

Abstract
One of the major hurdles for cancer immunotherapy is the host’s innate antiviral defense mechanisms. They include innate immune cells, such as natural killer (NK) cells and macrophages, which can be recruited within hours to the site of injection to clear the introduced oncolytic viruses. Here, we report a strategy to redirect these infiltrating innate immune cells to attack tumor cells instead by arming herpes simplex virus (HSV)-derived oncolytic viruses with secreted chimeric molecules that can engage these innate immune cells with tumor cells to kill the latter. These chimeric molecules have, at their N terminus, a custom-binding moiety for a tumor-associated antigen (TAA) and at their C terminus, protein L (PL) that binds to immunoglobulins (Igs). The binding of PL to Igs exposes the Fc to the Fc receptors on the surface of the innate immune cells, trigging them to attack the engaged tumor cells. In vitro and in vivo evaluation in a murine tumor model with limited permissiveness to oncolytic HSVs showed that arming the viruses with these chimeric molecules significantly boosts the killing effect and therapeutic activity. Moreover, our data also showed that the combined killing effect from the engaged innate immune cells and the oncolytic virus resulted in a more efficient stimulation of neoantigen-specific antitumor immunity than the virotherapy alone. Our data suggest that arming an oncolytic virus with this strategy represents a unique and pragmatic way of potentiating the oncolytic and immunotherapeutic effect of virotherapy.
Graphical Abstract
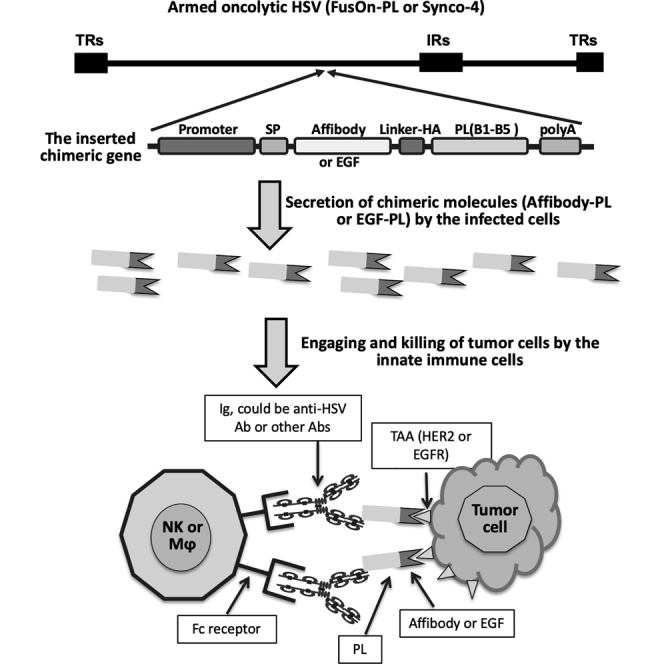
One major hurdle facing cancer immunotherapy is the host’s innate antiviral defense mechanisms. We report a strategy to redirect innate immune cells to attack tumor cells instead. Our data suggest that arming oncolytic viruses with this strategy represents a unique way of potentiating the oncolytic and immunotherapeutic effect of virotherapy.
Introduction
An oncolytic virus is defined by its ability to selectively replicate in and destroy tumor cells without harming normal cells. In order for an oncolytic virus to efficiently infect and lyse tumor cells, it has to overcome the host’s immune defense mechanisms that can be triggered by the introduced virotherapy. The innate immune system is the first line of the host’s defense against invading pathogens. It can be launched instantly as soon as an oncolytic virus is administered. As such, it presents as a significant barrier to cancer virotherapy.1 The major components of innate antiviral immunity include natural killer (NK) cells, macrophages, and interferons (IFNs). Indeed, studies have shown that depletion or functional inhibition of macrophages and NK cells during virotherapy can significantly improve the therapeutic activity from an oncolytic herpes simplex virus (HSV).2, 3, 4, 5 Studies by our own group have shown that arming an oncolytic HSV with the 18R gene of vaccinia virus, which can antagonize type I IFN activity, can boost the therapeutic effect of this virotherapy. NK cells were found to be recruited by oncolytic HSVs to the tumor site within hours after virus administration, leading to quick clearance of the introduced viruses and hence, a diminished therapeutic effect in a murine glioblastoma model.6 These and some other similar reports underscore the importance and necessity for curbing innate antiviral immunity during cancer virotherapy.5
The two major cellular components of innate antiviral immunity, NK cells and macrophages, also have the potential capability to kill malignant cells if properly activated and/or guided. Thus, it is plausible that a strategy could be developed to guide the infiltrating innate immune cells toward attacking tumor cells instead of clearing oncolytic viruses. With the consideration of that, for many patients, lack of a sufficient number of immune cells within tumor tissues is a major contributing factor to the inefficiency of cancer immunotherapy;7, 8, 9 it is particularly appealing to exploit the enhanced infiltration of these innate immune cells during virotherapy by converting them to tumor-targeted effector cells.
Antibody-dependent cell-mediated cytotoxicity (ADCC) is an important action mechanism of both NK cells and macrophages.10,11 ADCC is triggered by the binding of the Fc portion of immunoglobulins (Igs), which becomes exposed when multiple Ig molecules are in an aggregated multimeric form (e.g., within an immune complex), to the Fc receptors (FcRs) on the surface of innate immune cells, such as NK cells and macrophages. Protein L (PL) is an Ig-binding protein encoded by Peptococcus magnus.12 Unlike Proteins A and G, which bind to the Fc region of Ig, PL binds to the variable regions of the kappa light chain, and as such, it does not interfere with the engagement of the Fc fragment with its receptors.12 PL binds to the entire classes of Igs, including IgG, IgM, IgA, IgE, and IgD.12 Similar to Proteins A and G, PL also possesses multiple copies (up to five) of Ig-binding domains.13 This allows a single PL molecule to bind multiple units of Ig simultaneously, creating an aggregated multimeric form of Ig with the potential to induce FcR oligomerization and hence, the consequential activation of the innate immune cells though the FcR engagement.
We hypothesized that the above-mentioned features of PL could be exploited for the purpose of directing the infiltrating innate immune cells to attack tumor cells during virotherapy. To test our hypothesis, we armed both HSV-1- and HSV-2-based oncolytic viruses with a secreted chimeric molecule that can simultaneously engage innate immune cells with tumor cells through the incorporation of PL. The chimeric molecule is composed of an N-terminal binding moiety for a tumor-associated antigen (TAA) and a C-terminal PL. In vitro experiments demonstrate that the secreted chimeric molecule can actively engage NK cells and macrophages with TAA-expressing tumor cells, leading to efficient killing of the latter. In vivo evaluation in a murine tumor model with limited permissiveness to oncolytic HSV shows that oncolytic HSVs armed with the chimeric molecule can significantly enhance the therapeutic activity. Moreover, our data indicate that the combined killing effect from the engaged innate immune cells and the oncolytic virus resulted in a more efficient stimulation of the host’s antitumor immunity than the virotherapy alone. Together, our data suggest that arming an oncolytic virus with this strategy represents a viable way of potentiating the oncolytic and immunotherapeutic effect of virotherapy.
