

Abstract
Amyotrophic Lateral Sclerosis (ALS) is a devastating neurodegenerative disorder that results in death due to respiratory failure. Many genetic defects are associated with ALS; one such defect is a mutation in the gene encoding optineurin (OPTN). Using an optineurin null mouse (Optn−/−), we sought to characterize the impact of optineurin deficiency on respiratory neurodegeneration. Respiratory function was assessed at 6 and 12 mo of age using whole body plethysmography at baseline during normoxia (FiO2: 0.21; N2 balance) and during a respiratory challenge with hypoxia and hypercapnia (FiCO2: 0.07, FiO2: 0.10; N2 balance). Histological analyses to assess motor neuron viability and respiratory nerve integrity were performed in the medulla, cervical spinal cord and hypoglossal and phrenic nerves. Minute ventilation, peak inspiratory flow, and peak expiratory flow is significantly reduced during a respiratory challenge in 6 mo Optn−/−mice. By 12 mo, tidal volume is also significantly reduced in Optn−/− mice. Furthermore, 12mo Optn−/− mice exhibit hypoglossal motor neuron loss, phrenic and hypoglossal dysmyelination, and accumulated mitochondria in the hypoglossal nerve axons. Overall, these data indicate that Optn−/− mice display neurodegenerative respiratory dysfunction and are a useful model to study the impact of novel therapies on respiratory function for optineurin deficient ALS patients.
Keywords: respiration, motor neuron, ALS, mitophagy, optineurin, neurodegeneration
1. INTRODUCTION:
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder that results in paralysis and death from respiratory failure1. As ALS progresses, diaphragm and intercostal muscle weakness leads to inadequate ventilation and respiratory insufficiency. In addition to weakness of these respiratory muscles, the genioglossal muscle of the tongue progressively deteriorates2–4. This bulbar muscle regulates tongue shape, stiffness, and position and is crucial for upper airway patency. Thus, tongue muscle weakness results in dysarthria and dysphagia leading to recurrent aspiration, choking, and aggravation of respiratory disease5,6. The progressive bulbar and diaphragm weakness is a consequence of hypoglossal and phrenic motor unit degeneration5. As the disease progresses, motor neuron axons retract from tongue, diaphragm, and intercostal muscle fibers, which result in respiratory muscle weakness and atrophy7.
Throughout the last 25 years, genome analysis in ALS patients identified mutations in more than 40 genes8. In 2010, mutations in the gene OPTN, which encodes the protein optineurin (OPTN), were first identified in six Japanese ALS patients9. OPTN is a multifunctional adaptor protein involved in several, overlapping cellular pathways - mitophagy, NFκB regulated inflammation, and necroptosis10,11. By promoting mitophagy and inhibiting chronic inflammation, OPTN is a key protein in cell homeostasis12. Cellular homeostasis is particularly important for non-dividing cells such as motor neurons. The loss of OPTN also promotes necroptosis through its interaction with Receptor Interacting Serine/Threonine Kinase 1 (RIPK1) resulting in dysmyelination and axonal degeneration13. Patients with OPTN mutations have a typical age of onset (30–60 years) for ALS, but most display a slower disease progression with delayed respiratory failure9,10. Since 2010, OPTN mutations have been identified in 3–4% of Japanese fALS patients and up to 14% of Moroccan patients10,14. Mutations that result in loss of the functional OPTN protein lead to disrupted autophagy, necroptosis, and inflammation which result in harmful effects that leads to ALS pathogenesis14,15. Interestingly, individuals with loss of function mutations in OPTN develop tongue weakness early in the disease course that leads to choking, failure to control secretions, upper airway obstruction, and aspiration pneumonia9,10,16. As the disease progresses, phrenic nerve and motoneurons are progressively affected and death is ultimately due to respiratory failure9,16–19.
While much is known about how OPTN behaves molecularly, its role in ALS-associated respiratory dysfunction is unknown. The goal of this study was to characterize respiratory deficiency in a novel Optn−/− mouse as a model of ALS20. The overall hypothesis is that OPTN-deficiency induces neurodegeneration resulting in respiratory neuropathology and insufficiency. Overall, our findings indicate that the Optn−/− mouse is an ideal animal model for studying mechanisms of respiratory failure in patients with optineurin deficient ALS.
2. METHODS:
2.1. Mice:
All mice were approved by the Duke University Institutional Animal Care and Use Committee (IACUC). C57BL6/J, wildtype (WT), mice were obtained from the Jackson Laboratory. Optn−/− mice were generated by Dr. Tseng at Duke University21. All mice were housed at the Duke University Division of Laboratory Animal Resources.
2.2. Respiratory Analysis:
Whole body plethysmography (WBP) was performed as previously described22 at 6 and 12 mo in the WT and Optn−/− mice (N=8–11 mice per group). Briefly, unanesthetized, unrestrained mice were placed in a Plexiglas chamber (DSI, St. Paul, MN); data was collected and analyzed using FinePointe Software. Ventilation was monitored under normoxia (FiO2: 0.21; N2 balance) for 1.5 hours, within which a five-minute period of regular breathing was selected as the baseline. Following the period of normoxia, mice were exposed to a hypercapnic and hypoxic (FiCO2: 0.07, FiO2: 0.10; N2 balance) respiratory challenge for 10 minutes. Mice were then returned to normoxic air. Apneic events were also measured during the baseline and represent averages of 15 second intervals. For breaths to be defined as an apnea they needed to meet one of the following two criteria: (1) be 1.5 seconds in duration or longer, AND/OR (2) be 200% longer than the previous breath.
2.3. Motor Neuron Immunohistochemistry:
Hypoglossal and phrenic motor neurons were labeled with the retrograde tracer, cholera toxin subunit β (CT-β), as previously described23,24. A 0.2% solution of CT-β (Millipore-Sigma, C9903) in Lactated Ringers was delivered through intralingual and intrapleural injections to 12 mo old mice (N = 2 – 3 per tissue per genotype). 72 hours after injection, the brainstem and spinal cords en bloc within the bone were harvested and fixed in 4% paraformaldehyde (PFA) for 24 hours. The brainstems and spinal cords were extracted from the soft tissue and bone, then fixed in 4% PFA for an additional 24 hours. Extracted brainstems and spinal cords were then cryopreserved in 30% sucrose then embedded in OCT. The medulla and cervical spinal cord were sectioned at 40 μm. Every second section from the hypoglossal region of the medulla and from C3–C5 within the cervical spinal cord was stained with an anti-cholera toxin antibody (List-Biologicals, 703). A biotinylated secondary antibody (VECTASTAIN ABC kit, Vector Laboratories, 1:200) exposed with 3,3’-diaminobenzidine (DAB) was applied. Sections were counterstained with cresyl violet, then imaged with a Leica DMRA2 Compound Microscope with Open Lab Software at 10x magnification. Motor neurons in the XII and phrenic motor pools were independently counted by two lab members who were blinded to the genotype of the mice.
