

Abstract
Many interesting solid-state targets for biological research do not form crystalline structures; these materials include intrinsically disordered proteins, plant biopolymer composites, cell-wall polysaccharides, and soil organic matter. The absence of aligned repeating structural elements and atomic-level rigidity presents hurdles to achieving structural elucidation and obtaining functional insights. We describe strategies for adapting several solid-state NMR methods to determine the molecular structures and compositions of these amorphous biosolids.
The main spectroscopic problems in studying amorphous structures by NMR are over/under-sampling of the spin signals and spectral complexity. These problems arise in part because amorphous biosolids typically contain a mix of rigid and mobile domains, making it difficult to select a single experiment or set of acquisition conditions that fairly represents all nuclear spins in a carbon-based organic sample. These issues can be addressed by running hybrid experiments, such as using direct excitation alongside cross polarization-based methods, to develop a more holistic picture of the macromolecular system. In situations of spectral crowding or overlap, the structural elucidation strategy can be further assisted by coupling 13C spins to nuclei such as 15N, filtering out portions of the spectrum, highlighting individual moieties of interest, and adding a second or third spectral dimension to an NMR experiment in order to spread out the resonances and link them pairwise through space or through bonds. We discuss practical aspects and illustrations from the recent literature for 1D experiments that use cross or direct polarization and both homo- and heteronuclear 2D and 3D solid-state NMR experiments.
Keywords: solid-state NMR, Magic Angle Spinning, amorphous biosolids, biopolymer, correlation spectroscopy
Graphical Abstract

1. Introduction
Solid-state nuclear magnetic resonance spectroscopy (ssNMR) is a powerful technique that can probe molecular structure and dynamics in a broad range of chemical substances, from aggregated proteins to whole microbial cells to lithium batteries [1–3]. Freed from limitations related to crystallinity, solubility, or molecular weight, ssNMR is often heralded as the ‘gold standard’ for defining the atomic-level structure and molecular composition of intriguing materials that are heterogeneous, resistant to degradation, and/or otherwise suboptimal candidates for traditional biophysical techniques. Current ssNMR methodologies also offer attractive routes to atomic-level information that complements and fills information gaps left by related approaches such as solution-state NMR, e.g., the through-space dipole-dipole (DD) couplings that are averaged out in solution by rapid molecular tumbling are preserved in the solid state and can be exploited using correlation experiments to estimate internuclear distances. The value of ssNMR is arguably best exemplified by analyses of biological Preprint submitted to Solid State Nuclear Magnetic Resonance systems under near-physiological conditions, for instance by reporting on the conformational and chemical dynamics of integral membrane proteins, which must be embedded within a lipid bilayer to achieve functional states [4]. Indeed, ssNMR is uniquely suited to probe structural details and molecular interactions that are present only in assembled biomolecular complexes. Owing to advancements in detection technology and the widespread availability of high-field magnets, the past decade has witnessed a substantial increase in the application of magic-angle spinning (MAS) ssNMR to study other-than-protein macromolecular assemblies that play essential roles in living organisms and engineered materials [5–7]. However, systematic consideration of specialized methodological approaches to optimally characterize these complex systems has been reported much less commonly [8]. Moreover, many routinely implemented ssNMR experiments have been adapted from well-established solution methods designed for the structural elucidation of proteins, natural products, or small-molecule drug candidates, investigative targets that in many ways have fundamentally different nuclear spin properties from large biomolecular assemblies in the solid state.
Amorphous biological solids (biosolids, ABS) are prime examples of ubiquitous natural materials that are excellent targets for ssNMR, yet there is no evident consensus regarding methodological approaches or validated experimental protocols for their study. In this review, we designate ABS as any biologically synthesized composite materials that are either available in fully intact form or have undergone modest enough chemical or enzymatic processing that their native structural integrity and heterogeneous molecular composition are preserved. ABS may comprise components with repeating structural units and/or regions of high crystallinity (i.e., local order), but in the context of a macromolecular assembly they lack long-range organization. Following this definition, examples of such materials that have been investigated using ssNMR include whole microbial cells and intact plant stems as well as near-native microbial cell wall preparations and partially purified plant polymers [2,9–13]. Other commonly studied ABS are microbial biofilms, insect exoskeletons, and tissues such as bone or brain matter [14–19]. Although the systems that comprise ABS may seem disparate in their chemical characteristics, they share architectural features that inform their structural investigation by ssNMR methods. For current purposes, we set aside noncrystalline proteins, which have been considered in other recent reviews [20,21].
The NMR practitioner faces two principal challenges in characterizing the structural framework or chemical composition of ABS, first and foremost of which is achieving uniform excitation and detection of sites that differ in molecular mobility. As biologically synthesized composites, ABS are assembled from a variety of structurally and compositionally distinct constituents. Consequently, the solids will often possess regions with diverse architectures that display differing degrees of molecular motion, which in turn influence the efficacy of their observation using particular ssNMR experiments. Examples include plant or fungal cell walls embedded with suberin, lignin, or melanin polymers (discussed below) and membrane proteins (reviewed elsewhere) [21–23]. For instance, rigid regions will be well represented by the use of cross polarization (CP) to detect 13C or 15N spins; direct polarization (DP) will favor the more mobile moieties if the recycle delay is short with respect to the lengthy spin-lattice relaxation times of nuclei within rigid structures, but it can sample all ABS regions with quantitative fidelity if the recycle delay is sufficiently long [24].
The second considerable hurdle to be overcome in ABS NMR is spectral complexity arising from broad resonances, signal overlap, and spectral crowding. In the solid state, the information-rich DD interactions, which depend on the orientation of the internuclear vector with respect to the magnetic field, are a primary source of spectral line broadening in the absence of rapid liquid-like molecular motion. Even though motional averaging in ABS reduces the magnitude of these DD couplings, and magic-angle spinning usually abolishes chemical shift anisotropy, the lack of long-range order allows nuclei in nominally identical chemical environments to resonate at slightly different frequencies, producing signal overlap and an appearance of broad spectral features. Spectral complexity is further exacerbated if the ABS targets include a constituent that can have subtle structural variations (e.g., polymorphic chitin polysaccharides) [2] or multiple constituent types with resonances that each span substantial spectral regions and are thus likely to display peaks at similar frequencies (e.g., triglycerides and sterol esters [25], both of which are present in fungal cell walls). Together, these factors lead to NMR spectra that can be quite difficult to interpret.
In this practical review, we illustrate several experimental strategies that can be used in concert to provide a more holistic, information-rich, and quantitatively reliable picture of amorphous biosolids. Their lack of aligned repeating structural elements and atomic-level rigidity can make structural determination a technically challenging, but ultimately rewarding, goal. We describe strategies to adapt several solid-state NMR methods to elucidate both chemical composition and macromolecular structure in a variety of ABS spanning the kingdoms of living organisms.