Results
Design and Assembly of Chimeric Molecules That Can Guide Innate Immune Cells to Attack Tumor Cells through a Series of Intermolecular Engagements
The composition of the chimeric molecules is depicted in Figure 1A. We initially designed the human epidermal growth factor receptor (EGFR) type 2 (HER2) affibody-PL construct, which contains a copy of the HER2 affibody as the TAA-binding moiety.14,15 Affibody molecules are short peptides of approximately 58 amino acids and are based on a three-α-helical Z-domain scaffold that can be selected from combinatorial libraries to bind to a particular protein target with strong affinity and specificity.16 The EGF-PL was designed subsequently, after positive data were obtained from the HER2 affibody-PL in both in vitro and in vivo studies. The purpose was to test if the positive results obtained from HER2 affibody-PL could be recapitulated with a similarly designed chimeric molecule that targets a different TAA and is inserted to a different oncolytic HSV. The TAA-binding moiety in EGF-PL is EGF, which can bind to the EGFR with high specificity. Previous studies have shown that a mutant form of EGF (m123) has a significantly enhanced affinity and faster binding kinetic than wild-type EGF.17 We thus decided to use this mutant form of EGF (mEGF) for the chimeric EGF-PL construct. In both constructs, the coding sequence from the five Ig-binding domains (B1–B5) was chosen from PL of Peptostreptococcus magnus.18 HER2 affibody or mEGF and PL sequences were fused together in frame for the construction of affibody-PL or EGF-PL, respectively. A signal peptide (Sp) was added to the N terminus to allow the chimeric molecules to be secreted in a soluble form.
Figure 1.

The Design of the Chimeric Molecules, Their Anticipated Action Mechanism, and Their Insertion into Viral Genomes
(A) The composition of the gene cassettes for the chimeric molecules HER2 affibody-protein L (PL) and EGF-PL. On the sequential order from left to right: Rous sarcoma virus long terminal repeat (RSV-LTR; as the promoter element), signal peptide (Sp), TAA-binding moiety (affibody for HER2 and EGF for EGFR), linker-HA tag (Linker-HA), the B1–B5 immunoglobulin (Ig)-binding domains of PL from Peptostreptococcus magnus (PL), and the polyadenylation signal (polyA). The actual length of the coding sequence of each component is not proportional to the size of the drawn box. (B) Perceived action mechanism of the chimeric molecules after being delivered to tumors by an oncolytic virus. The individual key components are labeled. The chimeric molecules can engage the infiltrating innate immune cells, including NK cells and macrophages (Mφ), with tumor cells through a series of intermolecular binding: affibody to HER or EGF to EGFR, PL to Igs (including anti-HSV Ab or other Abs), and Igs (via Fc region) to NK cells or macrophages (via Fc receptor). (C) Insertion of the chimeric genes into HSV-based oncolytic viruses. The affibody-PL chimeric gene was inserted into the region adjacent to the 5′ of the ICP10 gene of the HSV-2-based FusOn-H2, and the EGF-PL chimeric gene was inserted into the UL46 and UL47 intergenic region of the HSV-1-based Synco-2D. Both were carried out by homologous recombination, followed by plaque purification. (D) Comparison of virus replication in CT26, CT26-HER2, and CT26-EGFR cells. Cells were seeded in 24-well plates in triplicates and were infected with the viruses at either 0.1 or 1 PFU/cell. Cells were harvested 24 or 48 h later, and the virus yield was determined by a plaque assay on Vero cells. ∗p < 0.05 as compared with FusOn-H2.
The schematic diagram in Figure 1B illustrates the action mechanism of the chimeric molecules. Local administration of the armed oncolytic viruses will bring all of the components together within the tumor microenvironment. The linchpin to trigger the illustrated chain intermolecular reaction is the soluble form of the chimeric molecules—affibody-PL or EGF-PL, which can simultaneously bind to HER2- or EGFR-expressing tumor cells. The PL in the chimeric molecules can bind to the Igs, which can either be anti-HSV antibodies or any other Igs. The Fc region of the engaged Igs can then bind to the FcRs on the surface of NK cells or macrophages, resulting in the activation of these innate immune cells and the consequential selective killing of the tumor cells. This strategy is envisaged to potentiate the overall antitumor effect of an oncolytic virotherapy on two fronts. First, it produces additional bystander antitumor activity by engaging innate immune cells with tumor cells. Second, it diverts two of the major components of antiviral immunity away from clearing the introduced oncolytic virus—the innate immune cells and the neutralizing antibodies—allowing virotherapy to exhibit maximal oncolytic effects.
The genes encoding the chimeric molecules were inserted into the genome of oncolytic HSVs through standard homologous recombination, as we have used in our previous studies.19, 20, 21, 22, 23 The EGF-PL chimeric gene cassette was inserted into the intergenic region of UL46 and UL47 genes in the genome of Synco-2D, an HSV-1-based oncolytic virus.24 The affibody-PL chimeric gene cassette was inserted into the 5′ mutated region of the ICP10 gene (mICP10) in the genome of FusOn-H2, which is a HSV-2-based oncolytic virus and was constructed by deleting the N-terminal region of the ICP10 gene in the HSV-2 genome.20 These newly constructed oncolytic HSV-1 and HSV-2 are designed Synco-4 and FusOn-PL, respectively. Correct insertion of the transgene in Synco-4 and FusOn-PL was confirmed by PCR and DNA sequencing (data not shown).
The replication of Synco-4 and Syco-2D was compared in CT26 and CT26-EGFR cells, and the replication of FusOn-PL and FusOn-H2 was compared in CT26 and CT26-HER2 cells. Cells were infected with the viruses at 0.1 and 1 plaque-forming unit (PFU)/cell and harvested 24 and 48 h later for titration of the virus yield. The results in Figure 1D showed that stable transduction of CT26 cells with either HER2 or EGFR did not alter the replication of the respective virus pairs, i.e., comparison of Synco-2D and Synco-4 replication in CT26 and CT26-EGFR cells and FusOn-H2 and FusOn-PL replication in CT26 and CT26-HER2 cells. However, the new viruses seem to replicate less efficiently than the respective parental viruses in both CT26 and HER2- or EGFR-transduced cells. This is particularly so for FusOn-PL, when cells were infected with the virus at a lower multiplicity of infection. This observation is in line with our empirical experience that insertion of an additional genetic payload to a HSV genome usually impacts the virus replication efficiency.
