

Abstract
Per- and polyfluoroalkyl substances (PFAS) are ubiquitous drinking water contaminants of concern due to mounting evidence implicating adverse health outcomes associated with exposure, including reduced kidney function, metabolic syndrome, thyroid disruption, and adverse pregnancy outcomes. PFAS have been produced in the U.S. since the 1940s and now encompass a growing chemical family comprised of diverse chemical moieties, yet the toxicological effects have been studied for relatively few compounds. Critically, exposures to some PFAS in utero are associated with adverse outcomes for both mother and offspring, such as hypertensive disorders of pregnancy (HDP), including preeclampsia, and low birth weight. Given the relationship between HDP, placental dysfunction, adverse health outcomes, and increased risk for chronic diseases in adulthood, the role of both developmental and lifelong exposure to PFAS likely contributes to disease risk in complex ways. Here, evidence for the role of some PFAS in disrupted thyroid function, kidney disease, and metabolic syndrome is synthesized with an emphasis on the placenta as a critical yet understudied target of PFAS and programming agent of adult disease. Future research efforts must continue to fill the knowledge gap between placental susceptibility to environmental exposures like PFAS, subsequent perinatal health risks for both mother and child, and latent health effects in adult offspring.
1. Introduction
Per- and polyfluoroalkyl substances (PFAS) comprise a diverse family of compounds used in a wide variety of industrial processes and the production of consumer goods (ITRC 2020). Since their genesis in the 1940s, PFAS have been detected in the ambient environment, wildlife, and human serum around the globe (Calafat et al. 2007; Domingo and Nadal 2019; Hanssen et al. 2013; Olsen et al. 2017). The environmentally and biologically persistent qualities of PFAS have raised concerns, further heightened by mounting scientific and clinical evidence associating PFAS exposure to multiple adverse health outcomes across all life stages.
1.1. Physicochemical Properties and Characterization of PFAS
Naming conventions for PFAS have evolved over time due in part to the increasing complexity and number of unique congeners, leading to inconsistencies across the scientific literature. For example, the term perfluorinated chemical, or PFC, has been widely used in the scientific literature, but it is inaccurate as it does not describe polyfluorinated substances, which are an important component of the PFAS family (ITRC 2020). Additionally, many PFAS exist in multiple ionic states (e.g. acids, cations, anions), with different names (e.g. perfluorooctane sulfonic acid and perfluorooctane sulfonate), and different physicochemical properties. Consistent and unified naming conventions for PFAS would provide clarity, consistency, and improved scientific communication.
PFAS contain 1 or more carbon atoms with fluorine atoms in place of hydrogen atoms, such that the compound contains the moiety CnF2n+1-R, where R represents additional functional groups (e.g. sulfonate, carboxylic acid; Figure 1) (ITRC 2020). The per- or polyfluoroalkyl moiety is highly chemically and thermally stable with extremely strong carbon-fluorine bonds, and it exhibits both lipophilic and hydrophilic properties (ITRC 2020). Together, these physical and chemical properties make PFAS ideal as surfactants and surface protection products.
Figure 1. Basic structural features of a perfluoroalkyl substance (PFAS).
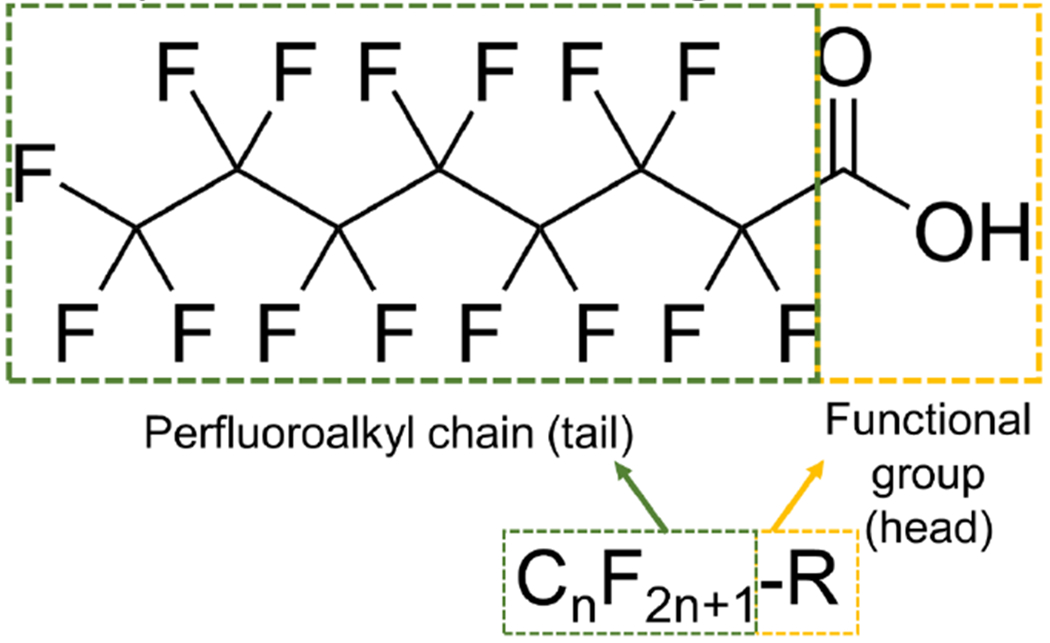
The compound perfluorooctanoic acid (PFOA) is shown here as an example. The perfluoroalkyl chain (tail) is indicated by a dashed green outline, while the functional group (head) is indicated by a dashed yellow outline. Legacy PFAS share these structural features while replacement, or “alternative chemistry” PFAS, contain substitutions along the carbon tail.
PFAS can be broadly divided into polymers and non-polymers. PFAS polymers are thought to pose less of a risk to human and ecological health than some non-polymers and include fluoropolymers, polymeric perfluoropolyethers, and side-chain fluorinated polymers (ITRC 2020). However, the production of PFAS polymers contributes to the presence of non-polymers in the environment through: (1) release of non-polymers as waste byproducts in the production of polymers, (2) degradation of polymers into non-polymers (e.g. PTFE forms PFOA and N-methyl perfluorooctane sulfonamide; ITRC 2020). Non-polymer PFAS can be further separated into perfluorinated (fully fluorinated) and polyfluorinated (partially fluorinated) chemicals.
PFAS are individually characterized by carbon chain length and side group structure as well as their history of use. “Legacy” PFAS include compounds with a longstanding history of use and/or long biologic/environmental persistence, whereas replacement and alternative chemistry PFAS are generally referred to as “alternative PFAS” or “replacement PFAS”. However, certain short-chain legacy PFAS are now substituted in place of longer chain compounds to comply with government-derived standards, hence such PFAS could be considered both a legacy and a replacement compound. For example, PFBS has been utilized as a substitute for PFOS in some manufacturing processes (Table 1 & Figure 2).
Table 1.
Common legacy per- and polyfluoroalkyl substances.
| Chemical name | CAS | Abbr | Family | # Carbons w/fluorine | Carbon chain length | Type |
|---|---|---|---|---|---|---|
| Perfluoorooctanoic acid | 335-67-1 | PFOA | PFCA | 7 | 8 | Legacy |
| Perfluorohexanoic acid | 307-24-4 | PFHxA | PFCA | 5 | 6 | Legacy |
| Perfluoropentanoic acid | 2706-90-3 | PFPeA | PFCA | 4 | 5 | Legacy |
| Perfluoroheptanoic acid | 375-85-9 | PFHpA | PFCA | 6 | 7 | Legacy |
| Perfluorononanoic acid | 375-95-1 | PFNA | PFCA | 8 | 9 | Legacy |
| Perfluorodecanoic acid | 335-76-2 | PFDA | PFCA | 9 | 10 | Legacy |
| Perfluorobutanoic acid | 375-22-4 | PFBA | PFCA | 3 | 4 | Legacy & replacement |
| Perfluorobutane sulfonic acid | 375-73-5 | PFBS | PFSA | 4 | 4 | Legacy & replacement |
| Perfluorohexane sulfonic acid | 355-46-4 | PFHxS | PFSA | 6 | 6 | Legacy |
| Perfluorooctane sulfonic acid | 1763-23-1 | PFOS | PFSA | 8 | 8 | Legacy |
| Perfluorooctane sulfonamide | 754-9-6 | PFOSA | PFSA | 8 | 8 | Legacy |
Abbr: Perfluorocarboxylic acid = PFCA, perfluorosulfonic acid or amide = PFSA
Figure 2. Structures of common legacy perfluoroalkyl substances (PFAS) and replacement PFAS.
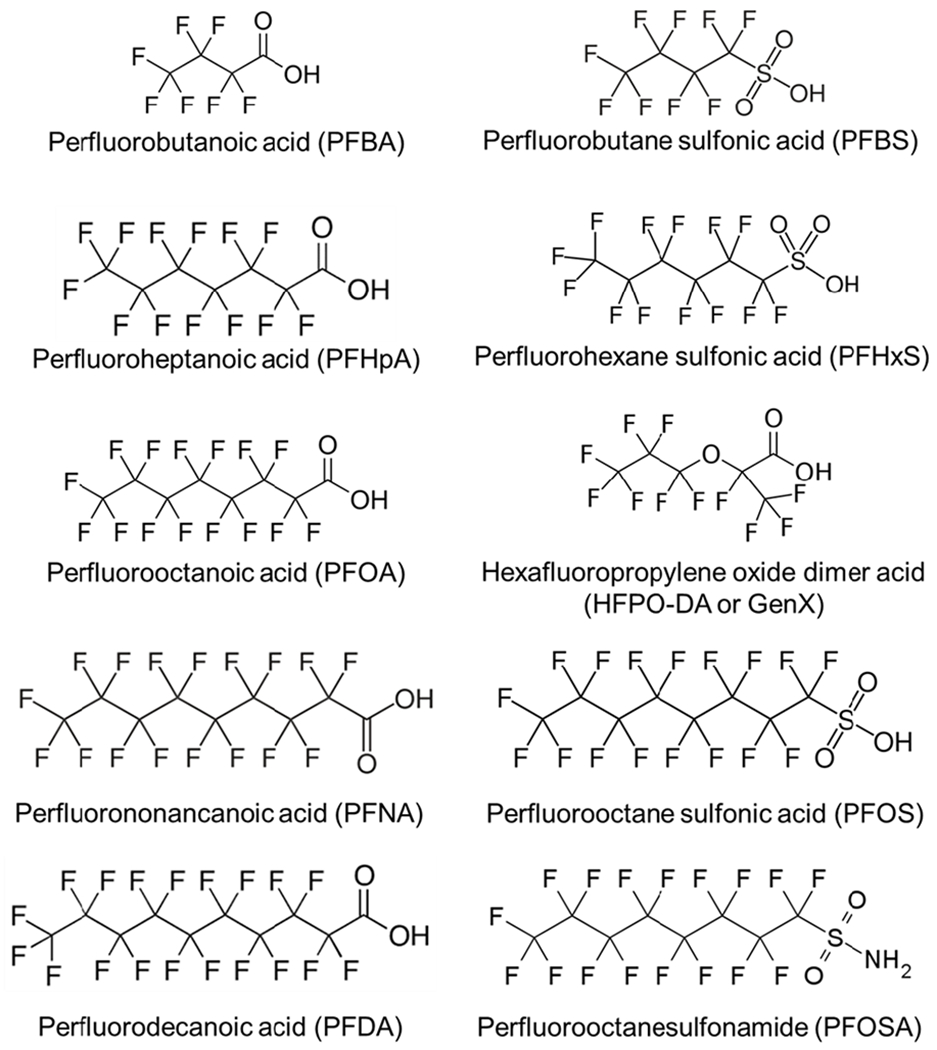
The ether substitution in the carbon tail of hexafluoropropylene oxide dimer acid (HFPO-DA, or GenX) is thought to favorably alter the toxicokinetic profile of the compound. Perfluorobutane sulfonic acid (PFBS) has classic structural features but has been selected as a replacement compound for longer-chain PFAS.
1.2. History of PFAS Usage in the U.S.
The earliest known PFAS compound in the U.S. was first synthesized as a fluoropolymer, called polytetrafluoroethylene (PTFE). Roy J. Plunkett is credited with the accidental discovery of PTFE in 1938 while employed by DuPont de Nemours, Inc. (Plunkett 1986). DuPont patented the discovery under the company Kinetic Chemicals in 1941 (Plunkett 1941) and registered the trademarked name, Teflon, in 1945 (Plunkett 1986). By 1948, DuPont was producing over 900 tons of PTFE per year at their facility in Parkersburg, West Virginia and another company, 3M, had begun production of their signature PFAS at their plant in Minnesota (Funderburg 2010).
Between the 1940s and 1950s, the 8-carbon perfluorooctane sulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) started to be used in the manufacturing of PTFE-based non-stick coatings. From the 1950s to the 1960s, PFOS and PFOA were also used to manufacture stain- and water-resistant products as well as protective coatings. By 1961, PTFE was applied to commercial cookware. From the 1960s to the 1970s, PFOS-based fire-fighting foam was manufactured, although it was later voluntarily phased out of Class A fire-fighting foams beginning in 2000 (US DOD 2000; 3M 2000).
Since their genesis in the mid-1900s, PFAS have since been incorporated into numerous other industrial and consumer products due to their excellent grease/water repellant properties, including: grease-resistant papers (e.g., fast food containers/wrappers, microwave popcorn bags, pizza boxes, candy wrappers); stain resistance either incorporated into or as spray-on coating applied to carpets and upholstery in home and vehicle products; water-resistant clothing and footwear, cleaning products, personal care products (e.g., shampoo, dental floss, cosmetics such as nail polish and eye makeup); paints, varnishes, ski wax and sealants; and Class B aqueous film forming foams (AFFF), which are used at airports and military facilities for firefighting and training exercises; and likely others (ATSDR 2019).
Now, in the 21st century, many consumer products and drinking water sources contain a mixture of PFAS. The specific mixture and volume of PFAS emitted into the environment varies based on the point source (Guelfo et al. 2019; Guelfo and Adamson 2018; Xu et al. 2016). For example, a water system near a military training area would likely have multiple species derived from AFFF present whereas a water system near a textile manufacturing plant would likely have a blend of PFAS precursor and legacy compounds present (e.g. both PFOSA, a PFOS precursor, and PFOS). Over time, the region-specific magnitudes and compositions of environmental contamination by PFAS has resulted in numerous U.S. states and communities with high levels of PFAS in their water, including states impacted by less well-studied alternative compounds [e.g. GenX and ADONA; for detailed reports see Hopkins et al. (2018) and Wang et al. (2013)].
