kindlin-2 works as a dimer, and any impairment of kindlin-2 dimerization affects cell adhesion (Li et al., 2017b). Interestingly, kindlin-1 and kindlin-2 have a specific affinity to each other, which may be due to high sequence homology among the kindlin protein family. But it might also suggest that, in several cell types in which kindlins-1 and −2 are co-expressed, kindlin-1/kindlin-2 heterodimers could be generated with potential downstream consequences in terms of FA stabilization and mechanosensing. Regular kindlin-2 partners include not only β1, β2, and β3 integrin cytosolic tails (Bledzka et al., 2010; Bouaouina et al., 2012; Li et al., 2017b; Liu et al., 2011), but also Integrin-linked Kinase and migfilin (Brahme et al., 2013), both involved in integrin signaling and actin cytoskeleton coupling (Fukuda et al., 2014) (Figure 2). Integrin-linked Kinase has been notably reported as a lifespan regulator (Kumsta et al., 2014), and its overactivation in young animals mimics aging-like cardiac defects (Nishimura et al., 2014). Sos (Son of sevenless) and Rac1 GTPase are two additional kindlin-2 binding proteins that can trigger the formation of the actin cytoskeleton (Jung et al., 2011; Wei et al., 2014). Kindlins work side-by-side with paxillin, another important FA scaffolding protein (Theodosiou et al., 2016).
Due to the presence of many phosphorylatable Ser/Thr and some tyrosine residues, paxillin plays a central yet dual role, since it can facilitate both FA assembling and dissociation in response to an interplay of kinases and phosphatases (including ERKs, JNK, PKA, PKC and GSK3) (Lopez-Colome et al., 2017; Mekhdjian et al., 2017; Zhou et al., 2017). As an adapter, paxillin recruits and indirectly favors the activation of Focal Adhesion Kinase (FAK) (Figure 2). Interestingly, the reduction of paxillin expression observed in dermal fibroblasts with aging resulted in fibroblasts is characterized by an abnormal phenotype and spreading pattern. Furthermore, inhibition of paxillin expression resulted in diminished myofibroblast transitioning, suggesting that this protein plays a central role between FA and cytoskeleton rearrangements leading to α-SMA expression (Zheng et al., 2012).
5.3. FAK
FAK is the main driver of mechano-induced cell signaling and is indispensable for fibroblast function (Gilmore and Romer, 1996). FAK has multiple abilities within a cell, including acting as a scaffolding protein complex, a bridge and/or as a tyrosine kinase (Ceccarelli et al., 2006; Hayashi et al., 2002; Nowakowski et al., 2002). FAK binds to integrins via its FERM domain, while its Focal Adhesion Targeting domain binds to paxillin, forming a bridge between the cytoskeleton and ECM receptors (Figure 2) (Hayashi et al., 2002). FAK membrane recruitment at the FA site promotes mechanical-induced conformational changes of FAK dimers, triggering its autophosphorylation, which in turn may favor the binding of Src and the consequent activation of FAK resulting in an active FAK/Src complex (Wu et al., 2015). Beyond the FA site, the FAK has an affinity for other growth factors or cytokine receptors (e.g.: Epidermal Growth Factor Receptor) that can reinforce FAK activity or promote its degradation (Chen and Chen, 2006; Sieg et al., 2000). In addition, phosphatidylinositol 4,5-bisphosphate has a higher avidity for FAK dimers (Brami-Cherrier et al., 2014), so FA sites become enriched in this phospholipid, providing substrates for actin polymerization and FAK-associated downstream cell signaling (Figure 2) (Goni et al., 2014). Therefore, FAK aberrant transduction might have a role in cardiac fibroblast aging; that is, the source of the aging defect might be on the FA dock (talin/paxillin/kindlin) or via Src recruitment (Figure 2).
5.4. Mechanosensing mechanism in fibroblasts
In female human lung fibroblasts, mechanical cell signaling, and activation of gene expression have been described as FAK-dependent through PI3K/AKT/mTOR mobilization (Xia et al., 2004). Mechanically induced biochemical responses driven by FAK include some GTPases (Rho/MDia, Rac, cdc42) and actin-binding proteins (N-WASP, Arp2/3), which regulates microtubules and actin cytoskeletal assembling/disassembling dynamics (Figure 2) (Romero et al., 2020; Shen et al., 2013). The myogenic transcriptional factor Myocyte Enhancer Factor 2a is also reported to be downstream from FAK activation (Peng et al., 2008). Although this specific FAK/Myocyte Enhancer Factor 2a axis might be disputable in fibroblast mechanosensing, other phosphorylation-dependent transcription or co-transcription factors involved in myofibroblast gene programs will follow this path, as will be discussed later in the nucleus section. Finally, MAP kinases, especially JNK and ERKs, represent main downstream effectors of FAK (Figure 2). JNK in turn, activates YAP/TAZ, SMAD4, and NFAT4, thus favoring cell survival, differentiation, and motility, and therefore the myofibroblast gene program (Zeke et al., 2016), as will be discussed below. Cardiac fibroblasts derived from the old heart have reduced activated JNK levels when compared with fibroblasts derived from the young mouse heart, supporting the notion that the FAK-JNK pathway is defective in the aging heart (Cieslik et al., 2011b).
In conclusion, FA ensures the connection between the ECM and the actin cytoskeleton, thus representing the main initiator of the intracellular signaling in response to ECM tension. As ECM composition changes in the aging heart, FA activity and assembly may be affected downstream. Yet, currently, only a few clues have been accumulated. For instance, an increase of vinculin, which bridges FA to the actin filament, seems to take part in an aging-remodeling process, while a decrease of paxillin is reported in aged skin fibroblasts. The altered JNK pathway in aging cells may support the concept of an affected FA signaling in aging cardiac fibroblasts, which requires further investigations. Considering the numbers of proteins within the integrome, the impact of FA is probably underestimated.
6. Cytoskeleton mechanotransduction
6.1. Actin, as a part of the cytoskeleton
The cytoskeleton rearrangement represents the mechanical response to ECM tension. In fibroblasts, this backbone includes a more-or-less dense meshwork of F-actin (also called microfilaments), a connecting network of microtubules and a polarizing frame of intermediate filaments of vimentin (Grabec et al., 2000; Ochsner et al., 2010). This specialized cytoskeleton thus provides mobility to the fibroblasts and contractile abilities to the mature myofibroblasts during wound resolution. Although vimentin is mentioned in a few cases of invading fibrosis, such as in idiopathic pulmonary fibrosis (Li et al., 2017a), and in aging human female dermal fibroblasts (Sliogeryte and Gavara, 2019), the mechanotransduction in cardiac fibroblasts involves mostly the actin cytoskeleton, on which microtubules play a supportive role (Omelchenko et al., 2002). Fibroblasts have a reduced ability to differentiate into myofibroblasts during aging, while their cytoskeleton exhibits a network of F-actin that is impaired (Table 2) (Cieslik et al., 2011b). Consequently, the deterioration of the actin cytoskeleton may be a part of the aging process in cardiac fibroblasts. Age-related changes can affect not only the actin dynamic treadmill but also actin(s) expression itself, as we have demonstrated before (Table 2) (Cieslik et al., 2011b) and both can apply a regulatory feedback to ECM, integrin and receptor signaling and gene transcriptions that can exacerbate age-related defects.
Table 2.
Actin Dynamics in Mechanosensing Downstream from the Focal Adhesion.
| The role of actin in mechanotransduction | ||
|---|---|---|
| Normal role in fibroblast physiology | Age-related changes | |
| Actin as part of the cytoskeleton | In mechanosensing, the actin cytoskeleton rearranges in response to ECM tension (Grabec et al., 2000) and takes part in fibroblast-myofibroblast differentiation (Omelchenko et al., 2002). | Aging is associated with drastic biochemical changes that may alter actin polymerization and expression (Cieslik et al., 2011; Lai and Wong, 2020). Oxidative stress can alter actin turnover and/or filament organization, due to ROS or methionine sulfoxidation imbalance (Aberle, 2013; Varland et al., 2019). |
| The impact of oxidative stress in actin polymerization has been demonstrated in eukaryotic cells (Farah et al., 2011) and adult cardiomyocytes (Angelini et al., 2020) but has not been shown in cardiac fibroblasts. | ||
| Actin in fibroblast maturation | During their maturation, myofibroblasts express a-SMA, a contractile actin isoform, in their cytoskeleton (Hinz et al., 2001, 2003); this favors wound healing (Hinz, 2006; Santiago et al., 2010). | Aged cardiac fibroblasts show decreased a-SMA isoform, along with a decreased and disorganized F-actin network leading to impaired cardiac fibroblast contractility (Cieslik et al., 2011). |
| Actin and mRNA “zip-coding” | The fibroblast is the only cell type in which mRNA zip-coding (cytoplasmic sorting of mRNA) persists after embryogenesis (Jansen, 2001; Lopez de Heredia and Jansen, 2004). | Modulation of mRNA turnover by age-related factors (Borbolis and Syntichaki, 2015) could potentially impair mRNA zip-coding. Its role remains unknown in the aging heart. |
| β-actin mRNA is reported to be extensively zip-coded and enriched at the FA site (Bassell et al., 1998). | ||
| Integrin signaling promotes ZBP phosphorylation, and therefore mRNA zipcoding in fibroblasts (Bassell et al., 1998; Chicurel et al., 1998; Kislauskis et al., 1994; Shestakova et al., 2001). | ||
| F-actin binds ZBP-1 and ensures the zip-coded mRNAs localization through polymerization (Nicastro et al., 2017). | ||
6.2. Actin in fibroblast maturation
The myofibroblast ensures tissue integrity and efficient healing processes, due to its ability to apply contractile force to a surface, as demonstrated in lung and cardiac fibroblasts from both male and female rats (Hinz, 2006; Santiago et al., 2010). Aside from the application of isometric force (Gardel et al., 2008), myofibroblasts exhibit a dense contractile apparatus that differs from the migratory structures. In fact, cytoplasmic β- and γ-actin isoforms are the ones involved in cell morphology and motility, whereas the myofibroblast cytoskeleton is highly enriched in α-SMA (Hinz et al., 2001; Hinz et al., 2003), an alpha-isoform that is usually found in contractile muscle cells such as striated myocytes (Clement et al., 2007) and smooth muscle cells (Chamley et al., 1977) in both male and female tissues. Despite extensive studies from the 1970s, it remains unclear whether α-SMA is the exclusive actin isoform within the myofibroblast contractile apparatus or if it co-polymerizes with β-/γ-actins. Yet, the importance of α-SMA in myofibroblast contractile force is reported both in vivo and in vitro: notably, the induction of Acta2 gene expression (encoding α-SMA) is enough to dramatically increase the traction force of the myofibroblast (by 2 to 3-fold compared with the isometric tension applied to quiescent cells) in cells originated from both male and female rats (Chen et al., 2007; Goffin et al., 2006; Hinz et al., 2001; Hinz et al., 2003). In the aging male mouse heart, the decrease in α-SMA level is one of the main aging features we discovered in cardiac fibroblasts (Table 2), which is paired with a lower amount of and disorganized F-actin network and consequently impaired contractility on a collagen matrix in vitro (Cieslik et al., 2011b).