In Vitro Characterization of the New Oncolytic HSVs and the Chimeric Molecules They Produced
First, we confirmed transgene expression from both viruses by western blotting. Both affibody-PL and EGF-PL chimeric molecules were readily detected in the supernatants (S/Ns) harvested from cells infected with these two viruses (Figure 2A), indicating that they are readily produced during virus infection and efficiently secreted into the milieu. Next, we measured the ability of the chimeric molecules to selectively bind to tumor cells that express the prospective TAAs. For affibody-PL, we tested the binding of the molecule to three human tumor cell lines that express different levels of HER2. Among them, SKOV3 and MCF7 have been shown to express high and medium levels of HER2, respectively, whereas MDA-MB-231 is a triple-negative breast cancer cell line that does not express any HER2.25, 26, 27 The results from flow cytometry analyses show that whereas no significant binding activity was detected in MDA-MB-231 cells (3.34%), affibody-PL binds to MCF7 and SKOV3 cells with increasing intensity (at 49.9% and 71.9%, respectively) (Figure 2B). We also examined the binding specificity of affibody-PL to a murine colon carcinoma cell line that was stably transduced with HER2.28 The result showed that whereas affibody-PL showed no detectable binding to the parental cell line (CT26), it bound to CT26-HER2 cells with high efficiency (76.6%) (Figure 2B). We tested binding of EGF-PL to a murine cell line (CT26) and its derivative, CT26-EGFR, which was established in our own laboratory by stably transducing the parental CT26 cells with the human EGFR gene. The graph on the left of Figure 2C confirmed that CT26-EGFR cells, but not the parental CT26 cells, express high levels of EGFR. The graph on the right of Figure 2C showed that the supernatant containing EGF-PL (from Synco-4) strongly binds to CT26-EGFR. These were in contrast to the control supernatant (from Synco-2D), which does not show any binding activity to the EGFR-expressing cells. Together, these results demonstrate that both chimeric molecules can bind to the prospective TAAs with strong selectivity and affinity.
Figure 2.

In Vitro Characterization of Chimeric Molecules Produced by the Armed Viruses
The supernatants harvested from virus-infected cells were passed through 0.22 μM filters before they were used for the experiments. (A) Western blot detection of transgene expression, with anti-HA tag IgG as the first antibody. (B and C) Flow cytometry analysis of the selective binding activity of affibody-PL (B) and EGF-PL (C) to tumor cells. Tumor cells were sequentially incubated with the following: (1) chimeric molecule-containing or control supernatant, (2) anti-HA antibody of mouse origin, and (3) FITC- or PE-conjugated goat anti-mouse IgG. The cells were then subject to flow cytometry analysis.
Next, we tested one of the two chimeric molecules, affibody-PL, to determine if it indeed can guide the innate immune cells to kill TAA-expressing tumor cells. To more closely mimic the actual in vivo situation, we chose to use peripheral blood mononuclear cells (PBMCs) as the source of innate immune cells, which contain NK cells and monocytes at the range of 5%–20% and 10%–30%, respectively.29 We mixed SKOV3 cells with PBMCs in the presence of Igs and affibody-PL-containing supernatant or the control supernatant. 24 h later, PBMCs were washed away, and the remaining adherent tumor cells were examined by direct visualization under a microscope after staining with 0.1% crystal violet-ethanol solution (Figure 3A). The killing effect on tumor cells was further quantitated by lysing the cells in 2% SDS and measuring the dye release (Figure 3B). The results showed that the presence of affibody-PL significantly increased the killing of tumor cells when compared to the mixture without this chimeric molecule. This effect was particularly obvious when the effector-to-target (E:T) ratio was relatively low (10:1). At this E:T ratio, the well with the control supernatants showed very little tumor cell killing, whereas nearly 60% of cells were killed in the well with the supernatant that contained the affibody-PL molecule. With the E:T ratio at 20 to 1, there was a notable increase in background killing in both wells with the two control supernatants. However, the tumor cell killing in the well with affibody-PL was further increased, to over 80 percent. A similar result was obtained from EGF-PL when it was assessed on CT26-EGFR cells (data not shown). Together, these results suggest the PL-containing chimeric molecules possess the anticipated capability to guide immune cells to kill tumor cells through a series of intermolecular engagements.
Figure 3.

The Ability of Affibody-PL Guides PBMCs to Kill HER2-Expressing Tumor Cells
PBMCs were mixed with SKOV3 tumor cells at the effector-to-target (E:T) ratio of either 10 (10:1) or 20 (20:1) in the presence of 5 μg/mL of IgG and medium, the supernatant harvested from a control vector (control S/N), or affibody-PL-containing supernatant (affibody-PL). The remaining living cells were stained with 0.1% crystal violet-ethanol solution 24 h later. (A) A representative micrograph from each well of the three different preparations. (B) Quantification of tumor cell killing. The stained tumor cells were lysed with 2% SDS for measurement of the released dye. The percentage of tumor cell killing was calculated by dividing the reading of cells in the well without adding PBMCs (and others) with the readings from each of the three wells. ∗p < 0.05 as compared with medium and control S/N.
Therapeutic Evaluation of the Armed Viruses In Vivo
To evaluate the therapeutic effect of the EGF-PL-armed Synco-4 and to compare it with that of the parental Synco-2D, we implanted CT26-EGFR tumor cells subcutaneously into the right flank of immune-competent BALB/c mice. Part of the reason for choosing this murine colon tumor model is that it is only marginally sensitive to the therapeutic effect of oncolytic HSV. This would allow the therapeutic benefit from the incorporated EGF-PL to be fully appreciated. Once tumors reached the approximate size of 5 mm in diameter, mice were grouped to receive intratumoral injection of either the parental Synco-2D or Synco-4 at the dose of 2 × 107 PFU. For the sake of fully evaluating the therapeutic benefit of the incorporated EGF-PL, only a single injection with a fixed virus dose was given. Another group received PBS as a mock control. The results from the periodic measurement of tumor growth showed that the parental Synco-2D had a negligible effect on the tumor growth, whereas administration of Synco-4 produced a significantly better therapeutic effect than the other two groups at days 14 and 21 after the initiation of oncolytic virus injection (Figure 4A).
Figure 4.

Therapeutic Evaluation of the Armed Viruses in a Murine Colon Cancer Model
(A) Therapeutic evaluation of Synco-4 and comparison with parental Synco-2D against CT26-EGFR tumor. Treatment groups include Synco-2D, Synco-4, and PBS. (B) Therapeutic evaluation of FusOn-PL and comparison with parental FusOn-H2 against CT26-HER2 tumor. Treatment groups include FusOn-H2, FusOn-PL, and PBS. ∗p < 0.05 as compared with the unarmed virus or PBS.
To evaluate the therapeutic effect of FusOn-PL and to compare it with that of the parental FusOn-H2, we again chose to use the murine CT26 colon tumor model. The only difference is that we used a CT26 cell line that was stably transduced with the HER2 gene for this experiment.28 Tumor implantation and subsequent treatment were performed in the same manner as the experiment in Figure 4A. To monitor NK cell infiltration during virotherapy, two mice from each group were euthanized at day 3 after treatment to collect tumor tissues for further analysis by immunohistochemical staining. The remaining animals were kept for 4 weeks to evaluate the therapeutic effect by monitoring tumor size. The results from therapeutic evaluation showed that administration of FusOn-H2 only slightly slowed down tumor growth when compared with the PBS control. In contrast, FusOn-PL treatment effectively halted tumor growth for an extended period of time. The treated tumors were significantly smaller than those in the FusOn-H2-treated group. By the end of the experiment, 50% of the mice in the FusOn-PL-treated group were essentially tumor free, whereas no tumor-free animals were observed in the other treatment groups. Together, these results demonstrate that incorporation of these chimeric molecules into oncolytic HSVs can enhance the antitumor effect of the virotherapy, and this potentiation strategy may be particularly beneficial to patients with the tumors that are relatively resistant to the direct oncolytic effect of the applied virus.
