

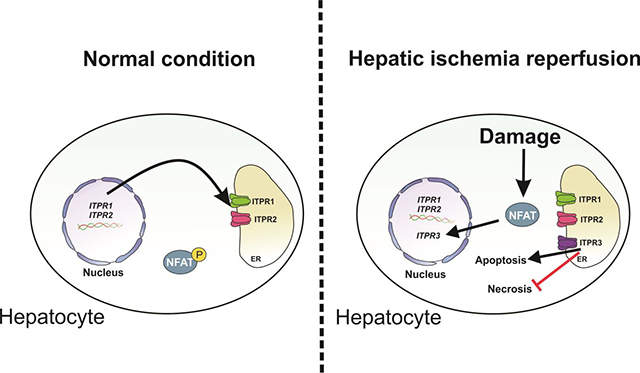
Abstract
Hepatic ischemia-reperfusion injury is seen in a variety of clinical conditions, including hepatic thrombosis, systemic hypotension, and liver transplantation. Calcium (Ca2+) signaling mediates several pathophysiological processes in the liver, but it is not known whether and how intracellular Ca2+ channels are involved in the hepatocellular events secondary to ischemia-reperfusion. Using an animal model of hepatic ischemia-reperfusion injury, we observed a progressive increase in expression of the type 3 isoform of the inositol trisphosphate receptor (ITPR3), an intracellular Ca2+ channel that is not normally expressed in healthy hepatocytes. ITPR3 expression was upregulated, at least in part, by a combination of demethylation of the ITPR3 promoter region and the increased transcriptional activity of the nuclear factor of activated T-cells (NFAT). Additionally, expression of pro-inflammatory interleukins and necrotic surface area were less pronounced in livers of control animals compared to liver-specific ITPR3 KO mice subjected to hepatic damage. Corroborating these findings, ITPR3 expression and activation of NFAT were observed in hepatocytes of liver biopsies from patients who underwent liver ischemia caused by thrombosis after organ transplant. Together, these results are consistent with the idea that ITPR3 expression in hepatocytes plays a protective role during hepatic injury induced by ischemia-reperfusion.
Keywords: Calcium signaling, nuclear factor of activated T-cells, necrosis, hepatocytes, transplantation
Graphical Abstract
1-. INTRODUCTION
Hepatic ischemia-reperfusion is due to the deprivation of nutrients and oxygen followed by reperfusion, which triggers oxidative stress, immune cell activation and cell death (1). During reperfusion, hepatocytes generate reactive oxygen species (ROS), causing lipid peroxidation, protein degradation, mitochondrial dysfunction, activation of inflammatory transcription factors, and apoptosis and necrosis (2), and these events are triggered by changes in intracellular calcium (Ca2+) levels (3–6). In hepatocytes, intracellular Ca2+ signaling is mediated by members of the inositol 1,4,5-trisphosphate receptor (ITPR) family, which includes three isoforms that share 60–80% homology in terms of amino acid sequence (7). Under normal conditions, hepatocytes express only ITPR1 and ITPR2 (8), which mediate metabolism and bile acid secretion, respectively (9–12). More recently, studies have shown the ITPR3 isoform becomes expressed in hepatocytes in a variety of liver diseases, including chronic viral hepatitis, cirrhosis, and hepatocellular carcinoma (13, 14).
Expression of the various ITPR isoforms is modulated by inflammatory transcription factors (15–17) as well as by epigenetic modification, including micro-RNAs (18) and methylation of the promoter region of the ITPR genes. These mechanisms play a causal role in a variety of liver diseases, including several forms of cholestasis (15, 17, 19), nonalcoholic fatty liver disease (NAFLD) (16), nonalcoholic steatohepatitis (NASH) (9), and hepatocellular carcinoma (HCC) (13). Because intracellular Ca2+ signaling plays a fundamental role in both normal and abnormal liver function, the aim of this study was to understand whether and how the three ITPR isoforms participate in the pathogenesis of hepatic ischemia-reperfusion injury.
2 -. MATERIAL AND METHODS
2.1 -. Animals and Human samples
Male C57/BL6 wild type or liver-specific ITPR3 knockout (ITPR3-LSKO) mice (8 weeks age) were housed under a 12-hour light–dark cycle, with ad libitum access to standard diet and water according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals. ITPR3-LSKO animals were generated by crossing FloxR3 with Albumin Cre mice (Jackson Laboratories, USA) (13). The procedures were approved by the Yale University Institutional Animal Care and Use Committee (n° 07602).
Human liver biopsies and clinical data (Supplementary Table A.1) from seven patients diagnosed with ischemia due to hepatic artery thrombosis diagnosed 72 hr after liver transplantation, were obtained from Hospital das Clínicas da Universidade Federal de Minas Gerais (UFMG) (2001 to 2016) under auspices of protocols approved by the Ethics Committee of UFMG (Brazil; number CAAE:06595912.2.20000.5125). Histologically normal tissues (n=3) were obtained from liver resections of metastatic colon cancer patients as determined by a review of pathology reports from 2010 to 2017 at Hospital das Clínicas (UFMG).
2.2 -. Partial hepatic Ischemia Reperfusion murine model
Mice (6 weeks old, n=5–7) were treated with NFAT activation inhibitor (Cyclosporine or VIVIT, 10 mg/kg for 2 weeks, 8 injections,i.p.) or saline (control) before surgery. Then, wild type and ITPR3-LSKO mice (8 weeks old) were anesthetized with xylazine (10 mg/kg,i.p.) and ketamine (100 mg/kg,i.p.), and a midline laparotomy was performed. The hepatic left lobe blood flow was transiently blocked using a straight micro clip (Roboz Surgical Instrument). After 75 min of ischemia, the clamp was removed to allow reperfusion (20). Mice were sacrificed after 3, 6, 24, 48 or 72 hours of reperfusion. Sham animals were not submitted to the ischemia procedure, whether they were pretreated or not with NFAT activation inhibitors (sham cyclosporine or VIVIT control groups). Liver samples and serum were collected for aminotransferase levels and histological analysis at each time point.
2.3 -. Histological evaluation
Liver samples were fixed in 10% formalin, embedded in paraffin and sectioned (4 μm-thick). Tissue sections were de-waxed and stained with hematoxylin and eosin (H&E). Histopathology was assessed and graded in a blinded fashion by an experienced liver pathologist using a 0–4 score, as follows: 0–absent, 1−low, 2−mild, 3-moderate, 4−severe, in terms of inflammation (presence of leukocyte infiltrate in the tissue) and cell death (amount of apoptotic and/or necrotic cells in the tissue). For analysis of necrotic areas, the regions of necrosis observed in photomicrographs were analyzed with Image J Software and results were expressed as percentage relative to control IR wild type mice.
2.4 -. RNA extraction, cDNA synthesis and RT-PCR
Total RNA was extracted from liver samples of sham and animals that underwent ischemia-reperfusion procedure, using the RNeasy Mini Kit (Qiagen). The reverse transcription reaction was performed with 1 μg of total RNA using the iScript cDNA Synthesis (BioRad). Relative messenger RNA (mRNA) expression of IL-1, IL-6, ITPR1, ITPR2, ITPR3 and β-actin were quantified in an ABI 7500 Real-time PCR system (Thermo Fisher Scientific) by the comparative Ct method (15).