6.3. The cytoskeleton of aging cells
Globally, aging impairs actin polymerization due to upstream changes in cell signaling (from FA and beyond) and local decrease in ATP availability, as well as downstream protein misfolding and a decrease in protein homeostatic expression (Lai and Wong, 2020). Additionally, oxidative stress can depolymerize oxidized actin, by a non-enzymatic process, leading to F-actin crosslinking and G-actin misfolding (Farah et al., 2011; Varland et al., 2019), but also by the catalytic activity of Molecule Interacting with CasL, a methionine oxygenase, and Methionine-R-sulfoxide reductases (Aberle, 2013). By these redox enzymes, actin can thus be sulfoxidized, which promotes its depolymerization and prevents new filament formation (Giridharan and Caplan, 2014; Grintsevich et al., 2017; Hung et al., 2011; Ilani and Fass, 2013). The activity of these Methionine-R-sulfoxide oxygenases and reductases is known to be involved both in aging (Lourenco Dos Santos et al., 2018) and in fibroblast and macrophage actin homeostasis (Giridharan and Caplan, 2014; Lee et al., 2013), but there is no current link established in the specific case of aging cardiac fibroblasts. However, oxidative stress is elevated in cardiac fibroblasts derived from aged hearts (Cieslik et al., 2014) and a similar mechanism has been recently demonstrated in cardiomyocytes (Angelini et al., 2020). Considering the high conservation of actin function, we hypothesize that an oxidative stress/actin double axis might also be involved in the dysfunction of aging cardiac fibroblasts. This is the reason why redox-signaling and metabolic function gauged by AMP Kinase (Gu et al., 2018) are foreseen as potential target(s) in injury-caused deleterious myofibroblast differentiation (Sampson et al., 2012). But, in the aging heart, since myofibroblast transition is impaired (rather than intensively triggered), this kind of drug therapy may not be helpful. In human skin fibroblasts (from both sexes), aging-mimicking artificial F-actin disorganization rapidly decreases TGFβ-RII in those cells, leading to a consequent reduction of collagen expression (Qin et al., 2018). We have found in the aging fibroblasts of the male mouse heart a reduced expression of both TGF-β receptors and disorganized F-actin (Cieslik et al., 2011b), which may suggest additional roles of actin.
6.4. Actin and mRNA “zip-coding”
The role of actin extends beyond the cytoskeleton and it includes transcriptional and post-transcriptional activities that may be equally affected by aging. Actin is notably involved in the mRNA “zip-coding”, a recently described mechanism that regulates the subcellular localization and translation of mRNAs (Jansen, 2001; Lopez de Heredia and Jansen, 2004). First, F-actin binds to zip-code-binding protein 1 (ZBP-1), a highly-conserved RNA-binding protein that recognizes a specific motif in the 3’UTR of these “zip-coded” mRNAs and ensures their mobility through an actin polymerization pushing force (Nicastro et al., 2017). Interestingly, fibroblasts represent one of the few cell types in which such a mechanism persists after embryogenesis, which may suggest an essential role in its specific function. Notably, β-actin mRNA is one of the most extensively zip-coded mRNAs and it is specifically enriched at the FA site during lamellipodia formation and directionality, but also along actin stress fibers (Bassell et al., 1998; Kislauskis et al., 1994; Shestakova et al., 2001). During the past few years, clear feedback has been demonstrated between integrin signaling, F-actin and mRNA zip-coding in fibroblasts, which could suggest mRNA zip-coding to be a concrete part of the mechanosensing in this cell (Chicurel et al., 1998). While integrin signaling promotes ZBP-1 phosphorylation by Src and so its further activation, β-actin mRNA zip-coding is ZBP-1/F-actin-dependent, and both processes enhance FA assembling and fibroblast mobility (Chicurel et al., 1998; Huttelmaier et al., 2005; Katz et al., 2012). For years, mRNA processing has been suspected to be affected by aging (as reviewed in (Borbolis and Syntichaki, 2015); therefore, impairment of mRNA zip-coding may be involved during aging (Table 2).
6.5. Actin at the core of the transcription machinery
Apart from its role in the cytoplasmic sorting of mRNA, actin can also be engaged in nuclear transcription. It has been demonstrated that nuclear actin can associate with all the three RNA polymerases (I, II and III) and is thus necessary for RNA transcription (Table 2) (Fomproix and Percipalle, 2004; Hu et al., 2004; Percipalle et al., 2003; Philimonenko et al., 2004; Serebryannyy et al., 2016; Visa, 2005). Therefore, the impairment of actin turnover in aging cardiac fibroblasts may also affect the activity of these different RNA polymerases, hence the whole gene expression machinery, although the mechanism has not been well explored. The role of actin in gene expression is not limited to its docking function for RNA polymerases (Table 2). In fact, it is also well-known that the actin treadmill takes an active part in the regulation of the SRF/MRTFA axis, as will be described below.
Actin represents the mechanical core of the cardiac fibroblast, ensuring both cellular function and signaling (Table 2). During the myofibroblast maturation and healing process, the actin cytoskeleton transits to a network of α-SMA enriched contractile fibers, and we previously demonstrated that this ability is specifically impaired in cardiac fibroblasts of the aging heart. In addition, albeit no direct link has been established in the specific case of aging cardiac fibroblasts, many features of aging, such as oxidative stress, are known to have an impact on the actin treadmill (F-/G-actin imbalance) (Table 2). The role of actin is extremely complex within the cells since it can be involved in RNA polymerases complex formation, but also in gene transcription and both are known to be affected in aging.
7. Nucleus and gene transcription
Myofibroblast gene program transcription is the ultimate step of mechanosensing. In fibroblasts from the aging heart, the alterations that are accumulated from mechanosensing lead to a decrease in α-SMA expression and extensive changes in ECM production. Therefore, a combination of alterations within different transcriptional axes may be involved. Since many of them are interconnected, the story in the nucleus is complex and remains elusive for some parts. Here we will focus on the ones that are important for mechanosensing and aging, such as Serum Response Factor (SRF), Myocardin-Related Transcription Factor-A (MRTF-A), Yes-associated protein (Yap) and Transcriptional co-Activator with a PDZ motif (Taz) (as summarized in Figure 3 and Table 3). Several age-related epigenetic and post-transcriptional modifications will also be discussed below.
Figure 3: Signaling in the nucleus that regulates the myofibroblast gene program.
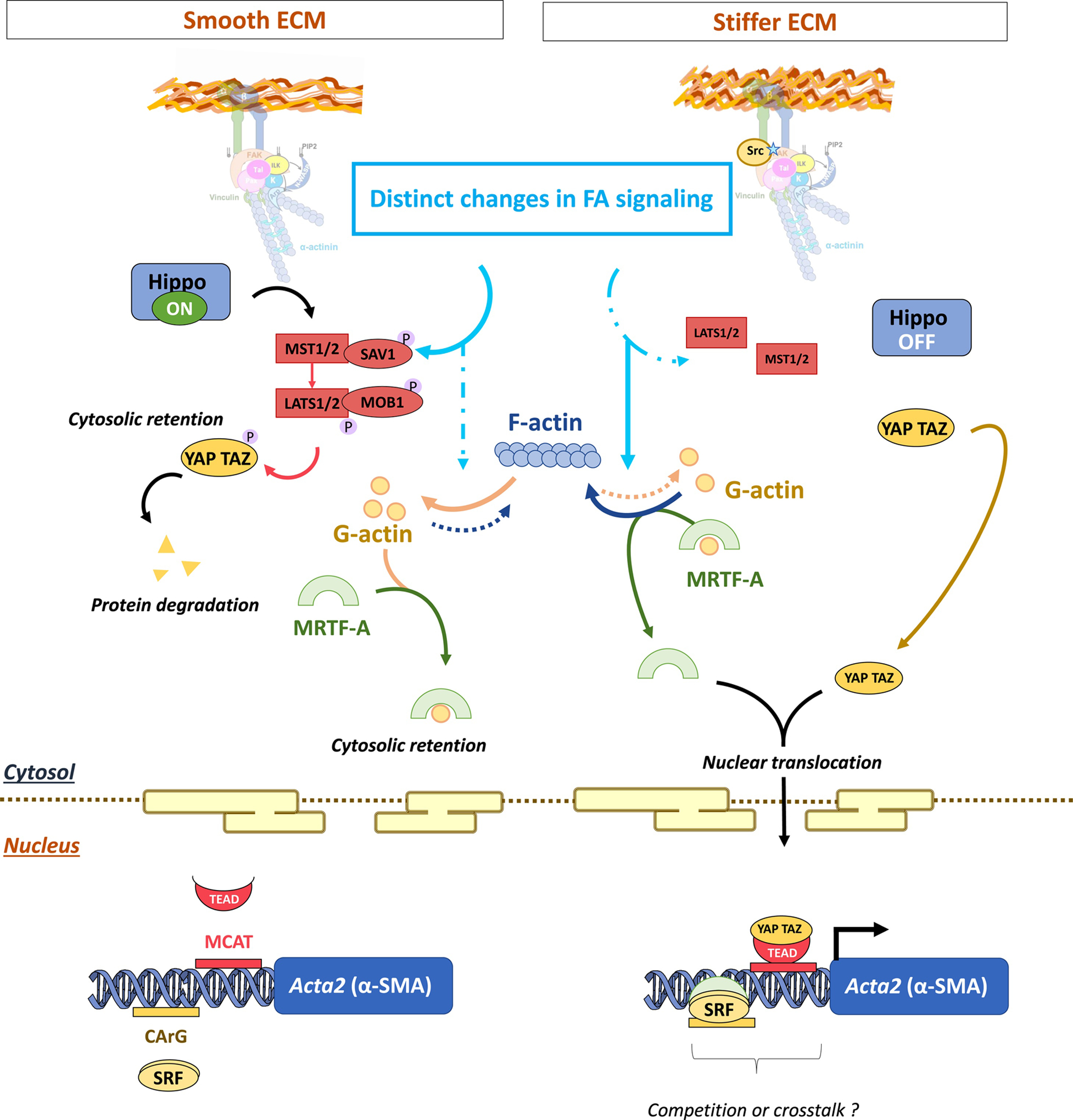
The mechanosensing differs between an elastic (left) or a stiffer (right) ECM, leading to downstream impacts on MRTF-A and Yap/Taz shuttling dynamics. The DNA binding sites of SRF and TEAD transcription factors (CArG and MCAT boxes, respectively) can overlap within the promoter region. So, both MRTF-A and Yap/Taz share common target genes, suggesting crosstalk or competitions. This crosstalk seems to be notably involved in the α-SMA expression.
Table 3.
