

Abstract
The extracellular accumulation of amyloid β (Aβ) fragments of amyloid precursor protein (APP) in brain parenchyma is a pathological hallmark of Alzheimer’s disease (AD). APP can be cleaved into Aβ on late endosomes/multivesicular bodies (MVBs). E3 ubiquitin ligases have been linked to Aβ production, but specific E3 ligases associated with APP ubiquitination that may affect targeting of APP to endosomes have not yet been described. Using cultured cortical neurons isolated from rat pups, we reconstituted APP movement into the internal vesicles (ILVs) of MVBs. Loss of endosomal sorting complexes required for transport (ESCRT) components inhibited APP movement into ILVs and increased endosomal Aβ42 generation, implying a requirement for APP ubiquitination. We identified an ESCRT-binding and APP-interacting endosomal E3 ubiquitin ligase, ubiquitination factor E4B (UBE4B), that regulates APP ubiquitination. Depleting UBE4B in neurons inhibited APP ubiquitination and internalization into MVBs, resulting in increased endosomal Aβ42 levels and increased neuronal secretion of Aβ42. When we examined AD brains, we found levels of the UBE4B-interacting ESCRT component, hepatocyte growth factor–regulated tyrosine kinase substrate (Hrs), were significantly decreased in AD brains. These data suggest that ESCRT components critical for membrane protein sorting in the endocytic pathway are altered in AD. These results indicate that the molecular machinery underlying endosomal trafficking of APP, including the ubiquitin ligase UBE4B, regulates Aβ levels and may play an essential role in AD progression.
Keywords: endocytosis, ubiquitination factor E4B (UBE4B), membrane trafficking, multivesicular body (MVB), Alzheimer’s disease (AD), amyloid β (Aβ)
Summary Statement.
This work identifies an endosome-associated E3 ubiquitin ligase, UBE4B, that regulates APP ubiquitination and is involved in a discrete step in endocytic APP trafficking that affects its pathogenic cleavage and secretion of Aβ fragments via a noncanonical pathway.
Introduction
Amyloid precursor protein (APP)1 and its cleavage product, amyloid beta (Aβ), have been implicated in the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease (AD), amyloid angiopathy, and vascular dementia 1–4. These diseases present a significant burden to patients, caregivers, and healthcare systems 1,5,6. Despite decades of investigation, there are currently no disease-modifying treatments 5–7 and only a few established biomarkers 8.. Post-mortem examination of AD brains often reveals marked neuronal death and pathology that includes extracellular accumulation of protein aggregates (i.e. plaques). These plaques, predominantly composed of Aβ, are thought to precede neuronal loss 2–4 and may correlate with cognitive decline 9–11. Aβ is produced in neurons 12–14 and can accumulate both intra- and extra-neuronally. The relative contributions of Aβ intraneuronal accumulation, increased Aβ secretion/parenchymal deposition, and decreased extraneuronal Aβ clearance to disease-producing amyloid burden are the subject of active investigation 3,15.
Endocytic trafficking of APP has been linked to the production and secretion of Aβ 15–21. The abnormal endosomal morphology and increased endosomal Aβ accumulation observed in post-mortem examination of AD brains suggests that endosomal dysfunction may play a role in the pathogenesis of AD 22–24. APP resides on the cell surface and is internalized into endosomal compartments. The fate of membrane proteins that move through this pathway depends on whether they enter the internal vesicles (ILVs) of the late endosome/multi-vesicular body (MVB) or remain on the limiting MVB membrane 25–30. Membrane proteins that are internalized into ILVs of MVBs can be degraded upon MVB-lysosome fusion 30,31 or can be secreted extracellularly upon MVB-PM fusion 32–34. APP can be internalized into ILVs of the MVB 17,18,20,35,36. The efficiency of APP movement into ILVs likely determines the time it spends in proximity to the endosome-associated β- and γ-secretases and therefore the propensity for Aβ fragment formation in endosomes.
Ubiquitination of membrane protein cargo is required for engagement of the Endosomal Sorting Complexes Required for Transport (ESCRT), a molecular machinery that allows for membrane protein movement into ILVs 30,31,37–40. The ESCRT machinery has been implicated in APP trafficking at endosomes, although evidence for their role in Aβ generation is conflicting17–19. ESCRT complex proteins contain ubiquitin-interaction motifs (UIM) that enable engagement with ubiquitin moieties present on cargo proteins for sorting into ILVs of MVBs 30,37,41,42. Therefore, involvement of ESCRTs in APP endosomal trafficking suggests that APP is ubiquitinated to allow for ESCRT interactions. APP ubiquitination is associated with APP accumulation on the endosomal membrane 17,43–45 and changes in the production of Aβ species. Although E3 ubiquitin ligases have been previously linked with amyloid production 43,44,46, a specific E3 ligase associated with APP ubiquitination that would affect its endosomal disposition has not been isolated.
We utilized a cell-free assay that reconstitutes ILV formation to identify an E3 ligase responsible for ubiquitinating APP. We confirmed that the ESCRT machinery is required for movement of APP into ILVs of MVBs and that loss of ESCRT components results in enhanced endosomal generation of Aβ42. More importantly, we identified UBE4B as an E3 ubiquitin ligase that interacts with APP and can regulate APP ubiquitination. We observed that loss of UBE4B results in enhanced Aβ generation in endosomes and increased Aβ secretion from neurons. Furthermore, we found that levels of the UBE4B-interacting ESCRT component Hrs are significantly decreased in mild-moderate AD brains and that levels of UBE4B show a decreasing trend in AD patient samples. These data suggest that efficient endocytic trafficking of APP may be important for regulating intra- and extra-neuronal Aβ levels, that UBE4B can ubiquitinate APP and enable its endocytic trafficking and secretion, and that Hrs and UBE4B levels may be altered in aging and age-related diseases.
Experimental Procedures
Materials—
Antibodies were purchased from the following commercial sources: C-terminal APP (Sigma; A8717; 5 μg/mL for immunoblotting (IB); 50 μg/mL for immunocytochemistry (ICC), N-terminal APP (Millipore; 22C11; 10 μg/mL for ICC), MAP2 (Millipore, AB5622; 5 μg/mL); Rab7 (Invitrogen; # D95F2; 2.5 μg/μL for ICC, 0.25 μg/mL for IB), EEA1 (Thermo Fisher; Cat. # PA5–17228; 2.5 μg/μL for ICC, 0.25 μg/mL for IB), BACE-1 (Cell Signaling; D10E5; 1 μg/mL), Presenilin 2 (Abcam; ab51249; l:1000),quan Actin (Sigma; A2066; 0.3 μg/mL), UBE4B (Abcam; ab126759; 0.3 μg/mL for IB; ab97697), Ubiquitin (Enzo; P4D1; 1 μg/μL), Hrs (Axxora; A-5; 1 μg/μL), EGFR (Invitrogen; Cat # PA1–1110; 1:200), phospho Tau (Abcam; ab109390; 1:1000).
DNA Constructs—
Plasmids encoding for short hairpin RNAs directed against UBE4B (CCGGTGGACCAACTGACGGATATTTCTCGAGAAATATCCGTCAGTTGGTCCATTTTTG; TRCN0000338294) and a scrambled shRNA (CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTT; SHC002) were purchased from Mission shRNA (Sigma).
Cell Culture—
Mammalian Cell Culture—
TLA-HEK 293T cells (ATCC) were cultured as a monolayer in 10-cm dishes as previously described in25. The cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Mediatech), containing 10% Fetal Bovine Serum (FBS, Sigma) under 5% CO2 at 37°C. To passage the cells, cells were removed from the plate using 0.05 mM trypsin/EDTA. Similarly, SK-N-AS (ATCC) cells were cultured as a monolayer in 10-cm dishes. SK-N-AS cells were maintained in Roswell Park Memorial Institute (RPMI, Mediatech), containing 10% FBS and 1% L-Glutamine (Life Technologies) under 5% CO2 at 37°C. Before each experiment, cells were passaged by removing them from the plate using 0.05 mM EDTA.
Primary Neuron Culture—
Primary cortical neurons were obtained from E21 rat embryos (both male and female), as described 47–50. Animal procedures were performed in accordance with National Institutes of Health Guidelines for the care and use of laboratory animals and approved by the Animal Welfare Committee at the University of Texas Health Science Center at Houston, TX, USA. Briefly, cortical brain tissue was extracted from rat embryonic brains, enzymatically dissociated, and seeded (8 million cells/plate) on 10-cm plates coated with 50 μg/mL Poly-D-Lysine (Millipore). Neurons were maintained in Neurobasal medium (Life Technologies), 2% B27 (Life Technologies), 1% Glutamax (Life Technologies), and 1% Penicillin-Streptomycin (Sigma) under 5% CO2 at 37°C. Neuron medium was replenished every 4 days. Experiments were carried out using neurons at 5-7 days in vitro unless otherwise specified.
Immunocytochemistry—
Neurons (1e6) were plated on coverslips coated with Poly-D-Lysine. After 5 days, neurons were fixed with 4% paraformaldehyde for 15 minutes at room temperature, and treated with 100mM glycine in PBS for 10 minutes. Fixed neurons were permeabilized with 0.1% Triton X-100 in PBS for 10 minutes, blocked with 10% normal goat serum in PBS for 1 hour, and subsequently incubated with primary antibodies overnight at 4 °C. The following day cells were washed with PBS (3X for 5 min) and incubated with secondary antibodies for 1 hour at room temperature. Slides were washed 3x with PBS and mounted using DAPI mounting medium. Images were obtained by fluorescent microscopy with a Leica TCS SPE microscope and analyzed using ImageJ (NIH).