Immunohistochemical staining on the collected tumor tissues shows that NK cells were only scarcely detected in the PBS-treated control tumors (Figure 5A), a finding consistent with the reports in the literature that NK cells were mostly detected at the low frequency of 1–3 per microscopic intratumoral field.30 However, NK cells were detected at a much higher frequency in tumors treated with both oncolytic viruses (Figure 5A), consistent with the reports that HSV infection can trigger significant infiltration of NK cells to the infection site.31, 32, 33 Moreover, the presence of NK cells in tumor tissues treated with FusOn-PL was particularly profound, indicating that local release of the armed chimeric molecule might have led to a further increase of NK cells in the treated tumor. To determine the proliferation status of NK cells in the collected tumor tissues, we doubly stained the tumor tissues for both an NK cell marker (NCR1) and Ki67 protein. The results showed that there was no significant Ki67 staining on NK cells in tumor tissues treated either with FusOn-H2 or FusOn-PL (Figure 5B). Thus, the increased presence of NK cells during FusOn-PL treatment is probably due to a positive feedback loop on their recruitment, rather than stimulation on NK proliferation.
Figure 5.

NK Cell Infiltration during Virotherapy
Tumor tissues were collected from two mice from the experiment described in Figure 4B at day 3 after therapeutic injection. (A and B) The tumor tissues were used for either staining for NCR1 with an HRP polymer kit (A) or immunofluorescent staining for NCR1 and Ki67, either individually or in combination (B). Positively stained NK cells are indicated with white arrowheads, and cells positively stained for Ki67 are indicated with white arrows. Quantitative enumeration on the positively stained NK cells in (A) showed that there is a significant difference on NK cell count between FusOn-PL- and FusOn-H2-treated tumor tissues.
Tumor Destruction by FusOn-PL Can Stimulate Neoantigen-Specific Antitumor Immunity
We subsequently challenged the tumor-free mice in the FusOn-PL-treated group in the experiment presented in Figure 4B with fresh CT26-HER2 tumor cells by implanting the cells in the left flank. All mice were completely protected, and no trace of tumor growth was detected from the challenge for more than 4 weeks. The tumor challenge was not done to mice in the other two treatment groups, as all of the mice had to be euthanized due to large tumor burden. The complete absence of tumor formation from the challenge of fresh tumor cells indicated that a robust antitumor immunity might have been generated from FusOn-PL treatment.
Tumor cells contain frequent point mutations that can result in neoantigen formation.34,35 The induction of immune responses to these neoantigens is particularly appealing for cancer immunotherapy, as theoretically, it is strictly tumor specific. The neoantigen profile of CT26 cells has recently been reported.36 To determine if FusOn-PL-mediated tumor cell killing could induce anti-neoantigen immunity, we chose 3 of the mutated peptides that were predicted by Kreiter et al.36 to contain major histocompatibility complex (MHC) class I neoantigen epitopes (Figure 6A) and examined if any cytotoxic T cells specific for these neoantigens could be detected in these protected mice. The splenocytes collected from these three mice were stimulated, with or without these peptides. The neoantigen-specific T cell response was determined by enzyme-linked immune-absorbent spot (ELISpot) assay, as described in Materials and Methods. The results show that splenocytes from one mouse reacted to a single peptide from this assay (Figure 6B), indicating that induction of a T cell response to these MHC class I neoantigen epitopes during FusOn-PL virotherapy could be detected, albeit infrequently.
Figure 6.

Detection of T Cells Specific for MHC Class I or II Neoantigen Peptides
(A) The chosen MHC I neoantigen peptides for screening. The table contains information on the chosen MHC I neoantigen peptides, as reported by Kreiter et al.36 (B) IFN-γ ELISpot assay on T cells stained positive for an MHC I neoantigen peptide. The splenocytes were collected from the tumor-free mice in the experiment shown in Figure 4B that had received tumor cell challenge. The photos show representative areas of the wells in the ELISpot assay from the neoantigen peptides or the no-peptide control. (C) Therapeutic effect from an additional in vivo experiment on the CT26-HER2 tumor model. Treatment groups include FusOn-H2, FusOn-PL, and PBS. (D) The chosen MHC II neoantigen peptides for detection. The peptide information for the chosen MHC II neoantigen peptides is from the report by Kreiter et al.36. (E) Quantification of IFN-γ ELISpot assays on T cells stained positive for the indicated MHC II neoantigen peptides. The controls include no peptide or an unrelated peptide (the ovalbumin MHC II peptide [OVA 323–339]). Mix, all four peptides mixed together. ∗p < 0.05 as compared with FusOn-H2; +p < 0.01 as compared with PBS.
To further characterize the neoantigen-specific immune response during virotherapy, we essentially repeated the in vivo animal experiment shown in Figure 4B. The only difference was that we euthanized all animals on day 21 after virotherapy, when all animals were still alive, and collected the spleens from the animals for analysis of neoantigen-specific T cell immunity. The therapeutic data collected from this experiment showed that even with this relatively short treatment period, FusOn-PL was still significantly more effective than FusOn-H2 at inhibiting tumor growth at day 21 after the start of virotherapy (Figure 6C), with 40% of mice shown to be tumor free by the end of the experiment.
For measurement of neoantigen-specific immunity, we included the same three MHC class I peptides listed in Figure 6A. We also chose 4 peptides that contain MHC class II neoantigen epitopes (Figure 6D), as previous studies with three different murine tumor models (including the CT26 tumor model we used in this study) have shown that the majority of the immunogenic mutanomes could be efficiently recognized by CD4+ T cells.36 For the three MHC class I peptides, no significant increase in ELISpot staining was detected for any animals in this experiment (data not shown). This reinforces the observation made from the previous experiment shown in Figure 6B that T cell immunity to MHC class I neoantigens was only infrequently induced during virotherapy. In contrast, significant increases in ELISpot staining were detected for all four MHC class II neoantigen peptides in samples from animals treated with FusOn-PL, although the magnitude of the response varies among the individual neoantigen epitopes (Figure 6E). Detailed ELISpot results for each individual animal from all three groups can be found in Figure S1. Taken together, these data demonstrate that tumor destruction by FusOn-PL can induce poly-neo-epitope T cells against predominately MHC class II neoantigens, which is in agreement with the report by Kreiter et al.36 for similar findings in both B16 and CT26 tumor models.