1.4. Sources of PFAS Exposure
PFAS exposure is considered ubiquitous in the U.S., with PFOA and PFOS detectable in >90% of the population (Kato et al. 2011). Humans are exposed to PFAS through a variety of pathways, including through drinking water, air (both indoor and outdoor), diet, dust, through maternal to fetal transfer in utero, and through breastfeeding as neonates (Figure 3; Sunderland et al. 2019). For adults, diet and drinking water are the main sources of exposure, however this may vary depending on lifestyle, diet, proximity to point and nonpoint sources, and local drinking water contamination levels. Formula-fed infants are thought to be the most highly exposed members of the human population due to their high water intake to body weight ratio (Goeden et al. 2019; Goeden 2018). Drinking water may be the largest source of PFAS exposure for communities impacted by high levels of PFAS contamination.
Figure 3. Major sources of human exposure to per- and polyfluoroalkyl substances.
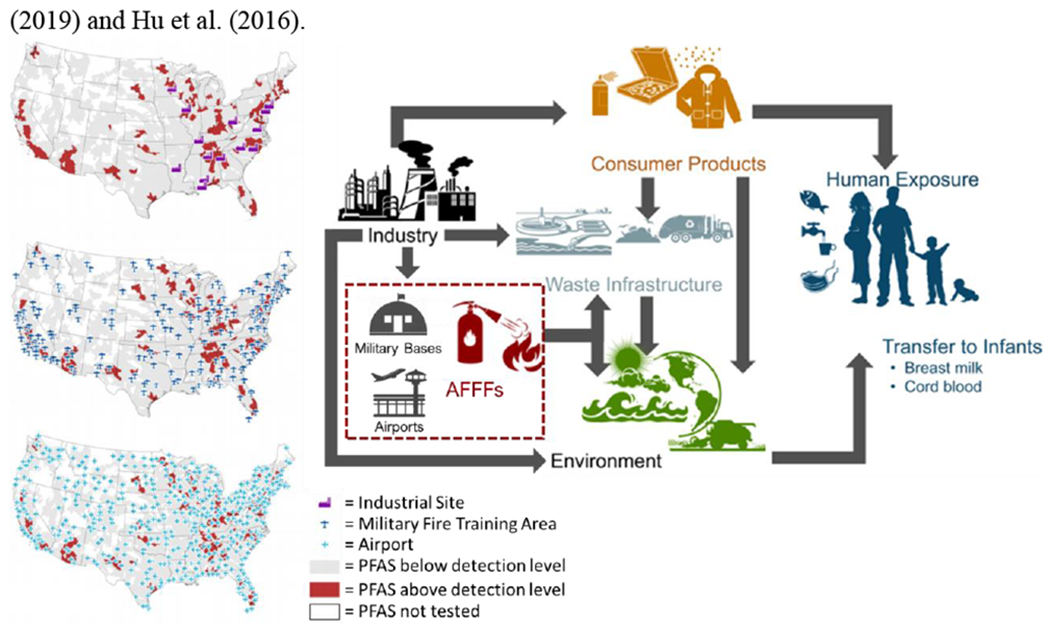
Humans are directly exposed to PFAS through the ambient environment (e.g. air), consumer products, house dust, drinking water, and diet. For developing humans, exposure can occur transplacentally in utero and through breastmilk. Environmental PFAS contamination is caused by waste and pollution generated by industrial complexes in the manufacturing or use of PFAS, including in the manufacturing of downstream products containing PFAS, such as aqueous filmforming foams (AFFFs). Environmental PFAS contamination is also caused by run-off of PFAS-containing AFFFs at military training bases and airports. Figure adapted from Sunderland et al. (2019) and Hu et al. (2016).
2. Adverse Health Effects Associated with PFAS
The desirable chemical properties of many PFAS paradoxically impart environmentally and biologically undesirable properties, including indefinite environmental persistence and long half lives in many living organisms, including humans. Unlike other common environmental pollutants, such as organochlorine pesticides, PFAS do not bioaccumulate in adipose tissue (Pérez et al. 2013). Some PFAS are structurally similar to natural fatty acids (Vanden Heuvel et al. 2006). Among the tissues evaluated, PFAS primarily accumulate in the serum, lungs, kidney, liver, and brain (Pérez et al. 2013). Human exposure to PFAS has been associated with adverse effects on the immune (NTP 2016), endocrine, metabolic, and reproductive systems (including fertility and pregnancy outcomes), and increased risk for cancer (ATSDR 2020). However, the weight of evidence supporting these associations varies by both outcome and specific PFAS examined.
2.1. History of PFAS-Related Human Health Concerns
Industrial usage of PFAS began in the 1940s, but exposure-related human health effects were not identified for another twenty years and were specific to occupationally exposed workers. These early concerns were largely contained within the industrial sphere for an additional forty years. Nearly six decades after PFAS began to be produced on an industrial scale, PFAS-related health concerns were raised by private citizens after the general public learned of connections between ecological observations and pollution potentially stemming from a nearby PFOA production site.
The first documented concerns regarding the toxicity of PFAS were mentioned in an internal memo at DuPont by Chief Toxicologist Dorothy Hood in 1961 (EWG 2019). By 1980, PFAS were measured in the serum of occupationally exposed workers and in 1981 concerns were raised internally at DuPont regarding birth defects in children born from occupationally exposed women (EWG 2019). Industrial and occupational exposure studies have since shown increased incidence of cancer including bladder (Alexander et al. 2003), kidney (Consonni et al. 2013), prostate (Gilliland and Mandel 1993; Lundin et al. 2009), and liver (Consonni et al. 2013), leukemia (Consonni et al. 2013), kidney disease (Steenland and Woskie 2012), and elevated cholesterol (Costa et al. 2009; Sakr et al. 2007).
Community-based exposure concerns in the U.S. were first raised in 1998 by Wilbur Tennant, a cattle farmer whose land was downstream from and bordered a landfill used by the DuPont Washington Works plant near Parkersburg, WV (Rich 2016). Tennant enlisted legal counsel from Rob Bilott after documenting unusual phenomenon, such as foamy, discolored water in the creek from which Tennant’s cattle drank, atypical behavior of the cattle, and a wide array of physical abnormalities in the cattle including black teeth, skin lesions and tumors, discolored organs, malformed hooves, and calves born with profound birth defects (Rich 2016). One hundred and fifty-three of Tennant’s original herd of two hundred cattle died, and Tennant believed the pollution dumped by DuPont into the landfill upstream of his cattle farm was the cause (Rich 2016). What began as a legal case on behalf of a private citizen ultimately led to a larger class-action lawsuit filed by Bilott on behalf of over 70,000 citizens in the Mid-Ohio Valley whose drinking water sources were affected by the contamination stemming from DuPont’s plant (Rich 2016).
The class-action lawsuit resulted in a settlement that funded a biomonitoring effort of unprecedented size and scale, called the C8 Health Study (Frisbee et al. 2009). The results of the C8 Health Study identified a probable link between exposure to PFOA and increased cholesterol, kidney cancer, testicular cancer, ulcerative colitis, thyroid disease, and pregnancy-induced hypertension (C8 Science Panel). Since the genesis of the C8 Health Study, numerous epidemiological studies of the adverse health effects associated with PFAS exposure have been conducted across the globe, including many other regions in the U.S., Europe, and China.
Multiple organizations and regulatory agencies in the U.S. have also generated statements based on systematic reviews of the available literature regarding the most well-supported human health effects associated with PFAS. The Center for Disease Control (CDC)/Agency for Toxic Substances and Disease Registry (ATSDR) states that PFAS might increase the risk of cancer, affect the immune system, increase cholesterol levels, act as endocrine disruptors, reduce female fertility, and adversely impact infant/early childhood growth and development (ATSDR 2019). The U.S. Environmental Protection Agency (EPA) states that elevated cholesterol in adults is the most consistent health effect associated with PFAS, but less consistent data suggest PFAS might be associated with low infant birth weight (Johnson et al. 2014; Koustas et al. 2014; Lam et al. 2014), effects on the immune system, increased risk for cancer (specific to PFOA), and disrupted thyroid hormones (specific to PFOS) (822-R-16-005 US EPA). The World Health Organization (WHO)/International Agency for Research on Cancer (IARC) states that PFOA is possibly carcinogenic to humans (IARC 2016). The National Toxicology Program (NTP) states that PFAS might have effects on metabolism, pregnancy, neurodevelopment, and the immune system, with both PFOA and PFOS presumed to be immune hazards (NTP 2016). Such statements only address the possible health effects associated with legacy PFAS as there are little to no data on health outcomes associated with replacement PFAS.
Although many biological systems are adversely impacted by PFAS, this review focuses primarily on PFAS-associated, placenta-based adverse pregnancy outcomes and how they may increase susceptibility for latent health effects including metabolic dyshomeostasis, thyroid disruption, and kidney effects (Figure 4).
Figure 4. Summary of adverse health outcomes associated with PFAS exposure, the target tissue implicated by the outcomes, and hypothesized mechanism(s) of PFAS toxicity.
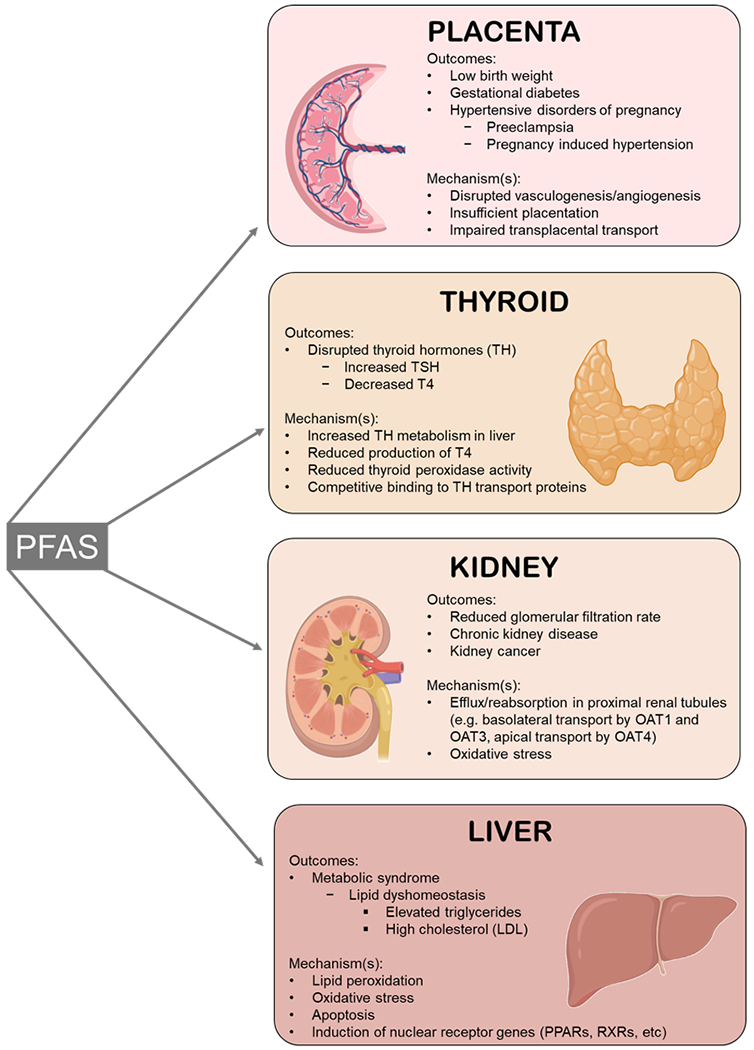
Image created using BioRender.
2.2. Challenges of Studying PFAS-Related Human Health Effects
There are many challenges in studying PFAS-related human health effects, including ones specific to studying PFAS exposure and effects in the human population as well as those that are specific to the study of PFAS toxicology in animal models.
First, humans are exposed to PFAS as complex mixtures that differ based on the source of exposure (e.g. near a military base or an industrial site, consumer product use) and differ temporally within the same geographic location (e.g. an industrial site phasing out PFOA and PFOS and utilizing alternative PFAS instead; seasonal patterns in rainfall/drought affecting the extent to which PFAS in water systems are diluted or concentrated). States have authority to develop statewide regulations on consumer product labels and contents, which also influences the extent to which individuals are potentially exposed to PFAS (e.g. consumers can choose to avoid PFAS-containing products under regulations such as Proposition 65). Thus, different individuals and populations are exposed to different levels and mixtures of PFAS. Little is known regarding the potential synergistic effects of PFAS mixtures. Human exposure to PFAS also varies by demographic factors, and they likely differ by individual PFAS congener within a given population. Lifelong exposure, combined with variable exposure to temporally stochastic mixtures and congener-specific demographic factors, presents a complex set of challenges to understanding the relationship between PFAS exposure and adverse health outcomes.
A second challenge in epidemiologic studies of PFAS-related health effects is the lack of an unexposed control population. All humans born after the year 1950, when PFAS were first introduced at the industrial scale, have potentially been exposed to PFAS their entire lives, including in utero. This precludes the ability to design and conduct cohort studies with a no-exposure comparison group. Instead, such cohort studies of PFAS must make comparisons between low and high exposure groups to estimate disease risk based on exposure status.
Third, it is possible that PFAS-related adverse health outcomes observed in adulthood are latent manifestations of perturbations during sensitive periods of development (Barker 2004). It is also possible that some PFAS-related adverse health outcomes observed in adulthood are due to chronic, lifelong exposure. In order to address these hypotheses, a population of humans without PFAS exposure during early development would be required. However, such a population only existed prior to 1950, making such a study extremely difficult if not impossible.
Fourth, while experimental toxicology studies provide invaluable information, the elimination kinetics, sensitive endpoints, and biological mechanisms of action in target tissue(s) demonstrate interspecies variation between experimental models (rats vs mice) as well as compared to humans (e.g. serum half-life elimination of PFHxS in mice is ~25-30 days, ~2-7 days in rats, and 5-8 years in humans; Fenton et al. 2020). Many of the interspecies differences regarding toxicological effects and toxicokinetics of PFAS are not fully understood and are further complicated by sex differences in elimination rates (e.g. the human serum half-life of GenX has yet to be fully characterized but it is 3 hours in male rats, 8 hours in female rats, and 18-20 hours in male or female mice; Fenton et al. 2020).
2.3. The Placenta as a Target of PFAS and Critical Driver of Adverse Pregnancy Outcomes
The placenta is a critical organ that exists exclusively during pregnancy and serves as a conduit between mother and developing offspring, mediating the maternal-fetal transfer of nutrients, oxygen, blood, waste, and xenobiotics. The healthy development and function of the placenta is paramount to not only the healthy development of the fetus, but also the health of the mother. Indeed, many adverse pregnancy outcomes are due to placental insufficiency (PI), a condition in which the functional capacity of the placenta is limited or deteriorates, resulting in reduced transplacental transfer of oxygen and nutrients to the fetus (Gagnon 2003). Previous studies have demonstrated that the placenta is vulnerable to environmental insults (Laine et al. 2015; Leclerc et al. 2014; Pedersen et al. 2014).