2. Amorphous biosolids have multiple spectroscopic ‘personalities’
Whether the material of interest comes from animal, vegetable, or fungal sources, many amorphous biosolids contain a mix of constituents as illustrated above. In analogy with “The Three Faces of Eve” of cinematic fame [26], these macromolecular entities can be rigid (‘solid-like,’ tumbling rate ≤10−5 sec−1) or flexible (‘liquid-like,’ tumbling rate ≥10−9 sec−1) or somewhere in between, resulting in dramatically different cross polarization build-up and rotating frame decay rates [24,27]. In practice, diminished dipole-dipole interactions for structural moieties that are flexible and/or proton poor prevent the full realization of sensitivity enhancements and time savings associated with cross polarization from e.g., 1H to 13C or 1H to 15N nuclei. The 1D CPMAS 13C spectrum of suberized cell walls in the periderm (skin) of wound-healing potato tubers (Figure 1 bottom, [28]) illustrates this issue: cell-wall polysaccharides (59–108 ppm) account for 46% of the integrated intensity across the spectrum, but are we overcounting these rigid constituents with respect to the long, flexible aliphatic chains of the suberin polyester (20–40 ppm)? Furthermore, the typical reliance on CP for the initial magnetization transfer in 2D through-space (13C-13C, 13C-15N) or through-bond (13C-13C) experiments (e.g., dipolar assisted rotational resonance (DARR) [29,30] or the Insensitive Natural Abundance DoublE QUAntum Transfer Experiment (INADEQUATE) [31,32]) will propagate the deficiencies of the 1D spectral acquisition: we may unintentionally edit the spectral data and preferentially detect correlations involving rigid and/or heavily protonated carbons.
Figure 1.
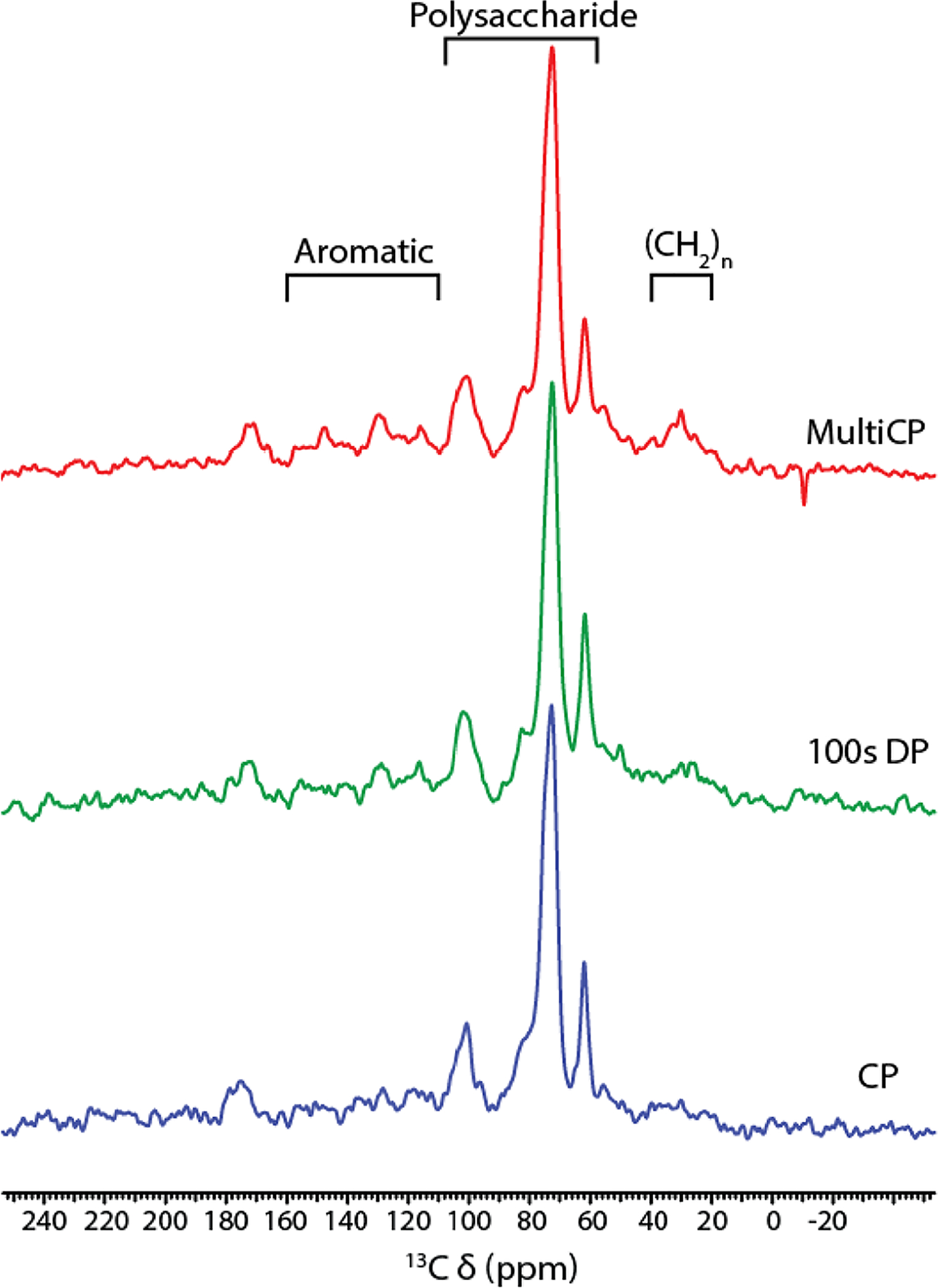
150 MHz 13C solid-state NMR of suberized cell walls from wound periderm of Norkotah Russet potato tubers, adapted from [28]. Bottom: 10 kHz CPMAS with a 10% linearly ramped 1H radiofrequency amplitude during a 1.0-ms contact time and a recycle delay of 3 s; middle: DPMAS with a 100-s recycle delay; top: multi-CP with 11 contact times of 1.0 ms each and separated by 1.0 s between each successive acquisition.
To count the 13C or other heteronuclei in a quantitatively reliable fashion, the traditional methods have included (1) acquiring a series of CPMAS spectra with varying mixing times, then extrapolating the set of peak intensities that diminish according to T1ρ(H) back to zero time [27,33,34]; or (2) running a direct polarization (DPMAS) experiment using a long enough recycle delay to accommodate spin relaxation of all 13C’s in the sample [35–37] (Figure 1 middle), but either method is time consuming. A more recently proposed experiment uses multiple CP [38,39], which yielded relative intensities within 5% of the DP-based “gold standard” for wound periderm suberin [28] (Figure 1 top). Nonetheless, this latter experiment must be optimized for each ABS sample; in our hands, reliable multi-CP data could not be obtained for related native skins from potato tubers or tomato fruits because the flexibility of the (CH2)n groups largely precluded cross polarization [35,40]. Caution is also advised if different molecular moieties vary in CP efficiency or are sparse in hydrogen nuclei from which polarization can be transferred. As outlined below, it can be necessary to embrace the several ‘personalities’ of the material, using a suite of tailored experiments to obtain a more holistic view of the system of interest.
2.1. Distinguishing through-bond 13C-13C connections in rigid vs. flexible ABS regions
Connections through covalent bonds are well-established elements of molecular structure determination, where correlating pairs of bonded carbons can strengthen otherwise ambiguous spectral assignments and several solution-state experiments have been demonstrated or adapted for 13C nuclear spins in the solid state. For example, the 2D J-INADEQUATE experiment [32,41] utilizes scalar (J) couplings to correlate directly bonded spin pairs, where a single-quantum (SQ) chemical shift of one nucleus in the direct frequency dimension is correlated with the sum of the shifts for a directly bonded pair in the indirect dimension. Alternatively, the sensitive, absorptive, refocused COSY (SAR-COSY) experiment is a scalar correlation experiment that retains the sensitivity of J-INADEQUATE while preserving the intuitively appealing symmetric 2D format of a single-quantum shift correlation COSY experiment [42]. Although SAR-COSY can exhibit small ‘relayed correlation’ artifacts and does not benefit from the spreading of signals into a double quantum (DQ) dimension, we have observed the corresponding spectral features to be ~65% sharper in the indirect dimension. And although the reduced spectral width in the indirect dimension of SAR-COSY would seem to require less spectrometer time, this advantage will be lost if slower decay of the SQ coherence demands a longer period of signal digitization.