Discussion
The sole oncolytic virus that has been approved by the US Food and Drug Administration (FDA) for clinical application, Imlygic (T-VEC), has shown measurable and in some cases, durable therapeutic efficacy in a relatively small percentage of melanoma patients.37 As such, it appears that the therapeutic efficacy of this and other oncolytic viruses should and can be further improved. One improvement strategy is to potentiate the ability of virotherapy to induce antitumor immunity. Several approaches have been reported in this attempt. One popular approach is to incorporate immune-stimulatory genes into the viral genome to enhance tumor antigen presentation for induction of T cell-mediated antitumor immunity.38, 39, 40 Other approaches include combining virotherapy with immune-checkpoint inhibitors41 or with adoptive transfer of tumor-specific T cells.42, 43, 44 Here, we report an alternative strategy designed to arm an oncolytic virus with a chimeric molecule that can guide the infiltrating innate immune cells to destroy tumor cells. One of the unique advantages of this strategy is that it exploits the extensive infiltration of the innate immune cells during virotherapy. Thus, this strategy directly addresses one of the major obstacles facing cancer virotherapy—lack of sufficient immune cell presence in solid tumors.45,46 Indeed, immunohistochemistry staining on tumor tissues treated with either FusOn-H2 or FusOn-PL revealed extensive infiltration of NK cells. Most interestingly, the presence of NK cells was further increased with FusOn-PL treatment, indicating the existence of a positive feedback loop of a chemoattractant effect on NK cells from the affibody-PL engagement activity. Consequently, our in vivo data from three separate experiments show that this arming strategy can contribute to additional and hence, enhanced antitumor activities. Indeed, over one-half of the tumor-bearing mice treated with FusOn-PL was completely tumor free by the end of the experiment, whereas no tumor-free mice were detected in other groups. Moreover, subsequent challenge of the tumor-free mice with fresh tumor cells failed to initiate tumor growth, indicating the possibility that a robust antitumoral immunity was generated by the applied virotherapy in these mice.
There has been increasing interest in recent years in exploring neoantigens for cancer immunotherapy.47 Unlike TAAs, neoantigens are derived from nonsynonymous mutations in the tumor cell genome and are thus strictly tumor specific.47, 48, 49 However, one of the challenges facing neoantigen-based immunotherapy is that the neoepitopes are usually not shared among cancer patients. The current approach of first identifying these neoantigens by exome sequencing, followed by synthesizing and delivering the antigenic epitopes to each individual patient, is cumbersome and costly and can only be applied to cancer patients on a case-by-case basis.47,49,50 In principle, oncolytic virotherapy would offer a simple means to release these neoantigens in individual patients, ensuring their efficient and timely presentation to the host’s immune system without the need to identify and then synthesize them in vitro. However, whether oncolytic virotherapy possesses such a capability and how to enable it to have such a capability have not yet been extensively investigated. We examined the tumor-free mice from the group treated with FusOn-PL for indication of T cells specific for potential neoantigen epitopes contained within CT26 tumor cells. Whereas T cells reacting to MHC class I neoantigen peptides were only detected sporadically, MHC class II neoantigen peptides were found to be more frequently recognized by T cells. These results are consistent with the report by Kreiter et al.,36 which showed that therapeutic immune responses to cancer were predominately toward MHC class II neoantigen epitopes when they were examined in several syngeneic murine tumor models, including the CT26 tumor.
One conceivable mechanism for the enhanced neoantigen presentation promoted by the chimeric molecule during FusOn-PL treatment is through the reported ability of NK cells in releasing C-C motif chemokine ligand 5 (CCL5) and XC-chemokine ligand 1 (XCL1), which can promote recruitment of cDC1 subset of dendritic cells (DCs) to the tumor site.51 Tumor-infiltrating NK cells are also found to be the predominant producers of Fms-related tyrosine kinase 3 ligand (FLT3L) in the tumor microenvironment,52 which can further promote the development and proliferation of the recruited cDC1.53 cDC1 is considered to have the unique capability in antigen cross-presentation and T cell cross-priming in cancer immunology and immunotherapy.54,55 Hence, incorporation of affibody-PL or EGF-PL into an oncolytic HSV forms a tactical combination, as the oncolysis by the virus can release abundant neoantigens that can be subsequently cross presented to T cells by the recruited cDC1 through the engagement of the recruited and activated NK cells. Obviously, the limited data we have collected and presented here on this observation are exploratory in nature, and clearly more investigation needs to be conducted on this in the near future, including confirmation of the actual contribution of the induced neoantigen-specific immunity to the overall efficacy of virotherapy and long-lasting therapeutic benefit.
Affibody-PL was the first chimeric molecule we designed for the intended purpose of diverting the infiltrating NK and other innate immune cells to attack tumor cells. The nature of this molecule, with the HER2 affibody as the targeting moiety, limits its application to tumor cells overexpressing HER2. However, this same strategy can be readily adapted to target other tumors by replacing the HER2 affibody with another binding moiety, such as a ligand or a single-chain antibody with binding specificity for other TAAs. To prove this, we subsequently designed a second chimeric molecule, EGF-PL, in which the HER2 affibody was replaced with the binding domain of EGF to EGFR. We incorporated this molecule into an HSV-1-based oncolytic virus. Although the potency of this HSV-1-based oncolytic virus (Synco-2D) is not quite as high as that of the HSV-2-based FusOn-H2,20 arming it with the EGF-PL molecule similarly enhanced its therapeutic effect in a similar manner as seen for the affibody-PL-armed FusOn-PL. Thus, our data suggest that this arming strategy can be applied to different designs of the chimeric molecule and to different backbones of oncolytic viruses, with the potential for treating a variety of malignant diseases.
One major hurdle facing virotherapy is the host’s innate antiviral immune mechanism that can be instantly triggered when virotherapy is given. The triggered antiviral innate immunity can severely limit oncolytic virus replication, which is the tenet of virotherapy. Consequently, it has been reported that ablating the function or directly depleting innate antiviral immune cells, such as macrophages and NK cells, can significantly improve the therapeutic activity of oncolytic virotherapy.2,4 Antibodies to the oncolytic virus represent another major limiting factor to the full therapeutic effect of virotherapy, as they can neutralize the oncolytic virus during its spread within tumor tissues. Our strategy of arming an oncolytic virus with the PL-containing chimeric molecules engages both the antiviral antibodies (as well as other Igs) and the innate immune cells and guides them to attack tumor cells. This action theoretically diverts these two immune components away from clearing the oncolytic virus, thus allowing it to fully replicate and spread within tumor tissues. As such, this strategy not only improves the killing effect but also potentiates the oncolytic effect of virotherapy per se.
Materials and Methods
Cell Lines
CT26 is a murine colon cancer cell line, and CT26-HER2 was derived from it by stable transduction of the gene encoding human HER2.28 CT26-EGFR cells were established from CT26 cells by stably transducing the cells with a lentiviral vector that contains EGFR extracellular and transmembrane domains without the intracellular sequence. These cells were cultured in in RPMI 1640 (Invitrogen, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS; purchased from Gemini Bioproducts, West Sacramento, CA, USA), 100 units/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). SKOV3 (HER2-high, ovarian cancer), MCF7 (HER2-medium, breast cancer), and MDA-MB-231 (HER2-negatve breast cancer), HEK293, and African green monkey kidney (Vero) cell lines were obtained from American Type Culture Collection (Rockville, MD, USA) and cultured in DMEM containing 10% FBS. All cells were incubated at 37°C in a humidified atmosphere saturated with 5% CO2.