Adverse pregnancy outcomes associated with PFAS exposure in humans include increased time to pregnancy (Bach et al. 2016; Lum et al. 2017), hypertensive disorders of pregnancy (which includes pregnancy induced hypertension [PIH] (Huang et al. 2019; Savitz et al. 2012b), and preeclampsia [PE] (Stein et al. 2009; Wikström et al. 2019)), gestational diabetes (GD) (Matilla-Santander et al. 2017; Zhang et al. 2015), excess gestational weight gain (Ashley-Martin et al. 2016), and low birth weight of the infant (Johnson et al. 2014; Li et al. 2017; Marks et al. 2019; Meng et al. 2018; Sagiv et al. 2018; Starling et al. 2017; Wikstrom et al. 2019; Xu et al. 2019). Independent of PFAS exposure, deficiencies in placental development and/or function are involved in the etiology of many of these adverse pregnancy outcomes (Hutcheon et al. 2012; Risnes et al. 2009; Thornburg et al. 2010). Thus, it is possible the placenta is a target of PFAS such that PFAS-induced reductions in placental function significantly contribute to adverse pregnancy and birth outcomes.
The placenta is a plausible target of PFAS as it is exposed to PFAS via maternal circulation, shares biologic features in common with other known target tissues of PFAS, such as kidney and liver, and plays a role in the etiology of pregnancy disorders related to PFAS exposure. It is well documented in humans and animal models that PFAS readily pass from maternal serum to the developing embryo via the placenta (Chen et al. 2017a; Chen et al. 2017b; Wang et al. 2019; Yang et al. 2016a; Yang et al. 2016b) and that PFOA transplacentally transfers to the mouse offspring (Fenton et al. 2009). During early stages of development, the placenta functions as the liver, lungs, and kidneys for the embryo; the placenta is responsible for xenobiotic metabolism, supplying oxygen to the fetus, and eliminating waste from embryonic/fetal circulation. Placental trophoblasts express multiple types of ATP-binding cassette (ABC) and organic anion transporter (OAT) proteins in order to shuttle nutrients, xenobiotics, and waste between the maternal and fetal compartments. Many of the transporter proteins expressed in placental trophoblasts are also expressed in liver or kidney, where they excrete substrates into the bile or urine, respectively (Joshi et al. 2016). An ex vivo study of human placenta showed the involvement of OAT4 in the placental transport of PFOA and PFOS (Kummu et al. 2015).
There is growing evidence of the mechanisms of PFAS toxicity towards the placenta. A study in mice showed dose-dependent necrotic changes in placenta after gestational exposure to 10 mg/kg/day and 25 mg/kg/day PFOA (Suh et al. 2011). In rats, gestational exposure to 20 mg/kg/day PFOS reduced fetal and placental weight, inhibited placental 11β-hydroxysteroid dehydrogenase 2 activity, and down-regulated 45 gene corresponding to growth factors and hormones, transporters, and signal transduces, among others (Li et al. 2016). More recently, it was demonstrated that gestational exposure to either PFOA or GenX induced placental lesions in CD-1 mice, with disrupted placental weights and fetal:placental weight ratios and histopathological changes in the placental labyrinth, including congestion and atrophy (Blake et al. 2020).
A study of ex vivo term placental cytotrophoblasts exposed to PFOS demonstrated concentration-dependent suppression of estradiol, human chorionic gonadotropin, and progesterone, reduced cell viability, and dysregulation of apoptotic proteins in favor of proapoptosis (Zhang et al. 2015). In vitro studies of JEG-3 human placental trophoblasts showed PFOS, PFOA, and PFBS inhibit aromatase (CYP19) activity and an exposure to a mixture of eight different PFAS caused an increase in certain lipid classes (Gorrochategui et al. 2014). In the JEG-3 human placental trophoblast line, exposure to PFOA, PFOS, or GenX altered gene expression profiles towards an anti-proliferative and pro-apoptotic state after exposure to 10 μg/mL in cell culture media (Bangma et al. 2020). However, JEG-3 internal cellular accumulation of GenX was far lower than that of PFOS or PFOA, which suggests a higher sensitivity of placental cells towards GenX as compared to PFOS or PFOA (Bangma et al. 2020). These findings suggest GenX, PFOA, and PFOS may adversely affect placental development and function via disrupted balance of apoptosis and proliferation of the trophoblasts.
Human-derived first trimester trophoblast HTR-8/SVneo cells have been used to demonstrate that PFOS, PFOA, and GenX disrupt inflammatory signaling, decrease trophoblast migration, and decrease trophoblast invasion (GenX only), which indicates a potential mechanism through which PFAS exposure could disrupt placental development and function (Szilagyi et al. 2020). Another study utilizing HTR-8/SVneo trophoblasts similarly showed concentration-dependent decreased migration and invasion after exposure to PFBS, in addition to altered expression of genes implicated in PE (Marinello et al. in press). Additionally, PFOS exposure in HTR-8/SVneo trophoblasts increased levels of miR29-b, a microRNA associated with PE, altered downstream epigenetic processes including global DNA hypomethylation and hyperacetylation of protein, and increased reactive oxidative species production, suggesting a mechanism for increased placental oxidative stress due to PFAS exposure which could ultimately lead to preeclampsia (Sonkar et al. 2019).
Taken together, these studies suggest PFAS induce adverse outcomes in the placenta via mechanisms involving endocrine/lipid/sterol disruption, oxidative stress, disrupted balance of pro- and anti-apoptotic signaling, impaired trophoblast function, and epigenetic alterations. It is possible that mechanisms of PFAS toxicity towards the placenta are compound-specific and may involve numerous overlapping aspects of biology. Improper development or function of the placenta is associated with an array of adverse pregnancy outcomes (including hypertensive disorders of pregnancy), many of which are also associated with maternal exposure to PFAS. It is possible that adverse pregnancy outcomes associated with PFAS exposure are mediated in part by the negative impact of PFAS on the development and function of the placenta. Understanding the risk of environmental insults on the health and development of the placenta is critical for protecting both short and long-term maternal and offspring health outcomes because the placenta is a driver of latent disease risk for both postpartum women and their offspring.
2.3.1. PFAS Exposure, Hypertensive Disorders of Pregnancy, and the Placenta
Hypertensive disorders of pregnancy (HDP) are a family of disorders that include PE, eclampsia, HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome, and PIH, all of which are related to deficiencies in placental development and/or function (Pijnenborg et al. 1991). This family of disorders not only endangers the health of both the mother and developing fetus during pregnancy, but also increases the risk of post-pregnancy hypertension, heart disease, and stroke in affected women as well as increased risk for adverse cardiometabolic outcomes in adult offspring (Pinheiro et al. 2016).
During normal pregnancy, at 13 weeks gestation the placental cytotrophoblasts begin to invade the maternal uterine wall and remodel the maternal spiral arteries to establish blood flow to the growing embryo (Shah 2020). This process of vasculogenesis and angiogenesis by placental cytotrophoblasts remodels the maternal spiral arteries to become low-resistance, low-capacitance vessels so that blood flow and sufficient nutrients and oxygen are supplied to the growing embryo (Shah 2020). This carefully coordinated process requires an appropriate balance of proangiogenic and antiangiogenic signals (Shah 2020). Disruption of this process can lead to abnormal placental vascularization, resulting in suboptimal pregnancy hemodynamics which can give rise to HDP as well as impaired fetal growth (Shah 2020).
There is growing evidence from human studies implicating maternal PFAS exposure in the development of HDP. Multiple epidemiologic studies have found positive associations between maternal exposure to PFAS and PE (Huang et al. 2019; Savitz et al. 2012a; Savitz et al. 2012b; Stein et al. 2009; Wikström et al. 2019a) as well as PFAS and PIH (C8 Science Panel; Darrow et al. 2013; Holtcamp 2012). It is possible that reduced or impaired placental function as a consequence of PFAS exposure results in or significantly contributes to the development of HDP. Indeed, a recent study in mice showed increased placenta weight, reduced fetal:placental weight ratios, and placental lesions following gestational exposure to PFOA or GenX, suggesting a role for placental toxicity by PFAS (Blake et al. 2020).
Furthermore, it is well documented that the placenta itself is strongly associated with cardiometabolic disease risk in adults. Indeed, the size of the placenta is strongly associated with both perinatal and adult outcomes. In a study of 87,600 Canadian singleton births, fetuses with abnormally small placentas were at increased risk for stillbirth (OR 2.0, 95% CI 1.4-2.6) while fetuses with abnormally large placentas were at increased risk for adverse neonatal outcomes, including seizures, respiratory morbidity, and low Apgar score (odds ratio [OR] 1.4, 95% confidence interval [CI] 1.2-1.7) (Hutcheon et al. 2012). Bigger is not necessarily better; it has been hypothesized that an enlarged placenta is a biomarker for impaired nutrient supply to the fetus. This reduced capacity could be explained by restricted or impaired blood flow through the placenta. It is possible that impaired blood flow, which would in turn disrupt the normal flow of nutrients, oxygen, and waste between maternal and fetal compartments, during critical windows of in utero development may result in long-term adverse function of the cardiometabolic system. A U.S. cohort study of nearly 30,000 births showed that large placenta relative to birth weight, but not birth weight itself, is associated with high blood pressure in childhood (Hemachandra et al. 2006). In a study of 31,307 Norwegian adults, disproportionately large placentas at birth were associated with increased risk of death associated with cardiovascular disease (CVD), and this association remained positive when considering placenta weight alone (Risnes et al 2009).
While mounting epidemiologic evidence suggests PFAS exposure as a risk factor for the development of HDP, a specific mechanism has yet to be validated in experimental settings. Such experimental systems are critical in addressing the hypothesis that maternal exposure to PFAS increases the risk of developing HDP via disruption of the placenta. Due to the complex and multifactorial nature of HDPs, and interspecies differences in maternal spiral artery structure and formation, current animal models are unable to adequately recapitulate the multiple manifestations of this complex group of disorders (Cushen and Goulopoulou 2017; Marshall et al. 2017). Indeed, there is no “standard” or preferred animal model of HDP, and it is unlikely that such an animal model will ever exist (Cushen and Goulopoulou 2017; Marshall et al. 2017). Thus, the development of sensitive laboratory tools for assessing clinically relevant biomarkers of HDPs in animal models is a necessary next step towards determining mechanisms of PFAS toxicity.
2.3.2. PFAS Exposure and Low Birth Weight
Low birth weight is the most consistently reported adverse pregnancy outcome associated with gestational exposure to PFAS in human epidemiologic and animal studies. It is also well established in the literature that placental insufficiency contributes to the etiology of low birth weight (Audette and Kingdom 2018; Cuffe et al. 2017; Henriksen and Clausen 2002). What remains unaddressed by the current body of literature is the relationship between gestational exposure to PFAS, placental function, and birth weight.
In 2014, a three-paper series performed systematic reviews of the existing human and animal literature on the relationship between in utero exposure to PFOA and low birth weight (Johnson et al. 2014; Koustas et al. 2014; Lam et al. 2014). After gathering primary data sets and detailed descriptions of study designs from numerous research teams, the authors were able to calculate estimates corresponding to the predicted reduction in birth weight in humans [−18.9 g birth weight per 1 ng PFOA/mL maternal serum, 95% CI: −29.8, −7.9; (Johnson et al. 2014)] and in mice [−23.0 mg birth weight per 1 mg PFOA/kg maternal body weight/day, 95% CI: −29.0, −16.0; (Koustas et al. 2014)]. The authors concluded from their systematic analyses of the literature that PFOA is indeed associated with reduced birth weight in both humans and mice. The effect estimate corresponding to birth weight in mice after gestational exposure to 1 mg PFOA/kg maternal body weight/day generated by Koustas et al. (2014) was consistent with findings in a recent study that measured near-term fetal body weights after gestational exposure to 1 mg PFOA/kg maternal body weight/day (−28 mg fetal weight at embryonic day 17.5, 95% CI: −114, 59; Blake et al. 2020).
Since the publication of these systematic reviews in 2014, the human epidemiologic literature on the relationship between PFAS and birth weight has expanded to include understudied PFAS (e.g. PFNA, PFDA, PFHxS). In a recent report, an analysis of 3,535 mother-infant pairs in the Danish National Birth Cohort examined six different PFAS and reported increased risk for preterm birth associated with maternal serum levels of PFOA, PFOS, PFNA, PFDA, and perfluoroheptane sulfonate (Meng et al. 2018). Low birth weight risk was also elevated, but estimates were less precise in this analysis (Meng et al. 2018). In a study of 1,533 Swedish mother-infant pairs, increased maternal exposure to PFOS, PFOA, PFNA, PFDA, and perfluorooundecanoic acid were associated with lower birth weight and size, but associations were only significant in girls (Wikstrom et al. 2019). Conversely, a study of 457 British motherson dyads showed inverse associations between maternal PFOS and measures of both birth weight and size (Marks et al. 2019), which had previously been shown in mother-daughter dyads in the same cohort (Maisonet et al. 2012). A study of 1,645 mother-infant pairs in Massachusetts showed that PFOS and PFNA were weakly inversely associated with birth weight and positively associated with higher odds of preterm birth (Sagiv et al. 2018). In another U.S. cohort, maternal serum PFOA and PFNA was inversely associated with birth weight in an analysis of 628 mother-infant pairs (Starling et al. 2017). A smaller study of 98 Chinese mother-infant pairs also found PFOS was negatively associated with birth weight, and that both PFOS and PFHxS were negatively associated with birth size (small for gestational age, SGA) (Xu et al. 2019). Taken together, these epidemiological studies further underscore the potential for PFAS other than PFOA to adversely affect fetal growth in utero.
There are limited animal studies supporting the association between PFAS, other than PFOA and PFOS, and reduced birth weight. Studies of both mice (Chang et al. 2018) and rats (Ramhoj et al. 2018) have shown reduced offspring body weight after developmental exposure to PFHxS. In rats, developmental exposure to perfluoroundecanoic acid has been shown to reduce offspring weight (Takahashi et al. 2014). Previous animal studies of the developmental effects of PFNA have reported adverse neonatal outcomes but did not report effects on birth weight (Das et al. 2015; Wolf et al. 2010). Similarly, a previous study of the developmental toxicity of PFDA in mice found no reductions in birth weight (Harris and Birnbaum 1989). Studies in mice have demonstrated a modest effect of prenatal exposure to GenX on near-term (Blake et al. 2020) and early postnatal offspring body weights, with effects becoming more significant throughout lactation (DuPont-18405-1037).
The association between PFAS exposure and low birth weight is well supported in humans, as is the causal link between placental insufficiency and low birth weight. This, in combination with the biologically plausible proposed mechanisms of PFAS toxicity towards the placenta, further underscores the need to test the hypothesis that the PFAS-mediated placental effects are a critical driver of adverse pregnancy and birth outcomes.