2.5 -. Protein extraction and Western blotting
Total liver proteins from ITPR3-LSKO and WT mice were extracted with RIPA buffer supplemented with inhibitors of proteases and phosphatases followed by centrifugation at 10000 rpm for 10 min at 4° C. For extraction of cytosolic and nuclear proteins, liver samples (50 mg) were homogenized in cold phosphate-buffered saline (PBS, pH 7.4), as described previously (21). For the western blotting, proteins were separated by SDS-PAGE in 12% Bis-Tris gels and transferred onto 0.45 μm nitrocellulose membranes (BioRad). The blots were incubated overnight at 4°C with anti-ITPR3 (BD transduction Laboratories), anti-catalase (Santa Cruz), anti-superoxide dismutase (Santa Cruz), anti-histone-h3 (Thermo Fisher Scientific), anti-GAPDH (Thermo Fisher Scientific), and anti-actin (Santa Cruz) antibodies followed by incubation with horseradish peroxidase (HRP)-conjugated secondary IgGs (GE Healthcare). Chemiluminescence signals were quantified using Image J software. β -actin, Histone H3 or GAPDH were used as a loading control (13).
2.6 -. Immunohistochemistry
Immunohistochemistry was performed with the Novolink Polymer Detection Kit (Leica Biosystems) using formalin-fixed, paraffin-embedded tissue slices of mice and human liver specimens from control and ischemia-reperfusion groups. Specimens were incubated with anti-ITPR3 (BD transduction Laboratories), anti-NFAT (Santa Cruz), 5 methylcytosine (5mc) (Zymo Research) and anti-ssDNA (Abcam) antibodies overnight at room temperature. The reactions were developed by applying 3,3’-diaminobenzidine (DAB). To compare ITPR3 staining in hepatic tissue, digital images were analyzed in a blinded fashion. The average pixel intensity was quantified in specimens using Image J software. The specificity of anti-ITPR3 was confirmed by observing apical labeling of cholangiocytes in all analyzed liver specimens, because this is the subcellular distribution of ITPR3 that has been established previously in these cells (19). The ITPR3 intensity value of IR groups was normalized according to control. For NFAT and ssDNA labeling analysis, cells with positive nuclear staining also were counted in a blinded fashion, being expressed as percentage of positive cells (15).
2.7 -. Immunofluorescence
Liver slices were dewaxed, and antigen retrieval was performed in citrate buffer 10mM containing 0.6% hydrogen peroxide. The slices were incubated with anti-ITPR3 (BD Transduction Laboratories) and NFAT (Santa Cruz) overnight at 4° C, followed by 1-hour incubation at room temperature with anti-mouse and anti-rabbit secondary antibody conjugated with Alexa Fluor 488 and 555 (Life Technologies). DAPI was used for nuclear staining. Controls in which primary antibodies were omitted showed no specific staining. Images were collected using a Nikon C2 confocal microscope (17).
2.8 -. Analysis of NFAT binding sites and transfection
The NFAT binding sites on human ITPR3 promoter sequence was analyzed using the database MatInspector (www.genomatix.de). To validate the in silico analysis, normal human cholangiocytes were co-transfected with luciferase ITPR3 construct (2kb 5`-upstream regions of the human ITPR3 gene (Gene ID: 3710) including the proximal promoter) and plasmids encoding nuclear factor of activated T cells (NFAT) family (Gene ID: 4772 for NFAT2 and ID:4775 for NFAT4). Luciferase activity was analyzed 48 hours later using the Dual Luciferase kit (Promega) (17).
2.9 -. DNA methylation analysis of ITPR3 promoter region of mouse primary hepatocytes
Mouse hepatocytes were isolated after hepatic ischemia followed by 3, 6 and 24 hours of reperfusion. Livers were perfused with Hanks A and then Hanks B medium supplemented with 0.05% collagenase (Roche Applied Science). The cells were filtrated in a 40-μm mesh nylon filter to collect primary hepatocytes in suspension. Genomic DNA extracted with DNeasy Blood & Tissue Kit (Qiagen) was treated with bisulfite using the EZ DNA Methylation Kit (Zymo Research), according to the manufacturer’s instruction. Mouse ITPR3 promoter region was amplified by conventional PCR using the forward primer 5’-AAGCCGTCTAGAGAACGCCC-3’ and reverse primer 5’ CCACACACATGCAAATCCCG-3’. Sequencing data were analyzed using Sequence Scanner Software (Applied Biosystems). The efficiency of DNA amplification was monitored using a 1% agarose gel electrophoresis and capillary electrophoresis sequencing in an ABI3730 apparatus with POP7 polymer and BigDye v3.1 (13).
2.10 -. Analysis of reperfusion with FITC-conjugated albumin
Mice were anesthetized and the livers were positioned on the stage of a Nikon A1 confocal microscope. Albumin fluorescein isothiocyanate was injected (0.1 mL at 5 mg/mL, i.v.) and the fluorescence intensity was monitored for 5 minutes under 10x magnification calibrated by central vein position (22).
2.11 -. In vivo Ca2+ measurements
C57/BL6 mice were anesthetized with xylazine (10 mg/kg) and ketamine (100 mg/kg), and a midline laparotomy was performed. Livers were loaded for 10 minutes with either 6 μM of the Ca2+ indicator Fluo-4/AM (Thermo Fisher Scientific) or Rhod-2/AM (Thermo Fisher Scientific), to monitor cytosolic or mitochondrial Ca2+ signaling, respectively, and placed onto the stage of a Nikon A1 confocal microscope. Vasopressin (AVP, 100 ng/mL,i.v.) was used to trigger InsP3-dependent Ca2+ release in vivo. Pericentral hepatocytes were selected based on central vein localization and within a radius of 2 times the average size of individual hepatocytes. The remaining cells were classified as periportal hepatocytes. For nuclear Ca2+ signaling, the nuclei of hepatocytes were identified (13). Data were expressed as percentage of responding cells for AVP-stimulus and fluorescence intensity according to baseline fluorescence × 100% (13). The rise time of each Ca2+ signal was calculated as the time required for the signal to increase from 25% to 75% of the maximum response (15). Therefore, a ‘faster’ or ‘shorter’ rise time both were taken to mean that it would take less time for a Ca2+ signal to rise from its baseline to its peak value. For mitochondrial Ca2+ in vivo studies, we evaluated 4 fields in each animal and the data are expressed as mean of these groups. The baseline fluorescence was defined as the mean fluorescence intensity during the 1 minute before adding the agonist. Microscope settings were the same for all experimental groups.
2.12 -. X-ray Ca2+ fluorescence
The dissected intact livers from ischemia-reperfusion protocol were fixed with paraformaldehyde 4% PBS for 24 hours and then sectioned (100-μm-thick) in a Vibratome (Leica). Values of X-ray Ca2+ fluorescence intensities were obtained in the Synchrotron Light National Laboratory (Laboratório Nacional de Luz Síncrotron) at the XRF beamline. Using these counting live-times, errors for each data by SR-μXRF methodologies are <0.1% (24).