For ABS that contain both rigid and mobile chemical constituents, J-INADEQUATE is run commonly with variants that use either CP or DP for initial excitation. Use of both methods, each applied with a range of experimental parameters, is expected to sample the full range of domain types. This strategy has proven useful in a detailed study of intact hydrated Aspergillus fumigatus cell walls [2]: CP-INADEQUATE favors resonances from the rigid cell wall constituents whereas DP-INADEQUATE favors mobile cell-wall domains. In particular, CP excitation permitted identification of both α and β anomers of 1,3 glucans and also minor amounts of β−1,4 and β−1,6 glucans (Figure 2). By contrast, DP-INADEQUATE allowed the unambiguous identification of mannan and arabinan. Since mannan is a major constituent of the glycoproteins used to form the outer layer of fungal cell walls, this result showed that the outer shell of the cell wall is very dynamic and distinct from the rigid inner region. Although α−1,3 glucan was found as expected in the stiff, hydrophobic core of the cell wall, its additional appearance in the DP-INADEQUATE spectrum supported a hypothesis that α−1,3 glucans form the outermost layer of the cell wall where they can block the immune recognition of β glucan receptors in a host.
Figure 2.
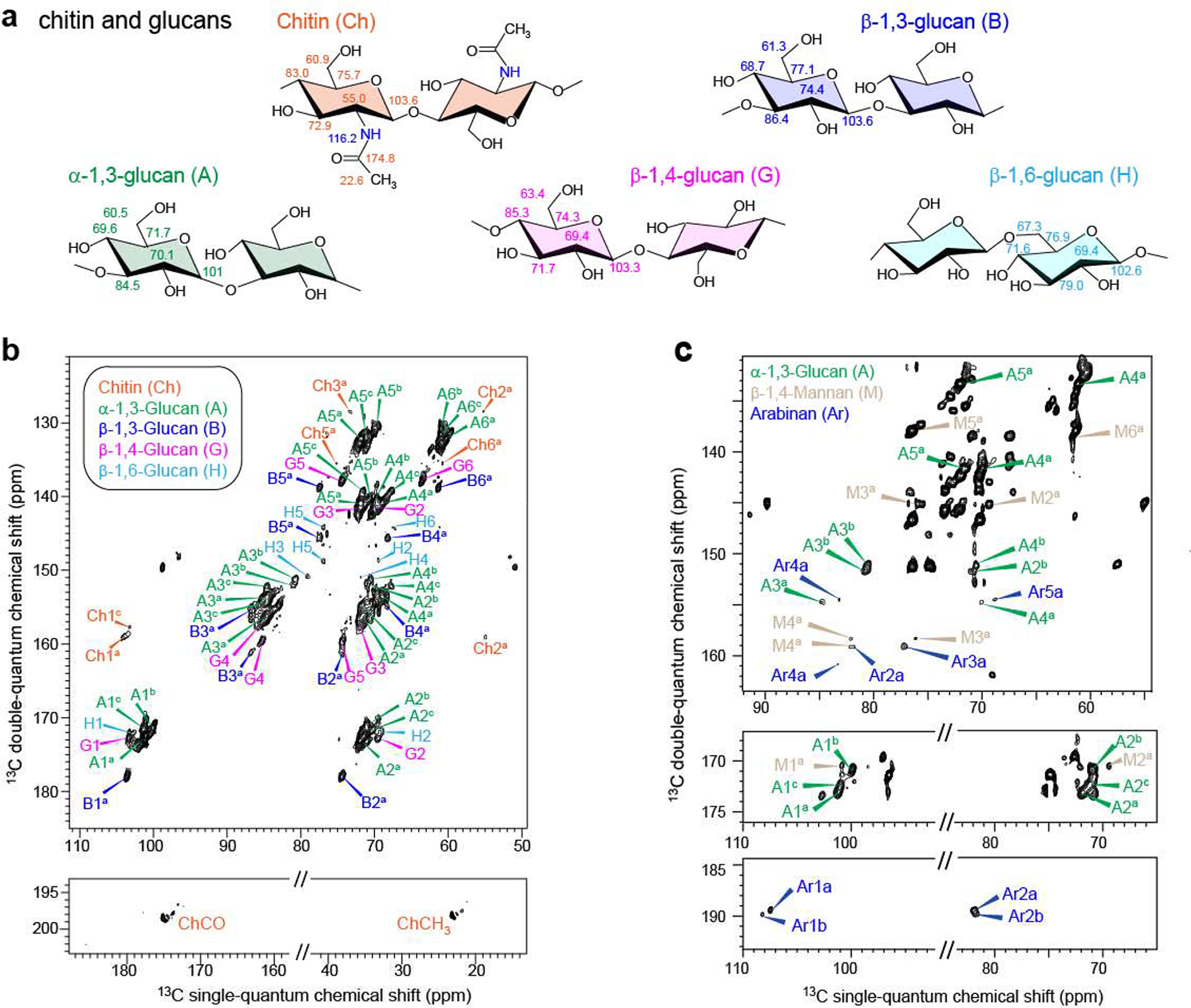
200 MHz 2D 13C CP- and DP-J-INADEQUATE spectra of A. fumigatus cell walls, adapted from [2]. a: molecular structures and representative chemical shifts of polysaccharide units; b: CP J-INADEQUATE contour plot showing through-bond connectivities for the rigid domain; c: DP J-INADEQUATE showing through-bond connectivities for polysaccharides of the mobile domain.
2.2. Distinguishing through-space 13C-13C connections in rigid or flexible ABS regions
Through-space proximities between NMR-active nuclei are another key component of chemical structure determination. In addition to their well-established usefulness to constrain the fold of a protein with known amino acid sequence, these typically pairwise spatial relationships represent critical tools that can identify molecular fragments present in ABS and map out the spatial relationships within and between their chemical constituents -- even if they lack regular repeating units. In cases such as indole-based melanin pigments [6] or phenylpropanoid-based lignins in wood [9], for instance, the chemical classes of the building blocks may be known but their sequential and stacking arrangements can be irregular. Appropriate ssNMR experiments can provide constraints for short-range intramolecular proximities (~2 Å) within a molecular unit or long-range through-space connections that span intermolecular distances (up to 6 Å for carbon-carbon pairs), depending on the length of the nuclear spin mixing times. One of the most commonly used 13C-13C through-space correlation experiments is dipolar assisted rotational resonance (DARR) [29,30]. The DARR mixing is achieved similarly to proton-driven spin diffusion (PDSD, which has no 1H decoupling during mixing): by setting the 1H field strength equal to an integer multiple of the rotor spinning speed, 1H-13C DD interactions are recoupled and can thus facilitate polarization transfer to nearby 13C nuclei.
However, DARR methods can become inefficient if the DD interactions are modest: at spinning rates above 30 kHz that make the 13C-13C matching conditions difficult to fulfill [29], when long distance constraints are sought in the presence of short-range connections (dipolar truncation), or if DD interactions are partially averaged by the region-specific ABS molecular motion described above. In these situations, third spin assisted recoupling (TSAR) methods that enhance the efficiency of homonuclear correlations can be optimized for a particular sample using published guidelines as a starting point [43].