Mechanosensing and Gene Regulation in the Maturation of Cardiac Fibroblasts.
| Transcription factors in the regulation of α-SMA expression | ||
|---|---|---|
| Normal role in fibroblast physiology | Age-related changes | |
| SRF/MRTF-A | SRF/MRTF-A is involved in the myofibroblast maturation gene program (Esnault et al., 2014; Small, 2012) and in α-SMA expression (Medjkane et al., 2009; Olson and Nordheim, 2010; Selvaraj and Prywes, 2004). | A two-fold increase in SRF level in the heart has been seen in aging (Lu et al., 1998; Takahashi et al., 1992). The upregulation of SRF remains unknown in aging cardiac fibroblasts. |
| MRTF-A is actin-regulated: G-actin promotes its cytosolic retention (Miralles et al., 2003; Percipalle et al., 2003; Posern et al., 2004, 2002). | SRF promoter-binding capacity is impaired with aging and pressure overload (Lu et al., 1998; Zhang et al., 2011). | |
| F-actin formation from mechanical/biochemical stimuli promotes MRTF-A translocation to the nucleus and dimerization with SRF (Miralles et al., 2003; Posern et al., 2004, 2002). | Competition with other transcription factors or diminished co-factor availability may alter the final gene transcript (Esnault et al., 2014; Lin et al., 2007, 2008; Prywes and Zhu, 1992; Sun et al., 2009). | |
| Its role is documented in cardiomyopathy models (Chang et al., 2003; Davis et al., 2002) as well as in fibrosis and scar formation (Gary-Bobo et al., 2005; Parlakian et al., 2005). | Age-related actin depolymerization (Farah et al., 2011; Varland et al., 2019) may lead to MRTF-A cytosolic retention. | |
| Yap/Taz | Yap/Taz integrates the FA signaling as part of the mechanosensing response (Ma et al., 2019; Piccolo et al., 2014). | The role of Yap/Taz remains unknown in the aging cardiac fibroblasts. Stiff ECM, as seen in the aging heart, promotes Yap/Taz nuclear translocation and competition with SRF/MRTF-A (Liu et al., 2015). |
| Yap/Taz is sensitive to ECM tension via the Hippo pathway; an elastic matrix favors Yap/Taz degradation (Ma et al., 2016). | ||
| Yap/Taz activates myofibroblast differentiation in a model of dilated cardiomyopathy (Jin et al., 2018). | ||
| Yap/Taz leads to repression of TGF-β signaling via beta-catenin degradation affecting myofibroblast gene program (Azzolin et al., 2014; Qin et al., 2018; Xu et al, 2014). | ||
| Yap/Taz antagonizes the SRF/MRFT-A transcriptional axis on α-SMA expression (Speight et al., 2016). | ||
7.1. SRF/MRTF-A
SRF/MRTF-A represents a major transcriptional axis that translates mechanosensing into gene expression, triggering multiple possible cell fates from survival to differentiation and migration. SRF is a ubiquitous and highly conserved transcription factor that affects an extensive list of more than 8000 target genes, including its own gene as a main target (Khanday et al., 2016; Sun et al., 2006). MRTF-A is an SRF co-factor that is actin-regulated (Miralles et al., 2003; Posern et al., 2004; Posern et al., 2002). In resting cells, monomers of actin (G-actin) bind to MRTF-A, leading to MRTF-A cytosolic retention, while mechanical or biochemical stimuli (either ECM tension or growth factor signaling) promote F-actin formation, and thus the release of MRTF-A from G-actin, finally promoting its nuclear translocation and local binding to the SRF dimer (Figure 3). Altogether, the SRF/MRTF-A axis notably regulates the cytoskeleton gene program, including the expression of all the actin isoforms (from β-/γ- to α-SMA) (Medjkane et al., 2009; Olson and Nordheim, 2010; Selvaraj and Prywes, 2004). Therefore, there is constant feedback between actin and SRF/MRTF-A. The preponderant role of SRF/MRTF-A in stress-induced myofibroblast differentiation is well-known and affected by RhoGTPase-associated actin dynamics (Esnault et al., 2014; Small, 2012). In addition, MRTF-A deficient adult mice develop reduced fibrosis and scar in response to isoproterenol stress or myocardial infarction (Small et al., 2010). In the literature, many studies have revealed the critical role of SRF (activity or protein level) in clinical cases and experimental models of cardiomyopathies (Chang et al., 2003; Davis et al., 2002), both leading to deleterious fibrosis (Gary-Bobo et al., 2005; Parlakian et al., 2005). The involvement of SRF during myocardial aging is also well documented, but currently, studies in the field have been mainly focused on whole-heart or cardiomyocyte-specific approaches. Therefore, the link between SRF/MRTF-A remains hypothetical in cardiac fibroblast aging. In the heart of old male and female rats, SRF basal protein level is greater than 2-fold increased, but it cannot be subsequently increased in response to myocardial infarction or pressure overload as it usually is in the young rat heart (Lu et al., 1998; Takahashi et al., 1992). This establishes a correlation between aging, maladaptive cardiac remodeling and SRF activity. In addition, mild overexpression of Srf gene in the male mouse heart, either total or cardiomyocyte-specific, leads to mild age-related features, including myocardial dysfunction and higher collagen deposition (Angelini et al., 2015; Zhang et al., 2003). Interestingly, the fact of artificially maintaining a youthful level of SRF in the heart of old mice trends to be beneficial against the long-term cardiac alterations due to isoproterenol stress (Azhar et al., 2007). Hence, SRF level and activity can be considered as a homeostatic gauge in cardiac cells. Noteworthy, SRF binding capacity to the promoters of its target genes is also affected during aging (Table 3) (Lu et al., 1998; Zhang et al., 2011). This specific alteration can be due to extensive defects in terms of cofactor availability or competition with other transcription factors. In fact, on the one hand, the inhibitory competition of co-factors (“squelching”) and MRTF-A retention are indeed reported (Lin et al., 2007; Prywes and Zhu, 1992). On the other hand, many SRF binding sites overlap with docking sites for other transcription factors (such as Yap/Taz or Nuclear factor of activated T-cells 4) that may thus compete for binding (Esnault et al., 2014; Sun et al., 2009), which could finally alter or even antagonize the global gene response. In the context of the aging heart, as previously mentioned, the defective mechanosensing leads to a higher rate of depolymerized actin, which may thus favor MRTF-A cytosolic retention, to the detriment of downstream SRF transcriptional balance. We hypothesize that age-related increased expression of SRF may be due to complex compensatory feedback, as an ultimate attempt to switch the transcriptional program back into an optimal adaptive range.
7.2. Yap/Taz
Paired with SRF/MRTF-A, Yap/Taz coactivators represent indispensable sensors and integrators of mechanosensing. A dual modulation exists for this dimer, through Hippo pathway-dependent or -independent pathways (extensively reviewed in (Ma et al., 2019; Piccolo et al., 2014) and (Totaro et al., 2018), respectively). Briefly, in resting cells, Yap/Taz can be phosphorylated by the Hippo pathway cascade of kinases (MST1/2, then LATS1/2), leading to their cytosolic retention and consequent degradation (Figure 3) (Ma et al., 2016). The absence of the Hippo pathway is thus reported to promote the nuclear translocation of the dimer in different cell lines (Zhang et al., 2017). But, as previously mentioned, Yap/Taz is also a substrate of FA downstream signaling via RhoGTPase (Rac1/p21 Activated Kinase) and Src, proteins that are both activated in response to FA formation and subsequent FAK activation (Lamar et al., 2019; Ma et al., 2015). However, since Rac1 and Src are also involved in F-actin formation and maintenance, Yap/Taz activation is thus equally dependent on cytoskeleton reorientation in response to ECM tension. In accordance with this concept, it has been demonstrated that a stiffer ECM, as it is developed in the aging heart, trends to promote Yap/Taz translocation, while a more elastic ECM favors their degradation (Figure 3) (Liu et al., 2015a). The involvement of Yap/Taz in myofibroblast differentiation is reported in the liver, skin and lung (reviewed in (Noguchi et al., 2018)), whereas a recent study suggests that increased activation of Yap/Taz promotes myofibroblast differentiation in a mouse model of dilated cardiomyopathy (Jin et al., 2018). Little is known about their involvement in aging cardiac fibroblasts, but recent reports suggest the importance of several complex crosstalks. First, Yap/Taz can facilitate the degradation of β-catenin or the repression of TGF-β signaling (through the expression of inhibitory SMAD7) (Azzolin et al., 2014; Qin et al., 2018; Xu et al., 2014), that are both involved in the myofibroblast gene program. In addition, this dimer can synergistically together with SRF/MRTF-A bind to promoters in cancer-associated fibroblasts (Foster et al., 2017), as well as in pro-fibrotic processes (Bialik et al., 2019). Recently, the role of crosstalk between SRF/MRTF-A and Yap/Taz has been specifically described as a critical modulator of α-SMA expression (Figure 3) (Speight et al., 2016). In this specific model, there is reciprocal inhibition between MRTF-A and Taz, leading to antagonistic effects on α-SMA expression that are context-dependent but mechanosensitive. It remains unclear whether such an interplay between MRTF-A and TAZ does exist in cardiac fibroblasts, and perhaps even plays a role during aging (Table 3). However, this should represent an interesting subject of investigation for the future.
7.3. Sirtuins
In addition to acute transcriptional effects, the persistence of a triggered gene program requires longer-term modification of the chromatin opening frame shift (Lai and Pugh, 2017; Sartorelli and Puri, 2018; Stillman, 2018). In multiple models of fibrosis, it has been indeed demonstrated that fibroblasts accumulate histone modifications, such as methylation or acetylation (Chelladurai et al., 2019; O’Reilly, 2017). Sirtuins represent a metabolic-sensitive family of histone deacetylases that are specifically involved in myofibroblast maturation and fibrotic development (Cencioni et al., 2015; Ponnusamy et al., 2015; Wyman and Atamas, 2018). Among the different Sirtuin family members, Sirtuin-1 plays a preponderant role in α-SMA and ECM gene expression although through a chemical TGF-β/SMAD3 pathway in fibroblasts (Zerr et al., 2016). The use of resveratrol (activator of Sirtuin 1) mitigates doxorubicin-induced fibrosis and diastolic dysfunction, notably by promoting the decrease in α-SMA and metalloproteinase 2, an ECM modifying enzyme (Cappetta et al., 2016). Interestingly, some of these dysfunctions are alleviated by the activation of the Sirtuin1/AMP kinase axis (Gu et al., 2018). Therefore, a feedback between ECM stiffness modulation, mechanosensing response and Sirtuins may exist, potentially contributing to the phenotype of the aging cardiac fibroblast.
Cell senescence is also associated with metabolism imbalance and oxidative stress generation (Gu et al., 2018), to which Sirtuins are particularly sensitive (Gambini et al., 2011). Therefore, there may be a continuous combination of crosstalk and feedback between both metabolic and mechanical aspects in the aging fibroblast that could be orchestrated through the Sirtuins.
In the nucleus, mechanosensing is translated into a transcriptional program that initiates the myofibroblast maturation. Here, we focused our attention on two main transcriptional axes that seem to be relevant to mechanosensing. In fact, the SRF/MRTF-A axis is actin regulated, and SRF expression and regulation is known to be affected in the aging heart, while Yap/Taz has been described as being sensitive to ECM stiffness (Figure 3). SRF/MRTF-A and Yap/Taz share promoter regions (Figure 3); therefore they may inhibit each other’s binding. In addition, here we discussed the chemical impact of the metabolic-sensitive deacetylases Sirtuins, which are known to be particularly involved in aging. Sirtuins, and notably Sirtuin-1, can have an impact on α-SMA transcription and ECM composition via the TGF-β/SMAD pathway, but the role of Sirtuins remains unknown in the aging cardiac fibroblast and thus represents an interesting research topic for future investigations.