2.4. Nerve g-Ratio Quantification:
Phrenic and hypoglossal nerves of 12 mo old mice (N=3–4 mice per group) were harvested and placed in 2.5% glutaraldehyde and 0.10M sodium cacodylate. Samples were subsequently washed in 1.0% osmium tetroxide and then placed into en bloc stain (1% uranyl acetate). Next, they were dehydrated in acetone and epoxy resin (EPON). Finally, the samples were embedded in Beem capsules and semi-thin sectioned (1 μm), and stained with 1% toluidine blue-1% sodium borate solution by the Duke University Electron Microscopy Core. Slides were imaged on a Leica DMRA2 Compound Microscope with Open Lab Software at 20x magnification. G-ratios of the cross-sections were quantified using a public downloadable ImageJ plugin25. The g-ratio was determined to be the ratio of the inner axonal diameter to the total outer axonal diameter with surrounding myelin sheath of each axon fiber. Each area was manually outlined on 100 randomly selected axons for the hypoglossal nerve, and 50 randomly selected axons for the phrenic nerve.
2.5. Mitochondrial Analysis
Processed and blocked phrenic and hypoglossal nerves from above (2.4) were ultrathin sectioned (~60 nm) on a Reichert Ultracut E ultramicrotome, and post-stained in 2% uranyl acetate and SATO’s lead stain. Images were captured using a Philips CM12 transmission electron microscope. Ultrastructural analysis was conducted using ImageJ 2.0 to quantify mitochondria per axon, axonal area, and mitochondrial area. Mitochondria were required to have a minimum diameter of 0.16 μm to be counted26.
2.6. Statistical Analysis:
All data were analyzed in GraphPad Prism Software. The WBP breathing parameters were analyzed using a two way ANOVA with repeated measures and post-hoc analysis was performed using a Bonferonni correction. Apnea, body weight, G-ratio, and mitochondrial size quantification, were analyzed using a Student’s t-test. Data are reported as mean ± SEM. For all statistical analyses, significance is defined as *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
3. RESULTS:
3.1. Respiratory deficiency in Optn−/− mice is present by 6 months of age.
Awake, spontaneous ventilation was assessed at baseline during normoxic breathing (FiO2: 0.21; nitrogen balance) and during a respiratory challenge with hypoxia and hypercapnia (FiO2: 0.10; FiCO2: 0.07; nitrogen balance). At baseline, there are no differences in frequency (f), tidal volume (TV), minute ventilation (MV), peak inspiratory frequency (PIF) or peak expiratory frequency (PEF) between WT and Optn−/− mice at 6 or 12 mo of age (p>0.05). However, during the respiratory challenge, 6 mo old Optn−/− mice have decreased MV (p=0.0004), PIF (p=0.0007), and PEF (p=0.003) (Figure 1), compared to WT mice. Furthermore, frequency is decreased (p=0.05) but TV is not significantly decreased (p=0.07). At 12 mo of age, Optn−/− mice develop a progressive deterioration in TV (p=0.02); and maintain a significant decrease in MV (p=0.003), PIF (p=0.01), and PEF (p<0.0001) (Figure 2). Frequency is not different between WT and Optn−/− mice at 12 mo of age (p=0.21).
FIGURE 1: Respiratory Deficits Present by 6 Months of Age in Optn−/− Mice.
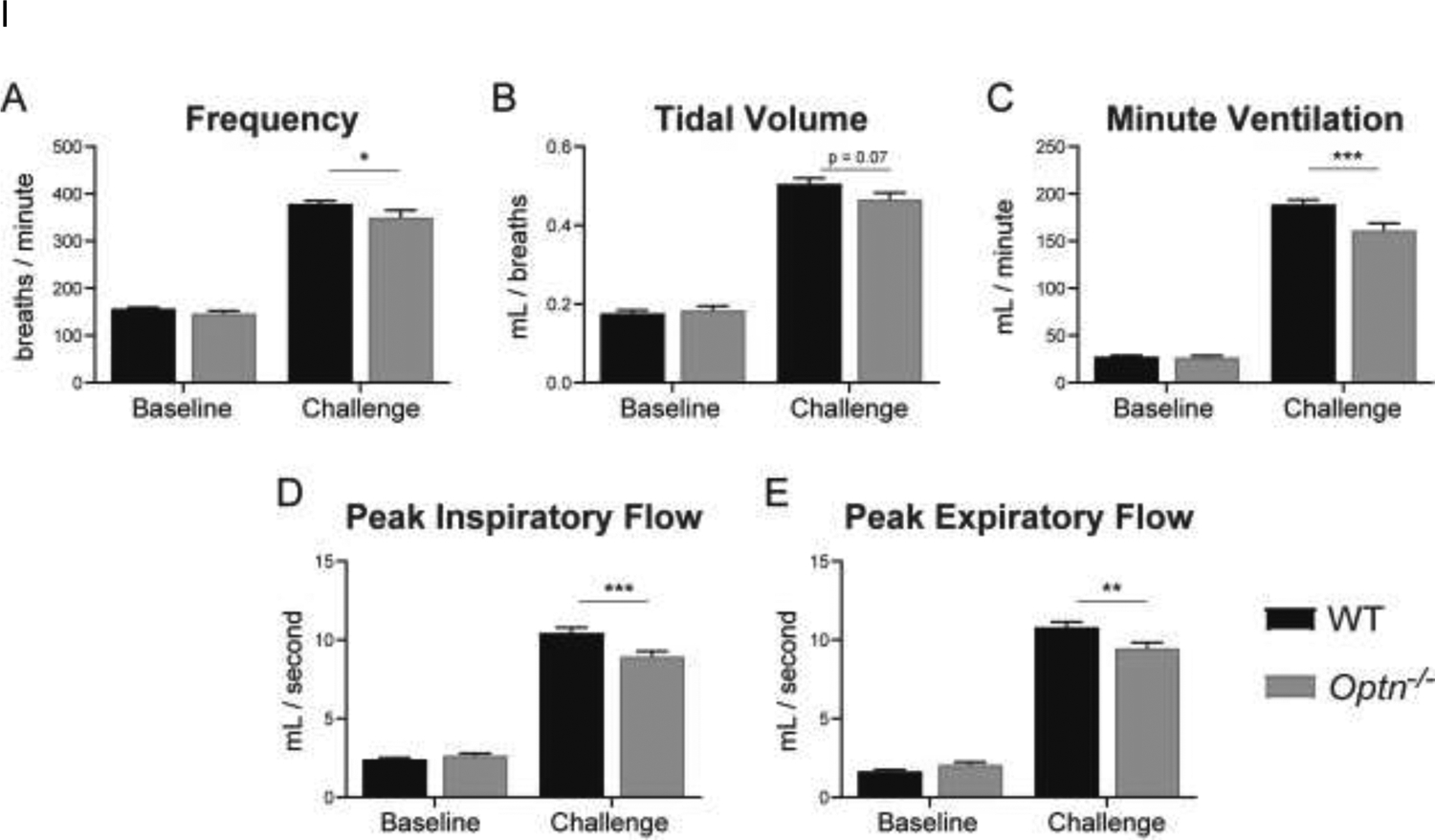
Measures of respiratory function – frequency (A), tidal volume (B), minute ventilation (C), peak inspiratory flow (D), peak expiratory flow (E) – evaluated under normoxia (FiO2: 0.21, N2 balance) (“Baseline”) and a maximal respiratory challenge with hypoxia + hypercapnia (FiCO2: 0.7, FiO2: 0.10, N2 balance) (“Challenge”). At 6 mo of age, Optn−/− mice do not display respiratory insufficiency at baseline. However, when exposed to the respiratory challenge, Optn−/− mice have decreased frequency, minute ventilation, peak inspiratory and peak expiratory flow as compared to C57BL/6J wild type (WT) controls. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
FIGURE 2: Respiratory Deficits Persist Through 12 Months of Age in Optn−/− mice.