How to proceed if an interesting amorphous biosolid contains both rigid and mobile domains? A subset of nuclear spins can be chosen before applying the recoupling sequence, as illustrated by the use of both CP- and DP-PDSD to distinguish the helical screw conformational folds formed by xylan as a function of cellulose level in the secondary cell walls of Arabidopsis stems [44]. In wild-type stems, xylan adopts a two-fold screw ‘ribbon’ conformation (one 360° twist per two glycosidic bonds) similar to the cellulose that is also present. The cellulose-deficient irx3 mutant instead forms a three-fold screw conformation. This distinction was evidenced by a 2.6-ppm change in the anomeric carbon chemical shift of the xylan (which was assigned by INADEQUATE methods and supported by DFT calculations). This conformational change was also found to alter the mobility of the polysaccharide: CP-PDSD of the WT stems showed the through-space connectivities of the rigid two-fold screw xylan (Figure 3, black contours), whereas DP-PDSD of irx3 stems revealed the mobile three-fold screw xylan (Figure 3, orange contours). In each case, experiments with mixing times ≥ 1 second revealed long-range through-space correlations, particularly the proximities of acetate substituents to sites on the xylan rings.
Figure 3.

214 MHz 2D 13C PDSD spectra of Arabidopsis stems (adapted from [44]). CP-PDSD (1-s mixing) spectrum of wild type (black) stems overlaid with DP-PDSD (1.5-s mixing) spectrum of irx3 (orange) stems shows two distinct xylan conformations. The wild type two-fold xylan resonates at 105.2 ppm, and the irx3 three-fold xylan resonates at 102.6 ppm.
2.3. Using through-space and through-bond experiments as structural complements
Whereas through-bond information from INADEQUATE or SAR-COSY is often used to make spectral assignments in known molecular frameworks or to compare a proposed structure to reference spectral data, through-space experiments such as DARR can offer a longer reach and richer conformational or architectural information for complex macromolecular assemblies if long mixing times are used. On the other hand, running short-mixing-time DARR experiments that span distances corresponding to about one bond length alongside SAR-COSY or INADEQUATE allows spectral comparisons that can identify which 13C-13C DARR correlations are also directly bonded. Caution is required in making such comparisons: a rigid molecular framework can exhibit efficient DD polarization transfer and produce CP-DARR cross-peaks that link 13C nuclei separated by unexpectedly long distances. Conversely, the presence of flexible structures in ABS argues for the importance of using both DP-DARR and DP-INADEQUATE strategies to ensure that essential spin connectivities are not missed.
3. The challenges of 13C NMR spectral complexity can be resolved by 15N-based editing and correlation experiments
The challenge of spectral complexity described in Section 1 for amorphous biosolid samples can be addressed in several ways. This section illustrates strategies to leverage the presence of the 15N-containing polysaccharides chitin and chitosan among a group of otherwise magnetically similar polymers in fungal cell walls, considering methods of progressively increasing complexity to select, resolve, and connect the structural moieties of interest to their architecturally associated ABS constituents. As an example, we consider melanized cell-wall samples (melanin ‘ghosts’) from the fungal pathogen Cryptococcus neoformans, which contain glucan-and chitin-based polysaccharides as well as residual lipids among their constituents [45]. Based on 13C chemical shifts, 13C-13C CP-DARR, and CP-SAR-COSY results [46], it had been proposed that melanin pigment virulence factors for this fungus form covalent bonds specifically with a chitin polysaccharide constituent [46], but the involvement of chitin was not established definitively.
3.1. 15N CPMAS selects sparse chemical moieties of interest
A simple way to check the proposed identification of chitin involves acquiring the 15N CPMAS spectrum of an isotopically 15N-enriched melanin ghost sample (Figure 4). The compositionally sparse chitin and chitosan constituents stand out in the 15N spectrum because only these polysaccharides contain nitrogen [25]; the 15N spectrum shows only peaks at 123 ppm and 35 ppm, which are diagnostic for nitrogens of the chitin amido and chitosan amino groups, respectively.
Figure 4.

60 MHz 1D 15N CPMAS spectrum of the H99 strain of C. neoformans melanin ghosts generated from cell cultures containing [U-13C6]-D-glucose and 15N-glycine as the sole carbon and nitrogen sources, respectively (adapted from [25]). Shown are the characteristic 15N peaks corresponding to the chitin amido (123 ppm) and chitosan amino (35 ppm) nitrogens, respectively.
3.2. 15N-13C through-space interactions confirm identifications in complex ABS
Moreover, through-space 13C-15N correlations obtained via 2D double cross polarization [27], Rotational Echo DOuble Resonance (REDOR) [47], or Transferred Echo Double Resonance (TEDOR) [48] experiments can link the 15N NMR findings with previously assigned 13C resonances and confirm the identification of proposed structures. 15N-15N correlation experiments are often precluded by the fact that 15N is chemically dilute even if the nitrogen nuclei are isotopically enriched. However, these 15N-13C correlation experiments can be broadly useful, because they work well for both crystalline and amorphous solids: the magnetization is completely refocused at the end of each rotor cycle [49], and it is possible distinguish magnetically similar molecular constituents of the 13C spectrum by observing only the 13C nuclei that are close in space to 15N nuclei.
REDOR and TEDOR are magic-angle spinning experiments that each use a number of ‘loops’ of π pulse trains to partially dephase rotational echoes based on reintroduction of DD couplings for each pair of coupled nuclear spins. Whereas Rotational Echo DOuble Resonance (REDOR) was originally designed to measure internuclear distances for isolated 15N-13C spin pairs, multiply labeled macromolecular systems are accessible using current implementations of Transferred Echo DOuble Resonance (TEDOR) that suppress homonuclear J-couplings [48], allowing for collection of 2D spectra that display cross-peaks exclusively for 13C’s that are near 15N’s [50] and have signal intensities that discriminate between short- and long-range correlations. If a more selective experiment is desired, double cross polarization (DCP) can be performed to select, for example, only carbonyl or only aliphatic carbons that are proximal to nitrogen.
The contour plot shown in Figure 5 demonstrates the usefulness of 2D TEDOR experiments for our investigations of cell-wall architecture in the pathogenic fungus C. neoformans [25,36]. In this illustration, the provisional identifications of chitin and chitosan made from the 15N CPMAS spectrum of Figure 4 could be confirmed definitively via 13C-15N correlations, using either 4 TEDOR loops to favor pairwise distances corresponding to ~1 bond (1 ms coherence transfer, dark purple) or 14 loops to favor ~2 bonds (4 ms, light purple) and thereby map out their N-acetylglucosamine and glucosamine molecular skeletons. These types of correlation experiments have also proven valuable to uncover numerous polymorphic forms of chitin in Aspergillus fumigatus cell walls [2] and to verify otherwise ambiguous 13C resonance assignments in other types of fungal ABS systems [25,46].
Figure 5.