8. Impact of cardiac cell senescence on fibroblast mechanosensing
During cardiac aging, some alterations to the fibroblast phenotype are in response to changes in other cell types. For example, changes in the cardiomyocyte phenotype and function can result in functional and structural changes in the heart. Cardiomyocytes in the aging heart gradually become dysfunctional, and they die (Bernhard and Laufer, 2008; Sheydina et al., 2011). The decreased ratio of cardiomyocytes to non-cardiomyocytes in the aging heart (Anversa et al., 1990) results from changes in three different mechanisms: proliferation, apoptosis, and senescence. The low rate of postnatal cardiomyocyte proliferation is even lower in the aging heart, decreasing to 0.45% per year (Anversa et al., 1990). Resulting from stress, which comes from a life-long accumulation of damages, molecular changes in pathways governing survival are altered, resulting in increased cell death and reduced survival. Even though some components of the caspase cascade are decreased in aging, there is a consensus that cardiomyocyte death is increased in the aging heart (Goldspink et al., 2003). Cell death by necrosis induces inflammation (Rock and Kono, 2008) and leads to the generation of reactive oxygen species that particularly alter mitochondrial homeostasis, further leading to changes in energy generation and compromised function. To compensate for the cardiomyocyte loss and as a result of ongoing low-grade inflammation, fibroblasts increase their production and processing of ECM, which affects their mechanosensing pathway. Eventually, due to the paracrine effects of neighboring cells and as a response to the altered environment, fibroblasts acquire a senescent phenotype. The senescence-associated secretory phenotype is characterized by reduced proliferation and enhanced secretion of cytokines, ECM components, and proteases that further contribute to cardiac remodeling and dysfunction (Xie et al., 2017).
9. Innovation
We have recently examined fibroblast activation in the aging heart (Trial and Cieslik, 2018), where we concentrated on the role of fibroblasts in the pathological processes such as reparative fibrosis (healing after myocardial infarction) and adverse interstitial fibrosis. We also covered various pathways leading to activation of fibroblasts such as canonical TGF-β-Smad, ERK1/2, angiotensin II, and adrenergic β2 pathways and finally addressed the issue of fibroblast heterogeneity.
In this current review, we concentrated on the pathway leading to α-SMA activation, but instead of the TGF-β dependent pathway, here we described the differences in mechanosensing. Mechanosensing represents an essential component in response to ECM tension and is a critical effector of the fibrotic reaction in myofibroblasts. We have reviewed the effector mechanisms operative in this response and proposed the possible major modulators of fibrosis in the aged heart. We specifically detailed changes (that occur in the aging fibroblast or heart) that have an impact on FA, actin polymerization and transcription factor translocation to the nucleus that affect α-SMA transcriptional program. We caution that some of the studies cited include data from other biological models extrapolated to our model; our results represent a hypothesis that will serve in constructing future studies.
10. Conclusions
In the aging heart, the mechanosensing driving force is altered at critical checkpoints, which finally leads to an impaired myofibroblast maturation (Figure 4). Briefly, ECM composition undergoes an enrichment in collagen I, biglycan, decorin, glycoproteins and FNEDA variants and a decrease in elastin. These severe changes, resulting in a stiffer matrix, apply a drastically different mechanical tension to the cells (Figure 4). FA sites are at the frontline of ECM changes, and they are particularly sensitive to this tension shift. Hence, not surprisingly, downstream age-related modifications include the effectors of the FA core complex, as well as changes in integrin subunits, an increased protein level of vinculin, and a decrease in JNK activity (Figure 4). Regarding the complexity of the adhesome that coordinates FA assembling, dynamics and signaling, the role of the FA core certainly remains underestimated and may require more in-depth investigations in the future.
Figure 4: Alterations of mechanosensing in fibroblasts from aged hearts.
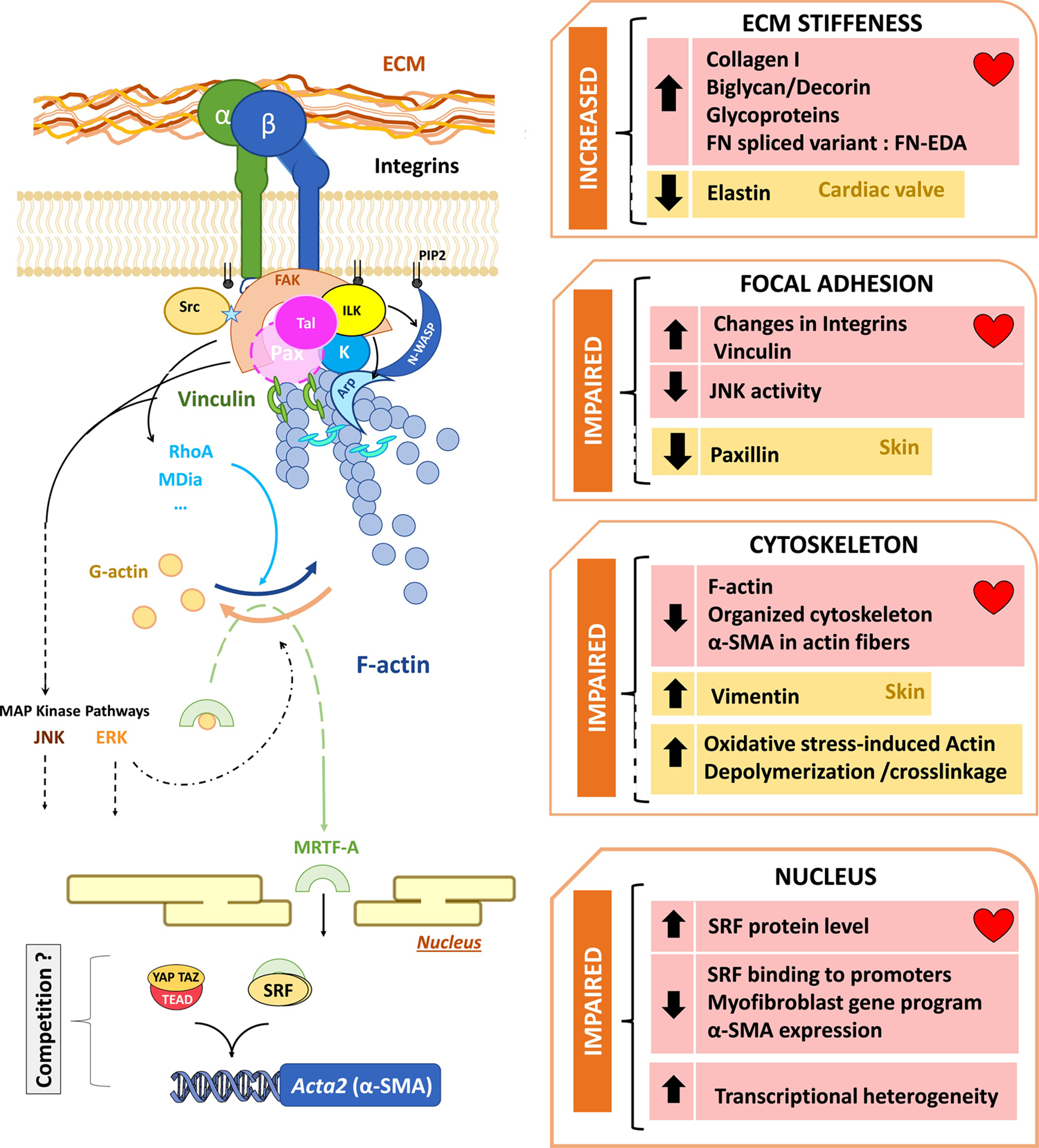
An altered extracellular matrix (ECM) composition is causally related to a changed phenotype in fibroblasts, leading to impairment at the FA, affecting the downstream cytoskeleton treadmill and myofibroblast gene program, which worsens the negative feedback of the mechanosensing. Evidence concerning the role of mechanosensing in aging fibroblasts has been accumulated both in the myocardium (pink squares) or other tissues (yellow squares).
The cytoskeleton, and especially the actin treadmill, represents the true mechanical part of the mechanosensing axis, but it also works as an integrative platform for both downstream and upstream signaling effectors. The decrease in F/G actin ratio is a typical age-related feature that has been reported by other teams (Lai and Wong, 2020) and by us (Cieslik et al., 2011b), in both cardiac fibroblasts and other cell types (Figure 4). Furthermore, we also demonstrated that the decreased α-SMA expression is one of the main deleterious features of impaired cardiac myofibroblast maturation, leading to weaker contractile abilities. Yet, these findings remain empirical, since little is known about the precise mechanism that promotes actin cytoskeleton disorganization. A link between FA signaling and actin can be strongly suspected, but it only remains extrapolated from the literature in other cell types or acute models. In this sense, there are also indirect clues about the impact of fibroblast aging on actin itself, in terms of post-translational modifications. The enzymatic-redox balance that has been recently described as a part of the actin dynamic treadmill could be a part of the aging-affected machinery as well. But this remains hypothetical and may require extensive clarifications in cardiac fibroblasts.
Beyond its role in the cytoskeleton, actin also takes an active part in transcriptional machinery and notably in the SRF/MRTF-A axis that is known to trigger the myofibroblast gene program and especially α-SMA gene expression. Consequently, an altered actin treadmill may affect SRF/MRTF-A transcriptional activity directly and also RNA transcription, thus leading to the rapid shutdown of myofibroblast transition. Noteworthy, SRF binding capacity to the promoter of its target genes is indeed altered in the aging human and rodent heart (Lu et al., 1998; Takahashi et al., 1992; Zhang et al., 2011), while SRF protein level is increased (Lu et al., 1998), suggesting feedback adaptation (Figure 4). Nonetheless, observations from the last decades have not provided a direct link between fibroblast aging and SRF activity. In addition, a stiffer matrix is also known to impact another crucial transcriptional axis; the Yap/Taz and Hippo pathways, that have been extensively studied in cardiomyocyte regeneration and reprogramming (Liu et al., 2015a). Yet, because this change in matrix composition after injury in young hearts resembles ECM modification in the aging heart (Figure 4), we could logically suspect an indirect impact of ECM on Yap/Taz transcriptional axis, which remains mainly unexplored. In addition, an interesting crosstalk/competition relationship has been recently described between Taz and MRTF-A (Speight et al., 2016). However, there is still a missing experimental link in the case of aging cardiac fibroblasts.
Last but not least, recent transcriptional data suggest heterogeneity in aging fibroblast populations (Ali et al., 2014; Farbehi et al., 2019; Mahmoudi et al., 2019; Vidal et al., 2019), in such a way that there might be not only a unique defective signature but a heterogeneous population of aged and young-like cells, which makes the whole complexity challenging to address by either in vitro or in vivo studies (Figure 4).
Finally, a better understanding of mechanosensing (in the specific case of cardiac fibroblasts in the aging heart) remains an important way to address and explore these recurrent questions about myocardial integrity and dysfunction in the elder heart.