2.4. PFAS Exposure and Metabolic Effects
Metabolic syndrome (also referred to as cardiometabolic syndrome) describes a group of disorders that affects approximately one third of the U.S. adult population and includes obesity, dyslipidemia, elevated blood pressure, and impaired glucose tolerance (Eckel et al. 2005; Ervin 2009). The liver is a central hub in the regulation of pathways controlling systemic metabolic homeostasis, and liver dysfunction is a major component of metabolic syndrome (D’Amore et al. 2014). While the liver regulates metabolic homeostasis, lifelong metabolic health is influenced by placental health and function during critical periods of in utero fetal programming (Nugent & Blake, 2016; Rinaudo & Wang, 2012). Therefore, environmental exposures impacting placental health and function, such as PFAS, may prime the system for increased susceptibility to metabolic syndrome, which could be further exacerbated by continued PFAS exposure across the lifespan.
A recent review of the literature examined 69 epidemiological studies evaluating the association between PFAS exposure and a variety of metabolic outcomes, including lipid homeostasis, diabetes, overweight/obesity, and cardiovascular disease (Sunderland et al. 2019). Across the human literature, Sunderland et al. (2019) reported relatively consistent and modest positive associations between PFAS exposure and serum lipids, such as total cholesterol and triglycerides. Although associations were less consistent across studies, Sunderland et al. (2019) also reviewed epidemiologic studies examining the adverse effect of PFAS exposure on insulin resistance, diabetes, hypertension, vascular disease, and stroke. Across these health outcomes, adverse effects were most consistently reported in association with exposure to PFOA (Sunderland et al. 2019). The effects of PFAS on metabolism of developmentally exposed offspring is the topic covered more in depth by Cope et al. (this issue).
More recently, additional studies of humans have explored the association between PFAS exposure and metabolic syndrome. Christensen et al. (2019) leveraged National Health and Nutrition Examination Survey (NHANES) data sets to investigate the relationship between PFAS exposure and metabolic syndrome in the general U.S. population between years 2007-2014 and found PFNA was consistently associated with increased risk of metabolic syndrome, while the highest levels of PFHxS exposure were associated with elevated triglycerides (Christensen et al. 2019). This study suggests that PFAS other than PFOA and PFOS may contribute to adverse metabolic health outcomes in humans.
Most animal studies investigating the effect of PFAS exposure on metabolic syndrome have focused on PFOA. Previous work in mice has shown that developmental exposure to PFOA disrupts weight gain, serum leptin, and insulin sensitivity later in life (Abbott et al. 2012; Hines et al. 2009). While data from adult-dosed animals initially suggested that peroxisome proliferator-activated receptor alpha (PPARα) was the mechanism through which PFOA exerted deleterious effects on the liver and metabolic system (Elcombe et al. 2012; Kennedy et al. 2004; Klaunig et al. 2003; Rosen et al. 2007), later work using mice null for PPARα revealed adverse liver effects persisted or worsened following perinatal exposures, suggesting PFOA can induce toxicity (e.g. tumor development) in the liver via PPARα-independent mechanisms such as glycogen depletion and mitochondrial dysfunction (Filgo et al. 2015; Mashayekhi et al. 2015; Quist et al 2015) and that effects may have been mediated in utero (Filgo et al 2015; Quist et al 2015). However, more recently PFOA, PFOS, PFNA, and PFHxS were evaluated in mice and found to induce expression of hepatic genes involved in fatty acid and triglyceride synthesis, potentially causing steatosis through disruption of the balance between fatty acid accumulation and oxidation (Das et al. 2017). Similarly, in primary human and rat hepatocytes, PFOA and PFOS were shown to activate multiple nuclear receptors and the metabolic response shifted from carbohydrate metabolism to fatty acid accumulation and oxidation (Bjork et al. 2011). In an in vitro 3D spheroid model of mouse liver AML12 cells, PFOA, GenX, and another PFOA alternative, 3,5,7,9-tetraoxadecanoic perfluoro acid (PF04DA), were shown to induce PPARα targets, oxidative stress, and lactate dehydrogenase (LDH) leakage, suggesting PFOA alternatives have the potential to induce liver injury (Sun et al. 2019). Across the human, animal, and in vitro literature, there is consistent evidence to suggest PFAS disrupt metabolic homeostasis through damaging the liver. Whether PFAS-mediated effects on the placenta may have a role in programming latent liver and metabolism-related outcomes is the focus of on-going work.
2.5. PFAS Exposure and Thyroid Effects
PFAS are hypothesized to target the thyroid by influencing multiple biological mechanisms involved in thyroid homeostasis, including thyroid hormone biosynthesis, transport, metabolism, and interference with thyroid receptors in target tissues (Boas et al. 2009). Proposed mechanisms of action of thyroid disruption by PFAS include reduced circulating levels of thyroxine (T4) due to competitive binding to thyroid hormone transporter (THT) proteins (Weiss et al. 2009), increased T4 metabolism in the thyroid and liver (Chang et al. 2009; Yu et al. 2009; Yu et al. 2011), reduced thyroid production of T4 (Webster et al. 2014), or reduced thyroid peroxidase (TPO) activity (Coperchini et al. 2015). During pregnancy, the placenta regulates the exchange of thyroid hormone (TH) from the maternal to fetal compartments and must maintain appropriate TH levels in the fetal circulation throughout gestation to ensure healthy development (Chan et al. 2009). Fetal TH levels are modulated by the placenta through plasma membrane THTs, metabolism, and binding to proteins in trophoblast cells (Chan et al. 2009). Thus, any PFAS implicated as TH disrupters may perturb the ability of the placenta to appropriately modulate fetal thyroid hormone balance during development.
Previous epidemiologic studies have examined the association between exposure to PFAS and THs, with inconsistent findings across the literature. Several studies leveraged the NHANES database and showed positive associations between PFOA and total triiodothyronine (T3) (Jain 2013; Webster et al. 2016; Wen et al. 2013), TSH (Jain 2013; Lewis et al. 2015), and self-reports of current thyroid disease (Melzer et al. 2010). PFHxS has been associated with increases in total T4 across the general U.S. population (Jain 2013) as well as with sex-specific positive associations in women (Wen et al. 2013). A recent meta-analysis of twelve epidemiologic studies found that PFAS are generally negatively associated with T4, but certain associations may be non-monotonic (Kim et al. 2018). For example, PFOS was positively associated with T4 and TSH only in the intermediate exposure group (Kim et al. 2018). Positive associations between serum PFOS and TSH were similarly reported in a longitudinal study of adults with serum levels of PFOS within the range of the general U.S. population (Blake et al. 2018), and PFAS were generally found to be positively associated with TSH in pregnant women in a systematic review (Ballesteros et al. 2017). However, it is possible that inconsistencies across the human literature are attributable to differences in the overall range of PFAS exposure across populations studied, which may help explain the non-monotonic dose-response described in Kim et al. (2018).
Pregnant women and their developing offspring are particularly sensitive to disruptions in THs, which are critical endocrine modulators of early neurodevelopment (de Escobar et al. 2004; Porterfield 1994). PFAS have been documented as thyroid disrupters in pregnant women in some epidemiologic studies (Ballesteros et al. 2017; Berg et al. 2015; Wang et al. 2014; Webster et al. 2014). Generally, maternal PFAS levels during pregnancy are associated with shifts in TH levels consistent with hypothyroidism (e.g. elevated TSH), which is associated with increased risk for low birth weight (Alexander et al. 2017; Belet et al. 2003). In rodent models, gestational exposure to GenX resulted in reduced maternal serum T3 and T4 levels in rats (Conley et al. 2019) and increased placental T4 levels in mice (Blake et al. 2020), demonstrating the potential for this replacement compound to act as a TH disruptor during pregnancy.
A current challenge in studying the effect of PFAS on THs is the inherent negative bias of common analog methods used to measure THs (e.g. radioimmunoassay, chemiluminescent assay), but this artifact is likely specific to rodent studies. Reduction in TH binding protein or displacement of TH via competitive binding, such as displacement of T4 by PFOS in rats, reduces serum binding capacity for TH and analog and result in negatively biased measurements of free THs (for an in-depth explanation, see Chang et al. 2007). In order to avoid such negative bias, equilibrium dialysis or high-performance liquid chromatography-based methods should be employed in animal studies. However, a follow up study conducted using human samples demonstrated no such negative bias for either PFOA or PFOS with respect to free T4 whether assayed using the analog or equilibrium dialysis method (Lopez-Espinosa et al. 2012).
Due to the critical role that THs play in human health and development, disruptions at any life stage warrant further study to understand potential underlying mechanisms. Both hyperthyroidism and hypothyroidism are suboptimal health outcomes and increase risk for adverse effects on the function of TH-responsive tissues, such as kidney and the cardiovascular system. Therefore, continued study of the effects of PFAS on thyroid function are warranted, especially as it pertains to other associated health effects. The role of placenta-specific TH action on fetal development and metabolism needs additional research; how PFAS may affect this target is poorly understood.
2.6. PFAS Exposure and Kidney Effects
The kidney is considered a target tissue of PFAS, evidenced by a growing body of human epidemiologic data further supported by animal studies and in vitro models. In humans, PFAS exposure has been associated with adverse kidney outcomes such as reduced kidney function, chronic kidney disease (CKD), and kidney cancer, including mortality from kidney cancer. Human epidemiologic studies have shown an association between low birth weight and adverse kidney outcomes in later life (Dotsch et al. 2011; Hershkovitz et al. 2007). It is possible that placental dysfunction due to PFAS resulting in low birth weight may result in lifelong increased susceptibility and reduced kidney function. However, similar to metabolic syndrome, it is difficult to determine what proportion of disease susceptibility is programmed during development and how much is exclusively a consequence of adult exposures. Due to lifelong PFAS exposure stemming from their ubiquity in the environment, it is likely that increased susceptibility resulting from exposure during critical periods of development and continued exposure through adulthood interact in complex ways to influence disease risk.
The primary elimination route for PFAS is via urinary excretion. Given that the most well-studied PFAS do not undergo biotransformation prior to urinary excretion, one hypothesized mechanism of PFAS-induced kidney injury is via reabsorption of PFAS across the renal tubules which causes localized damage, potentially through oxidative stress (Han et al., 2011). It has been demonstrated in humans that PFAS, once absorbed, distribute primarily to serum, liver, and kidney (Fabrega et al. 2014; Perez et al. 2013). It is yet to be determined if the kidney is a sensitive target of PFAS due to high accumulation of PFAS within the renal tissue, creating a high internal dose in kidney, or if the kidney is particularly sensitive to PFAS-induced effects. The extent to which PFAS distribute and/or accumulate in different tissues likely varies by individual congener. For example, perfluorododecanoic acid (PFDoA), perfluorodecanoic acid (PFDeA), and perfluorobutyrate have been shown to highly accumulate in kidney relative to other tissues (Perez et al. 2013).
Elimination rates also vary widely by individual congener with human serum half-lives spanning from about 24 hours to ~15 years which generally (though not always) corresponds to carbon chain length and functional groups (Fenton et al. 2020). Such differences in elimination rate and serum half-life are hypothesized to result from different rates of secretion and reabsorption by the kidney proximal tubules. This process, driven by renal tubule efflux transporters, actively transports PFAS back into systemic circulation, thus contributing to their long half-lives in the human body (Han et al., 2011). Uptake of PFAS into proximal tubules, both apically and basolaterally, is mediated by solute-carrier protein family transporters such as organic anion transporters (OAT). OAT1 and OAT3 have been shown to transport PFAS basolaterally in proximal tubules with apical transport mediated by OAT4 and urate transporter 1 (URAT1) (Worley et al. 2017; Yang et al. 2010; Zhang et al. 2013).
In a recent scoping review of the effect of PFAS on kidney health, Stanifer et al. (2018) identified 74 studies comprised of epidemiologic, pharmacokinetic, and toxicological studies of humans, animals, and in vitro models (Stanifer et al. 2018). Across 21 epidemiologic studies of occupationally and non-occupationally exposed populations, Stanifer et al. (2018) reported consistent associations between PFAS exposure and adverse kidney outcomes, which included reduced kidney function and kidney cancer (Stanifer et al. 2018). Studies published since this scoping review have similarly provided evidence suggesting human exposure to PFAS is detrimental to kidney health (Blake et al. 2018, Jain and Ducatman 2019a).
In addition to concerns over reduced kidney function, compensatory increases in kidney function (e.g. glomerular filtration rate) are associated with increased risk for cardiovascular morbidity and mortality. Such hyperfiltration has been observed in patients with prediabetes and prehypertension (Palatini 2012; Shastri and Sarnak 2011). A positive association between methyl-perfluorosulfonic acid (Me-PFOSA) and increased kidney filtration was reported (Blake et al. 2018). Indeed, the relationship between PFAS exposure and kidney function (glomerular filtration rate) is nonmonotonic (Jain and Ducatman 2019a). It has been hypothesized that altered balance between glomerular secretion of PFAS into the urine for excretion (e.g. via OAT1 and OAT3) and renal reabsorption (e.g. via OAT4) with increasing severity of kidney disease progression may explain the inverse U-shaped relationship. Based on these findings, Jain and Ducatman (2019a) hypothesized that PFAS reabsorption in renal tubules decreases with advancing stages of renal failure. In a follow up study, Jain and Ducatman (2019b) demonstrated that increased albuminuria may provide further explanation for decreasing serum PFAS concentrations with increasing kidney failure. Excessive albumin proteins in the urine may essentially off-load the body burden of PFAS with high binding affinity to albumin proteins (Jain and Ducatman 2019b). However, these studies were conducted using cross-sectional data from NHANES and the reported findings would be greatly strengthened if validated in longitudinal cohorts.
Animal studies and in vitro models have provided further evidence for the adverse effect of PFAS on kidney health (Stanifer et al. 2018). Studies conducted in rats and mice have reported increased kidney weight (Butenhoff et al. 2012; Curran et al. 2008; Ladics et al. 2005), increased blood urea nitrogen (BUN) (Butenhoff et al. 2012; Seacat et al. 2003; Takahashi et al. 2014), renal tubular atrophy (Klaunig et al. 2015) or hypertrophy (Ladics et al. 2005), and tubular epithelial hyperplasia (Kim et al. 2011), among other adverse kidney findings (see Stanifer et al. 2018 for a comprehensive review). In vitro systems have provided further evidence to suggest oxidative stress as a mechanism of PFAS-induced kidney damage (Chung 2015; Wen et al. 2016). A recent study by Gong et al. (2019) used rat mesangial cells as an in vitro model of diabetic kidney disease to determine the effect of PFAS exposure in the diabetic condition, and similarly reported PFOA or PFOS exposure resulted in increased oxidative stress, fibrosis, and inflammation in this in vitro model (Gong et al. 2019). Studies using rats must be interpreted with some caution due to interspecies differences in PFAS elimination rates, with PFAS excretion rates being more rapid in rats (especially female rats) compared to mice (Loccisano et al. 2013; Lou et al. 2007; Pizzurro et al. 2019; Russell et al. 2013).