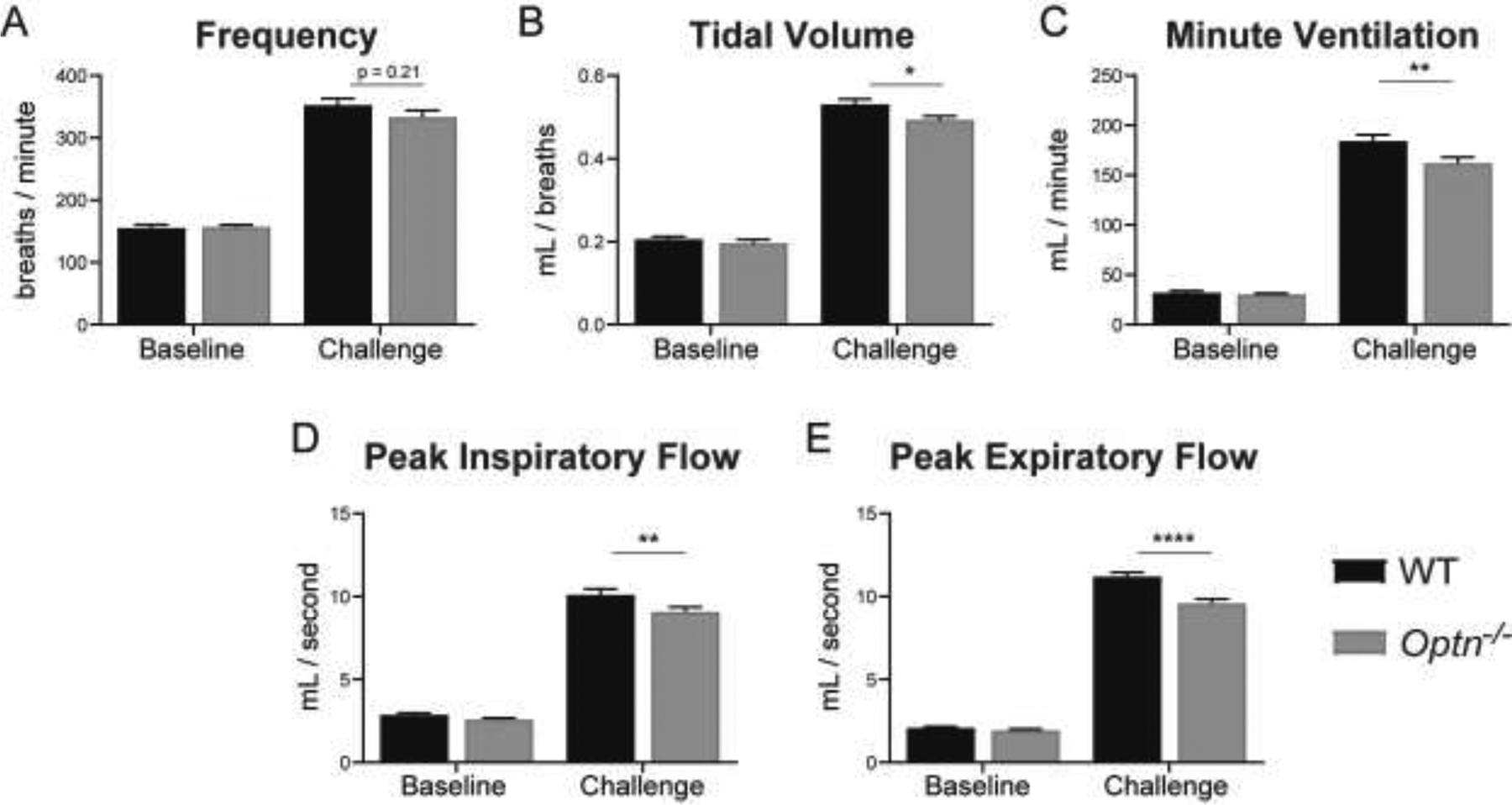
Measures of respiratory function – frequency (A), tidal volume (B), minute ventilation (C), peak inspiratory flow (D), peak expiratory flow (E) – evaluated under normoxia (FiO2: 0.21, N2 balance) (“Baseline”) and a maximal respiratory challenge with hypoxia + hypercapnia (FiCO2: 0.7, FiO2: 0.10, N2 balance) (“Challenge”). At 12 mo of age, Optn−/− mice still do not display respiratory insufficiency at baseline, but after a respiratory challenge have a decrease in tidal volume, minute ventilation, peak inspiratory flow and peak expiratory flow compared to WT controls. Frequency is no longer significantly different during the respiratory challenge at 12 mo of age. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
Since ALS patients also have significant respiratory apnea, we assessed the apneic events in Optn−/− mice and compared this to WT mice7. Both WT and Optn−/−mice had several apneic events which has been documented in C57BL6 mice27. However, compared to WT controls, 6-mo-old Optn−/− mice trend toward longer apneic events, which is lost in 12-mo-old mice (Figure 3A and 3B) (6-mo: p=0.053; 12-mo: p=0.29). In WT controls, apneas last the duration of 2 – 3 breaths, whereas, an apnea in Optn−/− mice can last up to 3 – 5 breaths, as observed in the representative tracing (Figure 3C).
FIGURE 3: Optn−/− mice do not have significantly longer apneas.