Highlights.
Mechanosensing is the ability of a cell to respond to mechanical signals such as force or rigidity
Fibroblasts transition into contractile myofibroblasts upon chemical and mechanical signals
Matrix, integrins, focal adhesions and cytoskeleton play essential parts in mechanosensing
In the aging heart, myofibroblast transition is impaired leading to dysfunction after infarct
Acknowledgements
We thank Dr. Mark Entman for helpful discussion. We also thank Sharon Malinowski for editorial assistance. This work was supported by a grant from the National Institute on Aging (AG059599).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
None.
References
- Aberle H, 2013. Redox switch for actin. Nat Cell Biol 15, 1403–1404. [DOI] [PubMed] [Google Scholar]
- Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM, Thodeti CK, 2013. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol 54, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alex L, Russo I, Holoborodko V, Frangogiannis NG, 2018. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol 315, H934–H949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Muller AM, Volz KS, Tang Z, Red-Horse K, Ardehali R, 2014. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res 115, 625–635. [DOI] [PubMed] [Google Scholar]
- Angelini A, Gorey MA, Dumont F, Mougenot N, Chatzifrangkeskou M, Muchir A, Li Z, Mericskay M, Decaux JF, 2020. Cardioprotective effects of alpha-cardiac actin on oxidative stress in a dilated cardiomyopathy mouse model. FASEB J 34, 2987–3005. [DOI] [PubMed] [Google Scholar]
- Angelini A, Li Z, Mericskay M, Decaux JF, 2015. Regulation of Connective Tissue Growth Factor and Cardiac Fibrosis by an SRF/MicroRNA-133a Axis. PLoS One 10, e0139858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antia M, Baneyx G, Kubow KE, Vogel V, 2008. Fibronectin in aging extracellular matrix fibrils is progressively unfolded by cells and elicits an enhanced rigidity response. Faraday Discuss 139, 229–249; discussion 309–225, 419–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anversa P, Palackal T, Sonnenblick EH, Olivetti G, Meggs LG, Capasso JM, 1990. Myocyte cell loss and myocyte cellular hyperplasia in the hypertrophied aging rat heart. Circ Res 67, 871–885. [DOI] [PubMed] [Google Scholar]
- Asif M, Egan J, Vasan S, Jyothirmayi GN, Masurekar MR, Lopez S, Williams C, Torres RL, Wagle D, Ulrich P, Cerami A, Brines M, Regan TJ, 2000. An advanced glycation endproduct cross-link breaker can reverse age-related increases in myocardial stiffness. Proc Natl Acad Sci U S A 97, 2809–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azhar G, Zhang X, Wang S, Zhong Y, Quick CM, Wei JY, 2007. Maintaining serum response factor activity in the older heart equal to that of the young adult is associated with better cardiac response to isoproterenol stress. Basic Res Cardiol 102, 233–244. [DOI] [PubMed] [Google Scholar]
- Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V, Fassina A, Cordenonsi M, Piccolo S, 2014. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 158, 157–170. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Zhang H, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, Kosik KS, 1998. Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci 18, 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard D, Laufer G, 2008. The aging cardiomyocyte: a mini-review. Gerontology 54, 24–31. [DOI] [PubMed] [Google Scholar]
- Bialik JF, Ding M, Speight P, Dan Q, Miranda MZ, Di Ciano-Oliveira C, Kofler MM, Rotstein OD, Pedersen SF, Szaszi K, Kapus A, 2019. Profibrotic epithelial phenotype: a central role for MRTF and TAZ. Sci Rep 9, 4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bledzka K, Bialkowska K, Nie H, Qin J, Byzova T, Wu C, Plow EF, Ma YQ, 2010. Tyrosine phosphorylation of integrin beta3 regulates kindlin-2 binding and integrin activation. J Biol Chem 285, 30370–30374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbolis F, Syntichaki P, 2015. Cytoplasmic mRNA turnover and ageing. Mech Ageing Dev 152, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaouina M, Goult BT, Huet-Calderwood C, Bate N, Brahme NN, Barsukov IL, Critchley DR, Calderwood DA, 2012. A conserved lipid-binding loop in the kindlin FERM F1 domain is required for kindlin-mediated alphaIIbbeta3 integrin coactivation. J Biol Chem 287, 6979–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahme NN, Harburger DS, Kemp-O’Brien K, Stewart R, Raghavan S, Parsons M, Calderwood DA, 2013. Kindlin binds migfilin tandem LIM domains and regulates migfilin focal adhesion localization and recruitment dynamics. J Biol Chem 288, 35604–35616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Gervasi N, Arsenieva D, Walkiewicz K, Boutterin MC, Ortega A, Leonard PG, Seantier B, Gasmi L, Bouceba T, Kadare G, Girault JA, Arold ST, 2014. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J 33, 356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG, 2008. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol 51, 1384–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, 2017. Focal adhesions: a personal perspective on a half century of progress. FEBS J 284, 3355–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscemi L, Ramonet D, Klingberg F, Formey A, Smith-Clerc J, Meister JJ, Hinz B, 2011. The single-molecule mechanics of the latent TGF-beta1 complex. Curr Biol 21, 2046–2054. [DOI] [PubMed] [Google Scholar]
- Cappetta D, Esposito G, Piegari E, Russo R, Ciuffreda LP, Rivellino A, Berrino L, Rossi F, De Angelis A, Urbanek K, 2016. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int J Cardiol 205, 99–110. [DOI] [PubMed] [Google Scholar]
- Ceccarelli DF, Song HK, Poy F, Schaller MD, Eck MJ, 2006. Crystal structure of the FERM domain of focal adhesion kinase. J Biol Chem 281, 252–259. [DOI] [PubMed] [Google Scholar]
- Cencioni C, Spallotta F, Mai A, Martelli F, Farsetti A, Zeiher AM, Gaetano C, 2015. Sirtuin function in aging heart and vessels. J Mol Cell Cardiol 83, 55–61. [DOI] [PubMed] [Google Scholar]
- Chamley JH, Groschel-Stewart U, Campbell GR, Burnstock G, 1977. Distinction between smooth muscle, fibroblasts and endothelial cells in culture by the use of fluoresceinated antibodies against smooth muscle actin. Cell Tissue Res 177, 445–457. [DOI] [PubMed] [Google Scholar]
- Chang J, Wei L, Otani T, Youker KA, Entman ML, Schwartz RJ, 2003. Inhibitory cardiac transcription factor, SRF-N, is generated by caspase 3 cleavage in human heart failure and attenuated by ventricular unloading. Circulation 108, 407–413. [DOI] [PubMed] [Google Scholar]
- Chelladurai P, Boucherat O, Stenmark K, Kracht M, Seeger W, Bauer UM, Bonnet S, Pullamsetti SS, 2019. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol. [DOI] [PubMed] [Google Scholar]
- Chen J, Li H, SundarRaj N, Wang JH, 2007. Alpha-smooth muscle actin expression enhances cell traction force. Cell Motil Cytoskeleton 64, 248–257. [DOI] [PubMed] [Google Scholar]
- Chen SY, Chen HC, 2006. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol Cell Biol 26, 5155–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicurel ME, Singer RH, Meyer CJ, Ingber DE, 1998. Integrin binding and mechanical tension induce movement of mRNA and ribosomes to focal adhesions. Nature 392, 730–733. [DOI] [PubMed] [Google Scholar]
- Chinthalapudi K, Rangarajan ES, Izard T, 2018. The interaction of talin with the cell membrane is essential for integrin activation and focal adhesion formation. Proc Natl Acad Sci U S A 115, 10339–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Erickson HP, 1997. Glycosaminoglycans modulate fibronectin matrix assembly and are essential for matrix incorporation of tenascin-C. J Cell Sci 110 (Pt 12), 1413–1419. [DOI] [PubMed] [Google Scholar]
- Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML, 2011a. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol 50, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslik KA, Taffet GE, Crawford JR, Trial J, Mejia Osuna P, Entman ML, 2013a. AICAR-dependent AMPK activation improves scar formation in the aged heart in a murine model of reperfused myocardial infarction. J Mol Cell Cardiol 63, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslik KA, Trial J, Carlson S, Taffet GE, Entman ML, 2013b. Aberrant differentiation of fibroblast progenitors contributes to fibrosis in the aged murine heart: role of elevated circulating insulin levels. FASEB J 27, 1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslik KA, Trial J, Crawford JR, Taffet GE, Entman ML, 2014. Adverse fibrosis in the aging heart depends on signaling between myeloid and mesenchymal cells; role of inflammatory fibroblasts. J Mol Cell Cardiol 70, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslik KA, Trial J, Entman ML, 2011b. Defective myofibroblast formation from mesenchymal stem cells in the aging murine heart rescue by activation of the AMPK pathway. Am J Pathol 179, 1792–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslik KA, Trial J, Entman ML, 2015. Mesenchymal stem cell-derived inflammatory fibroblasts promote monocyte transition into myeloid fibroblasts via an IL-6-dependent mechanism in the aging mouse heart. FASEB J 29, 3160–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslik KA, Trial J, Entman ML, 2017. Aicar treatment reduces interstitial fibrosis in aging mice: Suppression of the inflammatory fibroblast. J Mol Cell Cardiol 111, 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement S, Stouffs M, Bettiol E, Kampf S, Krause KH, Chaponnier C, Jaconi M, 2007. Expression and function of alpha-smooth muscle actin during embryonic-stem-cell-derived cardiomyocyte differentiation. J Cell Sci 120, 229–238. [DOI] [PubMed] [Google Scholar]
- Czubryt MP, 2019. Cardiac Fibroblast to Myofibroblast Phenotype Conversion-An Unexploited Therapeutic Target. J Cardiovasc Dev Dis 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FJ, Gupta M, Pogwizd SM, Bacha E, Jeevanandam V, Gupta MP, 2002. Increased expression of alternatively spliced dominant-negative isoform of SRF in human failing hearts. Am J Physiol Heart Circ Physiol 282, H1521–1533. [DOI] [PubMed] [Google Scholar]
- Dowling JJ, Gibbs E, Russell M, Goldman D, Minarcik J, Golden JA, Feldman EL, 2008a. Kindlin-2 is an essential component of intercalated discs and is required for vertebrate cardiac structure and function. Circ Res 102, 423–431. [DOI] [PubMed] [Google Scholar]
- Dowling JJ, Vreede AP, Kim S, Golden J, Feldman EL, 2008b. Kindlin-2 is required for myocyte elongation and is essential for myogenesis. BMC Cell Biol 9, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghbali M, Eghbali M, Robinson TF, Seifter S, Blumenfeld OO, 1989. Collagen accumulation in heart ventricles as a function of growth and aging. Cardiovasc Res 23, 723–729. [DOI] [PubMed] [Google Scholar]