Discontinuous sucrose gradient for endosome enrichment—
Neurons, [5-7 days in vitro (DIV)] were washed with PBS, scraped, and centrifuged (1500 x g, 10 min). Cell pellets were resuspended in 100 μL of homogenization buffer (HB; 20 mM HEPES pH 7.4, 0.25 M sucrose, 2 mM EGTA, 2 mM EDTA, and 0.1 mM DTT) containing protease inhibitors. Cells were then sonicated 3 times (10 pulses of 1 second at output control 3; Branson Sonifier 250, VWR Scientific), and centrifuged at 2000 x g for 10 minutes. The supernatant (Postnuclear supernatant, PNS) was collected, volume increased to 440 μL with HB, and loaded at the bottom of a 2ml ultracentrifugation tube and overlaid with three layers of sucrose: 35% sucrose (660 μL), 25% sucrose (440 μL), and 8% sucrose (500 μL) as described in 51. All sucrose-containing solutions contained imidazole (3mM) and EDTA (1mM, pH 7.4). The gradient was centrifuged (150,000 x g for 1 hour, model TLS55; Beckman Coulter). After centrifugation, 200 μL fractions were collected from the top of the gradient (10 steps per gradient). Fractions were diluted using at least 1:1 in HB and membranes were pelleted by centrifugation (150,000 x g for 30 min). Membrane fractions were used in cell-free reactions or for immunoblotting.
Cytosol preparation—
Mammalian cytosol:
Rat brain cytosol was produced as described 52. Neurons were scraped and centrifuged (2000 x g for 5 min at 4°C), resuspended in HB with protease inhibitors (112 μM PMSF, 3 μM aprotinin, 112 μM leupeptin, 17 μM pepstatin), and sonicated 3 times (8 pulses of 1 second at output control 2; Branson Sonifier 250, VWR Scientific). The resulting lysate was centrifuged (2000 x g for 10 min at 4°C) and the supernatant was further centrifuged (100,000 x g for 1 hour at 4°C). The supernatant was collected as cytosol and protein concentrations were calculated using a Bradford assay (Pierce). The cytosol was divided into 75 μg aliquots and stored at −80 °C.
For immunodepletion experiments, rat brain cytosol (100 μg) was incubated with antibody (1 μg) and a protease inhibitor cocktail overnight (end-over-end rotation at 4°C). Samples were then incubated with washed protein A agarose beads (15 μL) for 2 hours at 4°C. Following centrifugation (1000 x g for 3 seconds), immunodepleted supernatant was collected as cytosol and protein concentrations were calculated using a Bradford assay. The cytosol was divided into 75 μg aliquots and stored at −80 °C.
Yeast cytosol:
Saccharomyces cerevisiae deletion strains (Dharmacon) were plated on YPD plates (500 mL ddH20 containing: 10 g bactopeptone, 5 g yeast extract, 8 g agar, 25 mL 40% dextrose) with G418 (500 μg/mL), and incubated overnight at 30°C. The parental strain was cultured without G418. Colonies were inoculated in 5 mL of YPD medium (500 mL ddH20 containing: 10 g bactopeptone, 5 g yeast extract, 25 mL 40% dextrose) and incubated overnight on a shaker at 30 °C. A secondary culture of YPD medium (100 mL) was inoculated with the overnight culture and grown until OD600 reached 0.8-1.0. Cells were collected (3000 x g for 10 min) and washed with ddH2O, followed by TP buffer (20 mM Tris, pH 7.9; 0.5 mM EDTA; 10% glycerol; 50 mM NaCl, 112 μM leupeptin, 3 μM aprotinin, 112 μM PMSF, and 17 μM pepstatin). The final pellet was resuspended in 100 μL of TP buffer. Cells were lysed using acid-washed glass beads (1 min vortex-1 min incubation on ice, 5 times). Lysate was centrifuged (3000 x g for 10 min), and the supernatant was collected and further centrifuged (100,000 x g for 60 min) to collect cytosol in the supernatant. Protein concentrations were calculated using a Bradford assay. The cytosol was divided into 160 μg aliquots and stored at −80 °C.
Immunoprecipitation—
For immunoprecipitation experiments, neurons were scraped from a plate using a rubber scraper, and centrifuged (1500 x g for 10 mins). The cell pellet was resuspended in 100 μL of RIPA Buffer (10 mM Tris-C1 pH 8, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-1000, 0.1% sodium deoxycholate, 140 mM NaCl) containing protease inhibitors, lysed (1 hour at 4 °C with end-over-end rotation) and centrifuged (20,000 x g for 10min). Cell lysate (100 μg) was incubated with antibody (1 μg) and a protease inhibitor cocktail overnight (end-over-end rotation at 4 °C). Samples were subsequently incubated with washed protein A agarose beads (15 μL) for 2 hours at 4°C. After 2 hours, beads were washed (3x with PBS) prior to resuspension in sample buffer for immunoblotting.
Cell-free endosomal maturation assay—
Reconstitution of endosomal maturation (and formation of inwardly-budded vesicles) was performed as previously described 25,27,53, with modifications. Early endosomes from primary neurons were obtained from a discontinuous sucrose gradient (as described above) and resuspended in HB. Endosomal membranes (starting material, 5 μL) were either incubated on ice or were trypsin-treated (10 μL of 0.25 μg/μL trypsin; 4 °C for 30 min). For reactions containing rat brain cytosol, a standard reaction (50 μL) contained 15 μL endosomal membranes, 6 μL ATP regeneration system (2mM MgATP, 50 μg/mL creatine kinase, 8mM phosphocreatine and 1mM DTT of final concentrations), 75 μg of rat brain cytosol and HB to a total reaction volume of 50 μL. For reactions containing Saccharomyces cerevisiae cytosol, a standard reaction (50 μL) contained 15 μL membranes, 6 μL ATP regeneration system, 160 μg of Saccharomyces cerevisiae cytosol and HB to a total reaction volume of 50 μL. Saccharomyces cerevisiae cytosol did not contain detectable levels of APP (data not shown).
All experimental reactions were incubated for 3 h at 37 °C, followed by trypsin-treatment (10 μL of 0.25 μg/μL trypsin; 30 min at 4 °C). After trypsin treatment, experimental reactions were centrifuged (20,000×g: 30 min at 4 °C) while control reactions remained on ice. Control reactions were resuspended in sample buffer for SDS-PAGE. For experimental reactions, supernatant was aspirated, and the pellet was resuspended in sample buffer for biochemical examination by SDS-PAGE. Resultant blots were probed with an antibody that recognizes the C-terminal epitope of APP (Sigma). All experimental reactions were normalized to starting endosomal controls to obtain the reaction efficiency. To obtain reaction efficiency, the amount of APP on starting membranes added to each reaction was compared to the amount of APP in reactions that contained membranes, cytosol, ATP and had been subsequently cleaved by trypsin. Typical reaction efficiencies were 30–60% and are shown in the figures and/or their respective figure legends. For experiments examining the generation of Aβ42, Following the 3-hr incubation, experimental reactions were centrifuged (20,000×g for 30 min at 4 °C) and resulting pellets were resuspended in HB for analysis by ELISA (described below, section 4.10).
Cell Transfection and lentivirus production—
TLA-HEK 293T cells were grown at 37 °C as described in 4.3. They were transfected with third-generation lentiviral packaging plasmids (pMLg/pRRE, pRSV-Rev, pMD2.g; Addgene) and target DNA using Lipofectamine 3000 (Thermo Fisher), according to manufacturer’s instructions. After 48h, viral media was collected from TLA-HEK293T cells, passed through a 0.22 μm PVDF filter (VWR), and incubated with cortical neurons (DIV 1) at a 1:3 ratio with neuronal media. After 24h, viral media was removed and neurons were incubated with filtered conditioned media for 48h prior to experimentation.
ELISA—
The murine AB42 enzyme-linked immunosorbent assay (ELISA) kit (Life Technologies) was used according to manufacturer’s instructions. For analysis of endosomal Aβ42, endosomal membranes generated from each reaction of the cell-free assay were resuspended in 50 μL HB, sonicated (2 x 10 pulses), diluted 1:1 with buffer, and loaded onto the bottom of an ELISA well. Each experiment was performed at least 3 times. For experiments measuring Aβ42 secretion, 100 μL of conditioned medium was diluted 1:1 with buffer and loaded onto wells. Each conditioned media sample was measured in duplicate. Values generated by ELISA were normalized to the protein concentration present in lysate from each trial.
Preparation of human lysate—
All human tissue samples were kindly provided by Dr. Julia Kofler and obtained from the University of Pittsburgh neurodegenerative brain bank with appropriate ethics committee approval (Committee for Oversight of Research and Clinical Training Involving Decedents). The studies abide by the Declaration of Helsinki principles. Human Brain neocortex tissue from Control (n=8) or AD patients (n=13) (Supplemental Table 2) were homogenized and lysed in buffer containing 1% NP-40, 1 mM PMSF, cOmplete (Roche), and PhosSTOP (Roche). Following centrifugation, the supernatant was removed, and protein concentration was measured via a Bradford Assay. Protein (50μg) was diluted 1:1 in 2X Laemmli Sample Buffer (Bio-Rad, 161-0737) supplemented with 4% β-mercaptoethanol and then heated to 100 °C for 5 minutes. Following sample preparation, proteins were separated using a 4-15% Mini-PROTEAN TGX 15-well gels (Bio-Rad, cat. #4561083). Protein bands were subsequently transferred to a nitrocellulose membrane and probed with anti-UBE4B, anti-APP, anti-EGFR, anti-Bace-1, anti-Presenilin 2, anti-Hrs, anti-phsphoTau, and anti-Actin. Blots were then probed with HRP-conjugated goat anti-rabbit secondary antibody (1:5000, Vector Laboratories, cat. PI-1000) or goat antimouse secondary antibody (1:5000, Vector Laboratories, cat. PI-2000) and imaged using a Bio-Rad ChemiDoc Imager.