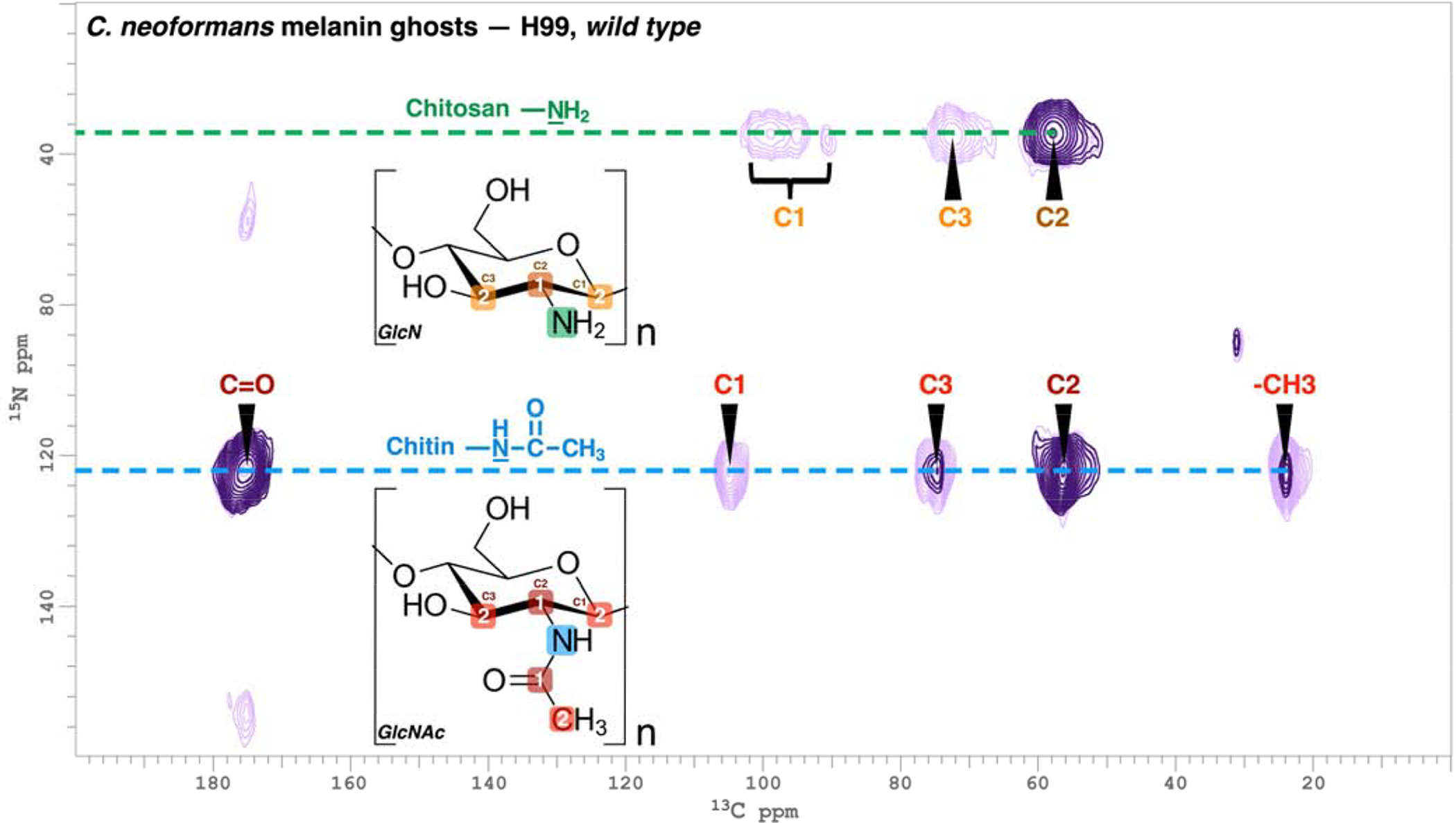
TEDOR spectra with 13C and 15N frequencies of 150 and 60 MHz, respectively for H99 C. neoformans melanin ghosts (adapted from [25]). Coherence transfer periods of 1 and 4 ms were used in separate experiments to obtain proximal carbon-nitrogen pairs separated by distances corresponding to ~1 bond length (dark purple contours) and ~2 bond lengths (light purple contours). The boxed numbers displayed on the chitin N-acetyl glucosamine (GlcNAc) and chitosan glucosamine (GlcN) monomeric units denote the number of bonds between each 13C-15N nuclear pair.
3.3. 3D 15N-13C-13C and 13C-13C-13C through-space interactions resolve ambiguities in 2D NMR spectra
Some ABS materials are so complex in terms of macromolecular architecture or number of chemical constituents that interpretation of their spectra can benefit from higher dimensional NMR strategies. Adding additional dimensions allows the experimenter to simplify the spectra by further editing based on bonding or proximity to a particular nuclear spin species or to separate overlapping signals from a 2D spectrum. Naturally, such maneuvers come at the cost of increased experiment time and possible decreases in signal-to-noise ratio, making it important to decide if the trade-off between information and effort is worthwhile for a particular sample. Below we illustrate how the analysis of amorphous biosolids can be augmented by three-dimensional (3D) experiments, typically preceded by the 2D experiments already detailed in the previous sections.
3.3.1. TEDOR-DARR
In addition to the TEDOR experiments discussed above, 3D TEDOR-DARR experiments have been used to investigate the through-space proximities in C. neoformans cell walls, focusing on the network of carbons that are nearby a carbon that has been linked (through space) with a nitrogen. Adding a DARR mixing period to a TEDOR experiment provides a convenient probe of the network of carbons that are proximal to each 13C-15N pair.
When run on a [U-13C6]glucose, [15N]glycine-enriched C. neoformans melanin ghost sample (Figure 6), the resulting TEDOR-DARR spectrum shows all of the ring carbons in the rigid semicrystalline chitin polymer, including those that are furthest away from the nitrogen and thus not visible in the 2D TEDOR spectrum (C4 and C6). The more mobile chitosan polymer now shows the C4 and C5 carbons, which are missing or very broad, respectively, in the TEDOR spectrum. The C1 of chitosan also shows up as a single peak at ~98 ppm; the other two TEDOR cross-peaks are likely to arise from amorphous chitosan C1’s with partially averaged DD interactions [25]), so those spectral features are further disfavored in the TEDOR-DARR experiment.
Figure 6.