- Esnault C, Stewart A, Gualdrini F, East P, Horswell S, Matthews N, Treisman R, 2014. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev 28, 943–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Murphy AJ, Dart AM, 2017. A Clinical Perspective of Anti-Fibrotic Therapies for Cardiovascular Disease. Front Pharmacol 8, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah ME, Sirotkin V, Haarer B, Kakhniashvili D, Amberg DC, 2011. Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton (Hoboken) 68, 340–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JW, Nordon RE, Harvey RP, 2019. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomproix N, Percipalle P, 2004. An actin-myosin complex on actively transcribing genes. Exp Cell Res 294, 140–148. [DOI] [PubMed] [Google Scholar]
- Foster CT, Gualdrini F, Treisman R, 2017. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev 31, 2361–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG, 2019. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med 65, 70–99. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Bledzka K, Yang J, Perera HD, Plow EF, Qin J, 2014. Molecular basis of kindlin-2 binding to integrin-linked kinase pseudokinase for regulating cell adhesion. J Biol Chem 289, 28363–28375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahmberg CG, Gronholm M, Madhavan S, Jahan F, Mikkola E, Viazmina L, Koivunen E, 2019. Regulation of cell adhesion: a collaborative effort of integrins, their ligands, cytoplasmic actors, and phosphorylation. Q Rev Biophys 52, e10. [DOI] [PubMed] [Google Scholar]
- Gambini J, Gomez-Cabrera MC, Borras C, Valles SL, Lopez-Grueso R, Martinez-Bello VE, Herranz D, Pallardo FV, Tresguerres JA, Serrano M, Vina J, 2011. Free [NADH]/[NAD(+)] regulates sirtuin expression. Arch Biochem Biophys 512, 24–29. [DOI] [PubMed] [Google Scholar]
- Gardel ML, Sabass B, Ji L, Danuser G, Schwarz US, Waterman CM, 2008. Traction stress in focal adhesions correlates biphasically with actin retrograde flow speed. J Cell Biol 183, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary-Bobo G, Huet A, Li Z, Mericskay M, Parlakian A, Paulin D, 2005. [Development and cardiomyopathy: serum response factor, a key protein]. Arch Mal Coeur Vaiss 98, 655–660. [PubMed] [Google Scholar]
- Gazoti Debessa CR, Mesiano Maifrino LB, Rodrigues de Souza R, 2001. Age related changes of the collagen network of the human heart. Mech Ageing Dev 122, 1049–1058. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E, 1999. Integrin signaling. Science 285, 1028–1032. [DOI] [PubMed] [Google Scholar]
- Gilmore AP, Romer LH, 1996. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell 7, 1209–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giridharan SS, Caplan S, 2014. MICAL-family proteins: Complex regulators of the actin cytoskeleton. Antioxid Redox Signal 20, 2059–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin JM, Pittet P, Csucs G, Lussi JW, Meister JJ, Hinz B, 2006. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J Cell Biol 172, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldspink DF, Burniston JG, Tan LB, 2003. Cardiomyocyte death and the ageing and failing heart. Exp Physiol 88, 447–458. [DOI] [PubMed] [Google Scholar]
- Goni GM, Epifano C, Boskovic J, Camacho-Artacho M, Zhou J, Bronowska A, Martin MT, Eck MJ, Kremer L, Grater F, Gervasio FL, Perez-Moreno M, Lietha D, 2014. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc Natl Acad Sci U S A 111, E3177–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabec D, Batista U, Zeks B, Svetina S, 2000. The influence of the successive depolymerization and polymerization of cytoskeleton components on the fibroblast shapes. Pflugers Arch 440, R204–205. [PubMed] [Google Scholar]
- Grintsevich EE, Ge P, Sawaya MR, Yesilyurt HG, Terman JR, Zhou ZH, Reisler E, 2017. Catastrophic disassembly of actin filaments via Mical-mediated oxidation. Nat Commun 8, 2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Li T, Jiang S, Yang Z, Lv J, Yi W, Yang Y, Fang M, 2018. AMP-activated protein kinase sparks the fire of cardioprotection against myocardial ischemia and cardiac ageing. Ageing Res Rev 47, 168–175. [DOI] [PubMed] [Google Scholar]
- Hayashi I, Vuori K, Liddington RC, 2002. The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat Struct Biol 9, 101–106. [DOI] [PubMed] [Google Scholar]
- He Y, Esser P, Schacht V, Bruckner-Tuderman L, Has C, 2011. Role of kindlin-2 in fibroblast functions: implications for wound healing. J Invest Dermatol 131, 245–256. [DOI] [PubMed] [Google Scholar]
- Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD, 2017. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol Biol Cell 28, 1871–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderer S, Schenke-Layland K, 2019. Cardiac fibrosis - A short review of causes and therapeutic strategies. Adv Drug Deliv Rev 146, 77–82. [DOI] [PubMed] [Google Scholar]
- Hinz B, 2006. Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur J Cell Biol 85, 175–181. [DOI] [PubMed] [Google Scholar]
- Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C, 2001. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell 12, 2730–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Dugina V, Ballestrem C, Wehrle-Haller B, Chaponnier C, 2003. Alpha-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol Biol Cell 14, 2508–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortells L, Johansen AKZ, Yutzey KE, 2019. Cardiac Fibroblasts and the Extracellular Matrix in Regenerative and Nonregenerative Hearts. J Cardiovasc Dev Dis 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton ER, Astudillo P, Humphries MJ, Humphries JD, 2016. Mechanosensitivity of integrin adhesion complexes: role of the consensus adhesome. Exp Cell Res 343, 7–13. [DOI] [PubMed] [Google Scholar]
- Horton ER, Byron A, Askari JA, Ng DHJ, Millon-Fremillon A, Robertson J, Koper EJ, Paul NR, Warwood S, Knight D, Humphries JD, Humphries MJ, 2015. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol 17, 1577–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Wu S, Hernandez N, 2004. A role for beta-actin in RNA polymerase III transcription. Genes Dev 18, 3010–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung RJ, Pak CW, Terman JR, 2011. Direct redox regulation of F-actin assembly and disassembly by Mical. Science 334, 1710–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttelmaier S, Zenklusen D, Lederer M, Dictenberg J, Lorenz M, Meng X, Bassell GJ, Condeelis J, Singer RH, 2005. Spatial regulation of beta-actin translation by Src-dependent phosphorylation of ZBP1. Nature 438, 512–515. [DOI] [PubMed] [Google Scholar]
- Ilani T, Fass D, 2013. Now at the Met: fine art of reversible sulfoxidation. Mol Cell 51, 281–282. [DOI] [PubMed] [Google Scholar]
- Inoue R, Kurahara LH, Hiraishi K, 2019. TRP channels in cardiac and intestinal fibrosis. Semin Cell Dev Biol 94, 40–49. [DOI] [PubMed] [Google Scholar]
- Jansen RP, 2001. mRNA localization: message on the move. Nat Rev Mol Cell Biol 2, 247–256. [DOI] [PubMed] [Google Scholar]
- Jin B, Zhu J, Shi HM, Wen ZC, Wu BW, 2018. YAP activation promotes the transdifferentiation of cardiac fibroblasts to myofibroblasts in matrix remodeling of dilated cardiomyopathy. Braz J Med Biol Res 52, e7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JL, Peana D, Veteto AB, Lambert MD, Nourian Z, Karasseva NG, Hill MA, Lindman BR, Baines CP, Krenz M, Domeier TL, 2019. TRPV4 increases cardiomyocyte calcium cycling and contractility yet contributes to damage in the aged heart following hypoosmotic stress. Cardiovasc Res 115, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung GY, Park YJ, Han JS, 2011. Mediation of Rac1 activation by kindlin-2: an essential function in osteoblast adhesion, spreading, and proliferation. J Cell Biochem 112, 2541–2548. [DOI] [PubMed] [Google Scholar]
- Kadler KE, Hill A, Canty-Laird EG, 2008. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol 20, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz ZB, Wells AL, Park HY, Wu B, Shenoy SM, Singer RH, 2012. beta-Actin mRNA compartmentalization enhances focal adhesion stability and directs cell migration. Genes Dev 26, 1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik G, Spenlehauer A, Sessions AO, Trujillo AS, Fuhrmann A, Fu Z, Venkatraman V, Pohl D, Tuler J, Wang M, Lakatta EG, Ocorr K, Bodmer R, Bernstein SI, Van Eyk JE, Cammarato A, Engler AJ, 2015. Vinculin network-mediated cytoskeletal remodeling regulates contractile function in the aging heart. Sci Transl Med 7, 292ra299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kechagia JZ, Ivaska J, Roca-Cusachs P, 2019. Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol 20, 457–473. [DOI] [PubMed] [Google Scholar]
- Khanday I, Das S, Chongloi GL, Bansal M, Grossniklaus U, Vijayraghavan U, 2016. Genome-Wide Targets Regulated by the OsMADS1 Transcription Factor Reveals Its DNA Recognition Properties. Plant Physiol 172, 372–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Wirtz D, 2013. Focal adhesion size uniquely predicts cell migration. FASEB J 27, 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislauskis EH, Zhu X, Singer RH, 1994. Sequences responsible for intracellular localization of beta-actin messenger RNA also affect cell phenotype. J Cell Biol 127, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapholz B, Brown NH, 2017. Talin - the master of integrin adhesions. J Cell Sci 130, 2435–2446. [DOI] [PubMed] [Google Scholar]
- Klingberg F, Chow ML, Koehler A, Boo S, Buscemi L, Quinn TM, Costell M, Alman BA, Genot E, Hinz B, 2014. Prestress in the extracellular matrix sensitizes latent TGF-beta1 for activation. J Cell Biol 207, 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan M, Muro AF, White ES, Berkman N, 2010. EDA-containing cellular fibronectin induces fibroblast differentiation through binding to alpha4beta7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J 24, 4503–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong P, Christia P, Frangogiannis NG, 2014. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 71, 549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD, 2015. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16, 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumsta C, Ching TT, Nishimura M, Davis AE, Gelino S, Catan HH, Yu X, Chu CC, Ong B, Panowski SH, Baird N, Bodmer R, Hsu AL, Hansen M, 2014. Integrin-linked kinase modulates longevity and thermotolerance in C. elegans through neuronal control of HSF-1. Aging Cell 13, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WF, Wong WT, 2020. Roles of the actin cytoskeleton in aging and age-associated diseases. Ageing Res Rev 58, 101021. [DOI] [PubMed] [Google Scholar]
- Lai WKM, Pugh BF, 2017. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol 18, 548–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamar JM, Xiao Y, Norton E, Jiang ZG, Gerhard GM, Kooner S, Warren JSA, Hynes RO, 2019. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J Biol Chem 294, 2302–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Peterfi Z, Hoffmann FW, Moore RE, Kaya A, Avanesov A, Tarrago L, Zhou Y, Weerapana E, Fomenko DE, Hoffmann PR, Gladyshev VN, 2013. MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol Cell 51, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FJ, Surolia R, Li H, Wang Z, Kulkarni T, Liu G, de Andrade JA, Kass DJ, Thannickal VJ, Duncan SR, Antony VB, 2017a. Autoimmunity to Vimentin Is Associated with Outcomes of Patients with Idiopathic Pulmonary Fibrosis. J Immunol 199, 1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Deng Y, Sun K, Yang H, Liu J, Wang M, Zhang Z, Lin J, Wu C, Wei Z, Yu C, 2017b. Structural basis of kindlin-mediated integrin recognition and activation. Proc Natl Acad Sci U S A 114, 9349–9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Liu X, Wei J, 2014. Ageing related periostin expression increase from cardiac fibroblasts promotes cardiomyocytes senescent. Biochem Biophys Res Commun 452, 497–502. [DOI] [PubMed] [Google Scholar]