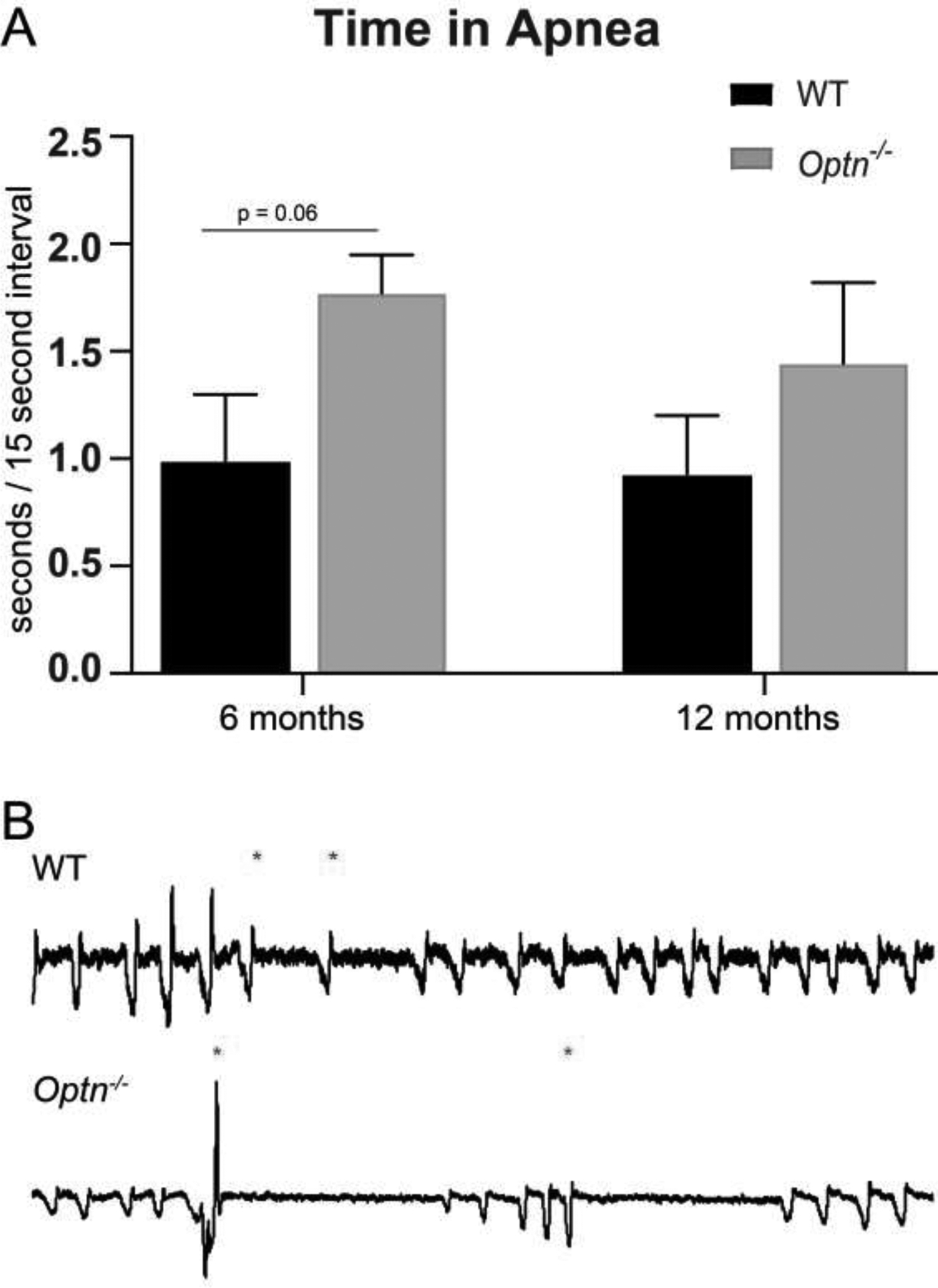
At 6 mo of age, Optn−/− mice have a trend of longer apneic events than the WT controls, which is lost at 12 mo of age. Panel A: Average apnea length over a 5 minute period of baseline breathing at 6 mo of age. Panel B: Average apnea length over a 5 minute period of baseline breathing at 12 mo of age. Panel C: Representative tracings of 10 seconds of respiration from 6 mo old C57BL/6J wild type control mice and Optn−/− mice during baseline breathing. Asterisks (*) denote beginning of apneic event.
3.2. Fewer motor neurons are present in respiratory centers in Optn−/− mouse brainstem.
Mice were euthanized at 12 mo of age for analysis of respiratory motor neuron and nerve axon health. Compared to WT mice, Optn−/− mice have fewer motor neurons in the hypoglossal motor nucleus, which controls the genioglossius muscle, at 12 mo of age (Figure 4A and 4C). We also quantified motor neurons in the putative phrenic motor nucleus in the ventral horn of the cervical spinal cord which control the diaphragm (Figure 3B). Unlike changes in the hypoglossal motor nucleus, phrenic motor neuron survival was unaffected (p=0.45).
FIGURE 4: Motor Neuron Loss in the Hypoglossal Motor Nucleus of Optn−/− mice.