2.13 -. Statistical analysis
Data were expressed as mean ± SEM of 5–6 animals per group, and statistical analysis was performed using GraphPad Prism 6.01 and Microsoft Excel. Differences between experimental groups were assessed for significance using one-way ANOVA followed by Bonferroni post hoc-tests or Student’s t-test. Statistical significance was considered as p<0.05.
3-. RESULTS
3.1 -. Hepatic ischemia-reperfusion injury transiently induces ITPR3 expression in hepatocytes
To investigate alterations of the Ca2+ signaling machinery after hypoxia followed by reperfusion in the liver and the consequences of these changes, we performed an ischemia-reperfusion protocol using a rodent animal model. Analysis of H&E staining (Figure 1a–b) revealed that ischemia-reperfusion induced a progressive, transient liver injury, which included an increase in leukocyte infiltration (p<0.01), cell death (p<0.001), serum AST (p<0.01) and ALT (p<0.001). Hypoxia also was evaluated by nuclear translocation of hypoxia-inducible factor 1α (HIF-1α) (Figure 1c). Similar to the histological damage, nuclear translocation of HIF-1α peaked in the initial hours of reperfusion, as previously reported (25) (Figure 1c). Moreover, oxidative stress was increased in ischemia-reperfusion as evidenced by reduced expression of catalase (p<0.01) and superoxide dismutase (p<0.05), two key antioxidant enzymes in ischemia-reperfusion liver injury (1), (Figure 1d). These findings demonstrate that this mouse model of liver injury recapitulates essential features of ischemia-reperfusion process such as induction of hypoxia, tissue damage, oxidative stress, cell death, and inflammatory infiltration (1).
Figure 1 – Ischemia-reperfusion injury induces progressive liver damage in a mouse model.

(a) Histological analysis was performed according to a score that evaluates cell death and inflammation in control (sham) and ischemia-reperfusion (IR) animals. (b) Serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT). (c) Immunohistochemistry for HIF-α (in brown). (d) Western blot analysis for catalase and superoxide dismutase (SOD-1). Scale bars, 50 and 25 μm. Values expressed as mean ± SEM, n=5–7, *p<0.05 **p<0.01*** p<0.001.
Dysregulation of intracellular Ca2+ signaling is involved in the pathogenesis of ischemia-reperfusion-liver injury (3, 6). The only intracellular Ca2+ channels expressed in hepatocytes are ITPRs, which are modulated in several liver diseases (15–17, 19). To understand whether and how ITPR expression changes during liver ischemia-reperfusion injury, we analyzed mRNA levels of all ITPR isoforms. ITPR1 and ITPR2 expression were unchanged in ischemia-reperfusion livers (Figure 2a and 2b). However, there was a transient but marked increase in ITPR3 mRNA, which peaked at 3 hours of reperfusion (p<0.05) (Figure 2c). Immunohistochemical staining of liver sections confirmed the increased ITPR3 expression at earlier time points after ischemia-reperfusion (p<0.01) (Figure 2d). ITPR3 was expressed diffusely throughout the liver, with a discrete increase in zone 2 of the hepatic lobule. This altered ITPR3 expression coincided with the early onset of cellular and molecular events related to the histological progression of ischemia-reperfusion injury shown in Figure 1. Together, these results demonstrate new expression of ITPR3 without significant change in ITPR1 or ITPR2 during hepatic ischemia-reperfusion.
Figure 2 – Hepatic ischemia-reperfusion injury leads to ITPR3 expression in hepatocytes.

qPCR of whole liver for ITPR isoforms 1 (a), 2 (b) and 3 (c), showing that there is transient expression of ITPR3. (d) Immunohistochemistry for ITPR3 (in brown) confirms the transient expression profile of ITPR3. Scale bars, 50 μm and 25 μm. Values expressed as mean ± SEM, n=5–7, *p<0.05 **p<0.01*** p<0.001.
3.2 -. ITPR3 expression in hepatocytes decreases the amplitude of Ca2+ signaling in the liver
Since ITPR1 and ITPR2 are the Ca2+ channel isoforms that normally regulate hepatocyte functions (26), we investigated how the de novo ITPR3 expression in hepatocytes would affect Ca2+ signaling in whole liver. To address this, we performed in vivo Ca2+ signal measurements using Fluo-4. This permitted observation of the increase in free Ca2+ in the cytosolic and nuclear compartments, relative to basal levels, elicited by an agonist that directly activates the IP3-dependent cascade. Although Fluo-4 is a non-ratiometric dye, it was chosen due to several advantages for in vivo studies, including high affinity for calcium, low background absorbance, and lower dye concentration requirement, which permits a shorter incubation time compared to other Ca2+ dyes (27). Analysis of Ca2+ signals showed a decrease in the rise time in IR hepatocytes, compared to the sham group (Figure 3a). The faster (shorter) rise time in IR hepatocytes compared to hepatocytes in the sham group (Figure 3b) may be due to the expression of ITPR3 (Figure 2c, d), because this isoform lacks feedback inhibition when higher amounts of Ca2+ are released (28). Although the amplitude of Ca2+ signals reached its peak twice as quickly in livers of animals subjected to ischemia-reperfusion (Sham: 3.6 ± 0.2 sec; IR 6h 1.75 ± 0.1 sec; p<0.01), both the amplitude (p<0.05) and the number of cells responding to arginine vasopressin (AVP) were decreased (p<0.05), albeit without changes in nuclear Ca2+ signals (Figure 3a–b and Supplementary Fig. A1). The amplitude of mitochondrial Ca2+ signals was also reduced in the IR group, relative to controls, in whole livers stimulate with AVP (Supplementary Fig. A2).
Figure 3 – Ca2+ signaling is impaired by hepatic ischemia-reperfusion.

(a) Representative tracings (left graph) of Ca2+signaling kinetics in whole livers of sham and ischemia-reperfusion injured animals, relative to baseline. Amplitude of vasopressin-induced Ca2+ signals (middle graph) and percentage of vasopressine-responsive cells (right graph). (b) Representative tracing (left) and summary bar graph (right) illustrating the faster rise time of the cytosolic Ca2+ signal in an hepatic ischemia-reperfusion mouse, compared to a sham control. The rise time of each Ca2+ signal was calculated as the time required for the signal increase from 25% to 75% of the maximum response. For the graph, the representative tracings were normalized to the same baseline and peak values to facilitate direct comparison of the rise times (23). The blue and red lines illustrate the rise from 25% to 75% of the peak value in the control and IR tracings, respectively. Note the steeper slope in the IR tracing. The rise time is significantly faster (shorter) in the IR group (right; **p<0.01). (c-e) Ca2+ signaling was decreased in both pericentral and periportal zones of the hepatic lobule. (f) X-ray Ca2+ fluorescence spectroscopy images. (g) Albumin-FITC injection (5 mg/mL, i.v.) in livers of ischemia-reperfused and sham animals. Scale bar, 25 μm. Values expressed as mean ± SEM, n=5, *p<0.05 **p<0.01*** p<0.001.