Overall, there is consistent evidence across human epidemiologic, animal, and in vitro studies to support the claim that PFAS are damaging to kidney health, however the relationship between in utero exposure to PFAS, placental dysfunction, and latent kidney disease, failure, or cancer is less clear and should be investigated further.
2.7. The Interplay Between Biologic Systems Affected by PFAS Exposure
The adverse health effects of PFAS exposure on human metabolic homeostasis, thyroid function, kidney function, and pregnancy outcomes are likely interrelated due to the extensive overlap between these biologic systems and how each of these systems are influenced by placental health in the context of pregnancy. For example, the liver is responsible for thyroid hormone metabolism and transport, both kidney and liver express type 1 deiodinase (an enzyme that converts T4 to T3) (Sanders et al. 1997), and both the liver and placenta express enzymes of the type 3 deiodinase system, which converts active T4 to inactive rT3, and/or active T3 or inactive rT3 to inactive T2 (Bianco et al. 2002; Darras et al. 1999). During pregnancy, the placenta regulates the degree to which maternal thyroid hormones pass to the developing fetus and maintains the optimal balance of thyroid hormones throughout in utero development (Chan et al. 2009).
There is human clinical evidence to suggest shared pathogenic mechanisms for non-alcoholic fatty liver disease and chronic kidney disease (Musso et al. 2016), thyroid disease (both hyperthyroidism and hypothyroidism) and liver injury (Malik and Hodgson 2002), and hypothyroidism and chronic kidney disease (Rhee 2016). The proposed shared pathogenic mechanisms for these overlapping biologic systems also coincide with hypothesized mechanisms of PFAS toxicity, including disruption of lipid metabolism (e.g. cholesterol and triglycerides), nuclear receptor activation (e.g. peroxisome proliferator-activated receptors, retinoid X receptor), and oxidative stress (Malik and Hodgson 2002; Musso et al. 2016; Rhee 2016), among others. In the context of pregnancy, chronic kidney disease increases the risk of PE and low birth weight (Fischer 2007), and non-alcoholic fatty liver disease increases risk of gestational diabetes and pregnancy-induced hypertension (Hershman et al. 2019).
The clinical evidence underscores the careful balance between multiple biologic systems, and the shared pathogenic mechanisms affecting these systems coincides with toxicologic mechanisms through which PFAS affect these same systems. The complex interplay between the placenta, liver, kidney, and thyroid is further complicated by the physiological demands of pregnancy, such as increased metabolic rate, oxygen consumption, blood volume, weight gain and fluid retention. Furthermore, in utero exposure to PFAS occurring during critical periods of development may increase susceptibility for latent onset of chronic adult diseases, such as obesity, diabetes, problems in kidney function, and fatty liver disease. Some of these latent effects have been demonstrated in the context other exposures, such as maternal malnutrition, to be directly associated with poor fetal growth and placental abnormalities (Barker 2004), suggesting a potential shared mechanism across different maternal/developmental/placental stressors and latent health outcomes. Given the complex interplay between placenta, PFAS-sensitive organ systems influencing maternal health, and PFAS-sensitive latent health outcomes in adulthood, pregnant women and their developing offspring should continue to be considered the most sensitive populations when conducting PFAS health and risk assessments.
3. Health Effects-Based Risk Evaluations of PFAS in the U.S.
Human health risk assessments of PFAS have been hindered by a paucity of data, which was first remedied by reports and publications resulting from the C8 Health Study. These data provided the foundation for preliminary risk evaluations and subsequent decision-making processes. The first U.S. actions taken to limit the use and emission of PFAS began in 2000 when the company 3M voluntarily phased PFOS out of production and use. Then in 2006, the U.S. EPA invited eight major fluoropolymer and fluorotelomer manufacturing companies to participate in a global stewardship program to commit to a 95% reduction of PFOA emissions and product by the year 2010 and a complete elimination by 2015 (EPA 2006). To meet these program goals, companies ceased manufacturing and importation of long-chain PFAS and/or transitioned to alternative chemicals while some companies exited the PFAS industry altogether. The impact of this phaseout can be appreciated by temporal declines in human serum PFOA and PFOS concentrations in datasets such as the NHANES; between 1999 and 2014, average PFOA and PFOS serum levels in the general U.S. population declined by ~60% and ~80%, respectively (ATSDR 2017).
Despite the reduction in serum burden of PFOA and PFOS since the genesis of the stewardship program, a recent biomonitoring effort (Hu et al. 2016) determined that drinking water sources for as many as six million U.S. citizens exceed the U.S. EPA-derived lifetime drinking water Health Advisory Level (HAL) for PFOA alone or in combination with PFOS of 70 parts per trillion (ppt) (822-R-16-005 US EPA). However, this number likely underestimates the number of U.S. citizens with drinking water exposure above the PFOA/PFOS HAL as the sampling method used by Hu et al. (2016) was conducted under the scope of the third Unregulated Contaminant Monitoring Rule (UCMR3), which limits drinking water PFAS measurements to public systems serving over 10,000 people. The exposure levels for U.S. citizens on private well water or public water systems serving under 10,000 people have yet to characterized.
The PFOA/PFOS HAL represents a non-enforceable exposure level under which no adverse health outcomes would be expected given a lifetime of exposure, based on the U.S. EPA’s risk assessment of PFOA, which used a weight of evidence approach to examine existing human epidemiologic and animal toxicity studies. Data from a carefully conducted developmental and reproductive toxicity study was selected to obtain the reference dose (RfD) used in calculation of the lifetime HAL for PFOA, in order to generate the most protective exposure level (822-R-16-005 US EPA; Lau et al. 2006).
The lack of federally enforceable regulations on any PFAS in the U.S., as well as increasing concerns over local contamination levels has led several states to implement their own regulatory actions based on varied health effects (Figure 5). The states of California, Connecticut, Massachusetts, Michigan, Minnesota, New Hampshire, New Jersey, New York, North Carolina, and Vermont have each proposed various drinking water actions to address PFAS contamination (ASDWA 2020). New Hampshire and New Jersey have formally adopted statewide regulations, mandating drinking water levels for PFOA (NH: 12 ppt), PFOS (NH: 15 ppt), PFHxS (NH: 18 ppt), and PFNA (NH: 11 ppt; NJ: 13 ppt) substantially lower than the U.S. EPA HAL of 70 ppt for PFOA alone or combined with PFOS (ASDWA 2020). Other states, such as North Carolina and Michigan, have proposed health advisories or maximum contaminant levels (MCLs) for replacement PFAS, such as GenX (NC: 140 ppt; MI: 370 ppt) (ASDWA 2020) while Ohio and Washington are in the process of developing draft health advisories for various PFAS (OH EPA 2019; WA DOH 2020). In August 2020, Michigan set new limits for several more PFAS: PFNA (6-ppt); PFOA (8-ppt); PFOS (16-ppt); PFHxS (51-ppt); PFBS (420-ppt); PFHxA (400,000-ppt).
Figure 5. Responses of U.S. states to PFAS contamination compared to nationwide PFAS levels (inset).
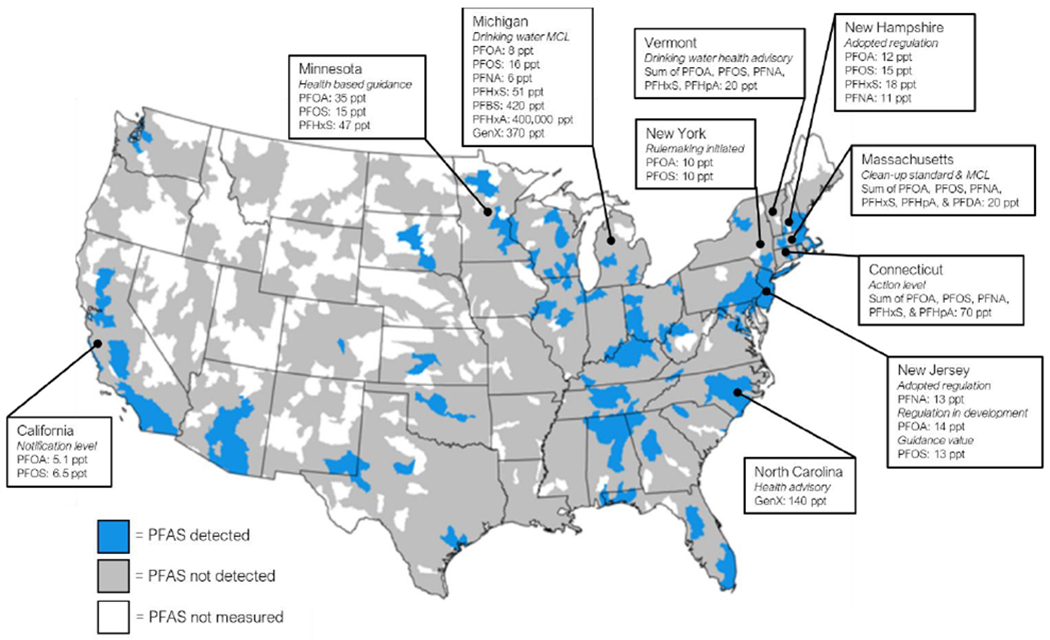
Most state actions are non-enforceable and include notification levels, maximum contamination limits (MCLs), health advisories or guidance, and action levels. Several states have adopted enforceable regulation, such as New Hampshire and New Jersey, while other states are actively in pursuit of enforceable legislation, such as New York. There are some states with considerable PFAS contamination but no statewide action, including Alabama, New Mexico, and Kentucky. Figure adapted from Hu et al. (2018).
Regulation of PFAS as the statewide level has resulted in disparate regulatory standards with regards to both the drinking water limit and extent to which regulations may be enforced. PFAS contamination in the environment and drinking water is driven by point sources of pollution, such as industrial/manufacturing sites, airports, biosolids fields, and military training bases. At each site, different levels of a multitude of PFAS are used in complex mixtures, depending on the application. The heterogeneity of the exposure mixture as well as relative concentrations of individual PFAS adds to the complexity of both studying PFAS effects of human health and setting appropriate standards to adequately protect the most vulnerable members of society. There is little to no toxicity data for the vast majority of PFAS currently in use, therefore there is great need for efficient methods to evaluate these compounds.
4. Conclusions
The complex family of PFAS compounds presents unique challenges to toxicologists and risk assessors. The effects of PFAS on human health differ based on compound, impact multiple overlapping biological systems, affect health outcomes at all life stages, and exposure levels (and mixtures) differ temporally and geographically. Importantly, early life PFAS insults may in fact disrupt placental growth and function, thereby increasing susceptibility for later life chronic health conditions, which may be further exacerbated by lifelong PFAS exposure. Thus, it is critical to improve our understanding of the developmental and adult health consequences associated with PFAS exposure over time, identify emerging PFAS threats to sensitive subpopulations, and develop tools to efficiently evaluate and characterize PFAS toxicity on sensitive targets such as the placenta. Future research is needed to determine if latent health effects of PFAS exposure are programmed or mediated by the placenta. Going forward, experimental toxicology studies designed to formally assess the placenta are needed to determine the sensitivity of this tissue towards PFAS and explore the molecular mechanisms of placental toxicity.
Acknowledgments
Funding
The authors declare they have no actual or potential competing financial interests. This research was supported in part by the NIH, National Institute of Environmental Health Sciences. This work was funded under NIEHS Z0ES102785 (SEF) and UNC-Chapel Hill T32 ES007126 (BEB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 3M 2000. 26-week capsule toxicity study with perfluorooctane sulfonic acid potassium salt (PFOS; T-6295) in cynomolgus monkeys. Administrative Record AR226-0145. URL: https://www.documentcloud.org/documents/4346742-US00004524.html
- 822-R-16-005 US EPA. 2016. Drinking water health advisory for perfluorooctanoic acid (pfoa). URL: https://www.epa.gov/sites/production/files/2016-05/documents/pfoa_health_advisory_final_508.pdf
- Abbott BD, Wood CR, Watkins AM, Tatum-Gibbs K, Das KP, Lau C. 2012. Effects of perfluorooctanoic acid (pfoa) on expression of peroxisome proliferator-activated receptors (ppar) and nuclear receptor-regulated genes in fetal and postnatal cd-1 mouse tissues. Reproductive Toxicology 33:491–505. [DOI] [PubMed] [Google Scholar]
- Agency for Toxic Substances and Disease Registry (ATSDR). 2015. Toxicological profile for perfluoroalkyls. Atlanta, GA: Agency for Toxic Substances and Disease Registry, U.S. Department of Health and Human Services, Public Health Service; URL: https://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=1117&tid=237 [acessed July 7, 2019]. [Google Scholar]
- Agency for Toxic Substances and Disease Registry (ATSDR). 2017. Perfluoroalkyl and polyfluoroalkyl substances (pfas) in the u.s. population. URL: https://www.atsdr.cdc.gov/pfas/pfas-in-population.html [accessed July 7, 2019].
- Agency for Toxic Substances and Disease Registry (ATSDR). 2019. Agency for toxic substances and disease registry: Pfas exposure assessments. Available: https://www.atsdr.cdc.gov/pfas/PFAS-Exposure-Assessments.html [accessed July 7, 2019].
- Agency for Toxic Substances and Disease Registry (ATSDR). 2020. Per- and polyfluoroalkyl substances (PFAS) and your health. Available: https://www.atsdr.cdc.gov/pfas/health-effects.html [accessed May 15, 2020].
- Alexander BH, Olsen GW, Burris JM, Mandel JH, Mandel JS. 2003. Mortality of employees of a perfluorooctanesulphonyl fluoride manufacturing facility. Occupational and Environmental Medicine 60:722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander EK, Pearce EN, Brent GA, Brown RS, Chen H, Dosiou C, et al. 2017. 2017 guidelines of the american thyroid association for the diagnosis and management of thyroid disease during pregnancy and the postpartum. Thyroid 27:315–389. [DOI] [PubMed] [Google Scholar]
- ASDWA. 2020. Per- and polyfluoroalkyl substances (pfas): Per- and polyfluoroalkyl substances (pfas) state drinking water program challenges. Available: https://www.asdwa.org/pfas/ [accessed Jan 01 2020].