Cryo-electron Microscopy—
Cryo-electron microscopy was performed as previously described in 25. Briefly, endosomal fractions 3-5 (containing late endosomal membranes) and endosomal fractions 7-9 (containing early endosomal membranes) were isolated as described above and collected. Endosomal Fractions (n=1 sample/group) were applied to freshly glow-discharged (30 s) 2/2 Quantifoil on 200 mesh copper grids. After 30 s, excess buffer was blotted and the sample was immediately plunged into ethane cooled to liquid N2 temperature. Cryo-preserved grids were stored in liquid N2 until use. Cryo-electron microscopy was performed on a FEI Polara G2 equipped with a Gatan K2 Summit direct electron detector. Multiple areas of the grid were chosen at random and 8 x 8 montages were collected at 4700x in low dose/photon counting mode using SerialEM. Endosomes were examined for fragmentation and to ensure they were intact.
Statistical Analysis—
Statistical analysis was performed by a blinded investigator using Prism 7 software, similar to described in 25. Prior to analysis, samples were tested for normality using the Shapiro-Wilk test, and found to be normal. If the samples followed a normal distribution, then either a Paired t-test, unpaired t-test, or a one-way ANOVA followed by post-hoc analysis (Tukey or Dunnet’s test) were performed. If the samples were not normal, then the Kruskal-Wallis analysis was performed. A p-value of < 0.05 was considered statistically significant with an n= >3.
Results
Measuring APP movement into the endosomal lumen
Endogenous APP expression was confirmed in cultured cortical neurons isolated from E17 rat pups (Fig. 1a). Endosomal subpopulations were separated from these neurons using a discontinuous sucrose gradient (Fig. 1b) 51,54. The Refractive index (RI) of each fraction was measured and is shown below the blot. Immunoblotting was used to examine the gradient fractions for markers for endosomal compartments (EEA1 for early endosomes, Rab7 for late endosomes) as well as APP, β-site amyloid precursor protein cleaving enzyme 1 (BACE-1), and the γ-secretase component Presenilin-2 (PS2). We observed two distinct endosomal peaks that localized with either markers for early endosomes (Fig. 1b, fractions 7-9, enriched in EEA1) or mixed/late endosomal markers (Fig. 1b, fractions 3-5, enriched in Rab7). APP was enriched in both of these peaks, while the APP-modifying secretases BACE-1 and PS2 were enriched in the mixed endosome peak (Fig. 1b). In addition, we probed for other markers on our gradient, including a marker for the plasma membrane (Na/K ATPase), an endoplasmic reticulum marker (GRP78/BiP), and a mitochondria marker (Tomm20) (Supplemental Fig. 1a). Our findings indicate that minimal levels of Na/K ATPase and Tomm20 are detected in early endosome fractions. In contrast, the ER marker, Grp78 is present in early endosome fractions. These data suggest our early endosome fractions co-fractionate with ER, but not plasma membrane-derived membranes and only minimally co-fractionate with mitochondria.
Figure 1. APP movement through endosomal compartments can be reconstituted in vitro.
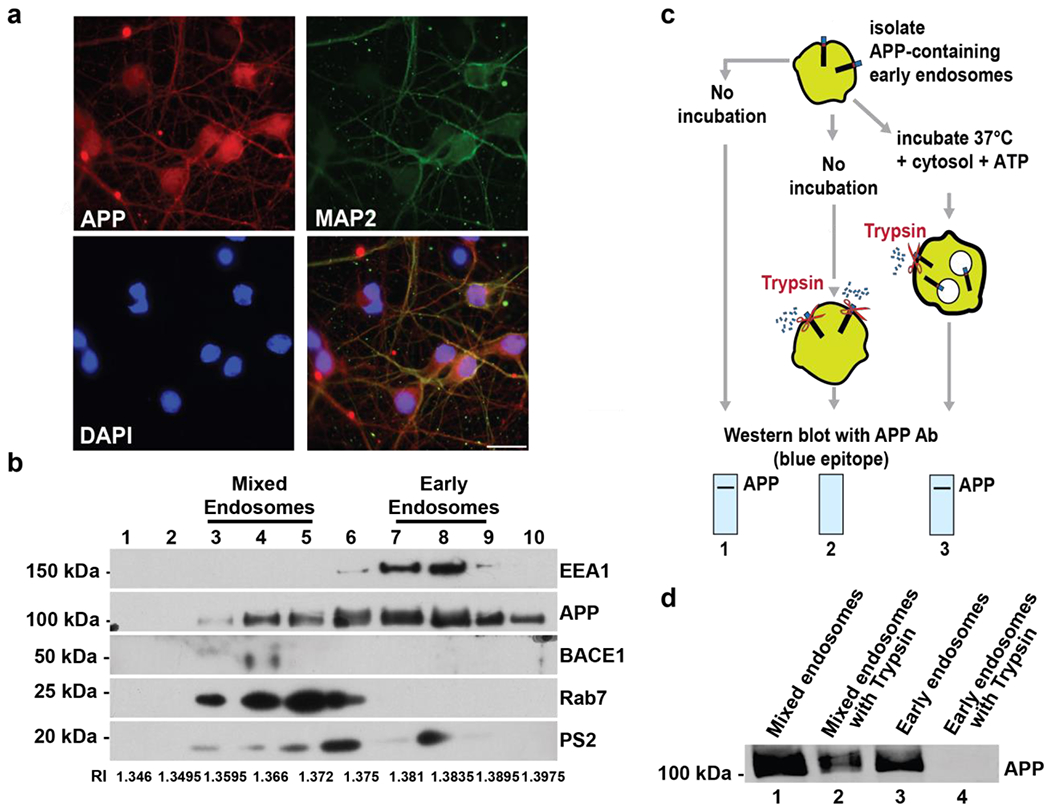
Primary cortical neurons from E17 rat pups were isolated and cultured for 5-7 days, a) 5-7 DIV rat cultured neurons were stained with fluorescent antibodies against APP, MAP2 (a neuronal marker), and DAPI (a nuclear stain). APP colocalizes with MAP2 in cortical neurons. Scale bar = 25 μm. b) Post-nuclear supernatant was loaded at the bottom of a discontinuous sucrose gradient. After centrifugation, fractions were pelleted and analyzed by immunoblot. The Refractive index (RI) of each fraction was also measured and is shown below the blot. Fractions were probed for an early endosome marker (EEA1), a late endosome marker (Rab7), APP, BACE-1, and Presenilin 2 (Ps2). Gels were cut to probe for EEA1 and APP above 75kDA, between 75kDA and 37kDA to probe for BACE1, and below 37kDA to probe for Rab7 and PS2 c) Schematic of the reconstitution of APP protein sorting from endosomal membranes. (1) Partially purified endosomes isolated from fractions 7-9 of the discontinuous sucrose gradient can be detected by immunoblotting using an intracellular epitope-specific antibody. (2) Incubation of these endosomes with trypsin removes the intracellular epitope of the receptor that protrudes from the plasma membrane, resulting in a loss of signal for that epitope on an immunoblot. Incubation of endosomes with ATP and cytosol (isolated from rat brain [75μg] or Saccharomyces cerevisiae [160μg]) at 37 °C results in formation of internal vesicles and protection of the intracellular APP epitope from subsequent trypsin d) APP in the mixed endosomal populations (fractions 3-5) was resistant to trypsin digestion. In contrast, APP present on early endosomal fractions (fractions 7-9) was susceptible to trypsin cleavage. Representative blots are shown.
To determine whether APP in early and mixed/late endosome populations was localized on the endosomal limiting membrane or in IFVs of MVBs, we used a cell-free assay that reconstitutes MVB formation and membrane protein movement from the limiting endosomal membrane into IFVs of MVBs (Fig. 1c) 25,27. To determine the localization of APP with respect to the endosomal limiting membrane, early endosome fractions (fractions 7-9, Fig. 1b) or mixed/late endosome fractions (fractions 3-5, Fig. 1b) were incubated with or without trypsin, as this protease digests the exposed intracellular fragment of APP that has not been internalized into MVBs (Fig. 1c, lane 2). Samples were analyzed via immunoblotting using APP antibodies directed against the intracellular APP epitope. We observed that APP was resistant to trypsin digestion in the mixed/late endosome fraction (Fig. 1d, lane 2 compared to lane 1), suggesting that some APP had accumulated inside the membrane-bound endosome. In contrast, the intracellular epitope of APP was no longer detected upon trypsin treatment in early endosomal fractions (Fig. 1d, lane 4 compared to lane 3), suggesting that APP is mostly localized on the early endosomal limiting membrane. Incubation of APP-containing early endosomes with ATP and cytosol at 37°C results in formation of IFVs 25,27 and internalization of APP into IFVs results in protection of the intracellular epitope of APP from subsequent trypsin cleavage (Fig. 1c, lane 3). Ultrastructural examination of mixed and early endosomal membrane factions was performed to ensure the endosomes were intact and not disrupted/fragmented during the homogenization process of the cell-free sorting assay (Supplemental Fig. 1b). We next used this approach with early endosome-enriched fractions to investigate the requirements for APP movement into endosomal IFVs.