A more heterogenous pattern was seen among responding cells in the IR group as well (Supplementary Fig. A1). The reduction in Ca2+ signaling amplitude was similar across the hepatic lobule (p<0.05) (Figure 3c–e). Because impaired Ca2+ signaling might be secondary to reduced cellular Ca2+ content, we analyzed the concentration of total Ca2+ in liver slices using synchrotron-based X-ray fluorescence spectroscopy. The Ca2+ content in the liver was higher in the ischemia-reperfusion group compared to sham (p<0.001) (Figure 3e), indicating that the smaller Ca2+ signaling amplitude observed in ischemia-reperfusion liver was not a consequence of depletion of Ca2+ from intracellular Ca2+ stores. We then investigated whether the impaired Ca2+ signaling resulted from poor blood perfusion, which could prevent access of AVP to hepatocytes in ischemia-reperfusion-injured livers. Intravenously injected FITC-albumin was similarly distributed in the liver among the experimental groups, demonstrating a preserved distribution of perfused blood. Therefore, the decreased AVP-induced Ca2+ signal in the ischemia-reperfusion animal model was not due to structural impairments in delivery of AVP to hepatocytes (Figure 3f). Together, these results demonstrate that de novo expression of ITPR3 in ischemia-reperfusion livers is associated with a reduced amplitude of intracellular Ca2+ signaling in hepatocytes.
3.3 -. Promoter demethylation and NFAT activation induce ITPR3 expression
ITPR3 expression can be induced in hepatocytes by hypomethylation of its promoter (13, 14). Therefore, we evaluated DNA methylation levels in liver slices of mice subjected to ischemia-reperfusion injury (Figure 4a). Global DNA methylation levels, as assessed by 5mC labeling of liver tissue, were significantly decreased in the initial hours after liver reperfusion (p<0.001). The time course of this coincides with the time course of ITPR3 mRNA expression and precedes ITPR3 protein expression (Figure 2). Analysis of ITPR3 promoter methylation status in particular demonstrated a significant hypomethylation in ischemia-reperfusion livers (p<0.05) (Figure 4b). Together, these findings suggest a link between methylation level and ITRP3 expression during ischemia-reperfusion injury. Because gene expression requires hypomethylation and the recruitment of transcription factors (29), we also investigated transcription factors that might regulate ITPR3 expression in ischemia-reperfusion injury. An initial in silico analysis revealed that the proximal 1 Kb of the human ITPR3 promoter region contains 16 predicted binding sites for Nuclear factor of activated T-cells (NFAT; Figure 4c); a transcription factor associated with ischemia-reperfusion damage (30). NFAT localization in the nucleus of hepatocytes, an index of protein activation, was increased within 3–6 hours after ischemia-reperfusion-induced liver injury (p<0.05) (Figure 4d). In addition, ITPR3 promoter activity was significantly increased in cells transfected with the 1Kb ITPR3 promoter together with NFAT2 (p<0.05) or NFAT4 (p<0.001), the two NFAT isoforms expressed in hepatocytes (Figure 4e). Together, these results suggest that the de novo expression of ITPR3 in ischemia-reperfusion is in part due to NFAT activation.
Figure 4 – NFAT induces ITPR3 expression after hepatic ischemia reperfusion.

(a) Immunohistochemistry for 5-methylcytosine (5mC; brown staining) in liver slices of control and ischemia-reperfusion animals at different time points. (b) DNA methylation levels (percentage) of ITPR3 promoter region. (c) Immunohistochemistry for NFAT (in brown) showing its activation. (d) Binding sites for NFAT in human 1 kb ITPR3 promoter region. (e) Luciferase assay shows that NFAT increases human ITPR3 promoter activation. Scale bars, 50 and 25 μm. Values expressed as mean ± SEM, n=5–7, *p<0.05 **p<0.01*** p<0.001.
Several approaches were taken to determine the relevance in vivo of NFAT activation to induce ITPR3 expression during ischemia-reperfusion injury. NFAT activation was inhibited by pretreatment with cyclosporine, a calcineurin inhibitor that prevents NFAT de-phosphorylation and translocation to the nucleus (31). Cyclosporine (CsA) was effective in reducing NFAT activity, as shown by both western blot analysis of nuclear protein fractions (p<0.05) (Figure 5A) and immunofluorescence (p<0.001) (Figure 5B), compared to sham-CsA and sham saline. Cyclosporine pretreatment also prevented the upregulation of ITPR3 (p<0.001) (Figure 5B). To more directly probe the role of NFAT in the induction of ITPR3, mice were pretreated with the specific NFAT inhibitor VIVIT. VIVIT abolished NFAT translocation to the nucleus relative to control animals (p<0.01), compared to sham VIVIT and sham saline animals (Figure 5C–D). The induction of ITPR3 expression also was decreased in animals pretreated with VIVIT (Figure 5D). In line with its effects on NFAT activation and ITPR3 expression after ischemia-reperfusion injury, VIVIT pretreatment restored the impaired Ca2+ signaling and number of responding cells to AVP-stimuli (Figure 5e). Taken together, these results suggest that NFAT activation is a key event controlling ITPR3 expression in hepatocytes in ischemia-reperfusion injury.
Figure 5 – Inhibition of NFAT activation prevents ITPR3 expression induced by hepatic ischemia-reperfusion.

Western blot of nuclei/cytosol fraction for NFAT in ischemia-reperfusion or sham mice pretreated or not with Cyclosporin (a) and VIVIT (c). Immunofluorescence for NFAT (in red) and ITPR3 (in green) after cyclosporin (b) and VIVIT (d) pretreatment. (e) Amplitude of Ca2+ signaling in vivo and number of responding cells to AVP-stimuli in animals pretreated or not with VIVIT after ischemia-reperfusion. Scale bar of 25 μm. Values expressed as mean ± SEM, n=5, *p<0.05 **p<0.01*** p<0.001. Sham: mice treated for 2 weeks with saline that were not submitted to the ischemia procedure; IR 6h: mice treated for 2 weeks with saline that were submitted to the ischemia procedure; Sham CsA: mice treated for 2 weeks with cyclosporin that were not submitted to the ischemia procedure; IR 6h CsA: mice treated for 2 weeks with cyclosporin that were submitted to the ischemia procedure; Sham VIVIT: mice treated for 2 weeks with VIVIT that were not submitted to the ischemia procedure; IR 6h VIVIT: mice treated for 2 weeks with VIVIT that were submitted to the ischemia procedure.