- Ashley-Martin J, Dodds L, Arbuckle TE, Morisset AS, Fisher M, Bouchard MF, et al. 2016. Maternal and neonatal levels of perfluoroalkyl substances in relation to gestational weight gain. International Journal of Environmental Research Public Health 13:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audette MC, Kingdom JC. 2018. Screening for fetal growth restriction and placental insufficiency. Seminars in Fetal and Neonatal Medicine 23:119–125. [DOI] [PubMed] [Google Scholar]
- Bach CC, Vested A, Jrgensen KT, Bonde JPE, Henriksen TB, Toft G 2016. Perfluoroalkyl and polyfluoroalkyl substances and measures of human fertility: A systematic review. Critical Reviews in Toxicology 46:735–755. [DOI] [PubMed] [Google Scholar]
- Ballesteros V, Costa O, Iñiguez C, Fletcher T, Ballester F, Lopez-Espinosa M-J. 2017. Exposure to perfluoroalkyl substances and thyroid function in pregnant women and children: A systematic review of epidemiologic studies. Environment International 99:15–28. [DOI] [PubMed] [Google Scholar]
- Bangma J, Szilagyi J, Blake BE, Plazas C, Kepper S, Fenton SE, Fry R. 2020. An assessment of serum-dependent impacts on intracellular accumulation and genomic response of per- and polyfluoroalkyl substances (PFAS) in a placenta trophoblast model. Environmental Toxicology. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. 2004. The developmental origins of chronic adult disease. Acta Paediatrica 93:26–33. [DOI] [PubMed] [Google Scholar]
- Belet N, Imdat H, Yamk F, Küçüködük Ş. 2003. Thyroid function tests in preterm infants born to preeclamptic mothers with placental insufficiency. Journal of Pediatric Endocrinology & Metabolism 16:1131–1136. [DOI] [PubMed] [Google Scholar]
- Berg V, Nost TH, Hansen S, Elverland A, Veyhe A-S, Jorde R, et al. 2015. Assessing the relationship between perfluoroalkyl substances, thyroid hormones and binding proteins pregnant women; a longitudinal mixed effects approach. Environment International 77:63–69. [DOI] [PubMed] [Google Scholar]
- Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. 2002. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocrine Reviews 23:38–89. [DOI] [PubMed] [Google Scholar]
- Bjork JA, Butenhoff JL, Wallace KB. 2011. Multiplicity of nuclear receptor activation by pfoa and pfos in primary human and rodent hepatocytes. Toxicology 288:8–17. [DOI] [PubMed] [Google Scholar]
- Blake BE, Cope HA, Hall SM, Keys RD, Mahler BW, McCord J, et al. 2020. Evaluation of maternal, embryo, and placental effects in cd-1 mice following gestational exposure to perfluorooctanoic acid (pfoa) or hexafluoropropylene oxide dimer acid (hfpo-da or genx). Environmental Health Perspectives 128:027006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake BE, Pinney SM, Hines EP, Fenton SE, Ferguson KK. 2018. Associations between longitudinal serum perfluoroalkyl substance (pfas) levels and measures of thyroid hormone, kidney function, and body mass index in the fernald community cohort. Environmental Pollution 242:894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butenhoff JL, Chang S-C, Olsen GW, Thomford PJ. 2012. Chronic dietary toxicity and carcinogenicity study with potassium perfluorooctanesulfonate in sprague dawley rats. Toxicology 293:1–15. [DOI] [PubMed] [Google Scholar]
- C8 Science Panel. URL:http://www.c8sciencepanel.org/.
- Centers for Disease Control (CDC). 2013. Fourth national report on human exposure to environmental chemicals. Atlanta (ga): CDC; 2009 URL: https://www.cdc.gov/biomonitoring/pdf/FourthReport_UpdatedTables_Feb2015.pdf [Google Scholar]
- Chan SY, Vasilopoulou E, Kilby MD. 2009. The role of the placenta in thyroid hormone delivery to the fetus. Nature Clinical Practice Endocrinology & Metabolism 5:45–54. [DOI] [PubMed] [Google Scholar]
- Chang S, Butenhoff JL, Parker GA, Coder PS, Zitzow JD, Krisko RM, et al. 2018. Reproductive and developmental toxicity of potassium perfluorohexanesulfonate in cd-1 mice. Reproductive Toxicology 78:150–168. [DOI] [PubMed] [Google Scholar]
- Chang SC, Ehresman DJ, Bjork JA, Wallace KB, Parker GA, Stump DG, et al. 2009. Gestational and lactational exposure to potassium perfluorooctanesulfonate (k+pfos) in rats: Toxicokinetics, thyroid hormone status, and related gene expression. Reproductive Toxicology 27:387–399. [DOI] [PubMed] [Google Scholar]
- Chang SC, Thibodeaux JR, Eastvold ML, Ehresman DJ, Bjork JA, Froehlich JW, Lau CS, Singh RJ, Wallace KB, Butenhoff JL. 2008. Negative bias from analog methods used in the analysis of free thyroxine in rat serum containing perfluorooctanesulfonate (PFOS). Toxicology. 234:21–33. [DOI] [PubMed] [Google Scholar]
- Chang SC, Thibodeaux JR, Eastvold ML, Ehresman DJ, Bjork JA, Froehlich JW, Lau C, Singh RJ, Wallace KB, Butenhoff JL. 2008. Thyroid hormone status and pituitary function in adult rats given oral doses of perfluorooctanesulfonate (PFOS). Toxicology 243:330–339. [DOI] [PubMed] [Google Scholar]
- Chen F, Yin S, Kelly BC, Liu W. 2017a. Chlorinated polyfluoroalkyl ether sulfonic acids in matched maternal, cord, and placenta samples: A study of transplacental transfer. Environmental Science & Technology 51:6387–6394. [DOI] [PubMed] [Google Scholar]
- Chen F, Yin S, Kelly BC, Liu W. 2017b. Isomer-specific transplacental transfer of perfluoroalkyl acids: Results from a survey of paired maternal, cord sera, and placentas. Environmental Science & Technology 51:5756–5763. [DOI] [PubMed] [Google Scholar]
- Christensen KY, Raymond M, Meiman J. 2019. Perfluoroalkyl substances and metabolic syndrome. International Journal of Hygiene and Environmental Health 222:147–153. [DOI] [PubMed] [Google Scholar]
- Chung A 2015. Perfluorooctane sulfonate (pfos) promotes renal injury under diabetic condition in vitro. Hong Kong Journal of Nephrology 17:S3–S4. [Google Scholar]
- Conley JM, Lambright CS, Evans N, Strynar MJ, McCord J, McIntyre BS, et al. 2019. Adverse maternal, fetal, and postnatal effects of hexafluoropropylene oxide dimer acid (genx) from oral gestational exposure in sprague-dawley rats. Environmental Health Perspectives 127:37008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consonni D, Straif K, Symons JM, Tomenson JA, van Amelsvoort LG, Sleeuwenhoek A, et al. 2013. Cancer risk among tetrafluoroethylene synthesis and polymerization workers. American Journal of Epidemiology 178:350–358. [DOI] [PubMed] [Google Scholar]
- Cope HA, Blake BE, Fenton SE. 2020. Evaluation of metabolic dysfunction in CD-1 mouse offspring after gestational exposure to perfluorooctanoic acid (PFOA) or hexafluoropropylene oxide dimer acid (HFPO-DA or GenX) and high fat diet. Toxicologic Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coperchini F, Pignatti P, Lacerenza S, Negri S, Sideri R, Testoni C, et al. 2015. Exposure to perfluorinated compounds: In vitro study on thyroid cells. Environmental Science and Pollution Research 22:2287–2294. [DOI] [PubMed] [Google Scholar]
- Costa G, Sartori S, Consonni D. 2009. Thirty years of medical surveillance in perfluooctanoic acid production workers. Journal of Occupational and Environmental Medicine 51:364–372. [DOI] [PubMed] [Google Scholar]
- Cuffe JSM, Holland O, Salomon C, Rice GE, Perkins AV. 2017. Review: Placental derived biomarkers of pregnancy disorders. Placenta 54:104–110. [DOI] [PubMed] [Google Scholar]
- Curran I, Hierlihy SL, Liston V, Pantazopoulos P, Nunnikhoven A, Tittlemier S, et al. 2008. Altered fatty acid homeostasis and related toxicologic sequelae in rats exposed to dietary potassium perfluorooctanesulfonate (pfos). Journal of Toxicology and Environmental Health, Part A 71:1526–1541. [DOI] [PubMed] [Google Scholar]
- Cushen SC, Goulopoulou S. 2017. New models of pregnancy-associated hypertension. American Journal of Hypertension 30:1053–1062. [DOI] [PubMed] [Google Scholar]
- D’Amore S, Palasciano G, Moschetta A. 2014. The liver in metabolic syndrome In: A systems biology approach to study metabolic syndrome, (Orešič M, Vidal-Puig A, eds). Cham:Springer International Publishing, 27–61. [Google Scholar]
- Darras VM, Hume R, Visser TJ. 1999. Regulation of thyroid hormone metabolism during fetal development. Molecular and Cellular Endocrinology 151:37–47. [DOI] [PubMed] [Google Scholar]
- Darrow LA, Stein CR, Steenland K. 2013. Serum perfluorooctanoic acid and perfluorooctane sulfonate concentrations in relation to birth outcomes in the mid-ohio valley, 2005-2010. Environmental Health Perspectives 121:1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das KP, Grey BE, Rosen MB, Wood CR, Tatum-Gibbs KR, Zehr RD, et al. 2015. Developmental toxicity of perfluorononanoic acid in mice. Reproductive Toxicology 51:133–144. [DOI] [PubMed] [Google Scholar]
- Das KP, Wood CR, Lin MT, Starkov AA, Lau C, Wallace KB, et al. 2017. Perfluoroalkyl acids-induced liver steatosis: Effects on genes controlling lipid homeostasis. Toxicology 378:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Escobar GM, Obregon MJ, del Rey FE. 2004. Maternal thyroid hormones early in pregnancy and fetal brain development. Best Practice & Research: Clinical Endocrinology & Metabolism 18:225–248. [DOI] [PubMed] [Google Scholar]
- Domingo JL, Nadal M. 2019. Human exposure to per- and polyfluoroalkyl substances (pfas) through drinking water: A review of the recent scientific literature. Environmental Research 177:108648. [DOI] [PubMed] [Google Scholar]
- Dötsch J, Plank C, Amann K. 2012. Fetal programming of renal function. Pediatric Nephrology. 27:513–20. [DOI] [PubMed] [Google Scholar]
- Eckel RH, Grundy SM, Zimmet PZ. 2005. The metabolic syndrome. The Lancet 365:1415–1428. [DOI] [PubMed] [Google Scholar]
- Edwards TL. 2010a. An oral (gavage) prenatal developmental study of h-28548 in rats. Dupont-18405-1037.
- Elcombe CR, Elcombe BM, Foster JR, Chang S-C, Ehresman DJ, Butenhoff JL. 2012. Hepatocellular hypertrophy and cell proliferation in sprague–dawley rats from dietary exposure to potassium perfluorooctanesulfonate results from increased expression of xenosensor nuclear receptors pparα and car/pxr. Toxicology 293:16–29. [DOI] [PubMed] [Google Scholar]
- Environmental Working Group (EWG). 2019. For 50 years, polluters knew pfas chemicals were dangerous but hid risks from public. Environmental Working Group. URL: https://static.ewg.org/reports/2019/pfa-timeline/3M-DuPont-Timeline_sm.pdf [Google Scholar]
- Ervin RB. 2009. Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United states, 2003-2006. National Health Statistics Reports: 1-7. [PubMed]
- European Union (EU). 2017. Commission regulation (eu) 2017/1000 of 13 june 2017 amending annex xvii to regulation (ec) no 1907/2006 of the european parliament and of the council concerning the registration, evaluation, authorisation and restriction of chemicals (reach) as regards perfluorooctanoic acid (pfoa), its salts and pfoa-related substances.
- Fenton SE, Ducatman A, Boobis A, DeWitt JC, Lau C, Ng C, Smith JS, Roberts SM. 2020. Per- and Polyfluoroalkyl Substance Toxicity and Human Health Review: Current State of Knowledge and Strategies for Informing Future Research. Environmental Toxicology & Chemistry in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton SE, Reiner JL, Nakayama SF, Delinsky AD, Stanko JP, Hines EP, et al. 2009. Analysis of pfoa in dosed cd-1 mice. Part 2. Disposition of pfoa in tissues and fluids from pregnant and lactating mice and their pups. Reproductive Toxicology 27:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filgo AJ, Quist EM, Hoenerhoff MJ, Brix AE, Kissling GE, Fenton SE. 2014. Perfluorooctanoic acid (pfoa)–induced liver lesions in two strains of mice following developmental exposures: Pparα is not required. Toxicologic Pathology 43:558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MJ. 2007. Chronic kidney disease and pregnancy: maternal and fetal outcomes. Advances in Chronic Kidney Disease 14:132–145. [DOI] [PubMed] [Google Scholar]
- Funderburg AC. 2010. Teflon. American Heritage: Invention and Technology.