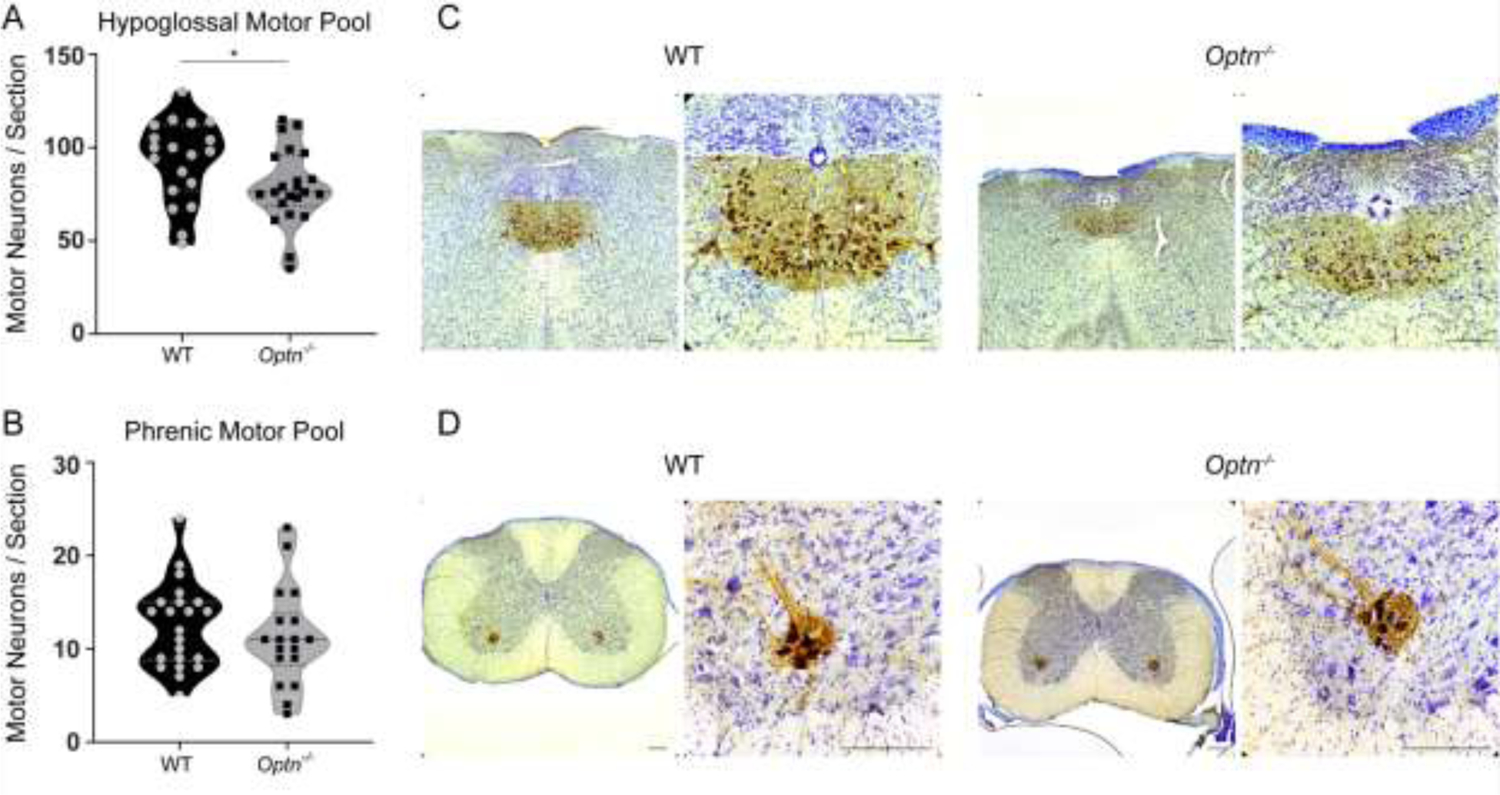
Hypoglossal and Phrenic motor neurons were labeled with CT-β (brown) – a retrograde tracer and counterstained with cresyl violet. Panels A and B: Quantification of motor neurons per 40μm section within the hypoglossal region of the medulla (A) and the phrenic region (B) from 12 mo old WT and Optn−/− mice. *p<0.05. Panels C and D: Representative images from WT and Optn−/− medulla sections (C) and cervical spinal cord (D), respectively. Scale Bar = 180 μm.
3.3. Optn−/− mice have disorganized myelin sheaths
The g-ratio, a measure of myelination in relation to axon size, is smaller in both the hypoglossal and phrenic nerves from Optn−/− mice (hypoglossal: p=0.0245; phrenic: p=0.0001), compared to WT mice. This decreased g-ratio indicates thicker myelin sheaths (Figure 5). Qualitative analysis of electron micrographs indicate that the smaller g-ratio is due to disorganized and frayed myelin, rather than an increase in myelin (Figure 6C and 6F). In comparison, WT mice exhibit thinner and more compact myelin sheaths and thus, larger g-ratios.
FIGURE 5: Myelin Disorganization in Respiratory Motor Neurons of Optn−/− mice.
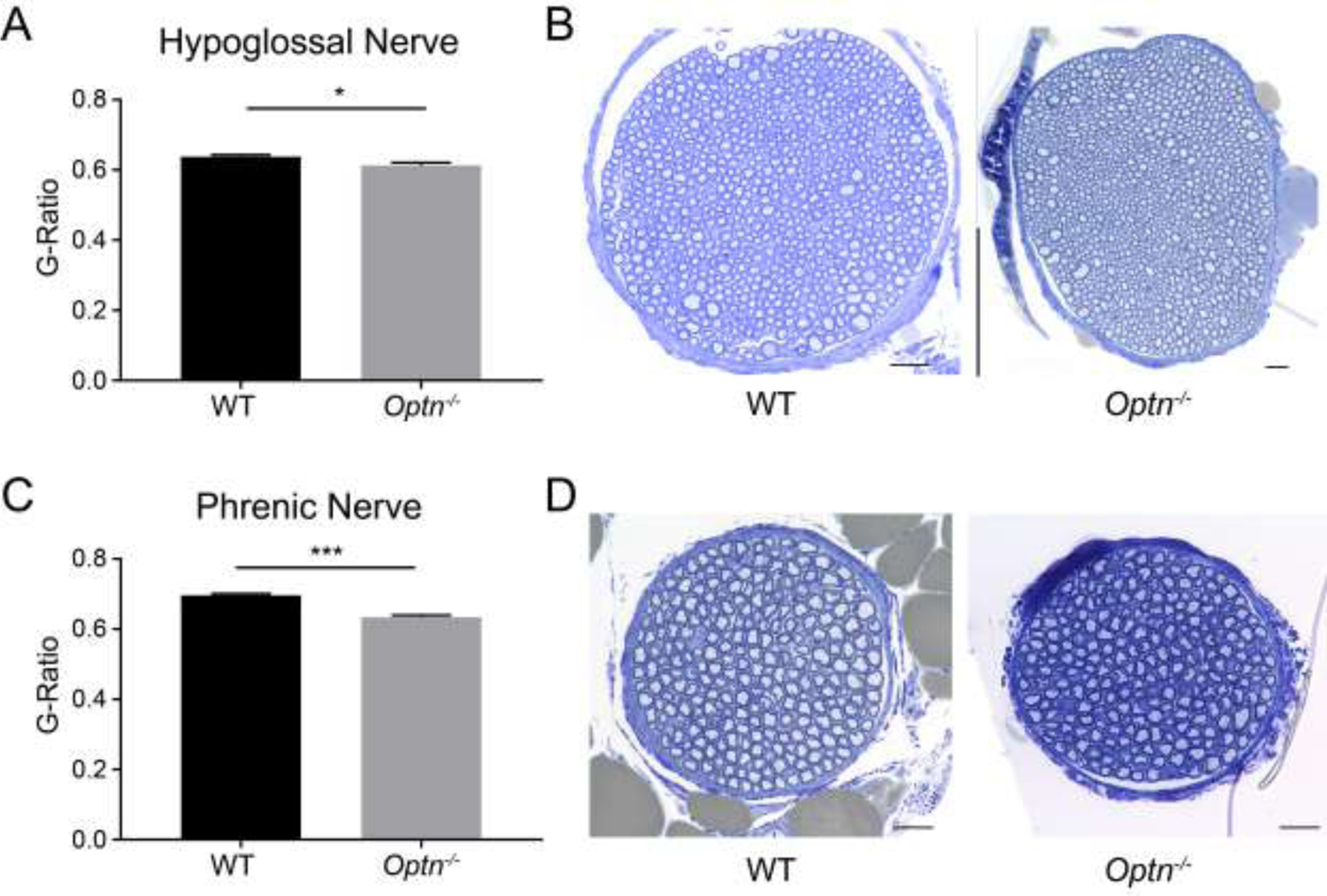
Panels A and C: G-ratio analysis, a measure of myelination, of axons within the hypoglossal and phrenic nerves, respectively, from 12 mo old mice. *p<0.05, ***p<0.001. Panels B and D: Representative images of WT and Optn−/− hypoglossal and phrenic nerve cross-sections from WT & Optn−/− mice stained with toluidine blue, Scale bar 20 μm.