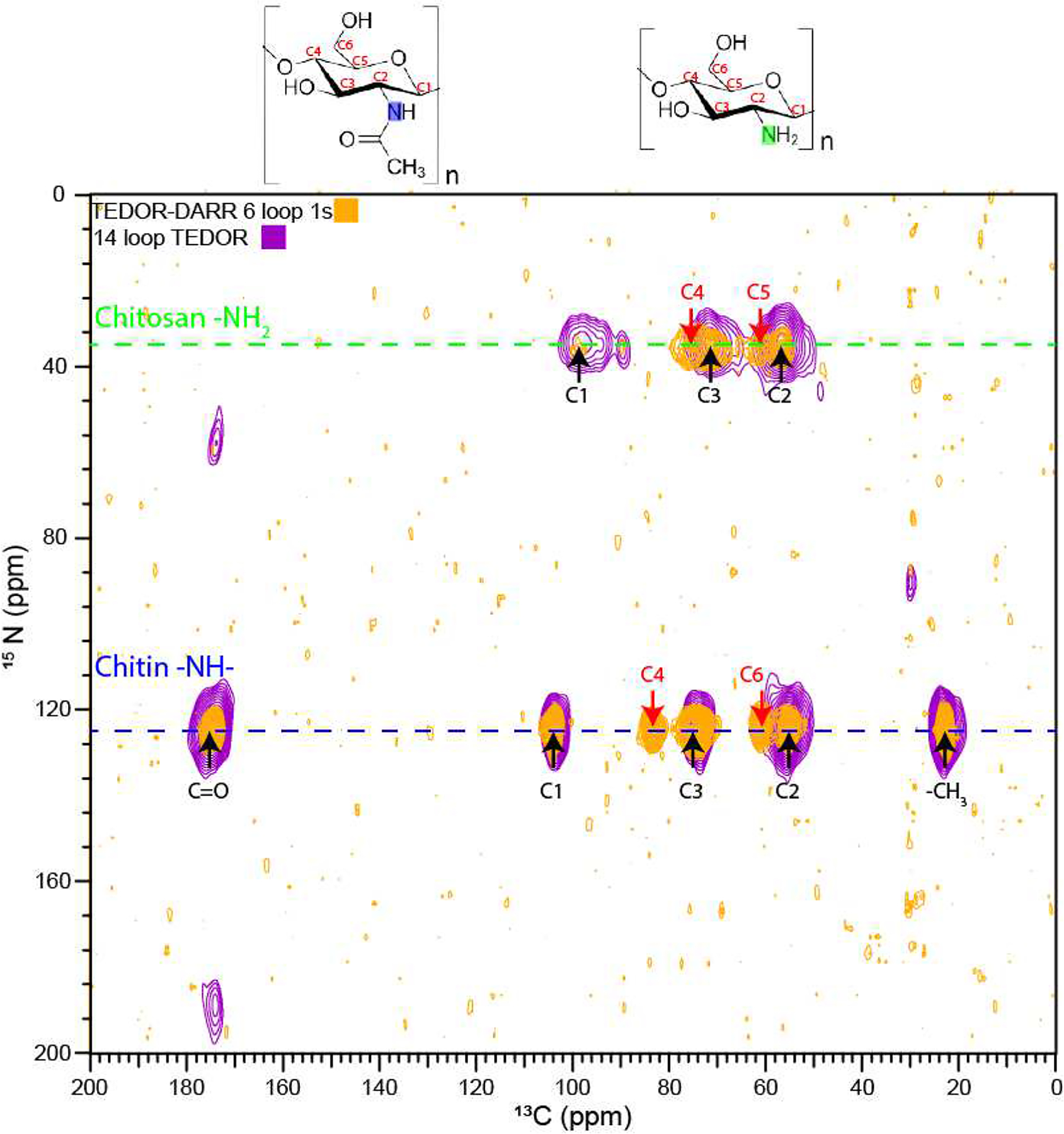
F3-F1 projection from a 3D TEDOR-DARR experiment obtained for H99 C. neoformans melanin ghosts with 13C and 15N frequencies of 150 and 60 MHz, respectively. A 1.6 ms coherence transfer period, corresponding to ~1 bond length, was followed by 1 s of DARR mixing. The peaks indicated with red arrows correspond to 15N-13C correlations that are not observed in the 2D TEDOR spectra of Figure 5.
3.3.2. DARR-DARR
Homonuclear 3D NMR can also be useful to resolve signals in crowded regions of an ABS spectrum; moreover, the experiments can be made more surgical by selecting for rigid or mobile moieties with otherwise similar molecular environments. For instance, Dick-Pérez, et al. used a combination of CP and DP 3D DARR-DARR experiments to investigate the structure and through-space interactions in the primary cell-wall polysaccharides of Arabidopsis thaliana plants [10]. This cell wall contains a complex mix of polysaccharides, and whereas many of these can be identified using 2D NMR experiments, the spectral crowding of the polysaccharide region (Fig. 3) makes it difficult to complete the spectral assignments. In addition, this crowding makes it difficult to distinguish the correlations that represent the close intramolecular spatial proximities from the relatively long-distance intermolecular interactions. The authors performed 3D DARR-DARR experiments with a short mixing time linking the first and second dimensions to view the correlations within the same molecule, and a longer mixing time linking the second and third dimensions to reveal any correlations between different polysaccharides. In order to detect both the rigid and mobile polysaccharides, they conducted parallel experiments in which the initial magnetization was prepared with either CP or DP, thus obtaining a “full picture” of these complex cell-wall composites.
The resulting structural information is richly detailed and informative regarding the means by which plants balance tensile strength and developmental growth. The 89-ppm plane of the CP DARR-DARR spectrum (Figure 7a, bottom) shows many cross-peaks between interior (C4, 89 ppm) and surface (C4, 85 ppm and C6, 62 ppm) sites of the cellulose microfibrils, signifying that these cell wall structures are small enough in diameter to allow for the close packing of surface and interior glucan chains. Additional interactions between the pectins and surface cellulose are also evident: the 69-ppm plane reveals several cross-peaks of rhamnose C5 and galacturonic acid C2 (69 ppm) with surface cellulose (75 ppm) (Figure 7b, bottom); likewise, the C1 carbon of rhamnose and galacturonic acid at 101 ppm exhibits clear cross-peaks with surface cellulose (not shown). Pairwise interactions are identified involving all three species of polysaccharides, suggesting that load bearing by the cell wall is achieved by a single network rather than being dominated by xyloglucan-cellulose correlations as previously proposed. Meanwhile, DP DARR-DARR on a xyloglucan-deficient mutant verifies that pectin-microfibril contacts remain present in the cell wall even in the absence of xyloglucan (not shown).
Figure 7.

Illustrative planes from a 3D DARR-DARR experiment obtained for wild-type A. thaliana cell walls at 150 MHz 13C (adapted from [10]). These experiments were run with preparation of the initial magnetization with either CP (tm2 = 300 ms) or DP (tm2 = 100 ms) to focus on the rigid or mobile polysaccharides, respectively. Cross-peaks marked in color correspond to intermolecular correlations.
4. Conclusion
Structural studies of amorphous biosolids can benefit greatly from the use of solid-state NMR. We have illustrated a group of experiments that, especially when used in concert, can provide a more holistic view of the biological targets from plant or fungal sources and allow the investigator to focus on a broad range of molecular constituents with divergent motional and order profiles or structures that contain distinctive dilute-spin species. It is straightforward to tailor the mode of spectral acquisition for 1D ssNMR (CPMAS or DPMAS; 13C or 15N), and each of these editing strategies can be extended to prepare the spin systems for through-bond and through-space multidimensional experiments. In this way, it is possible to accommodate the complexities of these materials while also completing more definitive structural analyses. We expect that these experiments and their applications to ABS will continue to evolve as we seek more detailed atomic-level information on increasingly challenging biological entities.
HIGHLIGHTS.
Structure elucidation is challenging for amorphous biomacromolecular solids.
Noncrystalline biological complexes can be studied structurally by solid-state NMR.
Solid-state NMR can distinguish mobile from rigid structures in biological solids.
Multidimensional solid-state NMR improves structural information for biosolids.
Solid-state NMR reveals molecular structure in microbial cells and plant polymers.