ESCRT components enable APP movement into the endosomal lumen
Early endosome membranes (fractions 7-9, Fig. 1b) were used as the donor membranes in cell-free MVB reconstitution assays (Fig. 1c) to examine the mechanisms underlying APP movement from the endosomal limiting membrane into IFVs. As previously described 25,27, membrane protein movement into IFVs depends on cytosolic components (supplemental Fig. 1c) and yeast cytosol is sufficient to support membrane protein internalization into MVBs 25. We took advantage of a yeast deletion strain library to examine whether each complex of the ESCRT machinery is required for efficient movement of APP into MVBs. Cytosol derived from yeast strains deleted of ESCRT proteins (hselΔ, vps23Δ, snf8Δ, and vps24Δ) was used in cell-free reactions in place of wild-type (parental) cytosol. Our data show that deletion of a component from each of the four ESCRT complexes (hselΔ for ESCRT-0, vps23Δ for ESCRT-I, snf8Δ for ESCRT-II, and vps24Δ for ESCRT-III, supplement table 1) significantly decreased APP protease protection compared to parental cytosol (Fig. 2a) suggesting that components of ESCRT-0-III are required for APP movement into ILVs.
Figure 2. Inward budding of APP into MVBs is dependent on ESCRT proteins.
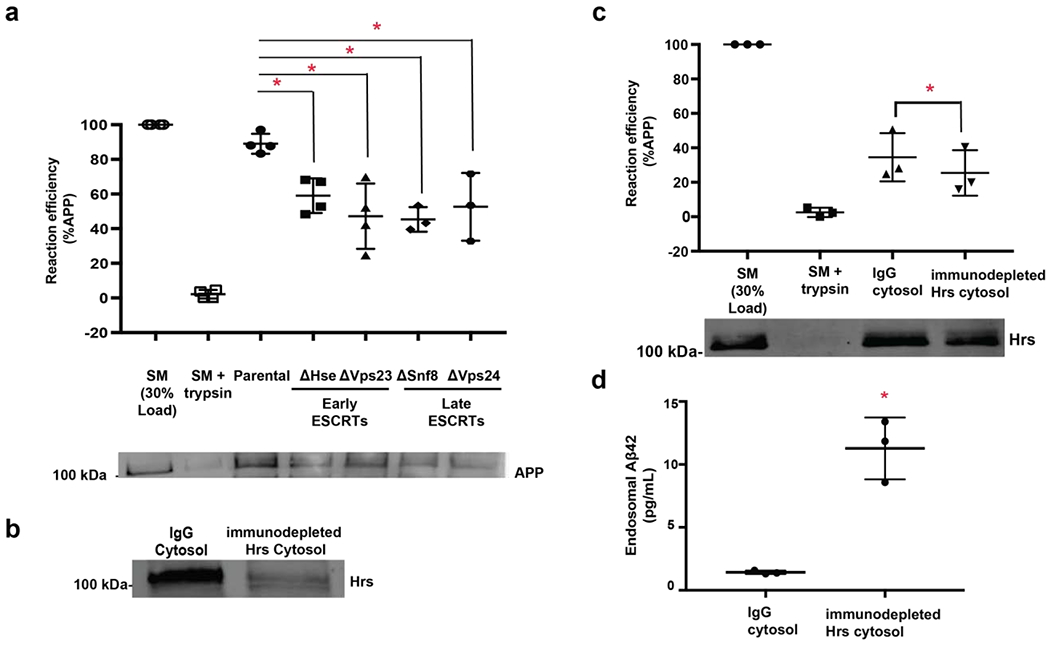
Early endosomal membranes were isolated from rat neurons as described in Fig. 1c. and cell-free reconstitution experiments were performed as in Fig. 1d. a) Reactions incubated with cytosol isolated from ESCRT-deficient strains (hseΔ, vps23Δ, snf8Δ, vps24Δ) significantly decrease protease protection of APP compared to cytosol isolated from a parental strain (lane 4-7 compared to lane 3). For these experiments, n=4 and P values were as follows: parental to hseΔ (p=0.0243), parental to vps23Δ (p=0.0024), parental to snf8Δ (p=0.0031), parental to vps24Δ (p=0.0116). Isolated early endosomal membranes (as in Fig. 1c, lane 1), known as starting material, (SM, Lane 1)) and SM digested with trypsin to remove the C-terminal epitope of the receptor (Lane 2) were used as controls, b) Rat brain cytosol was immunodepleted of mammalian Hrs or a non-specific IgG control by immunoprecipitation. c) Rab brain cytosol immunodepleted of Hrs or an IgG controls was incubated with APP-containing neuronal early endosomes. Depletion of Hrs decreased protease protection of APP compared to control reactions (n=3, p=0.0041). Endosomal membranes (Lane 1) and endosomal membranes digested with trypsin to remove the C-terminal epitope of the receptor (Lane 2) were used as controls. d) Rat brain cytosol was immunodepleted of mammalian Hrs or a non-specific IgG control by immunoprecipitation and incubated with APP-containing neuronal early endosomes. After incubation, endosomes were collected and analyzed for Aβ42 using ELISA. Depletion of Hrs increase Aβ42 levels within endosomes compared to control (n=3, p=0.0032). Data represents the mean +/− S.D. normalized to the control. *Denotes P < 0.05 (one-way ANOVA for A, paired t-test for b-c). Representative blots are shown.
To confirm that the mammalian version of the ESCRT-0 component, Hepatocyte growth factor regulated tyrosine kinase substrate (Hrs), is required for inward endosomal budding of APP, we immunodepleted Hrs from rat brain cytosol (Fig. 2b) and used this cytosol in place of control cytosol to examine APP protease protection in cell-free reactions. APP protease protection was significantly decreased in reactions containing cytosol immunodepleted of Hrs compared to reactions containing control cytosol immunodepleted with a nonspecific antibody 55 (Fig. 2c). These data suggest that the mammalian ESCRT-0 component Hrs is required for APP movement from the endosomal limiting membrane into IFVs of MVBs.
Because BACE1 and γ-secretase can be present on endosomes, we hypothesized that inefficient internalization from the MVB limiting membrane would result in increased APP cleavage and therefore increased levels of Aβ fragments in the endosome lumen. To determine whether the decrease in APP internalization into MVBs corresponds to an increase in endosomal Aβ42 levels, we measured Aβ42 levels in endosomal reactions incubated with control cytosol treated with a nonspecific antibody55 or with cytosol immunodepleted of Hrs using an EFISA. We observed significantly higher levels of Aβ42 within endosomes from reactions containing Hrs-immunodepleted cytosol compared to reactions containing IgG control cytosol (Fig. 2d). There is a significant correlation between APP internalization upon Hrs depletion using an anti-Hrs antibody and Aβ42 generation (r2=0.972; slope=−0.184; p<0.05) while control depletion using a non-specific IgG antibody resulted in no significant correlation between APP internalization and Aβ42 levels (r2=0.338; slope=0.006; p=0.30). These data suggest that inhibition of APP internalization into MVBs results in increased levels of Aβ42 within endosomes, presumably due to enhanced cleavage of APP into Aβ42 on the endosomal membrane.
The endosomally-associated E3 ligase, UBE4B, supports efficient movement of APP into the endosomal lumen.
ESCRT complexes are recruited to endosomal membranes to enable the sorting and movement of ubiquitinated membrane proteins into endosomal ILVs 26,31,37,38,42,56,57. ESCRT proteins contain UIMs that enable engagement with ubiquitin moieties present on membrane proteins, implying that APP is ubiquitinated prior to sorting into ILVs. We previously identified UBE4B as an endosomal-associated ubiquitin ligase 26,58,59 required for movement of the epidermal growth factor receptor (EGFR) into endosomal membranes. To determine whether UBE4B might play a role in APP movement into ILVs, we immunodepleted UBE4B from rat brain cytosol (Fig. 3a) and examined the protease protection of APP in cell-free reactions. Interestingly, immunodepletion of UBE4B significantly decreased the protease protection of APP compared to control reactions (Fig. 3b), suggesting that UBE4B is required for efficient movement of APP from the endosomal limiting membrane into ILVs.
Figure 3. Inward budding of APP into MVBs is dependent on UBE4B.