FIGURE 6: Enlarged Mitochondria Accumulate in Respiratory Motor Neuron Axons of Optn−/− Mice.
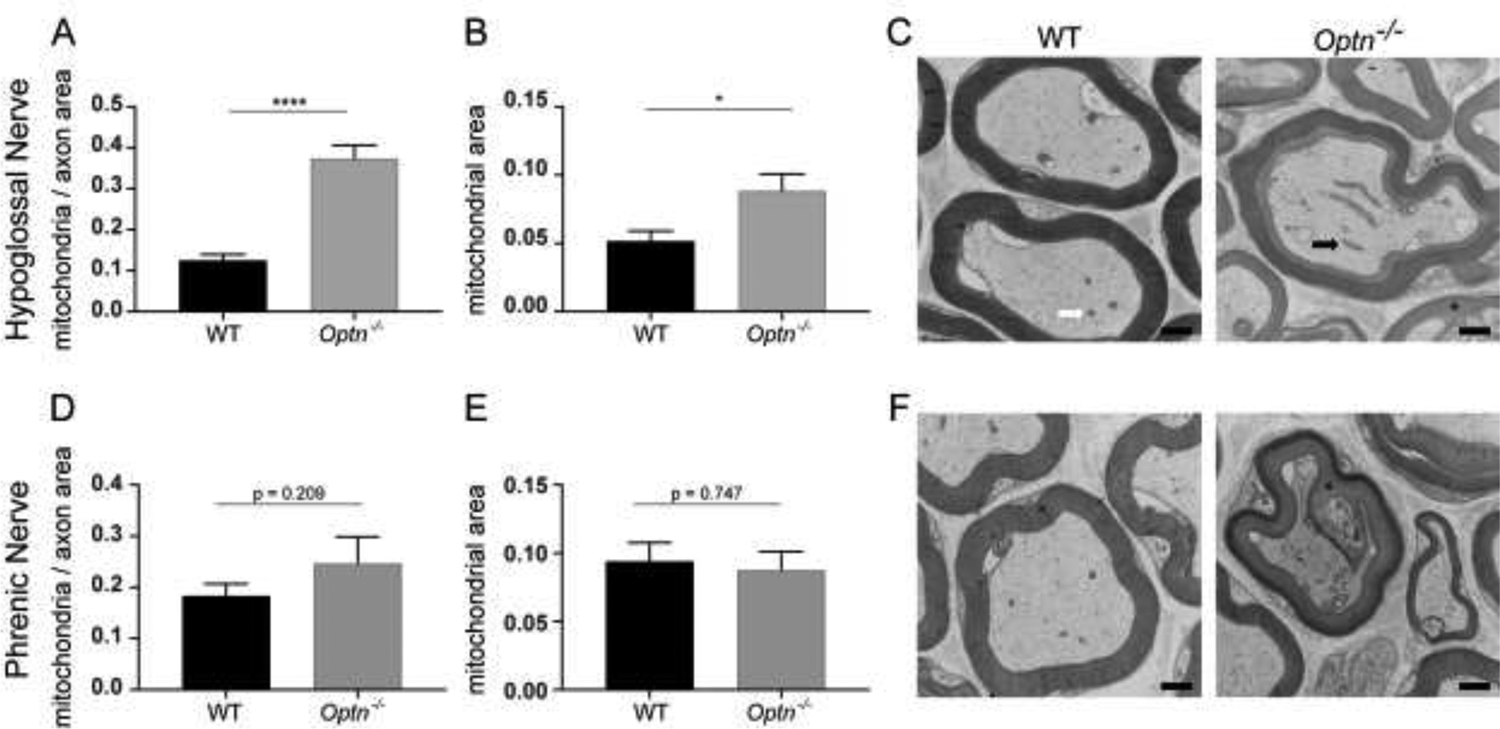
Panels A and D: Quantification of mitochondria within hypoglossal nerve axons and phrenic nerve axons, respectively, normalized to axonal area. ****p<0.0001. Panels B and E: Quantification of axonal mitochondrial size within the hypoglossal nerve axons and phrenic nerve axons, respectively. Panels C and F: Representative electron micrographs of hypoglossal nerve axons and phrenic nerve axons. The white arrow points to a representative healthy mitochondrion and the black arrow points to a representative enlarged mitochondrion. The blask asterisk identifies points of dysmyelination. Scale bar = 500nm.
3.4. Accumulation of mitochondria in Optn−/− axon at 12 mo
Mitochondrial health within hypoglossal and phrenic motor neuron axons was assessed using transmission electron microscopy. In 12-mo-old Optn−/− hypoglossal motor neuron axons, there are more mitochondria (p<0.0001), which qualitatively appear swollen, although not significantly (p=0.053) (Figure 6A, 6B, and 6C). In contrast, mitochondria in axons from WT mice demonstrate normal size and appearance. Interestingly, in the phrenic axons neither mitochondria accumulation (p=0.21), nor mitochondrial enlargement (p=0.75) in Optn−/− mice is observed (Figure 6D, 6E, and 6F).
4. DISCUSSION:
The primary findings of this study are that Optn−/− mouse exhibit respiratory insufficiency, hypoglossal motor neuron loss, and hypoglossal nerve abnormalities. Respiratory deficits in awake, unrestrained, spontaneously breathing mice are evident by 6 mo of age and continue through 12 mo of age. Interestingly, in the Optn−/− mouse there are fewer motor neurons in the hypoglossal motor nucleus but not in the phrenic motor nucleus. Furthermore, there is myelin disorganization along the hypoglossal and phrenic nerves, and accumulation of enlarged mitochondria in hypoglossal nerve axons. Our findings suggest that loss of optineurin leads to respiratory insufficiency in the Optn−/− mouse and that this pathology is more apparent in the hypoglossal motor pool.