- Lin H, McGrath J, Wang P, Lee T, 2007. Cellular toxicity induced by SRF-mediated transcriptional squelching. Toxicol Sci 96, 83–91. [DOI] [PubMed] [Google Scholar]
- Lin J, Lopez EF, Jin Y, Van Remmen H, Bauch T, Han HC, Lindsey ML, 2008. Age-related cardiac muscle sarcopenia: Combining experimental and mathematical modeling to identify mechanisms. Exp Gerontol 43, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lagares D, Choi KM, Stopfer L, Marinkovic A, Vrbanac V, Probst CK, Hiemer SE, Sisson TH, Horowitz JC, Rosas IO, Fredenburgh LE, Feghali-Bostwick C, Varelas X, Tager AM, Tschumperlin DJ, 2015a. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 308, L344–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Fukuda K, Xu Z, Ma YQ, Hirbawi J, Mao X, Wu C, Plow EF, Qin J, 2011. Structural basis of phosphoinositide binding to kindlin-2 protein pleckstrin homology domain in regulating integrin activation. J Biol Chem 286, 43334–43342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang Z, Thinn AM, Ma YQ, Zhu J, 2015b. The dual structural roles of the membrane distal region of the alpha-integrin cytoplasmic tail during integrin inside-out activation. J Cell Sci 128, 1718–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Heredia M, Jansen RP, 2004. mRNA localization and the cytoskeleton. Curr Opin Cell Biol 16, 80–85. [DOI] [PubMed] [Google Scholar]
- Lopez-Colome AM, Lee-Rivera I, Benavides-Hidalgo R, Lopez E, 2017. Paxillin: a crossroad in pathological cell migration. J Hematol Oncol 10, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco Dos Santos S, Petropoulos I, Friguet B, 2018. The Oxidized Protein Repair Enzymes Methionine Sulfoxide Reductases and Their Roles in Protecting against Oxidative Stress, in Ageing and in Regulating Protein Function. Antioxidants (Basel) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XG, Azhar G, Liu L, Tsou H, Wei JY, 1998. SRF binding to SRE in the rat heart: influence of age. J Gerontol A Biol Sci Med Sci 53, B3–10. [DOI] [PubMed] [Google Scholar]
- Ma B, Cheng H, Gao R, Mu C, Chen L, Wu S, Chen Q, Zhu Y, 2016. Zyxin-Siah2-Lats2 axis mediates cooperation between Hippo and TGF-beta signalling pathways. Nat Commun 7, 11123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Meng Z, Chen R, Guan KL, 2019. The Hippo Pathway: Biology and Pathophysiology. Annu Rev Biochem 88, 577–604. [DOI] [PubMed] [Google Scholar]
- Ma X, Chen Y, Xu W, Wu N, Li M, Cao Y, Wu S, Li Q, Xue L, 2015. Impaired Hippo signaling promotes Rho1-JNK-dependent growth. Proc Natl Acad Sci U S A 112, 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCannell KA, Bazzazi H, Chilton L, Shibukawa Y, Clark RB, Giles WR, 2007. A mathematical model of electrotonic interactions between ventricular myocytes and fibroblasts. Biophys J 92, 4121–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi S, Mancini E, Xu L, Moore A, Jahanbani F, Hebestreit K, Srinivasan R, Li X, Devarajan K, Prelot L, Ang CE, Shibuya Y, Benayoun BA, Chang ALS, Wernig M, Wysocka J, Longaker MT, Snyder MP, Brunet A, 2019. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 574, 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE, 2010. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol (Camb) 2, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald PC, Wilson JE, McNeill S, Gao M, Spinelli JJ, Rosenberg F, Wiebe H, McManus BM, 2002. The challenge of defining normality for human mitral and aortic valves: geometrical and compositional analysis. Cardiovasc Pathol 11, 193–209. [DOI] [PubMed] [Google Scholar]
- Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R, 2009. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol 11, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekhdjian AH, Kai F, Rubashkin MG, Prahl LS, Przybyla LM, McGregor AL, Bell ES, Barnes JM, DuFort CC, Ou G, Chang AC, Cassereau L, Tan SJ, Pickup MW, Lakins JN, Ye X, Davidson MW, Lammerding J, Odde DJ, Dunn AR, Weaver VM, 2017. Integrin-mediated traction force enhances paxillin molecular associations and adhesion dynamics that increase the invasiveness of tumor cells into a three-dimensional extracellular matrix. Mol Biol Cell 28, 1467–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior-Becker A, Dai G, Ding Z, Schafer L, Schrader J, Young MF, Fischer JW, 2011. Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype. J Biol Chem 286, 17365–17375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes AB, Ferro M, Rodrigues B, Souza MR, Araujo RC, Souza RR, 2012. Quantification of left ventricular myocardial collagen system in children, young adults, and the elderly. Medicina (B Aires) 72, 216–220. [PubMed] [Google Scholar]
- Meschiari CA, Ero OK, Pan H, Finkel T, Lindsey ML, 2017. The impact of aging on cardiac extracellular matrix. Geroscience 39, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalick L, Kuebler WM, 2020. TRPV4-A Missing Link Between Mechanosensation and Immunity. Front Immunol 11, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou AI, Treisman R, 2003. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113, 329–342. [DOI] [PubMed] [Google Scholar]
- Montanez E, Ussar S, Schifferer M, Bosl M, Zent R, Moser M, Fassler R, 2008. Kindlin-2 controls bidirectional signaling of integrins. Genes Dev 22, 1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM, 2014. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest 124, 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani T, Honda E, Hayakawa S, Sato M, Satoh K, Kudo M, Munakata H, 2008. Effects of decorin on the expression of alpha-smooth muscle actin in a human myofibroblast cell line. Mol Cell Biochem 308, 201–207. [DOI] [PubMed] [Google Scholar]
- Nicastro G, Candel AM, Uhl M, Oregioni A, Hollingworth D, Backofen R, Martin SR, Ramos A, 2017. Mechanism of beta-actin mRNA Recognition by ZBP1. Cell Rep 18, 1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M, Kumsta C, Kaushik G, Diop SB, Ding Y, Bisharat-Kernizan J, Catan H, Cammarato A, Ross RS, Engler AJ, Bodmer R, Hansen M, Ocorr K, 2014. A dual role for integrin-linked kinase and beta1-integrin in modulating cardiac aging. Aging Cell 13, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi S, Saito A, Nagase T, 2018. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, Trusk T, Potts JD, Goodwin RL, Davis J, Hoffman S, Wen X, Sugi Y, Kern CB, Mjaatvedt CH, Turner DK, Oka T, Conway SJ, Molkentin JD, Forgacs G, Markwald RR, 2007. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem 101, 695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski J, Cronin CN, McRee DE, Knuth MW, Nelson CG, Pavletich NP, Rogers J, Sang BC, Scheibe DN, Swanson RV, Thompson DA, 2002. Structures of the cancer-related Aurora-A, FAK, and EphA2 protein kinases from nanovolume crystallography. Structure 10, 1659–1667. [DOI] [PubMed] [Google Scholar]
- O’Reilly S, 2017. Epigenetics in fibrosis. Mol Aspects Med 54, 89–102. [DOI] [PubMed] [Google Scholar]
- Ochsner M, Textor M, Vogel V, Smith ML, 2010. Dimensionality controls cytoskeleton assembly and metabolism of fibroblast cells in response to rigidity and shape. PLoS One 5, e9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN, Nordheim A, 2010. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omelchenko T, Vasiliev JM, Gelfand IM, Feder HH, Bonder EM, 2002. Mechanisms of polarization of the shape of fibroblasts and epitheliocytes: Separation of the roles of microtubules and Rho-dependent actin-myosin contractility. Proc Natl Acad Sci U S A 99, 10452–10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlakian A, Charvet C, Escoubet B, Mericskay M, Molkentin JD, Gary-Bobo G, De Windt LJ, Ludosky MA, Paulin D, Daegelen D, Tuil D, Li Z, 2005. Temporally controlled onset of dilated cardiomyopathy through disruption of the SRF gene in adult heart. Circulation 112, 2930–2939. [DOI] [PubMed] [Google Scholar]
- Peng X, Wu X, Druso JE, Wei H, Park AY, Kraus MS, Alcaraz A, Chen J, Chien S, Cerione RA, Guan JL, 2008. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc Natl Acad Sci U S A 105, 6638–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percipalle P, Fomproix N, Kylberg K, Miralles F, Bjorkroth B, Daneholt B, Visa N, 2003. An actin-ribonucleoprotein interaction is involved in transcription by RNA polymerase II. Proc Natl Acad Sci U S A 100, 6475–6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philimonenko VV, Zhao J, Iben S, Dingova H, Kysela K, Kahle M, Zentgraf H, Hofmann WA, de Lanerolle P, Hozak P, Grummt I, 2004. Nuclear actin and myosin I are required for RNA polymerase I transcription. Nat Cell Biol 6, 1165–1172. [DOI] [PubMed] [Google Scholar]
- Piccolo S, Dupont S, Cordenonsi M, 2014. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 94, 1287–1312. [DOI] [PubMed] [Google Scholar]
- Ponnusamy M, Zhuang MA, Zhou X, Tolbert E, Bayliss G, Zhao TC, Zhuang S, 2015. Activation of Sirtuin-1 Promotes Renal Fibroblast Activation and Aggravates Renal Fibrogenesis. J Pharmacol Exp Ther 354, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posern G, Miralles F, Guettler S, Treisman R, 2004. Mutant actins that stabilise F-actin use distinct mechanisms to activate the SRF coactivator MAL. EMBO J 23, 3973–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posern G, Sotiropoulos A, Treisman R, 2002. Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Mol Biol Cell 13, 4167–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prywes R, Zhu H, 1992. In vitro squelching of activated transcription by serum response factor: evidence for a common coactivator used by multiple transcriptional activators. Nucleic Acids Res 20, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, Xia W, Fisher GJ, Voorhees JJ, Quan T, 2018. YAP/TAZ regulates TGF-beta/Smad3 signaling by induction of Smad7 via AP-1 in human skin dermal fibroblasts. Cell Commun Signal 16, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahaman SO, Grove LM, Paruchuri S, Southern BD, Abraham S, Niese KA, Scheraga RG, Ghosh S, Thodeti CK, Zhang DX, Moran MM, Schilling WP, Tschumperlin DJ, Olman MA, 2014. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest 124, 5225–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Kono H, 2008. The inflammatory response to cell death. Annu Rev Pathol 3, 99–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognoni E, Ruppert R, Fassler R, 2016. The kindlin family: functions, signaling properties and implications for human disease. J Cell Sci 129, 17–27. [DOI] [PubMed] [Google Scholar]