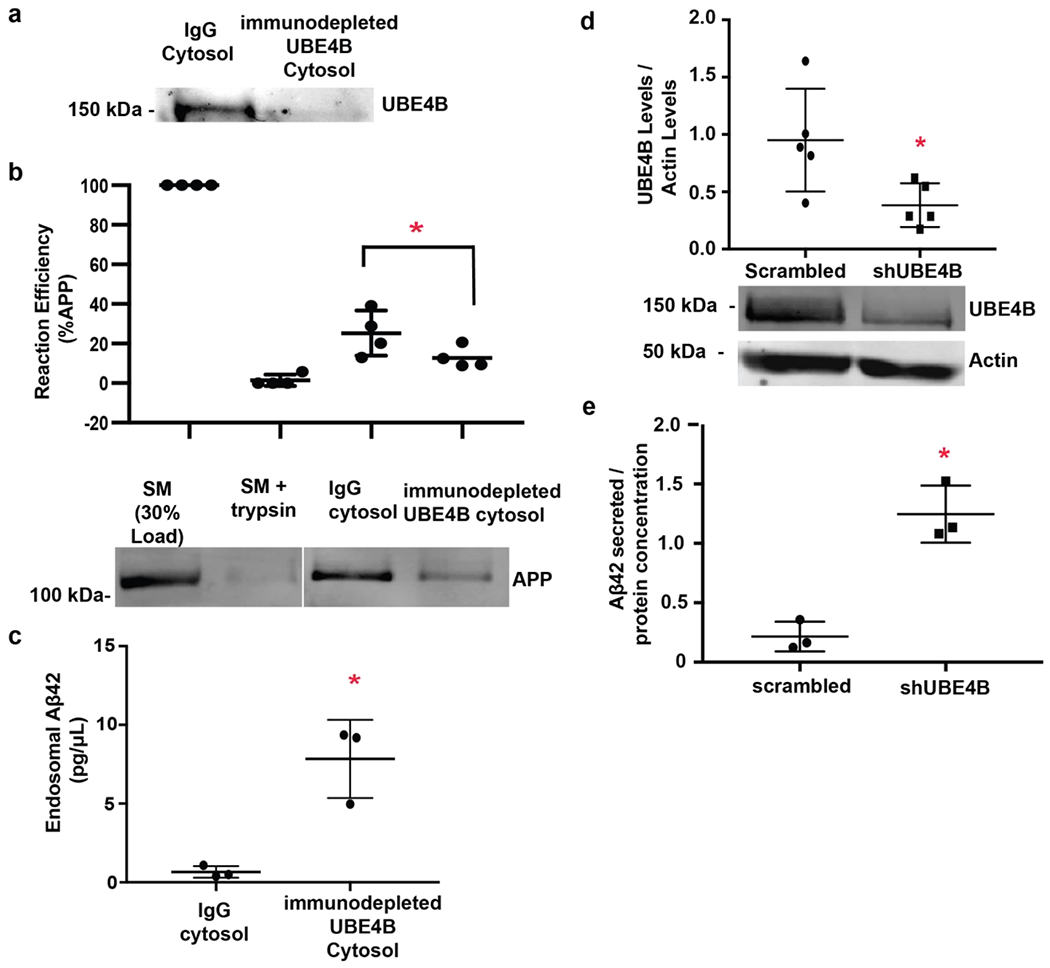
Early endosomal membranes were isolated from rat neurons as described in Fig. 1c. and cell-free reconstitution experiments were performed as in Fig. 1d. a) Rat brain cytosol was immunodepleted of mammalian UBE4B or a non-specific IgG control by immunoprecipitation. b) Rat brain cytosol immunodepleted of UBE4B or an IgG controls was incubated with APP-containing neuronal early endosomes. Depletion of UBE4B decreased protease protection of APP compared to control reactions (n=4, p=0.0121). Endosomal membranes (Lane 1) and endosomal membranes digested with trypsin to remove the C-terminal epitope of the receptor (Lane 2) were used as controls. c) Rat brain cytosol was immunodepleted of mammalian UBE4B or a non-specific IgG control by immunoprecipitation and incubated with APP-containing neuronal early endosomes. After incubation, endosomes were collected and analyzed for Aβ42 using ELISA. Depletion of UBE4B increases Aβ42 levels within endosomes compared to control (n=3, p=0.0082). d) shRNA against UBE4B was lentivirally delivered to primary cortical neurons. Protein levels for UBE4B were assessed via immunoblotting. UBE4B is significantly inhibited in neurons treated with shRNA against UBE4B compared to control (n=5, p= 0.0313). Gels were probed for UBE4B first followed by Actin. e) shRNA against UBE4B or a scrambled control was lentivirally delivered to primary cortical neurons. After 48 hours, conditioned media was analyzed by ELISA for Aβ42 levels. Depletion of UBE4B leads to increased levels of secreted Aβ42 compared to control cells (n=3, p=0.0042). Data represents the mean +/− S.D. normalized to the control. *Denotes P < 0.05 (paired t-test for a-e). Representative blots are shown.
To determine whether the decrease in APP internalization into ILVs, observed in the absence of UBE4B, corresponds to an increase in Aβ42 levels, we examined Aβ42 levels within endosomes incubated with cytosol immunodepleted of UBE4B or control cytosol treated with a nonspecific antibody 55. We observed significantly increased levels of endosomal Aβ42 (Fig. 3c) in reactions containing cytosol depleted of UBE4B. These data suggest that in the absence of UBE4B, like in the absence of ESCRT components, the decreased efficiency of internalization may result in increased levels of Aβ42 within endosomes presumably due to enhanced cleavage of APP into Aβ42 on the endosomal membrane.
While MVBs can fuse with lysosomes enabling degradation of ILV contents, MVB-plasma membrane 60 fusion is an alternate itinerary for MVBs that may underlie secretory events 15,32–34. Having observed increased endosomal Aβ42 in the absence of UBE4B, we examined whether decreased expression of UBE4B resulted in enhanced Aβ42 secretion from neurons. Cortical neurons were infected with either a lentivirally expressed shRNA directed against UBE4B or a scrambled control (Fig. 3d). After 48 hours, the neuronal media was collected and Aβ42 levels were measured using ELISA. UBE4B depletion resulted in significantly increased Aβ42 levels in the media surrounding neurons suggesting increased neuronal secretion of the amyloidogenic peptide (Fig. 3e). There is a significant correlation between APP internalization upon UBE4B depletion using an anti-UBE4B antibody and Aβ42 generation (r2=0.973; slope=−1.267; p<0.05) while control depletion using a non-specific IgG antibody resulted in no significant correlation between APP internalization and Aβ42 levels (r2=0.031; slope=0.008; p=0.44). Together, these results suggest that inhibition of UBE4B decreases APP movement into MVBs and may lead to enhanced cleavage of APP into Aβ on the limiting membrane of endosomes. Further, loss of UBE4B leads to significantly increased levels of Aβ42 within neuronal endosomes and increased secretion into the surrounding media.
UBE4B interacts with and ubiquitinates APP.
Because U-box E3 ubiquitin ligases physically interact with their substrates for ubiquitin transfer 61–65 we posited that UBE4B might interact with APP. We observed that UBE4B co-immunoprecipitated with APP in lysate from neuron-like SK-N-AS cell (Fig. 4a) suggesting that UBE4B interacts with APP and could therefore act to transfer ubiquitin to APP. To assess whether UBE4B expression can regulate APP levels in neurons, we depleted UBE4B in primary cortical neurons (Fig. 4c) and observed a significant increase in total APP levels in neuronal lysate (Fig. 4d). This result would be predicted if UBE4B is involved in efficient APP movement to the lysosome. To examine whether UBE4B can affect APP ubiqiuitination in situ, after depletion of UBE4B from primary neurons (Fig. 4c) we examined the levels of ubiquitin that were co-immunoprecipitated with APP (Fig. 4e). Since UBE4B depletion results in increased APP levels (Fig. 4d), we used a ratio measure to ensure that we accounted for the increase in APP levels produced by UBE4B depletion.. We observed significantly decreased levels of ubiquitin associated with immunoprecipitated APP in neurons depleted of UBE4B compared to control neurons (Fig. 4f). When we analyzed the raw ubiquitin levels after immunoprecipitation with APP, we found that ubiquitin levels are significantly decreased in ShUBE4B treated neurons compared to neurons treated with a scrambled control (Fig. 4g). These data suggest that UBE4B can regulate the ubiquitination and levels of APP in neurons.
Figure 4. UBE4B interacts with and ubiquitinates APP.
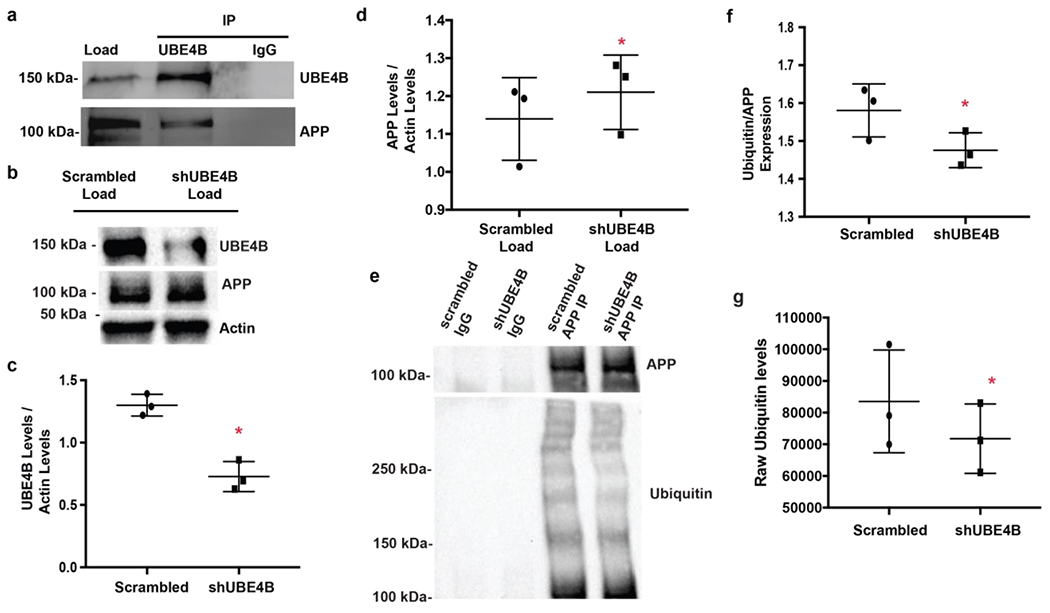
a) Lysate from SK-N-AS cells was incubated with anti-UBE4B antibody and protein A beads. APP co-immunoprecipitates with UBE4B, but not with a non-specific IgG control. b) shRNA against UBE4B was lentivirally delivered to primary cortical neurons. Protein levels for UBE4B, APP, and actin were assessed via immunoblotting. c) UBE4B is significantly inhibited in neurons treated with shRNA against UBE4B compared to control (n=3, p=0.0026). d) UBE4B depletion leads to significantly increased levels of APP (n=3, p= 0.0244). Gels were probed for UBE4B first, followed by APP and actin. e) Lysate obtained from cells infected with shUBE4B or scrambled shRNA (from Fig. 2b–d) were subjected to immunoprecipitation with an APP antibody. Protein attached to beads was analyzed via immunoblotting for APP and ubiquitin levels. IP gels were probed for APP followed by ubiquitin f) Depletion of UBE4B leads to decreased ubiquitination of APP compared to IgG control (n=3, p=0.0385). g) Raw ubiquitin levels are significantly decreased in ShUBE4B treated neurons compared to neurons treated with a scrambled control after immunoprecipitation with APP (n=3, p=0.0354). Data represents the mean +/− S.D. normalized to the control. *Denotes P < 0.05 (paired ratio t-test for b, d, and e). Representative blots are shown.