4.1. ALS & Respiratory Dysfunction:
Despite overwhelming evidence of respiratory distress in ALS patients, only a few studies report respiratory deficits in the most common mouse model, the SOD1G93A mouse28,29. In the SOD1G93A mouse, TV, MV, PIF, and PEF are maintained through 18 weeks of age, but drop off substantially compared to nontransgenic littermate controls at endstage (~20 weeks of age). Similar to our findings in the Optn−/− mouse, these differences are observed only when SODG93A mice are challenged, and not at baseline28. However, in contrast to the SOD1G93A mice, disease onset is much later and progression is much slower in the Optn−/− mice. Our data show that respiratory deficits are apparent by 6 mo of age. When exposed to a respiratory challenge, MV, PIF, and PEF are all reduced in the Optn−/− mice compared to the WT controls. A decrease in PIF indicates weakness of the inspiratory muscles – namely the diaphragm – whereas a decrease in PEF is a reflection of weakened expiratory muscles such as the internal intercostal and accessory respiratory muscles30. At 12 mo of age, significant differences between WT & Optn−/− mice are maintained for MV, PIF, and PEF, but TV deteriorates with age. This slow progression is also observed in other Optn−/− mouse models13. Another potential reason for this unexpected finding is that in ALS due to OPTN deficiency, patients show no symptoms initially and many then develop a slowly progressive disease until end stage when severe respiratory pathology occurs. At 12mo, the Optn−/− mice have not yet reached end stage and respiratory pathology may require additional environmental stressors to instigate disease progression. These stressors may include hypoxia, viral infections, or occupational stressors that can exacerbate and trigger further respiratory degeneration31. Further studies will need to investigate the impact of both environmental stressors and age on disease onset and progression.
To investigate the cause of the observed respiratory deficits, we examined respiratory motor neuron loss in the medulla and cervical spinal cord for loss of motor neurons which is a major hallmark of ALS. Specifically, we focused on hypoglossal motor neurons in the medulla, which are responsible for maintaining airway patency, and phrenic motor neurons in the cervical spinal cord, which innervate the diaphragm. These neurons and nerves are crucial for proper ventilation2. Loss of both hypoglossal and phrenic motor neurons is observed in the SOD1G93A mouse at endstage32,33. Here, we only saw loss of motor neurons in the hypoglossal motor nucleus and not in the phrenic motor pool. This preservation of phrenic motor neurons at 12 mo may explain the lack of severe respiratory decline from 6 to 12 mo. We hypothesize that this lack of phrenic motor neuron pathology at 12 mo occurs because Optn−/− mice are not yet at end stage. Thus, similar to patients with OPTN deficiency, the bulbur muscles are affected early, and phrenic motor neuron preservation delays respiratory failure for several years after diagnosis9,10. However, at 12 mo the phrenic nerve exhibits significant abnormalities in myelination which can explain the deteriorating tidal volume. Similar to our findings, Ito et al. did not find reduced numbers of motor neuron in the spinal cord of Optn−/− mouse; however, they only examined the lumbar motor neurons, and did not examine motor neurons rostral of the lumbar region.
Despite the preserved phrenic motor neuron counts, there was evidence of abnormal myelination in both the hypoglossal and phrenic nerve. This is congruent with the findings of Ito et al. who reported decompaction of the myelin sheath and abnormal myelination in the spinal cords of Optn−/− mice13. Together, these indicate that myelin disorganization occurs in both the phrenic and hypoglossal nerves, whereas the loss of motor neurons is more prominent in the hypoglossal motor pool. It is unclear why the hypoglossal motor neurons are more vulnerable early in disease, but these findings are consistent with the clinical phenotype34–36. Future studies that investigate instigating stressors might unmask phrenic motor neuron loss and further pathology.
ALS patients experience an increase in central sleep apnea. We examined the presence of apneas in these mice, and both the WT and the Optn−/− mice had increased apnea. Although there was a trend towards increased time in apnea in Optn−/− mice, we did not find significant differences between WT and the Optn−/− mice. This lack of significant difference in apnea is mostly likely secondary to the increased prevalence of apneas in C57BL/6J mice27.
4.2. Optineurin, Mitophagy, and ALS
Dysfunction of the autophagy pathway is a major cause of molecular ALS pathology15. OPTN plays several roles in autophagy as a scaffold for autophagosome elongation and maturation machinery, and as a cargo adaptor protein37–39. Reduced autophagy of mitochondria (mitophagy) results in accumulation of non-functional mitochondria and reactive oxidative species40–42. Interestingly, we discovered that Optn−/− hypoglossal motor neuron axons have an accumulation of mitochondria. The trend towards increased mitochondrial size indicates swelling, which is a hallmark of dysfunction in these organelles43 – suggests potential axonal transport deficiency or, more likely, impaired mitophagy44. Accordingly, mitochondrial abnormalities have been well-documented in the SOD1G93A mouse model of ALS as well45. Similar to the Optn−/− mice, motor axons in SOD1G93A contain swollen mitochondria that have been shown to contribute to the onset of motor neuron degeneration46.
4.3. Conclusion
Overall, we demonstrate that this novel Optn−/− mouse displays respiratory dysfunction which is similar to that observed in ALS patients with OPTN deficiency. This respiratory pathophysiology includes respiratory insufficiency during a hypoxia and hypercapnic challenge, hypoglossal and phrenic nerve myelin disorganization with loss of hypoglossal motor neuron cell bodies, and an accumulation of abnormal mitochondria in the hypoglossal nerve axons. Given the slow progression and mild ALS phenotype, Optn−/− mice provide an important opportunity and an ideal tool to evaluate novel therapeutics as well as identify stress-dependent mechanisms that exacerbate neurodegeneration and respiratory function in ALS.
Supplementary Material
HIGHLIGHTS:
The optineurin-deficient mouse model (Optn−/−) exhibits respiratory insufficiency during a hypoxic and hypercapnic challenge.
Motor neuron loss is evident in the hypoglossal motor nucleus of 12 months old Optn−/− mice.
The hypoglossal and phrenic nerves of Optn−/− mice demonstrate dysmyelination as evidenced by disorganized and frayed myelin.
An accumulation of mitochondria in hypoglossal nerve axons occurs in the absence of optineurin.
Ackowledgements:
The authors thank the Duke University Electron Microscopy Core, specifically Sara Miller, PhD, Ricardo Vancini, PhD, and Harold Makeel for their expertise and skill in the preparation of tissues.