- Romero S, Le Clainche C, Gautreau AM, 2020. Actin polymerization downstream of integrins: signaling pathways and mechanotransduction. Biochem J 477, 1–21. [DOI] [PubMed] [Google Scholar]
- Sabatier L, Chen D, Fagotto-Kaufmann C, Hubmacher D, McKee MD, Annis DS, Mosher DF, Reinhardt DP, 2009. Fibrillin assembly requires fibronectin. Mol Biol Cell 20, 846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson N, Berger P, Zenzmaier C, 2012. Therapeutic targeting of redox signaling in myofibroblast differentiation and age-related fibrotic disease. Oxid Med Cell Longev 2012, 458276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago JJ, Dangerfield AL, Rattan SG, Bathe KL, Cunnington RH, Raizman JE, Bedosky KM, Freed DH, Kardami E, Dixon IM, 2010. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev Dyn 239, 1573–1584. [DOI] [PubMed] [Google Scholar]
- Sartorelli V, Puri PL, 2018. Shaping Gene Expression by Landscaping Chromatin Architecture: Lessons from a Master. Mol Cell 71, 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato I, Shimada K, 2001. Quantitative analysis of tenascin in chordae tendineae of human left ventricular papillary muscle with aging. Ann Anat 183, 443–448. [DOI] [PubMed] [Google Scholar]
- Selvaraj A, Prywes R, 2004. Expression profiling of serum inducible genes identifies a subset of SRF target genes that are MKL dependent. BMC Mol Biol 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serebryannyy LA, Parilla M, Annibale P, Cruz CM, Laster K, Gratton E, Kudryashov D, Kosak ST, Gottardi CJ, de Lanerolle P, 2016. Persistent nuclear actin filaments inhibit transcription by RNA polymerase II. J Cell Sci 129, 3412–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Ma Y, Gao M, Lai Y, Wang G, Yu Q, Cui FZ, Liu X, 2013. Integrins-FAK-Rho GTPases pathway in endothelial cells sense and response to surface wettability of plasma nanocoatings. ACS Appl Mater Interfaces 5, 5112–5121. [DOI] [PubMed] [Google Scholar]
- Shestakova EA, Singer RH, Condeelis J, 2001. The physiological significance of beta -actin mRNA localization in determining cell polarity and directional motility. Proc Natl Acad Sci U S A 98, 7045–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheydina A, Riordon DR, Boheler KR, 2011. Molecular mechanisms of cardiomyocyte aging. Clin Sci (Lond) 121, 315–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde AV, Humeres C, Frangogiannis NG, 2017. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta Mol Basis Dis 1863, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD, 2000. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol 2, 249–256. [DOI] [PubMed] [Google Scholar]
- Sliogeryte K, Gavara N, 2019. Vimentin Plays a Crucial Role in Fibroblast Ageing by Regulating Biophysical Properties and Cell Migration. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small EM, 2012. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Transl Res 5, 794–804. [DOI] [PubMed] [Google Scholar]
- Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN, 2010. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res 107, 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speight P, Kofler M, Szaszi K, Kapus A, 2016. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFbeta-regulated Smad3. Nat Commun 7, 11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens EH, Chu CK, Grande-Allen KJ, 2008. Valve proteoglycan content and glycosaminoglycan fine structure are unique to microstructure, mechanical load and age: Relevance to an age-specific tissue-engineered heart valve. Acta Biomater 4, 1148–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman B, 2018. Histone Modifications: Insights into Their Influence on Gene Expression. Cell 175, 6–9. [DOI] [PubMed] [Google Scholar]
- Sun Q, Chen G, Streb JW, Long X, Yang Y, Stoeckert CJ Jr., Miano JM, 2006. Defining the mammalian CArGome. Genome Res 16, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Taurin S, Sethakorn N, Long X, Imamura M, Wang DZ, Zimmer WE, Dulin NO, Miano JM, 2009. Myocardin-dependent activation of the CArG box-rich smooth muscle gamma-actin gene: preferential utilization of a single CArG element through functional association with the NKX3.1 homeodomain protein. J Biol Chem 284, 32582–32590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen M, Vanhoutte D, Van Almen GC, Hamdani N, Schellings MW, D’Hooge J, Van der Velden J, Weaver MS, Sage EH, Bornstein P, Verheyen FK, VandenDriessche T, Chuah MK, Westermann D, Paulus WJ, Van de Werf F, Schroen B, Carmeliet P, Pinto YM, Heymans S, 2009. Absence of thrombospondin-2 causes age-related dilated cardiomyopathy. Circulation 120, 1585–1597. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Schunkert H, Isoyama S, Wei JY, Nadal-Ginard B, Grossman W, Izumo S, 1992. Age-related differences in the expression of proto-oncogene and contractile protein genes in response to pressure overload in the rat myocardium. J Clin Invest 89, 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallquist MD, Molkentin JD, 2017. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol 14, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker GD, Frangogiannis NG, Bujak M, Zymek P, Gaubatz JW, Reddy AK, Taffet G, Michael LH, Entman ML, Ballantyne CM, 2006. Effects of diet-induced obesity on inflammation and remodeling after myocardial infarction. Am J Physiol Heart Circ Physiol 291, H2504–2514. [DOI] [PubMed] [Google Scholar]
- Theodosiou M, Widmaier M, Bottcher RT, Rognoni E, Veelders M, Bharadwaj M, Lambacher A, Austen K, Muller DJ, Zent R, Fassler R, 2016. Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. Elife 5, e10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DP, McCormick RJ, Zimmerman SD, Vadlamudi RK, Gosselin LE, 1992. Aging- and training-induced alterations in collagen characteristics of rat left ventricle and papillary muscle. Am J Physiol 263, H778–783. [DOI] [PubMed] [Google Scholar]
- Totaro A, Panciera T, Piccolo S, 2018. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol 20, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trial J, Cieslik KA, 2018. Changes in cardiac resident fibroblast physiology and phenotype in aging. Am J Physiol Heart Circ Physiol 315, H745–H755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trial J, Heredia CP, Taffet GE, Entman ML, Cieslik KA, 2017. Dissecting the role of myeloid and mesenchymal fibroblasts in age-dependent cardiac fibrosis. Basic Res Cardiol 112, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varland S, Vandekerckhove J, Drazic A, 2019. Actin Post-translational Modifications: The Cinderella of Cytoskeletal Control. Trends Biochem Sci 44, 502–516. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Montell C, 2007. TRP channels. Annu Rev Biochem 76, 387–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal R, Wagner JUG, Braeuning C, Fischer C, Patrick R, Tombor L, Muhly-Reinholz M, John D, Kliem M, Conrad T, Guimaraes-Camboa N, Harvey R, Dimmeler S, Sauer S, 2019. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visa N, 2005. Actin in transcription. Actin is required for transcription by all three RNA polymerases in the eukaryotic cell nucleus. EMBO Rep 6, 218–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber KT, Janicki JS, Shroff SG, Pick R, Chen RM, Bashey RI, 1988. Collagen remodeling of the pressure-overloaded, hypertrophied nonhuman primate myocardium. Circ Res 62, 757–765. [DOI] [PubMed] [Google Scholar]
- Wei S, Chow LT, Shum IO, Qin L, Sanderson JE, 1999. Left and right ventricular collagen type I/III ratios and remodeling post-myocardial infarction. J Card Fail 5, 117–126. [DOI] [PubMed] [Google Scholar]
- Wei X, Wang X, Xia Y, Tang Y, Li F, Fang W, Zhang H, 2014. Kindlin-2 regulates renal tubular cell plasticity by activation of Ras and its downstream signaling. Am J Physiol Renal Physiol 306, F271–278. [DOI] [PubMed] [Google Scholar]
- Wu JC, Chen YC, Kuo CT, Wenshin Yu H, Chen YQ, Chiou A, Kuo JC, 2015. Focal adhesion kinase-dependent focal adhesion recruitment of SH2 domains directs SRC into focal adhesions to regulate cell adhesion and migration. Sci Rep 5, 18476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman AE, Atamas SP, 2018. Sirtuins and Accelerated Aging in Scleroderma. Curr Rheumatol Rep 20, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Nho RS, Kahm J, Kleidon J, Henke CA, 2004. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem 279, 33024–33034. [DOI] [PubMed] [Google Scholar]
- Xie J, Chen Y, Hu C, Pan Q, Wang B, Li X, Geng J, Xu B, 2017. Premature senescence of cardiac fibroblasts and atrial fibrosis in patients with atrial fibrillation. Oncotarget 8, 57981–57990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Zhang J, Ma D, 2014. [Crosstalk of Hippo/YAP and Wnt/beta-catenin pathways]. Yi Chuan 36, 95–102. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, Geiger B, 2010. The switchable integrin adhesome. J Cell Sci 123, 1385–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeke A, Misheva M, Remenyi A, Bogoyevitch MA, 2016. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol Mol Biol Rev 80, 793–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerr P, Palumbo-Zerr K, Huang J, Tomcik M, Sumova B, Distler O, Schett G, Distler JH, 2016. Sirt1 regulates canonical TGF-beta signalling to control fibroblast activation and tissue fibrosis. Ann Rheum Dis 75, 226–233. [DOI] [PubMed] [Google Scholar]
- Zhan J, Yang M, Chi X, Zhang J, Pei X, Ren C, Guo Y, Liu W, Zhang H, 2014. Kindlin-2 expression in adult tissues correlates with their embryonic origins. Sci China Life Sci 57, 690–697. [DOI] [PubMed] [Google Scholar]
- Zhan L, Li J, 2018. The role of TRPV4 in fibrosis. Gene 642, 1–8. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Meng F, Chen S, Plouffe SW, Wu S, Liu S, Li X, Zhou R, Wang J, Zhao B, Liu J, Qin J, Zou J, Feng XH, Guan KL, Xu P, 2017. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat Cell Biol 19, 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Azhar G, Furr MC, Zhong Y, Wei JY, 2003. Model of functional cardiac aging: young adult mice with mild overexpression of serum response factor. Am J Physiol Regul Integr Comp Physiol 285, R552–560. [DOI] [PubMed] [Google Scholar]
- Zhang X, Azhar G, Helms S, Burton B, Huang C, Zhong Y, Gu X, Fang H, Tong W, Wei JY, 2011. Identification of New SRF Binding Sites in Genes Modulated by SRF Over-Expression in Mouse Hearts. Gene Regul Syst Bio 5, 41–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Mu Y, Veevers J, Peter AK, Manso AM, Bradford WH, Dalton ND, Peterson KL, Knowlton KU, Ross RS, Zhou X, Chen J, 2016. Postnatal Loss of Kindlin-2 Leads to Progressive Heart Failure. Circ Heart Fail 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Chen S, Chen Y, Lyga J, Santhanam U, 2012. Critical role of paxillin in aging of human skin. J Invest Dermatol 132, 1290–1293. [DOI] [PubMed] [Google Scholar]
- Zhou DW, Lee TT, Weng S, Fu J, Garcia AJ, 2017. Effects of substrate stiffness and actomyosin contractility on coupling between force transmission and vinculin-paxillin recruitment at single focal adhesions. Mol Biol Cell 28, 1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]