Oncolytic HSV Construction
The composition of the chimeric molecules of HER2 affibody-PL and EGF-PL is depicted in Figure 1A. The sequence encoding affibody-PL (including Sp, affibody, linker-hemagglutinin [HA] tag, and PL B1–B5 domains) was codon optimized for human expression and synthesized by GenScript (Piscataway, NJ, USA). The synthesized affibody-PL coding sequence was then inserted into pNEB-PKF-4 to replace the GFP gene in this plasmid. After this cloning, the affibody-PL coding sequence is driven by the Rous sarcoma virus long terminal repeat (RSV-LTR) promoter with the simian virus 40 (SV40) polyadenylation signal at the 3′ end, and the entire gene cassette is flanked by approximately 1 kb of homologous sequences that enable homologous recombination for the insertion of the transgene into the 5′ region of the mICP10 gene in the genome of FusOn-H2, as indicated in Figure 1C. The parental FusOn-H2 is an HSV-2-based oncolytic virus and was constructed by deleting the N-terminal region of the ICP10 gene in the HSV-2 genome. The details of its construction were described in our previous publication.20 Synco-4 was constructed in a similar fashion, except that the affibody in affibody-PL was replaced with EGF for the assembly of EGF-PL, and the transgene cassette was inserted into the intergenic region of the UL46 and UL47 genes in the genome of Synco-2D, which is an HSV-1-based oncolytic virus.24 The details of its construction have been reported in our previous publication.24
Western Blot Analysis
Culture supernatants containing the chimeric molecules (from transfection or infection of FusOn-PL, pCR-EGF-PL, or Synco-4) or without the chimeric molecules (from transfection or infection of FusOn-H2, empty vector, or Synco-2D) were mixed with an equal amount (20 μL) of Laemmli 2× buffer before being loaded into the wells of a 4%–20% SDS-PAGE gel, along with molecular weight markers after boiling for 5 min. After running for 1 to 2 h at 120 V, the gel was transferred to a polyvinylidene fluoride (PVDF) membrane at 30 V overnight in a cold room. All of the above materials were purchased from Bio-Rad (Hercules, CA, USA ). The transferred membrane was blocked for 1 h at room temperature (RT) using 3% BSA-Tris-buffered saline 0.1% Tween 20 (TBST) blocking solution and then incubated with rabbit anti-HA tag antibody (1:1,000 dilution) (Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C, followed by a 1-h incubation with anti-rabbit horseradish peroxidase (HRP) antibody (1:2,000 dilution) (Cell Signaling Technology, Danvers, MA, USA) at RT. HRP activity was visualized by soaking the membrane in a SuperSignal West Pico chemiluminescence substrate (Thermo Scientific). Afterward, the luminescence signals on the membrane were quantified with an Amersham Imager 600 (GE, Pittsburgh, PA, USA).
Flow Cytometry Detection of Chimeric Molecule-Binding Specificity
Supernatants were collected from cells transfected or infected with plasmids or oncolytic HSVs that either contained or did not contain the chimeric genes. The supernatants were collected 48 h after transfection or infection and were filtered to remove cell debris and virus particles. For affibody-PL-binding detection, supernatants of 100 μL were incubated for 30 min at 4°C with tumor cells. After washing 3 times with PBS, 1 μL of mouse anti-HA tag IgG (Sigma, St. Louis, MO, USA) in 2% FBS-PBS was incubated for another 30 min at 4°C, which was followed by incubation with 2 μL of fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (Sigma, St. Louis, MO, USA) at 4°C for 30 min. For the EGF-PL binding assay, tumor cells were incubated with 100 μL of the collected supernatants for 30 min at 4°C and followed by wash and then incubation with 2 μL of phycoerythrin (PE)-conjugated mouse anti-HA tag IgG (BioLegend, San Diego, CA, USA). The cells were subjected to an analysis by flow cytometry (BD Biosciences, San Jose, CA, USA).
In Vitro ADCC Assay
Human PBMCs were prepared from white blood cell concentrate (buffy coat) obtained from a human blood bank (Gulf Coast Regional Blood Center, Houston, TX, USA). Briefly, the buffy coat was mixed with an equal amount of 1× PBS before it was loaded onto Lymphoprep (STEMCELL Technologies, Vancouver, BC, Canada) for centrifugation for 30 min at 800 × g with the brake off at RT. The layer containing PBMCs at the plasma:Lymphoprep interface was collected and washed three times with PBS. The purified PBMCs were cocultured in 12-well plates with HER2-positive SKOV3 tumor cells at the ratio of either 10 (E:T = 1:10) or 20 (E:T = 1:20) in the presence of 5 μg/mL of human IgG and the indicated supernatants. 24 h later, PBMCs and the dead cells floating in the medium were removed, and the remaining living cells were stained with 0.1% crystal violet-ethanol solution. After micrographs were taken, the stained tumor cells were lysed with 2% SDS, and the released dye was measured at a 595-nM wavelength with a SpectraMax M5 plate reader (Molecular Devices, San Jose, CA, USA). The percentage of tumor cell killing was calculated by dividing the reading of cells in the well without adding PBMCs with the readings from each of the other wells.
In Vivo Animal Experiments
Immune-competent female BALB/c mice (4–6 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All animal experiments were approved by the university’s Institutional Animal Care and Use Committee (IACUC). For evaluation of the therapeutic efficacy of the affibody-PL-armed FusOn-PL, freshly harvested CT26-HER2 cells (2 × 105) were injected into the right flank of BALB/c mice. Mice were then randomly grouped (n = 12 for the experiment presented in Figure 4B, and n = 5 for the experiment presented in Figure 6C). Once tumors reached approximately 5 mm in diameter, mice received an intratumoral injection of either PBS or 1 × 107 PFU of FusOn-H2 or FusOn-PL in a volume of 100 μL. Two mice from each group were euthanized at day 3 after virotherapy to collect tumor tissues for measurement of NK cell infiltration. The rest of the mice were kept for 3 or 4 weeks to monitor tumor growth by measuring two perpendicular tumor diameters with a caliper. Tumor volume was calculated by the formula: tumor volume (cubic millimeter) = (length [millimeter]) × (width [millimeter])2 × 0.52. The tumor growth ratio was calculated by dividing the tumor volume measured at the indicated time with the tumor volume immediately before the start of treatment.
For evaluation of the therapeutic effect of the EGF-PL-armed Synco-4, CT26-EGFR cells (2 × 105) were implanted into the right flank of BALB/c mice. The animals were then randomly grouped (n = 5 per group). When tumors were about 5 mm in size, the mice received an intratumoral injection of either PBS or 2 × 107 PFU of Synco-2D or Synco-4 in a volume of 100 μL. The tumor growth in the mice was monitored for 3 weeks, as described above.
In some experiments, spleens were collected after euthanasia of the animals for immunologic assays, as described in the following sections. In one experiment, the tumor-free mice from the FusOn-PL-treated group were rechallenged on the left flank with 3 × 105 CT26-HER2 tumor cells and were monitored for another 4 weeks for tumor growth on both flanks.