UBE4B and Hrs levels in AD patients.
To determine whether UBE4B levels are altered with disease progression in AD patients, protein levels for UBE4B, APP, EGFR, BACE-1, Presenilin 2, Hrs, phospho-Tau, and actin were assessed via immunoblotting in human AD patient brains (neocortex, n=13) compared to age-matched controls (n=8) (Figs. 5a and 5b, and supplemental Table 2 for patient information). In human AD patients, BACE-1 and Phospho-Tau levels were significantly increased compared to age-matched controls (Fig. 5a and 5b), consistent with pathology. APP, EGFR, and PS2 levels were not significantly altered compared to controls (Fig. 5a and 5b). Interestingly, Hrs levels were significantly decreased in AD patients compared to age-matched controls whereas UBE4B levels (p=0.18) were not significantly different in AD patients compared to age-matched controls (Fig. 5a and 5b). These data suggest that molecular components critical for membrane protein sorting in the endocytic pathway are altered in AD.
Figure 5. UBE4B and Hrs protein levels in AD patient samples. .
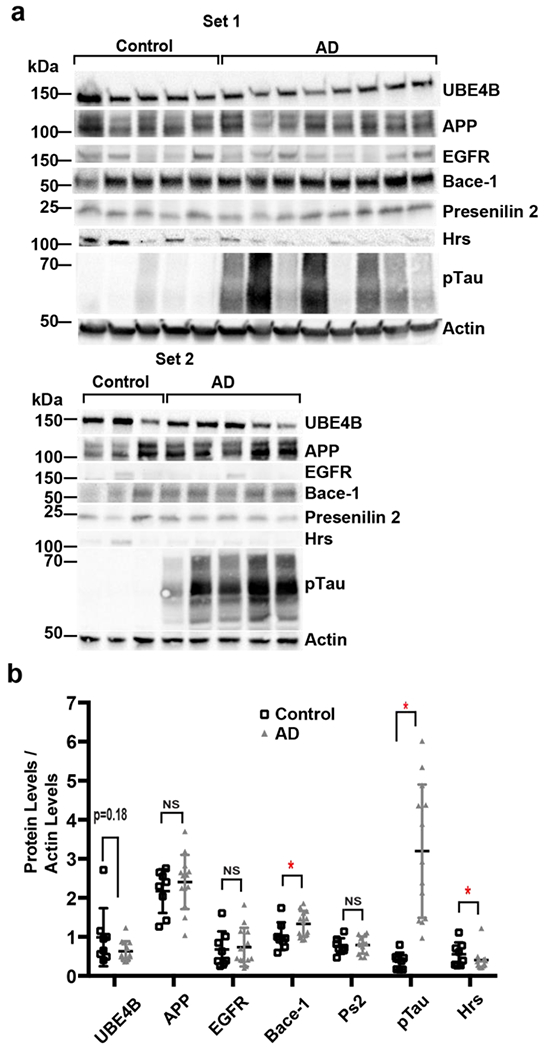
a,b)) Protein levels for UBE4B, APP, EGFR, Bace-1, Presenilin 2, Hrs, phospho-Tau, and actin were assessed via immunoblotting in human AD patient brains (n=13) compared to age-matched controls (n=8) (see supplemental Table 2 for patient information). Bace-1 (p=0.05) and phospho-Tau (p=0.0002) significantly increase in AD patients compared to age-matched controls. APP, EGFR, and PS2 levels show no change in AD patients compared to age-matched controls. Hrs levels (p=0.0306) are significantly decreased in AD patients compared to age-matched controls and UBE4B levels (p=0.18) are trending towards a decrease in AD patients compared to age-matched controls. Representative blots are shown. Gels were probed for UBE4B, followed by Bace-1, Actin, and pTau. Another gel was probed for APP, PS2, EGFR, and actin. Last gel was probed for Hrs and actin. Data represents the mean +/− S.D. normalized to the control. *Denotes P < 0.05 ( unpaired t-test for b).
Discussion
The identity of the subcellular compartment(s) involved in the disease-causing cleavage of APP into Aβ is controversial 3,66,67, however recent literature points to endosomes as a key cellular compartment in which APP is pathologically cleaved into Aβ 3,15,17,20,22–24,36,66–70. Endosomal dysfunction may be key to AD pathogenesis as abnormal endosomal morphology and increased endosomal Aβ burden has been observed in the brain of individuals with dementia22–24. The serial cleavage of APP by BACE-1 and the γ-secretase complex results in the production of multiple Aβ peptides 3,15,66. Both APP and BACE-1 can co-localize on endosomes 17,68. and BACE-1 and the γ-secretase complex are enzymatically active at this site 17,20,67,68. BACE-1 has optimal activity at the low pH found in endosomes 36, and BACE-1 inhibitors tethered to endosomes result in decreased APP processing into Aβ 70. These observations suggest that APP may be cleaved on endosomal membranes resulting in endosomal Aβ production. Given the topology of APP on the endosomal membrane, amyloidogenic APP cleavage into Aβ would result in Aβ accumulation in the lumen of the endosome 69. Our data show that a pool of Aβ is generated and secreted from MVBs, suggesting an important role for the endocytic pathway in AD, and providing a potential pathway for Aβ secretion into the extraneuronal space. Additionally, our results confirm the role of ESCRT proteins in APP trafficking and introduce the E3 ubiquitin ligase, UBE4B, as a new molecular player regulating APP trafficking.
Integral membrane proteins such as APP can exit neurons on membranes that bud from the plasma membrane 60 either following initial biosynthesis, or following internalization from the PM and subsequent retromer-dependent 71–74 trafficking to the trans-Golgi network (TGN) via the late endosome. Alternatively, membrane proteins may exit the cell following trafficking to late endosome/multivesicular body (MVB) and movement into the MVB’s internal vesicles, which can then be secreted as exosomes upon MVB-PM fusion 32–34. APP and its cleavage products have been identified on extracellular vesicles from a variety of cell lines 15,32–34, but these studies have not established whether these APP-containing vesicles originate from MVB-PM fusion as opposed to simple membrane budding from the PM into the extracellular space. Our data show that deletion of the molecular machinery involved in efficient APP trafficking into ILVs of MVBs results in increased generation of Aβ at endosomes and increased secretion of Aβ from neurons, suggesting that MVB-PM fusion is likely involved in the secretion of a pool of Aβ.
The ESCRTs have been previously implicated in APP trafficking, however there is conflicting evidence for a role of ESCRTs in the sorting of APP at endosomes and the secretion of Aβ 17–19. Depletion of early ESCRT components (e.g. ESCRT-0 or -I, Hrs and Tsg101, respectively) decreases Aβ secretion in some experimental models 18,19, whereas the same manipulation results in greater Aβ secretion in other models 17. These studies were performed in non-neuronal cell lines (HEK293 and HeLa cells, respectively) which may account for the differences detected. To clarify the role of the ESCRT proteins in APP trafficking, we used primary neuronal cultures and observed that components of all ESCRT complexes are required for efficient internalization of APP in ILVs of MVBs, and loss of the ESCRTs results in greater Aβ generation, consistent with the observations of Morel et al. 17. Additionally, we have found a significant decrease in Hrs levels in AD patient cortical biopsies, suggesting that the trafficking machinery is important for AD disease progression.
ESCRT-dependent endocytic trafficking implies that APP is ubiquitinated to enable ESCRT interaction. We have identified a specific endosome-associated E3 ubiquitin ligase, UBE4B, that can affect APP ubiquitination. Interestingly, the E3 ligase FBL2 has also been reported to ubiquitinate APP resulting in decreased internalization from the PM and increased proteasomal degradation 44,46. However, events required for PM internalization and/or proteasomal degradation may differ 40 from those required for endocytic trafficking. Our observation that UBE4B affects APP ubiquitination and facilitates its movement into ILVs suggests that APP ubiquitination by UBE4B is at least partly responsible for APP downregulation. Because we are aware that UBE4B depletion results in increased APP levels, we used a ratio measure to ensure that we accounted for the increase in APP levels produced by UBE4B depletion. However, as stated above UBE4B is not the only ubiquitin ligase capable of ubiquitinating APP. FBL2 ubiquitinates APP at the PM, leading to increased proteasomal degradation 44,46, while MGRN1 is involved in the ubiquitination of APP from endosomes into the Golgi 46. Therefore it is possible that the increased levels of APP produced by UBE4B depletion may lead to increased ubiquitination by other ubiquitin ligases that route APP into other compartments of the cell. Together, our data provide evidence for molecular mechanisms that may underlie the role of the endocytic pathway in Aβ generation and secretion.