3.4 -. Liver ischemia-reperfusion injury is potentiated in ITPR3 knockout mice
To understand the role of ITPR3 expression in hepatocytes during ischemia-reperfusion injury, partial liver ischemia followed by 6 hours of reperfusion was performed in liver-specific ITPR3 knockout mice (ITPR3-LSKO) (Figure 6a). Ischemia-reperfusion-induced liver damage was significantly enhanced in ITPR3 LSKO mice when compared to wild type (p<0.05) (Figure 6b). AST levels in ITPR3-LSKO mice also were significantly increased (p<0.05), although no difference was observed in ALT levels (Figure 6c). Because liver injury in ischemia-reperfusion may be mediated by a pro-inflammatory and pro-necrotic environment, we evaluated mRNA levels of the proinflammatory interleukins IL-1 and IL-6. Both cytokines were increased after ischemia-reperfusion, and both IL-1 (p<0.01) and IL-6 (p<0.05) were further elevated in KO animals (Figure 6d). Apoptosis, as evaluated by single-stranded DNA (ssDNA) immunolabeling, was decreased in ITPR3 LSKO mice compared to WT mice (p<0.05) (Figure 6e). These cellular alterations after ischemia-reperfusion are likely related to ITPR3 expression, because ITPR1 and ITPR2 mRNA levels were unchanged in ITPR3 LSKO mice in both baseline conditions and after ischemia-reperfusion (Supplementary Fig A.3). Taken together, these results indicate that absence of ITPR3 has a deleterious effect in this model of ischemia-reperfusion injury. This in turn suggests that ITPR3 plays a protective role in ischemia-reperfusion-induced liver damage.
Figure 6 – Hepatic ischemia-reperfusion injury is worse in ITPR3 LSKO mice.

(a) Western blot of total liver protein of ITPR3 in wild-type (WT) and liver-specific-knockout mice for ITPR3 (ITPR3 LSKO). (b) Photomicrograph of liver showing an increase in necrotic areas in LSKO relative to WT.Serum levels of (c) AST and ALT, relative mRNA levels for (d) IL-6 and IL-1. (e) Immunohistochemistry for ssDNA (in brown) and its quantification showing extent of apoptosis is attenuated in ITPR3 LSKO and WT mice. Scale bar, 50 μm. Values expressed as mean ± SEM, n=5, *p<0.05 **p<0.01 ***p<0.001.
3.5 -. NFAT and ITPR3 are increased in liver specimens from patients with hepatic ischemia
Ischemia-reperfusion injury is associated with increased risk for rejection after liver transplantation, due to local activation of inflammatory and oxidative stress machinery (1, 32). To investigate the clinical relevance of ITPR3 expression in this setting, human liver samples collected from patients diagnosed with acute hepatic artery thrombosis 72 hr after transplantation were stained for NFAT and ITPR3 (Figure 7). Hepatic artery thrombosis is a frequent complication in orthotopic liver transplantation, being one of the principal causes of liver graft failure (33, 34). Hepatocytes with NFAT labeling in the nucleus (Figure 7a and Supplementary Fig A.4) and cytoplasmic ITPR3 staining (Figure 7b) were observed all ischemia-injured human liver samples. This provides evidence that de novo ITPR3 expression occurs in response to hepatic ischemia in patients and that it is mediated at least in part by NFAT activation.
Figure 7 – NFAT is activated and ITPR3 expression is increased in liver biopsy specimens from patients with ischemic injury.

Representative immunohistochemistry for (a) NFAT and (b) ITPR3 in patients diagnosed with hepatic ischemia after blockage of the hepatic artery, showing that there is nuclear translocation (activation) of NFAT and increased ITPR3 expression (both in brown). Bile ducts in figure 7b represents a positive control for ITPR3 staining. Scale bars, 50 and 25 μm. Control: histologically normal tissues obtained from liver resections of metastatic colon cancer patients.
4 –. DISCUSSION
Ischemia-reperfusion injury is an important cause of morbidity and mortality in liver transplantation (32)and other disorders associated with diminished blood flow to the liver such as resection, shock and trauma (35). Primary graft dysfunction due to ischemia-reperfusion injury is responsible in approximately 80% of patients requiring re-transplantation (36). Here, we showed that ischemia-reperfusion injury induces ITPR3 expression in hepatocytes and that this effect is mediated by a combination of NFAT activation and promoter hypomethylation. Maximal demethylation of the ITPR3 promoter region and the nuclear translocation of NFAT coincide with the peak of ITPR3 mRNA transcription, and precede peak ITPR3 protein expression. Moreover, the absence of ITPR3 was associated with worse hepatic injury. NFAT inhibitors such as cyclosporine are an important tool to prevent organ rejection post-liver transplant (37), but this is thought to be because of its actions on the immune system rather than effects on hepatocytes. In fact, cyclosporine mitigated release of inflammatory cytokines and markers of hepatocellular damage in our animal model as well (Supplementary Fig. A.5). However, the de novo expression of a calcium channel unusual in hepatocytes is an unappreciated protective mechanism in ischemia-reperfusion injury. New expression of ITPR3 in hepatocytes was also recently recognized as a protective response to the acute liver injury that occurs in Yellow Fever (YF) viral infection. As in ischemia-reperfusion injury, demethylation of the ITPR3 promoter was implicated as a mechanism for sudden expression of ITPR3 in YF-infected hepatocytes. The protective mechanisms that resulted from ITPR3 expression in YF included enhanced nuclear Ca2+ signaling, leading to increased hepatocyte proliferation, and attenuation of adverse metabolic effects of YF infection, such as steatosis (14). In contrast to the findings in YF infection, the ITPR3 expression caused by ischemia-reperfusion did not increase nuclear Ca2+ signals (Supplementary Fig. A1), which suggests that the protective mechanisms related to ITPR3 is independent of cell proliferation in this type of liver injury. This differences in protective mechanisms between YF infection and ischemia-reperfusion injury may relate to ITPR2, because this isoform is in part responsible for increasing nuclear Ca2+ signals and hepatocyte proliferation (38), and its expression is increased in YF but not in ischemia-reperfusion.
The mechanism by which ITPR3 exerts a protective effect in ischemia-reperfusion injury is unclear. ITPR plays an important role in apoptosis because it regulates transmission of Ca2+ from the ER to mitochondria, and mitochondrial Ca2+ overload by this route induces formation of permeability transition pores in the mitochondrial membrane, which then leads to apoptosis (26). ITPR-mediated mitochondrial Ca2+ signaling has been linked to apoptotic and necrotic cell death in the liver in particular (39, 40), and new expression of ITPR3 in hepatocytes has been linked to resistance to apoptotic cell death in hepatocellular carcinoma (13). However, a similar mechanism is unlikely to be operative in ischemia-reperfusion injury because resistance to apoptosis in hepatocytes chronically expressing ITPR3, such as in cancer, is thought to reflect an adaptive response rather than an acute event (13). In acute types of hepatocyte injury, such as in ischemia-reperfusion, expression of ITPR3 is transient and may contribute to apoptosis, while the chronic ITPR3 expression that occurs in hepatocellular carcinoma may lead to adaptive mechanisms to evade apoptosis despite ongoing cell stress (41). Therefore, it may be that ITPR3 expressed in ischemia-reperfusion injury serves to shift from necrotic to apoptotic loss of hepatocytes, similar to what has been observed in cholangiocarcinoma (39). This explanation is supported by the current findings that ischemia-reperfusion injury is more severe in ITPR3-LSKO mice, as reflected by more extensive necrosis in these KO animals. The reduced mitochondrial Ca2+ signals observed in the liver post-ischemia-reperfusion also is consistent with the idea that ITPR3 expression leads to less hepatocellular damage (Supplementary Fig. A2). One possible explanation for ITPR3-mediated protection against necrosis involves the biophysical properties of ITPR3, which has a lower affinity for InsP3 than ITPR1 or ITPR2 (42), the two ITPR isoforms normally found in hepatocytes (19). New ITPR3 expressed in this cell type may compete for the channel ligand, altering the overall amplitude and/or pattern of Ca2+ signaling in hepatocytes. ITPR3 in hepatocytes may also form heterotetrameric ITPRs with ITPR1 or ITPR2, which would have intermediate affinity for InsP3, compared to the homotetrameric forms (43, 44). Therefore, the presence of ITPR3 subunits in the tetrameric channel formed by ITPR1 or ITPR2 may significantly decreases the affinity to InsP3. Finally, the presence of ITPR3 in hepatocytes might displace ITPR1 at the ER-mitochondria associated membrane, altering ITPR1 mediating apoptosis, since the ITPR1 is the main ITPR isoform in this specialized domain in the cells (9, 11). Thus, at any given agonist concentration, an hepatocyte expressing ITPR3 would be expected to display a less potent Ca2+ signal than hepatocytes lacking ITPR3.