ELISpot Assays
ELISpot assays were performed with a mouse IFN-γ ELISPOT Ready-SET-Go! kit (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. Briefly, 96-well filter plates (Millipore, Bedford, MA, USA) were precoated with anti-IFN-γ monoclonal antibody (mAb) and incubated overnight at 4°C. The plates were blocked with 10% FBS RPMI 1640 for 1 h at RT. Splenocytes were dispensed at 5 × 105 cells into duplicate wells in 10% FBS RPMI 1640 and stimulated with medium, nonspecific peptides (ovalbumin [OVA] 323–329) or neoantigen peptides, respectively. The peptide concentration was 5 μg/mL. After incubation at 37°C for 48 h, followed by washing, a detection antibody (Ab) was added to each well, and the plates were incubated for another 2 h at RT. An Avidin-HRP solution was added to the wells and incubated for another 45 min. Finally, freshly prepared 3-amino-9-ethylcarbazole (AEC) substrate solution was added. The color reaction was allowed to develop at RT for 10–60 min. After washing with tap water and drying, the spots were counted and imaged with an Olympus SZX7 Stereo Microscope Camera System (Olympus, Waltham, MA, USA).
Tumor Immunochemistry for NK Cell Infiltration
For immunohistochemistry staining of NK cells, paraffin sections from the collected tumor tissues were deparaffinized in xylene, rehydrated through graded alcohol, and processed for antigen retrieval by boiling in 10 mM citrate buffer (pH 6.0) for 15 min. Sections were incubated in 0.3% H2O2 in 50% methanol for 30 min at RT to quench endogenous peroxidase. To block nonspecific binding, sections were incubated in 3% BSA for 30 min, and then, a biotin-blocking system (Dako, Carpinteria, CA, USA) was used to block endogenous biotin. Sections were then incubated with 1:50 rabbit anti-NCR1 antibody (Abcam, Cambridge, MA, USA) overnight at 4°C. After washing, sections were incubated with an HRP polymer kit (Biocare Medical, Pacheco, CA, USA) for 30 min at RT, followed by 3, 3-diaminobenzidine tetrahydrochloride as the chromogen. The image was photographed with an Olympus BX41 microscope (Olympus, Waltham, MA, USA).
For immunofluorescence double staining, the sections were incubated overnight at 4°C with 1:50 rabbit anti-NCR1 and 1:200 mouse anti-Ki67 (BD Biosciences, San Jose, CA, USA) after blocking nonspecific binding in 3% BSA. Primary antibodies were detected with 1:400 donkey anti-rabbit Cy3 (Jackson ImmunoResearch, West Grove, PA) and 1:400 donkey anti-mouse FITC (Santa Cruz Biotechnology, Dallas, TX, USA). Sections were later counterstained with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA) to label nuclei. The image was photographed with an Olympus BX51 fluorescence microscope (Olympus, Waltham, MA, USA).
Statistical Analysis
All data were normally distributed, and Student’s t test (two-tailed) or one-way ANOVA was used to determine the statistical significance (p < 0.05) of various comparisons. The results are reported as mean ± standard deviations.
Author Contributions
L.T., W.W., and X.F. conducted the experiments. X.F. and X.Z. designed the experiments and wrote the paper.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Dr. M. Penichet for the gift of the CT26-HER2 cells and Dr. David M. Stewart for careful reading of the manuscript. This work was supported by the National Cancer Institute grant R01CA203852, CPRIT grant RP200464, and a grant from the William and Ella Owens Medical Research Foundation (to X.Z.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.09.002.
Supplemental Information
References
- 1.Wakimoto H., Johnson P.R., Knipe D.M., Chiocca E.A. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Ther. 2003;10:983–990. doi: 10.1038/sj.gt.3302038. [DOI] [PubMed] [Google Scholar]
- 2.Fulci G., Breymann L., Gianni D., Kurozomi K., Rhee S.S., Yu J., Kaur B., Louis D.N., Weissleder R., Caligiuri M.A., Chiocca E.A. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA. 2006;103:12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wakimoto H., Ikeda K., Abe T., Ichikawa T., Hochberg F.H., Ezekowitz R.A., Pasternack M.S., Chiocca E.A. The complement response against an oncolytic virus is species-specific in its activation pathways. Mol. Ther. 2002;5:275–282. doi: 10.1006/mthe.2002.0547. [DOI] [PubMed] [Google Scholar]
- 4.Fulci G., Dmitrieva N., Gianni D., Fontana E.J., Pan X., Lu Y., Kaufman C.S., Kaur B., Lawler S.E., Lee R.J. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007;67:9398–9406. doi: 10.1158/0008-5472.CAN-07-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez-Breckenridge C.A., Yu J., Price R., Wei M., Wang Y., Nowicki M.O., Ha Y.P., Bergin S., Hwang C., Fernandez S.A. The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J. Virol. 2012;86:4566–4577. doi: 10.1128/JVI.05545-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarez-Breckenridge C.A., Yu J., Price R., Wojton J., Pradarelli J., Mao H., Wei M., Wang Y., He S., Hardcastle J. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat. Med. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bindea G., Mlecnik B., Tosolini M., Kirilovsky A., Waldner M., Obenauf A.C., Angell H., Fredriksen T., Lafontaine L., Berger A. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Melero I., Rouzaut A., Motz G.T., Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014;4:522–526. doi: 10.1158/2159-8290.CD-13-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taube J.M., Klein A., Brahmer J.R., Xu H., Pan X., Kim J.H., Chen L., Pardoll D.M., Topalian S.L., Anders R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014;20:5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ochoa M.C., Minute L., Rodriguez I., Garasa S., Perez-Ruiz E., Inogés S., Melero I., Berraondo P. Antibody-dependent cell cytotoxicity: immunotherapy strategies enhancing effector NK cells. Immunol. Cell Biol. 2017;95:347–355. doi: 10.1038/icb.2017.6. [DOI] [PubMed] [Google Scholar]
- 11.Weiskopf K., Weissman I.L. Macrophages are critical effectors of antibody therapies for cancer. MAbs. 2015;7:303–310. doi: 10.1080/19420862.2015.1011450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Björck L. Protein L. A novel bacterial cell wall protein with affinity for Ig L chains. J. Immunol. 1988;140:1194–1197. [PubMed] [Google Scholar]
- 13.Graille M., Stura E.A., Housden N.G., Beckingham J.A., Bottomley S.P., Beale D., Taussig M.J., Sutton B.J., Gore M.G., Charbonnier J.B. Complex between Peptostreptococcus magnus protein L and a human antibody reveals structural convergence in the interaction modes of Fab binding proteins. Structure. 2001;9:679–687. doi: 10.1016/s0969-2126(01)00630-x. [DOI] [PubMed] [Google Scholar]
- 14.Orlova A., Magnusson M., Eriksson T.L., Nilsson M., Larsson B., Höidén-Guthenberg I., Widström C., Carlsson J., Tolmachev V., Ståhl S., Nilsson F.Y. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006;66:4339–4348. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 15.Steffen A.C., Orlova A., Wikman M., Nilsson F.Y., Ståhl S., Adams G.P., Tolmachev V., Carlsson J. Affibody-mediated tumour targeting of HER-2 expressing xenografts in mice. Eur. J. Nucl. Med. Mol. Imaging. 2006;33:631–638. doi: 10.1007/s00259-005-0012-3. [DOI] [PubMed] [Google Scholar]