Interestingly, the processes of ubiquitination/deubiquitination must be coordinated with the sorting machinery because removal of ubiquitin from cargo proteins occurs prior to cargo movement into internal MVB vesicles, but not before cargo recognition by ESCRTs 75. We have observed that UBE4B binds to APP and BACE-1 binds APP 20 such that BACE-1 may provide a mechanism by which one of its binding partners, the deubiquitinating enzyme USP8 76, may be brought into close proximity to APP for deubiquitination. We have previously shown that UBE4B and USP8 can work in concert to add and remove ubiquitin from a substrate 26. Thus, we hypothesize that USP8 and BACE-1 could act to deubiquitinate APP, allowing BACE-1 to cleave APP on the MVB limiting membrane and enable internalization of APP and/or APP fragments (depending on the length of APP exposure to BACE-1).
Previous studies suggest a neuroprotective role for UBE4B in spinocerebellar ataxia type 3, another neurodegenerative disease characterized by an increase in polyglutamine repeats 77. In this study, UBE4B expression promoted the degradation of a pathological form of ataxin-3 77, similar to what we observed for APP. Thus, if UBE4B acts as a neuroprotectant 59,78–80, the lack of UBE4B in disease may render neurons particularly sensitive to insult. While we have found no significant difference in UBE4B levels of AD patient cortical biopsies compared to controls, it is possible that since we only sampled a small part of the cortex, and UBE4B levels may be heterogeneously affected in disease state, we did not observe significantly altered protein levels. It would be interesting to determine whether UBE4B levels are specifically decreased in areas known to be highly affected by AD (e.g. hippocampus, cortex). Moreover, UBE4B may not be the only ubiquitin ligase that can affect APP trafficking. For example, MGRN1 is involved in the processing of APP in the Golgi 46 and, to the extent that the biosynthetic or retromer pathways play a role in in regulating APP and Aβ levels, this ligase may also play a role in APP trafficking.
We have identified UBE4B as an endosomally-associated E3 ligase that is present in the human brain and interacts with APP. We have shown that UBE4B down regulation leads to increases in Aβ generation and secretion. The requirement for UBE4B in APP sorting into internal MVB vesicles suggests that, in the presence of mutations that delay APP trafficking, such as ESCRT downregulation, or in mutations observed in familial AD that increases the interaction between APP and the secretases (e.g. APP Swe/Ind), enhanced UBE4B activity might promote internalization of full-length APP into MVBs before it is cleaved by the secretases into pathogenic Aβ. If a hyperactive UBE4B mutant81 can increase full-length APP sorting into the MVB, in the presence of mutations that delay APP trafficking into MVBs, this could represent a novel target and a potential therapeutic strategy.
Supplementary Material
Highlights.
APP can be cleaved into Aβ on late endosomes/MVBs
Loss of ESCRT components inhibited APP movement into internal vesicles of MVBs and increased endosomal Aβ42 generation
An endosome-associated E3 ubiquitin ligase, UBE4B, regulates APP ubiquitination
Depletion of UBE4B resulted in increased endosomal Aβ42 levels and increased neuronal secretion of Aβ42
Levels of UBE4B were trending towards a decrease and levels of the UBE4B-interacting ESCRT component, Hrs, were significantly decreased in AD brains.
Acknowledgements
We thank Dr. Kevin Morano for providing some Saccharomyces cerevisiae strains used in this study. We thank Max Odem for providing useful insight on the statistical analysis of this data. These studies were supported in part by National Institute of Health (CA166749). MGG was supported by the National Cancer Institute diversity-training program grant, National Cancer Institute RO1-CA 166749-02S1. The Russell and Diana Hawkins Family Foundation Discovery Fellowship supported SR. The National Institute of Neurological Disorders and Stroke, NS094543 (LM), supported studies in aging mice. JF was funded by the Aging and Disability Resource Center (ADRC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations
Alzheimer’s Disease (AD)
Amyloid Beta (Aβ)
Amyloid Precursor Protein (APP)
β-site amyloid precursor protein cleaving enzyme 1 (BACE-1)
Endosomal Sorting Complexes Required for Transport (ESCRTs)
Hepatocyte growth factor regulated tyrosine kinase substrate (Hrs)
Internal Vesicles (ILVs)
Multi-vesicular body (MVB)
Plasma Membrane (PM)
Presenilin-2 (PS2)
Ubiquitin-interaction motifs (UIM)
Conflicts of Interest
The authors have no competing financial interests.
References
- 1.Cummings JL Alzheimer’s disease. N Engl J Med 351, 56–67, doi: 10.1056/NEJMra040223 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ & Podlisny MB Deciphering the genetic basis of Alzheimer’s disease. Annu Rev Genomics Hum Genet 3, 67–99, doi: 10.1146/annurev.genom.3.022502.103022 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Haass C, Kaether C, Thinakaran G & Sisodia S Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2, a006270, doi: 10.1101/cshperspect.a006270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Brien RJ & Wong PC Amyloid precursor protein processing and Alzheimer’s disease. Annn Rev Neurosci 34, 185–204, doi: 10.1146/annurev-neuro-061010-113613 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tejada-Vera B Mortality from Alzheimer’s disease in the United States: data for 2000 and 2010. NCHS Data Brief 1–8 (2013). [PubMed] [Google Scholar]
- 6.Larson EB et al. Survival after initial diagnosis of Alzheimer disease. Ann Intern Med 140, 501–509 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Perrin RJ, Fagan AM & Holtzman DM Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature 461, 916–922, doi: 10.1038/nature08538 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blennow K & Zetterberg H Biomarkers for Alzheimer’s disease: current status and prospects for the future. J Intern Med 284, 643–663, doi: 10.1111/joim.l2816 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Nelson PT, Braak H & Markesbery WR Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 68, 1–14, doi: 10.1097/NEN.0b013e3181919a48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson PT et al. Acetylcholinesterase inhibitor treatment is associated with relatively slow cognitive decline in patients with Alzheimer’s disease and AD + DLB. J Alzheimers Dis 16, 29–34, doi: 10.3233/JAD-2009-0926 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson PT et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71, 362–381, doi: 10.1097/NEN.0b013e31825018f7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morley JE et al. A physiological role for amyloid-beta protein enhancement of learning and memory. J Alzheimers Dis 19, 441–449, doi: 10.3233/JAD-2009-1230 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Bishop GM & Robinson SR Physiological roles of amyloid-beta and implications for its removal in Alzheimer’s disease. Drugs Aging 21, 621–630, doi: 10.2165/00002512-200421100-00001 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Pearson HA & Peers C Physiological roles for amyloid beta peptides. J Physiol 575, 5–10, doi: 10.1113/jphysiol.2006.111203 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajendran L et al. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci USA 103, 11172–11177, doi: 10.1073/pnas.0603838103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cirrito JR et al. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58, 42–51, doi: 10.1016/j.neuron.2008.02.003 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morel E et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun 4, 2250, doi: 10.1038/ncomms3250 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edgar JR, Willen K, Gouras GK & Futter CE ESCRTs regulate amyloid precursor protein sorting in multivesicular bodies and intracellular amyloid-beta accumulation. J Cell Sci 128, 2520–2528, doi: 10.1242/jcs.170233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choy RW, Cheng Z & Schekman R Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid beta (Abeta) production in the trans-Golgi network. Proc Natl Acad Sci U S A 109, E2077–2082, doi: 10.1073/pnas.1208635109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das U et al. Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 79, 447–460, doi: 10.1016/j.neuron.2013.05.035 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Small SA & Gandy S Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to pathogenesis. Neuron 52, 15–31, doi: 10.1016/j.neuron.2006.09.001 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nixon RA Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 120, 4081–4091, doi: 10.1242/jcs.019265 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Jiang T et al. Temsirolimus promotes autophagic clearance of amyloid-beta and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol Res 81, 54–63, doi: 10.1016/j.phrs.2014.02.008 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Funk KE & Kuret J Lysosomal fusion dysfunction as a unifying hypothesis for Alzheimer’s disease pathology. Int J Alzheimers Dis 2012, 752894, doi: 10.1155/2012/752894 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gireud-Goss M et al. Distinct mechanisms enable inward or outward budding from late endosomes/multivesicular bodies. Exp Cell Res 372, 1–15, doi: 10.1016/j.yexcr.2018.08.027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sirisaengtaksin N et al. UBE4B protein couples ubiquitination and sorting machineries to enable epidermal growth factor receptor (EGFR) degradation. J Biol Chem 289, 3026–3039, doi: 10.1074/jbc.M113.495671 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun W et al. Cell-free reconstitution of multi vesicular body formation and receptor sorting. Traffic 11, 867–876, doi: 10.1111/j.1600-0854.2010.01053.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Futter CE, Pearse A, Hewlett LJ & Hopkins CR Multi vesicular endosomes containing internalized EGF-EGF receptor complexes mature and then fuse directly with lysosomes. J Cell Biol 132, 1011–1023 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felder S et al. Kinase activity controls the sorting of the epidermal growth factor receptor within the multivesicular body. Cell. 61, 623–634 (1990). [DOI] [PubMed] [Google Scholar]