Zonal damage to the liver may also contribute to the observed lower AVP-induced Ca2+ signals in post-ischemia-reperfusion livers. Ischemic damage is most severe in the pericentral region of the hepatic lobule. Hepatocytes in the pericentral region also have a higher density of the V1a AVP receptor, compared to periportal hepatocytes (45), which results in differences in AVP sensitivity and the formation of pericentral-to-periportal Ca2+ waves along the hepatic lobule (46). Therefore, the loss of these ‘pacemaker’ hepatocytes (47) from the pericentral region may also contribute to the reduction in Ca2+ signal amplitude and the percentage of responding cells throughout the hepatic lobule after ischemia-reperfusion. In addition to loss of hepatocytes that are more sensitive to AVP, expression of the V1a receptor is decreased by ischemia-reperfusion injury (48), which may further contribute to the decrease in AVP-induced Ca2+ signals. However, new expression of ITPR3 is likely to contribute at least in part to the decreased Ca2+ signaling in ischemia-reperfusion as well, because we found that the effect of ischemia-reperfusion was blocked by VIVIT, an NFAT inhibitor that prevented ITPR3 expression in hepatocytes (Figure 5e).
A final mechanism by which ITPR3 might exert its protective role in ischemia-reperfusion injury is through regulation of mitochondrial metabolism (49). ATP synthesis is associated with increases in mitochondrial Ca2+ coupled to the activation of the respiratory chain and generation of the proton motive force. Ca2+ signals from the ER to mitochondria may be preferentially transmitted by ITPR3 (50–52). Based on this idea, ITPR3 expression in hepatocytes might work to alleviate the ATP depletion that is a hallmark of ischemia-reperfusion injury (53). The current work also demonstrates that de novo expression of ITPR3 occurs as soon as several hours after hepatic arterial thrombosis in human liver.
5 –. CONCLUSION
Hepatocytes begin to express ITPR3 during ischemia-reperfusion injury. The effects of this are compatible with the idea that this protects the liver by shifting from a necrotic to an apoptotic response and by minimizing inflammatory infiltrate caused by ischemia-reperfusion injury.
Supplementary Material
Supplementary Figure A.1 – (a) Histogram of percentage of responding cells according to fluorescence intensity (% above baseline), and (b) nuclear Ca2+ signaling corresponding to Ca2+ signaling evoked by vasopressin (AVP) after hepatic ischemia of 75 min and reperfusion of 6 hours in vivo. (c) Ca2+ signaling in the liver in vivo after ischemia and 3 and 24 hours of reperfusion. (Values expressed as mean ± SEM, ns-non-significant, n=5, *p<0.05 **p<0.01 ***p<0.001.
Supplementary Figure A.2 – Mitochondrial Ca2+ signals in vivo are reduced post ischemia-reperfusion (IR) injury. Shown are the average of all tracings from in vivo measurements of mitochondrial Ca2+ signals in whole livers from sham (n=4 fields from each of 2 animals) and ischemia-reperfusion group (n=4 fields from each of 4 animals), normalized to baseline. The curves were smoothed using a second order polynomial and 5 nearest neighbors.
Supplementary Figure A.3 – Hepatic ischemia-reperfusion injury does not alter mRNA levels of ITPR1 or ITPR2 in ITPR3 LSKO animals. Values expressed as mean ± SEM, ns - non-significant, n=5, *p<0.05.
Supplementary Figure A.4 – ITPR3 is expressed and NFAT is activated in liver biopsy specimens from post-transplant patients with hepatic ischemia due to hepatic artery thrombosis. Immunohistochemistry for (a) NFAT and ITPR3(b).Scale bar, 50 and 25μm.
Supplementary Figure A.5 – Cyclosporine (CsA) treatment ameliorate ischemia-reperfusion injury. HE stainning, AST and ALT dosage (U/L) and relative mRNA levels of IL-1β and IL-6 after IR.Scale bar,50μm. Sham animal control groups were not submitted to the ischemic protocol, and were either treated or not with an inhibitor of NFAT activation. Values expressed as mean ± SEM, ns – non-significant, n=5, *p<0.05 **p<0.01 ***p<0.001.
Table A.1 - Human liver biopsies and clinical data of trombosis and control patients, including ALT (U/L), AST (U/L), bilirrubin total (mg/dL), bilirrubin indirect (mg/dL), bilirrubin direct (mg/dL), urea (mg/dL), creatinine (mg/dL), GGT (U/L), albumin, INR, LDH (U/L) and ALP (U/L).
Highlights.
There is de novo expression of ITPR3 in hepatocytes after hepatic ischemia reperfusion.
Hypoxia induced by hepatic ischemia-reperfusion activates NFAT, which leads to ITPR3 expression.
ITPR3 may play a protective role in hepatocytes after hepatic ischemia-reperfusion.
ITPR3 also is expressed in human hepatocytes after ischemia induced by thrombosis.
Acknowledgments
We thank the Brazilian Biosciences National Laboratory (LNBio), Liver Center at UFMG, the Yale Liver Center, CNPq, INCT, FAPEMIG, FAPESP, CAPES and NIH for their support.
Financial Support: CNPq, INCT, FAPEMIG, FAPESP (2018/20014-0), NIH (DK34989, DK57751, DK112797, and DK114041) and CAPES.