Acknowledgements
We thank Drs. Paul Dupree (U. of Cambridge), Mei Hong (M.I.T.), and Tuo Wang (Louisiana State U.) for permission to adapt figures from their previously published work. Drs. Hsin Wang (City College, CUNY) and Van Chanh Phan (Hostos College, CUNY) assisted with the development of pulse programs and provided technical support at City College and the CUNY Institute for Macromolecular Assemblies; additional infrastructural support for the 600 MHz NMR facilities was provided by National Institutes of Health Grant 3G12MD007603-30S2 from the National Institute on Minority Health and Health Disparities of the National Institutes of Health. This work was supported by grants from the U.S. National Science Foundation (MCB-1411984) and the U.S. National Institutes of Health (R01-AI052733). C.C. was the recipient of a fellowship from the U.S. Department of Education Graduate Assistance in Areas of National Need (GAANN) Program in Biochemistry, Biophysics and Biodesign at The City College of New York (PA200A120211 and PA200A150068).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Zhou DH, Nieuwkoop AJ, Berthold DA, Comellas G, Sperling LJ, Tang M, Shah GJ, Brea EJ, Lemkau LR, Rienstra CM, Solid-state NMR analysis of membrane proteins and protein aggregates by proton detected spectroscopy, J. Biomol. NMR 54 (2012) 291–305. 10.1007/s10858-012-9672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kang X, Kirui A, Muszyński A, Widanage MCD, Chen A, Azadi P, Wang P, Mentink-Vigier F, Wang T, Molecular architecture of fungal cell walls revealed by solid-state NMR, Nat. Commun 9 (2018). 10.1038/s41467-018-05199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Demers J-P, Fricke P, Shi C, Chevelkov V, Lange A, Structure determination of supra-molecular assemblies by solid-state NMR: Practical considerations, Prog. Nucl. Magn. Reson. Spectrosc 109 (2018) 51–78. 10.1016/j.pnmrs.2018.06.002. [DOI] [PubMed] [Google Scholar]
- [4].McDermott A, Structure and Dynamics of Membrane Proteins by Magic Angle Spinning Solid-State NMR, Annu. Rev. Biophys 38 (2009) 385–403. 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- [5].Serra O, Chatterjee S, Huang W, Stark RE, Mini-review: What nuclear magnetic resonance can tell us about protective tissues, Plant Sci. 195 (2012) 120–124. 10.1016/j.plantsci.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nosanchuk JD, Stark RE, Casadevall A, Fungal Melanin: What do We Know About Structure?, Front. Microbiol 6 (2015). 10.3389/fmicb.2015.01463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhao W, Fernando LD, Kirui A, Deligey F, Wang T, Solid-state NMR of plant and fungal cell walls: A critical review, Solid State Nucl. Magn. Reson 107 (2020) 101660. 10.1016/j.ssnmr.2020.101660. [DOI] [PubMed] [Google Scholar]
- [8].Wang T, Hong M, Solid-state NMR investigations of cellulose structure and interactions with matrix polysaccharides in plant primary cell walls, J. Exp. Bot 67 (2016) 503–514. 10.1093/jxb/erv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kang X, Kirui A, Dickwella Widanage MC, Mentink-Vigier F, Cosgrove DJ, Wang T, Lignin polysaccharide interactions in plant secondary cell walls revealed by solid-state NMR, Nat. Commun 10 (2019) 347 10.1038/s41467-018-08252-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dick-Pérez M, Zhang Y, Hayes J, Salazar A, Zabotina OA, Hong M, Structure and Interactions of Plant Cell-Wall Polysaccharides by Two- and Three-Dimensional Magic-Angle-Spinning Solid-State NMR, Biochemistry. 50 (2011) 989–1000. 10.1021/bi101795q. [DOI] [PubMed] [Google Scholar]
- [11].Phyo P, Hong M, Fast MAS 1H–13C correlation NMR for structural investigations of plant cell walls, J. Biomol. NMR 73 (2019) 661–674. 10.1007/s10858-019-00277-x. [DOI] [PubMed] [Google Scholar]
- [12].Romaniuk JAH, L., Peptidoglycan and Teichoic Acid Levels and Alterations in Staphylococcus aureus by Cell-Wall and Whole-Cell Nuclear Magnetic Resonance, Biochemistry. 57 (2018) 3966–3975. 10.1021/acs.biochem.8b00495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wisser D, Brückner SI, Wisser FM, Althoff-Ospelt G, Getzschmann J, Kaskel S, Brunner E, 1H–13C–29Si triple resonance and REDOR solid-state NMR—A tool to study interactions between biosilica and organic molecules in diatom cell walls, Solid State Nucl. Magn. Reson 66–67 (2015) 33–39. 10.1016/j.ssnmr.2014.12.007. [DOI] [PubMed] [Google Scholar]
- [14].Reichhardt C, Ferreira JAG, Joubert L-M, Clemons KV, Stevens DA, Cegelski L, Analysis of the Aspergillus fumigatus Biofilm Extracellular Matrix by Solid-State Nuclear Magnetic Resonance Spectroscopy, Eukaryot. Cell 14 (2015) 1064–1072. 10.1128/EC.00050-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reichhardt C, Fong JCN, Yildiz F, Cegelski L, Characterization of the Vibrio cholerae extracellular matrix: A top-down solid-state NMR approach, Biochim. Biophys. Acta BBA - Biomembr 1848 (2015) 378–383. 10.1016/j.bbamem.2014.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Andersen SO, Insect cuticular sclerotization: A review, Insect Biochem. Mol. Biol 40 (2010) 166–178. 10.1016/j.ibmb.2009.10.007. [DOI] [PubMed] [Google Scholar]
- [17].Merritt ME, Christensen AM, Kramer KJ, Hopkins TL, Schaefer J, Detection of Intercatechol Cross-Links in Insect Cuticle by Solid-State Carbon-13 and Nitrogen-15 NMR, J. Am. Chem. Soc 118 (1996) 11278–11282. 10.1021/ja961621o. [DOI] [Google Scholar]
- [18].Wise ER, Maltsev S, Davies ME, Duer MJ, Jaeger C, Loveridge N, Murray RC, Reid DG, The Organic-Mineral Interface in Bone Is Predominantly Polysaccharide, Chem. Mater 19 (2007) 5055–5057. 10.1021/cm702054c. [DOI] [Google Scholar]
- [19].Aime S, Bergamasco B, Casu M, Digilio G, Fasano M, Giraudo S, Lopiano L, Isolation and 13C‐NMR characterization of an insoluble proteinaceous fraction from substantia nigra of patients with parkinson’s disease, Mov. Disord 15 (2000) 5. [DOI] [PubMed] [Google Scholar]
- [20].van der Wel PCA, New applications of solid-state NMR in structural biology, Emerg. Top. Life Sci 2 (2018) 57–67. 10.1042/ETLS20170088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Siemer AB, Advances in studying protein disorder with solid-state NMR, Solid State Nucl. Magn. Reson 106 (2020) 101643. 10.1016/j.ssnmr.2020.101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mandala VS, Williams JK, Hong M, Structure and Dynamics of Membrane Proteins from Solid-State NMR, Annu. Rev. Biophys 47 (2018) 201–222. 10.1146/annurev-biophys-070816-033712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Matlahov I, van der Wel PCA, Hidden motions and motion-induced invisibility: Dynamics-based spectral editing in solid-state NMR, Methods. 148 (2018) 123–135. 10.1016/j.ymeth.2018.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kolodziejski W, Klinowski J, Kinetics of Cross-Polarization in Solid-State NMR: A Guide for Chemists, Chem. Rev 102 (2002) 613–628. 10.1021/cr000060n. [DOI] [PubMed] [Google Scholar]