- 30.Piper RC & Luzio JP Late endosomes: sorting and partitioning in multivesicular bodies. Traffic 2, 612–621 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Babst M, Katzmann DJ, Estepa-Sabal EJ, Meerloo T & Emr SD Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Developmental cell 3, 271–282 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Corrado C et al. Exosomes as intercellular signaling organelles involved in health and disease: basic science and clinical applications. Int J Mol Sci 14, 5338–5366, doi: 10.3390/ijms14035338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharpies RA et al. Inhibition of gamma-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J 22, 1469–1478, doi: 10.1096/fj.07-9357com (2008). [DOI] [PubMed] [Google Scholar]
- 34.Perez-Gonzalez R, Gauthier SA, Kumar A & Levy E The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J Biol Chem 287, 43108–43115, doi: 10.1074/jbc.M112.404467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smolarkiewicz M et al. Gamma-secretase subunits associate in intracellular membrane compartments in Arabidopsis thaliana. J Exp Bot 65, 3015–3027, doi: 10.1093/jxb/eru147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Strooper B, Vassar R & Golde T The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol 6, 99–107, doi: 10.1038/nrneurol.2009.218 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katzmann DJ, Babst M & Emr SD Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 106, 145–155 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Hurley JH & Emr SD The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu Rev Biophys Biomol Struct 35, 277–298, doi: 10.1146/annurev.biophys.35.040405.102126 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowers K et al. Protein-protein interactions of ESCRT complexes in the yeast Saccharomyces cerevisiae. Traffic 5, 194–210, doi: 10.1111/j.1600-0854.2004.00169.x (2004). [DOI] [PubMed] [Google Scholar]
- 40.Hicke L A new ticket for entry into budding vesicles-ubiquitin. Cell 106, 527–530 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Thrower JS, Hoffman L, Rechsteiner M & Pickart CM Recognition of the polyubiquitin proteolytic signal. EMBO J 19, 94–102, doi: 10.1093/emboj/19.1.94 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Katzmann DJ, Stefan CJ, Babst M & Emr SD Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Biol 162, 413–423, doi: 10.1083/jcb.200302136 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williamson RL et al. Disruption of amyloid precursor protein ubiquitination selectively increases amyloid beta (Abeta) 40 levels via presenilin 2-mediated cleavage. J Biol Chem 292, 19873–19889, doi: 10.1074/jbc.M117.818138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe T, Hikichi Y, Willuweit A, Shintani Y & Horiguchi T FBL2 regulates amyloid precursor protein (APP) metabolism by promoting ubiquitination-dependent APP degradation and inhibition of APP endocytosis. J Neurosci 32, 3352–3365, doi: 10.1523/JNEUROSCI.5659-11.2012(2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.El Ayadi A, Stieren ES, Barral JM & Boehning D Ubiquilin-1 regulates amyloid precursor protein maturation and degradation by stimulating K63-linked polyubiquitination of lysine 688. Proc Natl Acad Sci U S A 109, 13416–13421, doi: 10.1073/pnas.1206786109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benvegnu S, Wahle T & Doth CG E3 ligase mahogunin (MGRN1) influences amyloid precursor protein maturation and secretion. Oncotarget 8, 89439–89450, doi: 10.18632/oncotarget.20143 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pacifici M & Peruzzi F Isolation and culture of rat embryonic neural cells: a quick protocol. J Vis Exp, e3965, doi: 10.3791/3965 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moruno Manchon JF et al. Cytoplasmic sphingosine-1-phosphate pathway modulates neuronal autophagy. Sci Rep 5, 15213, doi: 10.1038/srep15213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsvetkov AS et al. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A 107, 16982–16987, doi: 10.1073/pnas.1004498107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arrasate M & Finkbeiner S Automated microscope system for determining factors that predict neuronal fate. Proc Natl Acad Sci U S A 102, 3840–3845, doi: 10.1073/pnas.0409777102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Araujo ME, Lamberti G & Huber LA Purification of Early and Late Endosomes. Cold Spring Harb Protoc 2015, pdb top074443, doi: 10.1101/pdb.top074443 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Sun W, Yan Q, Vida TA & Bean AJ Hrs regulates early endosome fusion by inhibiting formation of an endosomal SNARE complex. J Cell Biol 162, 125–137, doi: 10.1083/jcb.200302083 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gireud M, Sirisaengtaksin N, Tsunoda S & Bean AJ Cell-free reconstitution of multivesicular body (MVB) cargo sorting. Methods Mol Biol 1270, 115–124, doi: 10.1007/978-1-4939-2309-0_9 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Scott CC, Vacca F & Gruenberg J Endosome maturation, transport and functions. Semin Cell Dev Biol 31, 2–10, doi: 10.1016/j.semcdb.2014.03.034 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Hardy JA & Higgins GA Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185, doi: 10.1126/science.1566067 (1992). [DOI] [PubMed] [Google Scholar]
- 56.Raiborg C & Stenmark H The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452, doi: 10.1038/nature07961 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Bilodeau PS, Winistorfer SC, Kearney WR, Robertson AD & Piper RC Vps27-Hse1 and ESCRT-I complexes cooperate to increase efficiency of sorting ubiquitinated proteins at the endosome. J Cell Biol 163, 237–243, doi: 10.1083/jcb.200305007 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zage PE et al. UBE4B levels are correlated with clinical outcomes in neuroblastoma patients and with altered neuroblastoma cell proliferation and sensitivity to epidermal growth factor receptor inhibitors. Cancer 119, 915–923, doi: 10.1002/cncr.27785 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaneko-Oshikawa C et al. Mammalian E4 is required for cardiac development and maintenance of the nervous system. Mol Cell Biol 25, 10953–10964, doi: 10.1128/MCB.25.24.10953-10964.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hsiao K et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102, doi: 10.1126/science.274.5284.99 (1996). [DOI] [PubMed] [Google Scholar]
- 61.Borden KL RING domains: master builders of molecular scaffolds? J Mol Biol 295, 1103–1112, doi: 10.1006/jmbi.1999.3429 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Joazeiro CA et al. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science 286, 309–312 (1999). [DOI] [PubMed] [Google Scholar]
- 63.Levkowitz G et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cb1/Sli-1. Mol Cell 4, 1029–1040 (1999). [DOI] [PubMed] [Google Scholar]
- 64.Fang S, Jensen JP, Ludwig RL, Vousden KH & Weissman AM Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 275, 8945–8951 (2000). [DOI] [PubMed] [Google Scholar]
- 65.Honda R & Yasuda H Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene 19, 1473–1476, doi: 10.1038/sj.onc.1203464 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Bowers K et al. Degradation of endocytosed epidermal growth factor and virally ubiquitinated major histocompatibility complex class I is independent of mammalian ESCRTII. J Biol Chem 281, 5094–5105, doi: 10.1074/jbc.M508632200 (2006). [DOI] [PubMed] [Google Scholar]
- 67.Zhang X & Song W The role of APP and BACE1 trafficking in APP processing and amyloid-beta generation. Alzheimers Res Ther 5, 46, doi: 10.1186/alzrt211 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frykman S et al. Synaptic and endosomal localization of active gamma-secretase in rat brain. PLoS One 5, e8948, doi: 10.1371/journal.pone.0008948 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takahashi RH, Nagao T & Gouras GK Plaque formation and the intraneuronal accumulation of beta-amyloid in Alzheimer’s disease. Pathol Int 67, 185–193, doi: 10.1111/pin.12520 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Ben Halima S et al. Specific Inhibition of beta-Secretase Processing of the Alzheimer Disease Amyloid Precursor Protein. Cell Rep 14, 2127–2141, doi: 10.1016/j.celrep.2016.01.076 (2016). [DOI] [PubMed] [Google Scholar]
- 71.Gores GJ, Herman B & Lemasters JJ Plasma membrane bleb formation and rupture: a common feature of hepatocellular injury. Hepatology 11, 690–698 (1990). [DOI] [PubMed] [Google Scholar]
- 72.Fackler OT & Grosse R Cell motility through plasma membrane blebbing. J Cell Biol 181, 879–884, doi: 10.1083/jcb.200802081 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seaman MN The retromer complex - endosomal protein recycling and beyond. J Cell Sci 125, 4693–4702, doi: 10.1242/jcs.103440 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burd C & Cullen PJ Retromer: a master conductor of endosome sorting. Cold Spring Harb Perspect Biol 6, doi: 10.1101/cshperspect.a016774 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Richter C, West M & Odorizzi G Dual mechanisms specify Doa4-mediated deubiquitination at multivesicular bodies. EMBO J 26, 2454–2464, doi: 10.1038/sj.emboj.7601692 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yeates EF & Tesco G The Endosome-associated Deubiquitinating Enzyme USP8 Regulates BACE1 Enzyme Ubiquitination and Degradation. J Biol Chem 291, 15753–15766, doi: 10.1074/jbc.M116.718023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matsumoto M et al. Molecular clearance of ataxin-3 is regulated by a mammalian E4. EMBO J 23, 659–669, doi: 10.1038/sj.emboj.7600081 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mack TG et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci 4, 1199–1206, doi: 10.1038/nn770 (2001). [DOI] [PubMed] [Google Scholar]
- 79.Conforti L et al. Wld S protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J Cell Biol 184, 491–500, doi: 10.1083/jcb.200807175 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Okumura F, Hatakeyama S, Matsumoto M, Kamura T & Nakayama KI Functional regulation of FEZ1 by the U-box-type ubiquitin ligase E4B contributes to neuritogenesis. J Biol Chem 279, 53533–53543, doi: 10.1074/jbc.M402916200 (2004). [DOI] [PubMed] [Google Scholar]
- 81.Starita LM et al. Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc Natl Acad Sci U S A 110, E1263–1272, doi: 10.1073/pnas.1303309110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.