List of Abbreviation
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- AVP
arginine vasopressin
- Ca2+
calcium
- CAT
catalase
- CsA
Cyclosporine
- DAB
3,3’-diaminobenzidine
- HCC
hepatocellular carcinoma
- IL
interleukins
- IR
ischemia-reperfusion
- ITPR3
type 3 inositol 1,4,5-trisphosphate receptor
- LSKO
liver specific knockout
- NFAT
nuclear factor of activated T-cells
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- SOD-1
superoxide dismutase 1
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: Nothing to disclose
REFERENCES
- 1.Cannistra M, Ruggiero M, Zullo A, Gallelli G, Serafini S, Maria M, Naso A, et al. Hepatic ischemia reperfusion injury: A systematic review of literature and the role of current drugs and biomarkers. Int J Surg 2016;33 Suppl 1:S57–70. [DOI] [PubMed] [Google Scholar]
- 2.Jaeschke H Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J Gastroenterol Hepatol 2011;26 Suppl 1:173–179. [DOI] [PubMed] [Google Scholar]
- 3.Dhar DK, Takemoto Y, Nagasue N, Uchida M, Ono T, Nakamura T. FK506 maintains cellular calcium homeostasis in ischemia-reperfusion injury of the canine liver. J Surg Res 1996;60:142–146. [DOI] [PubMed] [Google Scholar]
- 4.Mellstrom B, Savignac M, Gomez-Villafuertes R, Naranjo JR. Ca2+-operated transcriptional networks: molecular mechanisms and in vivo models. Physiol Rev 2008;88:421–449. [DOI] [PubMed] [Google Scholar]
- 5.Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, et al. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999;284:339–343. [DOI] [PubMed] [Google Scholar]
- 6.Farber JL. The role of calcium in cell death. Life Sci 1981;29:1289–1295. [DOI] [PubMed] [Google Scholar]
- 7.Mikoshiba K IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J Neurochem 2007;102:1426–1446. [DOI] [PubMed] [Google Scholar]
- 8.Hirata K, Pusl T, O’Neill AF, Dranoff JA, Nathanson MH. The type II inositol 1,4,5-trisphosphate receptor can trigger Ca2+ waves in rat hepatocytes. Gastroenterology 2002;122:1088–1100. [DOI] [PubMed] [Google Scholar]
- 9.Feriod CN, Oliveira AG, Guerra MT, Nguyen L, Richards KM, Jurczak MJ, Ruan HB, et al. Hepatic Inositol 1,4,5 Trisphosphate Receptor Type 1 Mediates Fatty Liver. Hepatol Commun 2017;1:23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kruglov EA, Gautam S, Guerra MT, Nathanson MH. Type 2 inositol 1,4,5-trisphosphate receptor modulates bile salt export pump activity in rat hepatocytes. Hepatology 2011;54:1790–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perry RJ, Zhang D, Guerra MT, Brill AL, Goedeke L, Nasiri AR, Rabin-Court A, et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 2020;579:279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruz LN, Guerra MT, Kruglov E, Mennone A, Garcia CR, Chen J, Nathanson MH. Regulation of multidrug resistance-associated protein 2 by calcium signaling in mouse liver. Hepatology 2010;52:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guerra MT, Florentino RM, Franca A, Lima Filho AC, Dos Santos ML, Fonseca RC, Lemos FO, et al. Expression of the type 3 InsP3 receptor is a final common event in the development of hepatocellular carcinoma. Gut 2019;68:1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemos FdO, França A, Lima Filho ACM, Florentino RM, Santos ML, Missiaggia DG, Rodrigues GOL, et al. Molecular Mechanism for Protection Against Liver Failure in Human Yellow Fever Infection. Hepatology Communications;n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franca A, Carlos Melo Lima Filho A, Guerra MT, Weerachayaphorn J, Loiola Dos Santos M, Njei B, Robert M, et al. Effects of Endotoxin on Type 3 Inositol 1,4,5-Trisphosphate Receptor in Human Cholangiocytes. Hepatology 2019;69:817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khamphaya T, Chukijrungroat N, Saengsirisuwan V, Mitchell-Richards KA, Robert ME, Mennone A, Ananthanarayanan M, et al. Nonalcoholic fatty liver disease impairs expression of the type II inositol 1,4,5-trisphosphate receptor. Hepatology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weerachayaphorn J, Amaya MJ, Spirli C, Chansela P, Mitchell-Richards KA, Ananthanarayanan M, Nathanson MH. Nuclear Factor, Erythroid 2-Like 2 Regulates Expression of Type 3 Inositol 1,4,5-Trisphosphate Receptor and Calcium Signaling in Cholangiocytes. Gastroenterology 2015;149:211–222.e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ananthanarayanan M, Banales JM, Guerra MT, Spirli C, Munoz-Garrido P, Mitchell-Richards K, Tafur D, et al. Post-translational regulation of the type III inositol 1,4,5-trisphosphate receptor by miRNA-506. J Biol Chem 2015;290:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shibao K, Hirata K, Robert ME, Nathanson MH. Loss of inositol 1,4,5-trisphosphate receptors from bile duct epithelia is a common event in cholestasis. Gastroenterology 2003;125:1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan S, Zhou B, Zhang Q, Li Z, Shao Y, Chen H, Zheng S. Hepatic venous occlusion causes more impairment after reperfusion compared with portal clamping in a murine model. J Surg Res 2011;169:117–124. [DOI] [PubMed] [Google Scholar]
- 21.Dimauro I, Pearson T, Caporossi D, Jackson MJ. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res Notes 2012;5:513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB, Sousa Pereira RV, et al. Combination of Mass Cytometry and Imaging Analysis Reveals Origin, Location, and Functional Repopulation of Liver Myeloid Cells in Mice. Gastroenterology 2016;151:1176–1191. [DOI] [PubMed] [Google Scholar]
- 23.Leite MF, Thrower EC, Echevarria W, Koulen P, Hirata K, Bennett AM, Ehrlich BE, et al. Nuclear and cytosolic calcium are regulated independently. Proc Natl Acad Sci U S A 2003;100:2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rezende KM, Bonecker M, Perez CA, Mantesso A. Synchrotron radiation X-ray micro-fluorescence: Protocol to study mesenchymal stem cells. Microsc Res Tech 2016;79:149–154. [DOI] [PubMed] [Google Scholar]
- 25.Depping R, Steinhoff A, Schindler SG, Friedrich B, Fagerlund R, Metzen E, Hartmann E, et al. Nuclear translocation of hypoxia-inducible factors (HIFs): involvement of the classical importin alpha/beta pathway. Biochim Biophys Acta 2008;1783:394–404. [DOI] [PubMed] [Google Scholar]
- 26.Lemos FO, Florentino RM, Lima Filho ACM, Dos Santos ML, Leite MF. Inositol 1,4,5-trisphosphate receptor in the liver: Expression and function. World J Gastroenterol 2019;25:6483–6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paredes RM, Etzler JC, Watts LT, Zheng W, Lechleiter JD. Chemical calcium indicators. Methods 2008;46:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hagar RE, Burgstahler AD, Nathanson MH, Ehrlich BE. Type III InsP3 receptor channel stays open in the presence of increased calcium. Nature 1998;396:81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kenneth NS, Rocha S. Regulation of gene expression by hypoxia. Biochem J 2008;414:19–29. [DOI] [PubMed] [Google Scholar]