Funding: This work was supported by the National Institutes of Health NINDS R21 NS098131-02 (MKE) and 1R21NS112781-01A1 (MKE).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERNCES
- 1.Brown RH & Al-Chalabi A Amyotrophic Lateral Sclerosis. N Engl J Med 377, 162–172 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Fregosi RF & Fuller DD Respiratory-related control of extrinsic tongue muscle activity. Respir Physiol 110, 295–306 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Gestreau C, Dutschmann M, Obled S & Bianchi AL Activation of XII motoneurons and premotor neurons during various oropharyngeal behaviors. Respir Physiol Neurobiol 147, 159–176 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Remmers JE, deGroot WJ, Sauerland EK & Anch AM Pathogenesis of upper airway occlusion during sleep. J Appl Physiol Respir Environ Exerc Physiol 44, 931–938 (1978). [DOI] [PubMed] [Google Scholar]
- 5.Fusco AF, et al. The Respiratory Phenotype of Rodent Models of Amyotrophic Lateral Sclerosis and Spinocerebellar Ataxia. J Neuroinflamm Neurodegener Dis 3(2019). [PMC free article] [PubMed] [Google Scholar]
- 6.Lind LA, Murphy ER, Lever TE & Nichols NL Hypoglossal Motor Neuron Death Via Intralingual CTB-saporin (CTB-SAP) Injections Mimic Aspects of Amyotrophic Lateral Sclerosis (ALS) Related to Dysphagia. Neuroscience 390, 303–316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nichols NL, et al. Ventilatory control in ALS. 189, 429–437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alsultan AA, Waller R, Heath PR & Kirby J The genetics of amyotrophic lateral sclerosis: current insights. Degener Neurol Neuromuscul Dis 6, 49–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maruyama H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Markovinovic A, et al. Optineurin in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Progress in neurobiology 154, 1–20 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ying H & Yue BY Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol 294, 223–258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slowicka K, Vereecke L & van Loo G Cellular Functions of Optineurin in Health and Disease. Trends Immunol 37, 621–633 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Ito Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toth RP & Atkin JD Dysfunction of Optineurin in Amyotrophic Lateral Sclerosis and Glaucoma. Front Immunol 9, 1017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen DKH, Thombre R & Wang J Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci Lett 697, 34–48 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Del Bo R, et al. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. Journal of neurology, neurosurgery, and psychiatry 82, 1239–1243 (2011). [DOI] [PubMed] [Google Scholar]
- 17.van Blitterswijk M, et al. Novel optineurin mutations in sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging 33, 1016.e1011–1017 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Tumer Z, et al. Novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol Aging 33, 208.e201–205 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Weishaupt JH, et al. A novel optineurin truncating mutation and three glaucoma-associated missense variants in patients with familial amyotrophic lateral sclerosis in Germany. Neurobiol Aging 34, 1516.e1519–1515 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Wong SW, et al. Global deletion of Optineurin results in altered type I IFN signaling and abnormal bone remodeling in a model of Paget’s disease. Cell death and differentiation (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong S-W, et al. Global deletion of Optineurin results in altered type I IFN signaling and abnormal bone remodeling in a model of Paget’s disease. Cell Death & Differentiation (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeRuisseau LR, et al. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proceedings of the National Academy of Sciences of the United States of America 106, 9419–9424 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.ElMallah MK, et al. Retrograde gene delivery to hypoglossal motoneurons using adeno-associated virus serotype 9. Human gene therapy methods 23, 148–156 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falk DJ, et al. Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease. Molecular therapy : the journal of the American Society of Gene Therapy 21, 1661–1667 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goebbels S, et al. Elevated Phosphatidylinositol 3,4,5-Trisphosphate in Glia Triggers Cell-Autonomous Membrane Wrapping and Myelination. 30, 8953–8964 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer TD, Dash PK, Liu J & Waxham MN Morphology of mitochondria in spatially restricted axons revealed by cryo-electron tomography. PLoS Biol 16, e2006169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stettner GM, et al. Spontaneous central apneas occur in the C57BL/6J mouse strain. Respir Physiol Neurobiol 160, 21–27 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Stoica L, et al. Restrictive Lung Disease in the Cu/Zn Superoxide-Dismutase 1 G93A Amyotrophic Lateral Sclerosis Mouse Model. American journal of respiratory cell and molecular biology 56, 405–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tankersley CG, Haenggeli C & Rothstein JD Respiratory impairment in a mouse model of amyotrophic lateral sclerosis. Journal of applied physiology (Bethesda, Md. : 1985) 102, 926–932 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Huang P, et al. Impaired respiratory function in mdx and mdx/utrn(+/−) mice. Muscle & nerve 43, 263–267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vanacore N, Cocco P, Fadda D & Dosemeci M Job strain, hypoxia and risk of amyotrophic lateral sclerosis: Results from a death certificate study. Amyotroph Lateral Scler 11, 430–434 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Ferrucci M, et al. A systematic study of brainstem motor nuclei in a mouse model of ALS, the effects of lithium. Neurobiology of disease 37, 370–383 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Nichols NL, Satriotomo I, Harrigan DJ & Mitchell GS Acute intermittent hypoxia induced phrenic long-term facilitation despite increased SOD1 expression in a rat model of ALS. Experimental neurology 273, 138–150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Comley L, et al. Motor neurons with differential vulnerability to degeneration show distinct protein signatures in health and ALS. Neuroscience 291, 216–229 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Nimchinsky EA, et al. Differential vulnerability of oculomotor, facial, and hypoglossal nuclei in G86R superoxide dismutase transgenic mice. The Journal of comparative neurology 416, 112–125 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Haenggeli C & Kato AC Differential vulnerability of cranial motoneurons in mouse models with motor neuron degeneration. Neurosci Lett 335, 39–43 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Wong YC & Holzbaur EL Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proceedings of the National Academy of Sciences of the United States of America 111, E4439–4448 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bansal M, et al. Optineurin promotes autophagosome formation by recruiting the autophagy-related Atg12-5-16L1 complex to phagophores containing the Wipi2 protein. The Journal of biological chemistry 293, 132–147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryan TA & Tumbarello DA Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front Immunol 9, 1024 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shefa U, et al. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen Res 14, 749–756 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dutta D, Xu J, Kim JS, Dunn WA Jr. & Leeuwenburgh C Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity. Autophagy 9, 328–344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menzies FM, et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 93, 1015–1034 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Gerencser AA, Doczi J, Torocsik B, Bossy-Wetzel E & Adam-Vizi V Mitochondrial swelling measurement in situ by optimized spatial filtering: astrocyte-neuron differences. Biophys J 95, 2583–2598 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen H & Chan DC Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum Mol Genet 18, R169–176 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan W, Pasinelli P & Trotti D Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim Biophys Acta 1842, 1295–1301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kong J & Xu Z Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci 18, 3241–3250 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.