- Gagnon R 2003. Placental insufficiency and its consequences. European Journal of Obstetrics & Gynecology and Reproductive Biology 110:S99–S107. [DOI] [PubMed] [Google Scholar]
- Gilliland FD, Mandel JS. 1993. Mortality among employees of a perfluorooctanoic acid production plant. Journal of Occupational Medicine 35:950–954. [DOI] [PubMed] [Google Scholar]
- Goeden HM, Greene CW & Jacobus JA. 2019. A transgenerational toxicokinetic model and its use in derivation of Minnesota PFOA water guidance. Journal of Exposure Science & Environmental Epidemiology 29, 183–195. 10.1038/s41370-018-0110-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeden HM. 2018. Focus on chronic exposure for deriving drinking water guidance underestimates potential risk to infants. Int J Environ Res Public Health. 15:512 10.3390/ijerph15030512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Yang C, Hong Y, Chung ACK, Cai Z. 2019. Pfoa and pfos promote diabetic renal injury in vitro by impairing the metabolisms of amino acids and purines. Science of The Total Environment 676:72–86. [DOI] [PubMed] [Google Scholar]
- Gorrochategui E, Perez-Albaladejo E, Casas J, Lacorte S, Porte C. 2014. Perfluorinated chemicals: Differential toxicity, inhibition of aromatase activity and alteration of cellular lipids in human placental cells. Toxicology and Applied Pharmacology 277:124–130. [DOI] [PubMed] [Google Scholar]
- Guelfo JL, Adamson DT (2018). Evaluation of a national data set for insights into sources, composition, and concentrations of per-and polyfluoroalkyl substances (PFASs) in CIS drinking water. Environmental Pollution, 236, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelfo JL, Marlow T, Klein DM, Savitz DA, Frickel S, Crimi M, Suuberg EM (2018). Evaluation and management strategies for per-and polyfluoroalkyl substances (PFASs) in drinking water aquifers: perspectives from impacted US Northeast communities. Environmental health perspectives, 126(6), 065001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen L, Dudarev AA, Huber S, Odland JØ, Nieboer E, Sandanger TM. 2013. Partition of perfluoroalkyl substances (pfass) in whole blood and plasma, assessed in maternal and umbilical cord samples from inhabitants of arctic russia and Uzbekistan. Science of The Total Environment 447:430–437. [DOI] [PubMed] [Google Scholar]
- Harris MW, Birnbaum LS. 1989. Developmental toxicity of perfluorodecanoic acid in c57b1/6n mice. Fundamental and Applied Toxicology 12:442–448. [DOI] [PubMed] [Google Scholar]
- Hemachandra AH, Klebanoff MA, Duggan AK, Hardy JB, Furth SL. 2006. The association between intrauterine growth restriction in the full-term infant and high blood pressure at age 7 years: Results from the collaborative perinatal project. International Journal of Epidemiology 35:871–877. [DOI] [PubMed] [Google Scholar]
- Henriksen T, Clausen T. 2002. The fetal origins hypothesis: Placental insufficiency and inheritance versus maternal malnutrition in well-nourished populations. Acta Obstetricia et Gynecologica Scandinavica 81:112–114. [DOI] [PubMed] [Google Scholar]
- Hershkovitz D, Burbea Z, Skorecki K, Brenner BM. 2007. Fetal programming of adult kidney disease: cellular and molecular mechanisms. Clinical Journal of the American Society of Nephrology. 2:334–42. [DOI] [PubMed] [Google Scholar]
- Hershman M, Mei R, Kushner T. 2019. Implications of nonalcoholic fatty liver disease on pregnancy and maternal and child outcomes. Gastroenterology & Hepatology 15:221–228. [PMC free article] [PubMed] [Google Scholar]
- Hines EP, White SS, Stanko JP, Gibbs-Flournoy EA, Lau C, Fenton SE. 2009. Phenotypic dichotomy following developmental exposure to perfluorooctanoic acid (pfoa) in female cd-1 mice: Low doses induce elevated serum leptin and insulin, and overweight in midlife. Molecular and Cellular Endocrinology 304:97–105. [DOI] [PubMed] [Google Scholar]
- Holtcamp W 2012. Pregnancy-induced hypertension “probably linked” to pfoa contamination. Environmental Health Perspectives 120:a59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins ZR, Sun M, DeWitt JC, Knappe DR. 2018. Recently detected drinking water contaminants: GenX and other per- and polyfluoroalkyl ether acids. Journal-American Waterworks Association. 110(7): 13–28. [Google Scholar]
- Hu XC, Andrews DQ, Lindstrom AB, Bruton TA, Schaider LA, Grandjean P, et al. 2016. Detection of poly-and perfluoroalkyl substances (pfass) in us drinking water linked to industrial sites, military fire training areas, and wastewater treatment plants. Environmental Science & Technology Letters 3:344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Chen Q, Zhang L, Luo K, Chen L, Zhao S, et al. 2019. Prenatal exposure to perfluoroalkyl and polyfluoroalkyl substances and the risk of hypertensive disorders of pregnancy. Environmental Health 18:5–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheon JA, McNamara H, Platt RW, Benjamin A, Kramer MS. 2012. Placental weight for gestational age and adverse perinatal outcomes. Obstetrics & Gynecology 119:1251–1258. [DOI] [PubMed] [Google Scholar]
- International Agency for Research on Cancer (IARC) 2016. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. IARC monographs on the evaluation of carcinogenic risks to humans Volume 110 Perfluorooctanoic Acid, Tetrafluoroethylene, Dichloromethane, 1,2-Dichloropropane, and 1,3-Propane Sultone. URL: https://publications.iarc.fr/547 [Google Scholar]
- Interstate Technology and Regulatory Council (ITRC). 2020. Interstate technology and regulatory council: Naming conventions and physical and chemical properties of per- and polyfluoroalkyl substances (pfas). URL: https://pfas-1.itrcweb.org/fact-sheets/ [Google Scholar]
- Jain RB, Ducatman A. 2019a. Perfluoroalkyl acids serum concentrations and their relationship to biomarkers of renal failure: Serum and urine albumin, creatinine, and albumin creatinine ratios across the spectrum of glomerular function among us adults. Environmental Research 174:143–151. [DOI] [PubMed] [Google Scholar]
- Jain RB, Ducatman A. 2019b. Perfluoroalkyl substances follow inverted u-shaped distributions across various stages of glomerular function: Implications for future research. Environmental Research 169:476–482. [DOI] [PubMed] [Google Scholar]
- Jain RB. 2013. Association between thyroid profile and perfluoroalkyl acids: Data from nhnaes 2007–2008. Environmental Research 126:51–59. [DOI] [PubMed] [Google Scholar]
- Johnson PI, Sutton P, Atchley DS, Koustas E, Lam J, Sen S, et al. 2014. The navigation guide - evidence-based medicine meets environmental health: Systematic review of human evidence for pfoa effects on fetal growth. Environmental Health Perspectives 122:1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AA, Vaidya SS, St-Pierre MV, Mikheev AM, Desino KE, Nyandege AN, et al. 2016. Placental abc transporters: Biological impact and pharmaceutical significance. Pharmaceutical Research 33:2847–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Wong LY, Jia LT, Kuklenyik Z, Calafat AM. 2011. Trends in exposure to polyfluoroalkyl chemicals in the US population: 1999– 2008. Environmental science & technology. 45:8037–45. [DOI] [PubMed] [Google Scholar]
- Kennedy GL, Butenhoff JL, Olsen GW, O’Connor JC, Seacat AM, Perkins RG, et al. 2004. The toxicology of perfluorooctanoate. Critical Reviews in Toxicology 34:351–384. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Moon S, Oh B-C, Jung D, Ji K, Choi K, et al. 2018. Association between perfluoroalkyl substances exposure and thyroid function in adults: A meta-analysis. PLOS ONE 13 :e0197244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Choi K, Ji K, Seo J, Kho Y, Park J, et al. 2011. Trans-placental transfer of thirteen perfluorinated compounds and relations with fetal thyroid hormones. Environmental Science & Technology 45:7465–7472. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, et al. 2003. Ppara agonist-induced rodent tumors: Modes of action and human relevance. Critical Reviews in Toxicology 33:655–780. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Shinohara M, Iwai H, Chengelis CP, Kirkpatrick JB, Wang Z, et al. 2015. Evaluation of the chronic toxicity and carcinogenicity of perfluorohexanoic acid (pfhxa) in sprague-dawley rats. Toxiocologic Pathology 43:209–220. [DOI] [PubMed] [Google Scholar]
- Koustas E, Lam J, Sutton P, Johnson PI, Atchley DS, Sen S, et al. 2014. The navigation guide - evidence-based medicine meets environmental health: Systematic review of nonhuman evidence for pfoa effects on fetal growth. Environmental Health Perspectives 122:1015–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummu M, Sieppi E, Koponen J, Laatio L, Vahakangas K., Kiviranta H, Rautio A, Myllynen P (2015). Organic anion transporter 4 (OAT 4) modifies placental transfer of perfluorinated alkyl acids PFOS and PFOA in human placental ex vivo perfusion system. Placenta, 36(10), 1185–1191. [DOI] [PubMed] [Google Scholar]
- Ladies GS, Stadler JC, Makovec GT, Everds NE, Buck RC. 2005. Subchronic toxicity of a fluoroalkylethanol mixture in rats. Drug and Chemical Toxicology 28:135–158. [DOI] [PubMed] [Google Scholar]
- Laine JE, Ray P, Bodnar W, Cable PH, Boggess K, Offenbacher S, Fry RC (2015). Placental cadmium levels are associated with increased preeclampsia risk. PloS one, 10(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam J, Koustas E, Sutton P, Johnson PI, Atchley DS, Sen S, et al. 2014. The navigation guide - evidence-based medicine meets environmental health: Integration of animal and human evidence for pfoa effects on fetal growth. Environmental Health Perspectives 122:1040–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, Thibodeaux JR, Hanson RG, Narotsky MG, Rogers JM, Lindstrom AB, et al. 2006. Effects of perfluorooctanoic acid exposure during pregnancy in the mouse. Toxicological Sciences 90:510–518. [DOI] [PubMed] [Google Scholar]
- Leclerc F, Dubois M-F, Aris A. 2014. Maternal, placental and fetal exposure to bisphenol a in women with and without preeclampsia. Hypertension in Pregnancy 33:341–348. [DOI] [PubMed] [Google Scholar]
- Li M, Zeng XW, Qian ZM, Vaughn MG, Sauve S, Paul G, Lin S, Lu L, Hu LW, Yang BY, Zhou Y. 2017. Isomers of perfluorooctanesulfonate (PFOS) in cord serum and birth outcomes in China: Guangzhou Birth Cohort Study. Environment international. 102:1–8. [DOI] [PubMed] [Google Scholar]
- Li X, Ye L, Ge Y, Yuan K, Zhang Y, Liang Y, Wei J, Zhao C, Lian QQ, Zhu X, Ge RS. 2016. In utero perfluorooctane sulfonate exposure causes low body weights of fetal rats: A mechanism study. Placenta. 39:125–33 [DOI] [PubMed] [Google Scholar]
- Loccisano AE, Longnecker MP, Campbell JL Jr, Andersen ME, Clewell III. 2013. Development of pbpk models for pfoa and pfos for human pregnancy and lactation life stages. Journal of Toxicology and Environmental Health, Part A 76:25–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Espinosa MJ, Fitz-Simon N, Bloom MS, Calafat AM, Fletcher T. 2012. Comparison between free serum thyroxine levels, measured by analog and dialysis methods, in the presence of perfluorooctane sulfonate and perfluorooctanoate. Reproductive Toxicology. 33:552–5. [DOI] [PubMed] [Google Scholar]
- Lou I, Wambaugh JF, Lau C, Hanson RG, Lindstrom AB, Strynar MJ, et al. 2009. Modeling single and repeated dose pharmacokinetics of pfoa in mice. Toxicological Sciences 107:331–341. [DOI] [PubMed] [Google Scholar]
- Lum KJ, Sundaram R, Barr DB, Louis TA, Buck Louis GM. 2017. Perfluoroalkyl chemicals, menstrual cycle length, and fecundity: Findings from a prospective pregnancy study. Epidemiology 28:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin JI, Alexander BH, Olsen GW, Church TR. 2009. Ammonium perfluorooctanoate production and occupational mortality. Epidemiology 20:921–928. [DOI] [PubMed] [Google Scholar]
- Maisonet M, Terrell ML, McGeehin MA, Christensen KY, Holmes A, Calafat AM, et al. 2012. Maternal concentrations of polyfluoroalkyl compounds during pregnancy and fetal and postnatal growth in british girls. Environmental Health Perspectives 120:1432–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Hodgson H. 2002. The relationship between the thyroid gland and the liver. QJM: An International Journal of Medicine 95:559–569. [DOI] [PubMed] [Google Scholar]
- Marinello WP, Mohseni Z, Huang R, Cunningham SJ, Crute CE, Reddy TE, Zhang J, Feng L. 2020. Perfluorobutane sulfonate exposure disrupted human placental cytotrophoblast proliferation and invasion involving in dysregulating preeclampsia related genes. FASEB J. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks KJ, Cutler AJ, Jeddy Z, Northstone K, Kato K, Hartman TJ. 2019. Maternal serum concentrations of perfluoroalkyl substances and birth size in british boys. International Journal of Hygiene and Environmental Health 222:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, Hannan NJ, Jelinic M, Nguyen TPH, Girling JE, Parry LJ. 2017. Animal models of preeclampsia: Translational failings and why. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 314:R499–R508. [DOI] [PubMed] [Google Scholar]
- Mashayekhi V, Tehrani KHME, Hashemzaei M, Tabrizian K, Shahraki J, Hosseini MJ (2015). Mechanistic approach for the toxic effects of perfluorooctanoic acid on isolated rat liver and brain mitochondria. Human & experimental toxicology, 34(10), 985–996. [DOI] [PubMed] [Google Scholar]
- Matilla-Santander N, Valvi D, Lopez-Espinosa MJ, Manzano-Salgado CB, Ballester F, Ibarluzea J, et al. 2017. Exposure to perfluoroalkyl substances and metabolic outcomes in pregnant women: Evidence from the spanish inma birth cohorts. Environmental Health Perspectives 125:117004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q, Inoue K, Ritz B, Olsen J, Liew Z. 2018. Prenatal exposure to perfluoroalkyl substances and birth outcomes; an updated analysis from the danish national birth cohort. International Journal of Environmental Research and Public Health 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso G, Cassader M, Cohney S, De Michieli F, Pinach S, Saba F, et al. 2016. Fatty liver and chronic kidney disease: Novel mechanistic insights and therapeutic opportunities. Diabetes Care 39:1830–1845. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program (NTP). 2016. National toxicology program monograph on immunotoxicity associated with exposures to pfoa and pfos. URL: https://ntp.niehs.nih.gov/ntp/ohat/pfoa_pfos/pfoa_pfosmonograph_508.pdf [Google Scholar]
- North Carolina Department of Enviornmental Quality (NC DEQ). 2017. Notice of partial suspension and 60-day notice of intent to partially revoke NPDES permit NC0003573; The Chemours Company, Fayetteville Works. North Carolina Department of Environmental Quality; 16 November 2017 URL:https://files.nc.gov/ncdeq/GenX/Letter%20November%2011-16-17.pdf [Google Scholar]
- Nugent BM, Bale TL. 2015. The omniscient placenta: metabolic and epigenetic regulation of fetal programming. Frontiers in neuroendocrinology. 39:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohio Environmental Protection Agency (OH EPA). 2019. Ohio per- and polyfluoroalkyl substances (PFAS) action plan for drinking water. Ohio Environmental Protection Agency. December 2019 URL:https://epa.ohio.gov/Portals/28/documents/pfas/PFASActionPlan.pdf [Google Scholar]