- 30.Zhang YJ, Mei HS, Wang C, Wang YL, Zhang YJ. Involvement of nuclear factor of activated T-cells (NFATc) in calcineurin-mediated ischemic brain damage in vivo. Yao Xue Xue Bao 2005;40:299–305. [PubMed] [Google Scholar]
- 31.Macian F NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 2005;5:472–484. [DOI] [PubMed] [Google Scholar]
- 32.Casillas-Ramirez A, Mosbah IB, Ramalho F, Rosello-Catafau J, Peralta C. Past and future approaches to ischemia-reperfusion lesion associated with liver transplantation. Life Sci 2006;79:1881–1894. [DOI] [PubMed] [Google Scholar]
- 33.Pastacaldi S, Teixeira R, Montalto P, Rolles K, Burroughs AK. Hepatic artery thrombosis after orthotopic liver transplantation: a review of nonsurgical causes. Liver Transpl 2001;7:75–81. [DOI] [PubMed] [Google Scholar]
- 34.Pareja E, Cortes M, Navarro R, Sanjuan F, Lopez R, Mir J. Vascular complications after orthotopic liver transplantation: hepatic artery thrombosis. Transplant Proc 2010;42:2970–2972. [DOI] [PubMed] [Google Scholar]
- 35.Lu L, Zhou H, Ni M, Wang X, Busuttil R, Kupiec-Weglinski J, Zhai Y. Innate Immune Regulations and Liver Ischemia-Reperfusion Injury. Transplantation 2016;100:2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uemura T, Randall HB, Sanchez EQ, Ikegami T, Narasimhan G, McKenna GJ, Chinnakotla S, et al. Liver retransplantation for primary nonfunction: analysis of a 20-year single-center experience. Liver Transpl 2007;13:227–233. [DOI] [PubMed] [Google Scholar]
- 37.Taylor AL, Watson CJ, Bradley JA. Immunosuppressive agents in solid organ transplantation: Mechanisms of action and therapeutic efficacy. Crit Rev Oncol Hematol 2005;56:23–46. [DOI] [PubMed] [Google Scholar]
- 38.Khamphaya T, Chukijrungroat N, Saengsirisuwan V, Mitchell-Richards KA, Robert ME, Mennone A, Ananthanarayanan M, et al. Nonalcoholic fatty liver disease impairs expression of the type II inositol 1,4,5-trisphosphate receptor. Hepatology 2018;67:560–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ueasilamongkol P, Khamphaya T, Guerra MT, Rodrigues MA, Gomes DA, Kong Y, Wei W, et al. Type 3 Inositol 1,4,5-Trisphosphate Receptor Is Increased and Enhances Malignant Properties in Cholangiocarcinoma. Hepatology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guerra MT, Fonseca EA, Melo FM, Andrade VA, Aguiar CJ, Andrade LM, Pinheiro AC, et al. Mitochondrial calcium regulates rat liver regeneration through the modulation of apoptosis. Hepatology 2011;54:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 42.Ramos-Franco J, Bare D, Caenepeel S, Nani A, Fill M, Mignery G. Single-channel function of recombinant type 2 inositol 1,4, 5-trisphosphate receptor. Biophysical journal 2000;79:1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alzayady KJ, Wagner LE, Chandrasekhar R, Monteagudo A, Godiska R, Tall GG, Joseph SK, et al. Functional inositol 1,4,5-trisphosphate receptors assembled from concatenated homo- and heteromeric subunits. The Journal of biological chemistry 2013;288:29772–29784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chandrasekhar R, Alzayady KJ, Wagner LE, 2nd,, Yule DI. Unique Regulatory Properties of Heterotetrameric Inositol 1,4,5-Trisphosphate Receptors Revealed by Studying Concatenated Receptor Constructs. J Biol Chem 2016;291:4846–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nathanson MH, Burgstahler AD, Mennone A, Fallon MB, Gonzalez CB, Saez JC. Ca2+ waves are organized among hepatocytes in the intact organ. Am J Physiol 1995;269:G167–171. [DOI] [PubMed] [Google Scholar]
- 46.Tordjmann T, Berthon B, Jacquemin E, Clair C, Stelly N, Guillon G, Claret M, et al. Receptor-oriented intercellular calcium waves evoked by vasopressin in rat hepatocytes. Embo j 1998;17:4695–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leite MF, Hirata K, Pusl T, Burgstahler AD, Okazaki K, Ortega JM, Goes AM, et al. Molecular basis for pacemaker cells in epithelia. J Biol Chem 2002;277:16313–16323. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Luo G, Jiang J, Ma T, Lin X, Jiang L, Cheng J, et al. Signaling through hepatocyte vasopressin receptor 1 protects mouse liver from ischemia-reperfusion injury. Oncotarget 2016;7:69276–69290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, Urra F, et al. Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep 2016;14:2313–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proceedings of the National Academy of Sciences 1999;96:13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem 2005;280:40892–40900. [DOI] [PubMed] [Google Scholar]
- 52.Gaspers LD, Mémin E, Thomas AP. Calcium-dependent physiologic and pathologic stimulus-metabolic response coupling in hepatocytes. Cell calcium 2012;52:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gasbarrini A, Borle AB, Farghali H, Bender C, Francavilla A, Van Thiel D. Effect of anoxia on intracellular ATP, Na+i, Ca2+i, Mg2+i, and cytotoxicity in rat hepatocytes. J Biol Chem 1992;267:6654–6663. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure A.1 – (a) Histogram of percentage of responding cells according to fluorescence intensity (% above baseline), and (b) nuclear Ca2+ signaling corresponding to Ca2+ signaling evoked by vasopressin (AVP) after hepatic ischemia of 75 min and reperfusion of 6 hours in vivo. (c) Ca2+ signaling in the liver in vivo after ischemia and 3 and 24 hours of reperfusion. (Values expressed as mean ± SEM, ns-non-significant, n=5, *p<0.05 **p<0.01 ***p<0.001.
Supplementary Figure A.2 – Mitochondrial Ca2+ signals in vivo are reduced post ischemia-reperfusion (IR) injury. Shown are the average of all tracings from in vivo measurements of mitochondrial Ca2+ signals in whole livers from sham (n=4 fields from each of 2 animals) and ischemia-reperfusion group (n=4 fields from each of 4 animals), normalized to baseline. The curves were smoothed using a second order polynomial and 5 nearest neighbors.
Supplementary Figure A.3 – Hepatic ischemia-reperfusion injury does not alter mRNA levels of ITPR1 or ITPR2 in ITPR3 LSKO animals. Values expressed as mean ± SEM, ns - non-significant, n=5, *p<0.05.
Supplementary Figure A.4 – ITPR3 is expressed and NFAT is activated in liver biopsy specimens from post-transplant patients with hepatic ischemia due to hepatic artery thrombosis. Immunohistochemistry for (a) NFAT and ITPR3(b).Scale bar, 50 and 25μm.
Supplementary Figure A.5 – Cyclosporine (CsA) treatment ameliorate ischemia-reperfusion injury. HE stainning, AST and ALT dosage (U/L) and relative mRNA levels of IL-1β and IL-6 after IR.Scale bar,50μm. Sham animal control groups were not submitted to the ischemic protocol, and were either treated or not with an inhibitor of NFAT activation. Values expressed as mean ± SEM, ns – non-significant, n=5, *p<0.05 **p<0.01 ***p<0.001.
Table A.1 - Human liver biopsies and clinical data of trombosis and control patients, including ALT (U/L), AST (U/L), bilirrubin total (mg/dL), bilirrubin indirect (mg/dL), bilirrubin direct (mg/dL), urea (mg/dL), creatinine (mg/dL), GGT (U/L), albumin, INR, LDH (U/L) and ALP (U/L).