- [25].Chrissian C, Camacho E, Fu MS, Prados-Rosales R, Chatterjee S, Cordero RJB, Lodge JK, Casadevall A, Stark RE, Melanin deposition in two Cryptococcus species depends on cell-wall composition and flexibility, J. Biol. Chem 295 (2020) 1815–1828. 10.1074/jbc.RA119.011949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wikipedia contibutors, The Three Faces of Eve - Wikipedia, Wikipedia. (2020). https://en.wikipedia.org/wiki/The_Three_Faces_of_Eve (accessed April 10, 2020). [Google Scholar]
- [27].Schaefer J, Stejskal F, High-Resolution 13C NMR of Solid Polymers, in: Top. Carbon-13 NMR Spectrosc., Levy GC, Ed., 1979: pp. 283–324. [Google Scholar]
- [28].Dastmalchi K, Kallash L, Wang I, Phan VC, Huang W, Serra O, Stark RE, Defensive Armor of Potato Tubers: Nonpolar Metabolite Profiling, Antioxidant Assessment, and Solid-State NMR Compositional Analysis of Suberin-Enriched Wound-Healing Tissues, J. Agric. Food Chem 63 (2015) 6810–6822. 10.1021/acs.jafc.5b03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Takegoshi K, Nakamura S, Terao T, 13C–1H dipolar-assisted rotational resonance in magic-angle spinning NMR, Chem. Phys. Lett 344 (2001) 631–637. 10.1016/S0009-2614(01)00791-6. [DOI] [Google Scholar]
- [30].Morcombe CR, Gaponenko V, Byrd RA, Zilm KW, Diluting Abundant Spins by Isotope Edited Radio Frequency Field Assisted Diffusion, J. Am. Chem. Soc 126 (2004) 7196–7197. 10.1021/ja047919t. [DOI] [PubMed] [Google Scholar]
- [31].Lesage A, Auger C, Caldarelli S, Emsley L, Determination of Through-Bond Carbon-Carbon Connectivities in Solid-State NMR Using the INADEQUATE Experiment, J. Am. Chem. Soc 119 (1997) 7867–7868. 10.1021/ja971089k. [DOI] [Google Scholar]
- [32].Lesage A, Bardet M, Emsley L, Through-Bond Carbon-Carbon Connectivities in Disordered Solids by NMR, J. Am. Chem. Soc 121 (1999) 10987–10993. 10.1021/ja992272b. [DOI] [Google Scholar]
- [33].Zlotnik-Mazori T, Stark RE, Nuclear magnetic resonance studies of cutin, an insoluble plant polyester, Macromolecules. 21 (1988) 2412–2417. 10.1021/ma00186a019. [DOI] [Google Scholar]
- [34].Garbow JR, Stark RE, Nuclear magnetic resonance relaxation studies of plant polyester dynamics. 1. Cutin from limes, Macromolecules. 23 (1990) 2814–2819. 10.1021/ma00212a037. [DOI] [Google Scholar]
- [35].Chatterjee S, Matas AJ, Isaacson T, Kehlet C, Rose JKC, Stark RE, Solid-State 13C NMR Delineates the Architectural Design of Biopolymers in Native and Genetically Altered Tomato Fruit Cuticles, Biomacromolecules. 17 (2016) 215–224. 10.1021/acs.biomac.5b01321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chatterjee S, Prados-Rosales R, Tan S, Phan VC, Chrissian C, Itin B, Wang H, Khajo A, Magliozzo RS, Casadevall A, Stark RE, The melanization road more traveled by: Precursor substrate effects on melanin synthesis in cell-free and fungal cell systems, J. Biol. Chem 293 (2018) 20157–20168. 10.1074/jbc.RA118.005791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jin L, Cai Q, Huang W, Dastmalchi K, Rigau J, Molinas M, Figueras M, Serra O, Stark RE, Potato native and wound periderms are differently affected by down-regulation of FHT, a suberin feruloyl transferase, Phytochemistry. 147 (2018) 30–48. 10.1016/j.phytochem.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Johnson RL, Schmidt-Rohr K, Quantitative solid-state 13C NMR with signal enhancement by multiple cross polarization, J. Magn. Reson 239 (2014) 44–49. 10.1016/j.jmr.2013.11.009. [DOI] [PubMed] [Google Scholar]
- [39].Duan P, Schmidt-Rohr K, Composite-pulse and partially dipolar dephased multiCP for improved quantitative solid-state 13C NMR, J. Magn. Reson 285 (2017) 68–78. 10.1016/j.jmr.2017.10.010. [DOI] [PubMed] [Google Scholar]
- [40].Huang W, Serra O, Dastmalchi K, Jin L, Yang L, Stark RE, Comprehensive MS and Solid-State NMR Metabolomic Profiling Reveals Molecular Variations in Native Periderms from Four Solanum tuberosum Potato Cultivars, J. Agric. Food Chem 65 (2017) 2258–2274. 10.1021/acs.jafc.6b05179. [DOI] [PubMed] [Google Scholar]
- [41].Fayon F, Massiot D, Levitt MH, Titman JJ, Gregory DH, Duma L, Emsley L, Brown SP, Through-space contributions to two-dimensional double-quantum J correlation NMR spectra of magic-angle-spinning solids, J. Chem. Phys 122 (2005) 194313 10.1063/1.1898219. [DOI] [PubMed] [Google Scholar]
- [42].Lee D, Struppe J, Elliott DW, Mueller LJ, Titman JJ, Sensitive absorptive refocused scalar correlation NMR spectroscopy in solids, Phys. Chem. Chem. Phys 11 (2009) 3547 10.1039/b818867j. [DOI] [PubMed] [Google Scholar]
- [43].Lewandowski JR, De Paëpe G, Griffin RG, Proton Assisted Insensitive Nuclei Cross Polarization, J. Am. Chem. Soc 129 (2007) 728–729. 10.1021/ja0650394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Simmons TJ, Mortimer JC, Bernardinelli OD, Pöppler A-C, Brown SP, deAzevedo ER, Dupree R, Dupree P, Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR, Nat. Commun 7 (2016) 1–9. 10.1038/ncomms13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Free SJ, Fungal Cell Wall Organization and Biosynthesis, in: Adv. Genet., Elsevier, 2013: pp. 33–82. 10.1016/B978-0-12-407677-8.00002-6. [DOI] [PubMed] [Google Scholar]
- [46].Chatterjee S, Prados-Rosales R, Itin B, Casadevall A, Stark RE, Solid-state NMR Reveals the Carbon-based Molecular Architecture of Cryptococcus neoformans Fungal Eumelanins in the Cell Wall, J. Biol. Chem 290 (2015) 13779–13790. 10.1074/jbc.M114.618389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gullion T, Schaefer J, Rotational-echo double-resonance NMR, J. Magn. Reson. 1969 81 (1989) 196–200. 10.1016/0022-2364(89)90280-1. [DOI] [PubMed] [Google Scholar]
- [48].Jaroniec CP, Filip C, Griffin RG, 3D TEDOR NMR Experiments for the Simultaneous Measurement of Multiple Carbon-Nitrogen Distances in Uniformly 13C,15N-Labeled Solids, J. Am. Chem. Soc 124 (2002) 10728–10742. 10.1021/ja026385y. [DOI] [PubMed] [Google Scholar]
- [49].Schaefer J, REDOR and TEDOR, in: Harris RK (Ed.), Encycl. Magn. Reson, John Wiley & Sons, Ltd, Chichester, UK, 2007: p. emrstm0448 10.1002/9780470034590.emrstm0448. [DOI] [Google Scholar]
- [50].Hing AW, Vega S, Schaefer J, Transferred-echo double-resonance NMR, J. Magn. Reson. 1969. 96 (1992) 205–209. 10.1016/0022-2364(92)90305-Q. [DOI] [Google Scholar]