- Olsen GW, Mair DC, Lange CC, Harrington LM, Church TR, Goldberg CL, Herron RM, Hanna H, Nobiletti JB, Rios JA, Reagen WK. 2017. Per- and polyfluoroalkyl substances (pfas) in american red cross adult blood donors, 2000–2015. Environmental Research 157:87–95. [DOI] [PubMed] [Google Scholar]
- Palatini P 2012. Glomerular hyperfiltration: a marker of early renal damage in pre-diabetes and pre-hypertension, Nephrology Dialysis Transplantation 27(5): 1708–1714. [DOI] [PubMed] [Google Scholar]
- Pedersen M, Stayner L, Slama R, Sorensen M, Figueras F, Nieuwenhuijsen MJ, et al. 2014. Ambient air pollution and pregnancy-induced hypertensive disorders: A systematic review and meta-analysis. Hypertension 64:494–500. [DOI] [PubMed] [Google Scholar]
- Pérez F, Nadal M, Navarro-Ortega A, Fàbrega F, Domingo JL, Barceló D, et al. 2013. Accumulation of perfluoroalkyl substances in human tissues. Environment International 59:354–362. [DOI] [PubMed] [Google Scholar]
- Pijnenborg R, Anthony J, Davey DA, Rees A, Tiltman A, Vercruysse L, van Assche L. 1991. Placental bed spiral arteries in the hypertensive disorders of pregnancy. British Journal of Obsetrics and Gynaecology. 98:648–655. [DOI] [PubMed] [Google Scholar]
- Pinheiro TV, Brunetto S, Ramos JGL, Bernardi JR, Goldani ML. 2016. Hypertensive disorders during pregnancy and health outcomes in the offspring: A systematic review. Journal of Developmental Origins of Health and Disease 7:391–407. [DOI] [PubMed] [Google Scholar]
- Pizzurro DM, Seeley M, Kerper LE, Beck BD. 2019. Interspecies differences in perfluoroalkyl substances (pfas) toxicokinetics and application to health-based criteria. Regulatory Toxicology and Pharmacology 106:239–250. [DOI] [PubMed] [Google Scholar]
- Plunkett RJ. 1941. Tetrafluoroethylene polymers. Vol. US223065A. US:Kinetic Chemicals, Inc. [Google Scholar]
- Plunkett RJ. 1986. The history of polytetrafluoroethylene: Discovery and development In: High performance polymers: Their origin and development. Springer, 261–266. [Google Scholar]
- Porterfield SP. 1994. Vulnerability of the developing brain to thyroid abnormalities: Environmental insults to the thyroid system. Environmental Health Perspectives 102 Suppl 2:125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quist EM, Filgo AJ, Cummings CA, Kissling GE, Hoenerhoff MJ, Fenton SE (2015). Hepatic mitochondrial alteration in CD-I mice associated with prenatal exposures to low doses of perfluorooctanoic acid (PFOA). Toxicologic pathology, 43(4), 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramhøj L, Hass U, Boberg J, Scholze M, Christiansen S, Nielsen F, et al. 2018. Perfluorohexane sulfonate (pfhxs) and a mixture of endocrine disrupters reduce thyroxine levels and cause antiandrogenic effects in rats. Toxicological Sciences 163:579–591. [DOI] [PubMed] [Google Scholar]
- Rhee CM. 2016. The interaction between thyroid and kidney disease: An overview of the evidence. Current Opinions in Endocrinology Diabetes and Obesity 23:407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich N 2016. The lawyer who became dupont’s worst nightmare. The New York Times. [Accessed 16 January 2016], URL: https://www.nytimes.com/2016/01/10/magazine/the-lawyer-who-became-duponts-worst-nightmare.html
- Rijs KJ, Bogers RP. 2017. Pfoa and possible health effects: A review of scientific literature. RIVM Report 2017-0086. DOI: 10.21945/RIVM-2017-0086. URL: https://rivm.openrepository.com/handle/10029/620851 [DOI] [Google Scholar]
- Rinaudo P, Wang E. Fetal programming and metabolic syndrome. 2012. Annual review of physiology. 74:107–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risnes KR, Romundstad PR, Nilsen TI, Eskild A, Vatten LJ. 2009. Placental weight relative to birth weight and long-term cardiovascular mortality: Findings from a cohort of 31,307 men and women. American Journal of Epidemiology 170:622–631. [DOI] [PubMed] [Google Scholar]
- Rosen MB, Thibodeaux JR, Wood CR, Zehr RD, Schmid JE, Lau CJT. 2007. Gene expression profiling in the lung and liver of pfoa-exposed mouse fetuses. Toxicology 239:15–33. [DOI] [PubMed] [Google Scholar]
- Russell MH, Nilsson H, Buck RCJC. 2013. Elimination kinetics of perfluorohexanoic acid in humans and comparison with mouse, rat and monkey. Chemosphere 93:2419–2425. [DOI] [PubMed] [Google Scholar]
- Sagiv SK, Rifas-Shiman SL, Fleisch AF, Webster TF, Calafat AM, Ye X, et al. 2018. Early-pregnancy plasma concentrations of perfluoroalkyl substances and birth outcomes in project viva: Confounded by pregnancy hemodynamics? American Journal of Epidemiology 187:793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakr CJ, Kreckmann KH, Green JW, Gillies PJ, Reynolds JL, Leonard RC. 2007. Cross-sectional study of lipids and liver enzymes related to a serum biomarker of exposure (ammonium perfluorooctanoate or apfo) as part of a general health survey in a cohort of occupationally exposed workers. Journal of Occupational and Environmental Medicine 49:1086–1096. [DOI] [PubMed] [Google Scholar]
- Salafia CM, Charles AK, Maas EM. 2006. Placenta and fetal growth restriction. Clinical Obstetrics and Gynecology 49:236–256. [DOI] [PubMed] [Google Scholar]
- Sanders JP, Van der Geyten S, Kaptein E, Darras VM, Kuhn ER, Leonard JL, et al. 1997. Characterization of a propylthiouracil-insensitive type i iodothyronine deiodinase. Endocrinology 138:5153–5160. [DOI] [PubMed] [Google Scholar]
- Savitz DA, Stein CR, Bartell SM, Elston B, Gong J, Shin H-M, et al. 2012. Perfluorooctanoic acid exposure and pregnancy outcome in a highly exposed community. Epidemiology 23:386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitz DA, Stein CR, Elston B, Wellenius GA, Bartell SM, Shin HM, et al. 2012b. Relationship of perfluorooctanoic acid exposure to pregnancy outcome based on birth records in the mid-ohio valley. Environmental Health Perspectives 120:1201–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seacat AM, Thomford PJ, Hansen KJ, Clemen LA, Eldridge SR, Elcombe CR, et al. 2003. Subchronic dietary toxicity of potassium perfluorooctanesulfonate in rats. Toxicology 183:117–131. [DOI] [PubMed] [Google Scholar]
- Shah S 2020. Hypertensive disorders in pregnancy In: Obstetric and gynecologic nephrology: Women’s health issues in the patient with kidney disease, (Sachdeva M, Miller I, eds). Cham:Springer International Publishing, 11–23. [Google Scholar]
- Shastri S, Sarnak MJ. 2011. Chronic kidney disease: High egfir and mortality: High true gfr or a marker of frailty? Nature Reviews Nephrology 7:680–682. [DOI] [PubMed] [Google Scholar]
- Sonkar R, Kay MK, Choudhury M. PFOS modulates interactive epigenetic regulation in first-trimester human trophoblast cell line HTR-8/SVneo. 2019. Chemical research in toxicology. 32:2016–27. [DOI] [PubMed] [Google Scholar]
- Stanifer JW, Stapleton HM, Souma T, Wittmer A, Zhao X, Boulware LE. 2018. Perfluorinated chemicals as emerging environmental threats to kidney health: A scoping review. Clinical Journal of American Society of Nephrology 13:1479–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starling AP, Adgate JL, Hamman RF, Kechris K, Calafat AM, Ye X, et al. 2017. Perfluoroalkyl substances during pregnancy and offspring weight and adiposity at birth: Examining mediation by maternal fasting glucose in the healthy start study. Environmental Health Perspectives 125:067016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenland K, Woskie S. 2012. Cohort mortality study of workers exposed to perfluorooctanoic acid. American Journal of Epidemiology 176:909–917. [DOI] [PubMed] [Google Scholar]
- Stein CR, Savitz DA, Dougan M. 2009. Serum levels of perfluorooctanoic acid and perfluorooctane sulfonate and pregnancy outcome. American Journal of Epidemiology 170:837–846. [DOI] [PubMed] [Google Scholar]
- Suh CH, Cho NK, Lee CK, Lee CH, Kim DH, Kim JH, et al. 2011. Perfluorooctanoic acid-induced inhibition of placental prolactin-family hormone and fetal growth retardation in mice. Molecular and Cellular Endocrinology 337:7–15. [DOI] [PubMed] [Google Scholar]
- Sun M, Arevalo E, Strynar M, Lindstrom A, Richardson M, Kearns B, et al. 2016. Legacy and emerging perfluoroalkyl substances are important drinking water contaminants in the cape fear river watershed of north carolina. Environmental Science & Technology Letters 3:415–419. [Google Scholar]
- Sunderland EM, Hu XC, Dassuncao C, Tokranov AK, Wagner CC, Allen JG. 2019. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (pfass) and present understanding of health effects. Journal of Exposure Science & Environmental Epidemiology 29:131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilagyi JT, Freedman AN, Kepper SL, Keshava AM, Bangma JT, Fry RC. 2020. Per-and Polyfluoroalkyl Substances Differentially Inhibit Placental Trophoblast Migration and Invasion In Vitro. Toxicological Sciences. 175:210–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Ishida S, Hirata-Koizumi M, Ono A, Hirose A. 2014. Repeated dose and reproductive/developmental toxicity of perfluoroundecanoic acid in rats. The Journal of Toxicological Sciences 39:97–108. [DOI] [PubMed] [Google Scholar]
- Thornburg KL, O'Tierney PF, Louey S. 2010. Review: The placenta is a programming agent for cardiovascular disease. Placenta S54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United States Department of Defense (US DOD). 2000. Minutes of the DOD AFFF environmental meeting. URL: https://www.documentcloud.org/documents/4341546-DOC-605.html
- United States Environmental Protection Agency (US EPA). 2006. 2010/2015 pfoa stewardship program. URL: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/fact-sheet-20102015-pfoa-stewardship-program [accessed July 7, 2019].
- Vanden Heuvel JP, Thompson JT, Frame SR, Gillies PJ (2006). Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: a comparison of human, mouse, and rat peroxisome proliferator-activated receptor-α,-β, and-γ, liver X receptor-β, and retinoid X receptor-α. Toxicological Sciences, 92(2), 476–489. [DOI] [PubMed] [Google Scholar]
- Wang Z, Cousins IT, Scheringer M, Hungerbühler K. 2013. Fluorinated alternatives to long-chain perfluoroalkyl carboxylic acids (PFCAs), perfluoroalkane sulfonic acids (PFSAs) and their potential precursors. Environment international. 60:242–8. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang J, Du H, Xu L, Liu S, Yi J, et al. 2018. Perfluoroalkyl substances, glucose homeostasis, and gestational diabetes mellitus in Chinese pregnant women: A repeat measurement-based prospective study. Environment International 114:12–20. [DOI] [PubMed] [Google Scholar]
- Wang Y, Han W, Wang C, Zhou Y, Shi R, Bonefeld-Jorgensen EC, Yao Q, Yuan T, Gao Y, Zhang J, Tian Y. Efficiency of maternal-fetal transfer of perfluoroalkyl and polyfluoroalkyl substances. 2019. Environmental Science and Pollution Research. 26:2691–8. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rogan WJ, Chen P-C, Lien G-W, Chen H-Y, Tseng Y-C, et al. 2014. Association between maternal serum perfluoroalkyl substances during pregnancy and maternal and cord thyroid hormones: Taiwan maternal and infant cohort study. Environmental Health Perspectives 122:529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington Department of Health (WA DOH). 2020. PFAS. Washington State Department of Health; [Accessed 26 Feb 2020] URL:https://www.doh.wa.gov/CommunityandEnvironment/Contaminants/PFAS [Google Scholar]
- Washino N, Saijo Y, Sasaki S, Kato S, Ban S, Konishi K, et al. 2009. Correlations between prenatal exposure to perfluorinated chemicals and reduced fetal growth. Environmental Health Perspectives 117:660–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster GM, Rauch SA, Marie NS, Mattman A, Lanphear BP, Venners SA. 2016. Cross-sectional associations of serum perfluoroalkyl acids and thyroid hormones in us adults: Variation according to tpoab and iodine status (nhanes 2007–2008). Environmental Health Perspectives 124:935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster GM, Venners SA, Mattman A, Martin JW. 2014. Associations between perfluoroalkyl acids (pfass) and maternal thyroid hormones in early pregnancy: A population-based cohort study. Environmental Research 133:338–347. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Andersson PL, Lamoree MH, Leonards PE, van Leeuwen SP, Hamers T. 2009. Competitive binding of poly-and perfluorinated compounds to the thyroid hormone transport protein transthyretin. Toxicological Sciences 109:206–216. [DOI] [PubMed] [Google Scholar]
- Wen LL, Lin CY, Chou HC, Chang CC, Lo HY, Juan SH. 2016. Perfluorooctanesulfonate mediates renal tubular cell apoptosis through ppargamma inactivation. PLOS ONE 11 :e0155190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikström S, Lin PI, Lindh CH, Shu H, Bornehag CG. 2019. Maternal serum levels of perfluoroalkyl substances in early pregnancy and offspring birth weight. Pediatric Research. 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikström S, Lindh CH, Shu H, Bornehag C-G. 2019. Early pregnancy serum levels of perfluoroalkyl substances and risk of preeclampsia in Swedish women. Scientific Reports 9:9179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf CJ, Zehr RD, Schmid JE, Lau C, Abbott BD. 2010. Developmental effects of perfluorononanoic acid in the mouse are dependent on peroxisome proliferator-activated receptor-alpha. PPAR Research 2010:282896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Yin S, Liu Y, Chen F, Zhong Z, Li F, et al. 2019. Prenatal exposure to chlorinated polyfluoroalkyl ether sulfonic acids and perfluoroalkyl acids: Potential role of maternal determinants and associations with birth outcomes. Journal of Hazardous Materials 380:120867. [DOI] [PubMed] [Google Scholar]
- Yang L, Li J, Lai J, Luan H, Cai Z, Wang Y, et al. 2016a. Placental transfer of perfluoroalkyl substances and associations with thyroid hormones: Beijing prenatal exposure study. Scientific Reports 6:21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang Z, Shi Y, Li J, Wang Y, Zhao Y, et al. 2016b. Human placental transfer of perfluoroalkyl acid precursors: Levels and profiles in paired maternal and cord serum. Chemosphere 144:1631–1638. [DOI] [PubMed] [Google Scholar]
- Yu WG, Liu W, Jin YH. 2009. Effects of perfluorooctane sulfonate on rat thyroid hormone biosynthesis and metabolism. Environmental Toxicology and Chemistry 28:990–996. [DOI] [PubMed] [Google Scholar]
- Yu WG, Liu W, Liu L, Jin YH. 2011. Perfluorooctane sulfonate increased hepatic expression of oapt2 and mrp2 in rats. Archives of Toxicology 85:613–621. [DOI] [PubMed] [Google Scholar]
- Zhang C, Sundaram R, Maisog J, Calafat AM, Barr DB, Louis GM. 2015. A prospective study of prepregnancy serum concentrations of perfluorochemicals and the risk of gestational diabetes. Fertility and Sterility. 103:184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wang WS, Li WJ, Liu C, Wang Y, Sun K. 2015. Reduction of progesterone, estradiol and hCG secretion by perfluorooctane sulfonate via induction of apoptosis in human placental syncytiotrophoblasts. Placenta. 36:575–80. [DOI] [PubMed] [Google Scholar]