- 16.Feldwisch J., Tolmachev V. Engineering of affibody molecules for therapy and diagnostics. Methods Mol. Biol. 2012;899:103–126. doi: 10.1007/978-1-61779-921-1_7. [DOI] [PubMed] [Google Scholar]
- 17.Lahti J.L., Lui B.H., Beck S.E., Lee S.S., Ly D.P., Longaker M.T., Yang G.P., Cochran J.R. Engineered epidermal growth factor mutants with faster binding on-rates correlate with enhanced receptor activation. FEBS Lett. 2011;585:1135–1139. doi: 10.1016/j.febslet.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kastern W., Sjöbring U., Björck L. Structure of peptostreptococcal protein L and identification of a repeated immunoglobulin light chain-binding domain. J. Biol. Chem. 1992;267:12820–12825. [PubMed] [Google Scholar]
- 19.Fu X., Rivera A., Tao L., De Geest B., Zhang X. Construction of an oncolytic herpes simplex virus that precisely targets hepatocellular carcinoma cells. Mol. Ther. 2012;20:339–346. doi: 10.1038/mt.2011.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu X., Tao L., Cai R., Prigge J., Zhang X. A mutant type 2 herpes simplex virus deleted for the protein kinase domain of the ICP10 gene is a potent oncolytic virus. Mol. Ther. 2006;13:882–890. doi: 10.1016/j.ymthe.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 21.Fu X., Tao L., Zhang X. Genetically coating oncolytic herpes simplex virus with CD47 allows efficient systemic delivery and prolongs virus persistence at tumor site. Oncotarget. 2018;9:34543–34553. doi: 10.18632/oncotarget.26167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu X., Rivera A., Tao L., Zhang X. Incorporation of the B18R Gene of Vaccinia Virus Into an Oncolytic Herpes Simplex Virus Improves Antitumor Activity. Mol. Ther. 2012;20:1871–1881. doi: 10.1038/mt.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu X., Tao L., Jin A., Vile R., Brenner M.K., Zhang X. Expression of a fusogenic membrane glycoprotein by an oncolytic herpes simplex virus potentiates the viral antitumor effect. Mol. Ther. 2003;7:748–754. doi: 10.1016/s1525-0016(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 24.Nakamori M., Fu X., Meng F., Jin A., Tao L., Bast R.C.J., Jr., Zhang X. Effective therapy of metastatic ovarian cancer with an oncolytic herpes simplex virus incorporating two membrane fusion mechanisms. Clin. Cancer Res. 2003;9:2727–2733. [PubMed] [Google Scholar]
- 25.DeFazio-Eli L., Strommen K., Dao-Pick T., Parry G., Goodman L., Winslow J. Quantitative assays for the measurement of HER1-HER2 heterodimerization and phosphorylation in cell lines and breast tumors: applications for diagnostics and targeted drug mechanism of action. Breast Cancer Res. 2011;13:R44. doi: 10.1186/bcr2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siddiqa A., Long L.M., Li L., Marciniak R.A., Kazhdan I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer. 2008;8:129. doi: 10.1186/1471-2407-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gall V.A., Philips A.V., Qiao N., Clise-Dwyer K., Perakis A.A., Zhang M., Clifton G.T., Sukhumalchandra P., Ma Q., Reddy S.M. Trastuzumab Increases HER2 Uptake and Cross-Presentation by Dendritic Cells. Cancer Res. 2017;77:5374–5383. doi: 10.1158/0008-5472.CAN-16-2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penichet M.L., Challita P.M., Shin S.U., Sampogna S.L., Rosenblatt J.D., Morrison S.L. In vivo properties of three human HER2/neu-expressing murine cell lines in immunocompetent mice. Lab. Anim. Sci. 1999;49:179–188. [PubMed] [Google Scholar]
- 29.Kleiveland, C.R. (2015). Peripheral blood mononuclear cells. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models [Internet], K. Verhoeckx, P. Cotter, I. López-Expósito, C. Kleiveland, T. Lea, A. Mackie, T. Requena, D. Swiatecka, H. Wichers, eds. (Springer), pp. 161–167. [PubMed]
- 30.Coca S., Perez-Piqueras J., Martinez D., Colmenarejo A., Saez M.A., Vallejo C., Martos J.A., Moreno M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. 1997;79:2320–2328. doi: 10.1002/(sici)1097-0142(19970615)79:12<2320::aid-cncr5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 31.Ashkar A.A., Rosenthal K.L. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J. Virol. 2003;77:10168–10171. doi: 10.1128/JVI.77.18.10168-10171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nandakumar S., Woolard S.N., Yuan D., Rouse B.T., Kumaraguru U. Natural killer cells as novel helpers in anti-herpes simplex virus immune response. J. Virol. 2008;82:10820–10831. doi: 10.1128/JVI.00365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdul-Careem M.F., Lee A.J., Pek E.A., Gill N., Gillgrass A.E., Chew M.V., Reid S., Ashkar A.A. Genital HSV-2 infection induces short-term NK cell memory. PLoS ONE. 2012;7:e32821. doi: 10.1371/journal.pone.0032821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rizvi N.A., Hellmann M.D., Snyder A., Kvistborg P., Makarov V., Havel J.J., Lee W., Yuan J., Wong P., Ho T.S. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Allen E.M., Miao D., Schilling B., Shukla S.A., Blank C., Zimmer L., Sucker A., Hillen U., Foppen M.H.G., Goldinger S.M. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kreiter S., Vormehr M., van de Roemer N., Diken M., Löwer M., Diekmann J., Boegel S., Schrörs B., Vascotto F., Castle J.C. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 38.Andreansky S., He B., van Cott J., McGhee J., Markert J.M., Gillespie G.Y., Roizman B., Whitley R.J. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 1998;5:121–130. doi: 10.1038/sj.gt.3300550. [DOI] [PubMed] [Google Scholar]
- 39.Parker J.N., Gillespie G.Y., Love C.E., Randall S., Whitley R.J., Markert J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA. 2000;97:2208–2213. doi: 10.1073/pnas.040557897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song X.T. Combination of virotherapy and T-cell therapy: arming oncolytic virus with T-cell engagers. Discov. Med. 2013;16:261–266. [PubMed] [Google Scholar]
- 41.Sivanandam V., LaRocca C.J., Chen N.G., Fong Y., Warner S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncolytics. 2019;13:93–106. doi: 10.1016/j.omto.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu X., Rivera A., Tao L., Zhang X. An HSV-2 based oncolytic virus can function as an attractant to guide migration of adoptively transferred T cells to tumor sites. Oncotarget. 2015;6:902–914. doi: 10.18632/oncotarget.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ajina A., Maher J. Synergistic combination of oncolytic virotherapy with CAR T-cell therapy. Prog. Mol. Biol. Transl. Sci. 2019;164:217–292. doi: 10.1016/bs.pmbts.2019.06.015. [DOI] [PubMed] [Google Scholar]
- 44.Wing A., Fajardo C.A., Posey A.D., Jr., Shaw C., Da T., Young R.M., Alemany R., June C.H., Guedan S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol. Res. 2018;6:605–616. doi: 10.1158/2326-6066.CIR-17-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gajewski T.F., Schreiber H., Fu Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zumwalt T.J., Arnold M., Goel A., Boland C.R. Active secretion of CXCL10 and CCL5 from colorectal cancer microenvironments associates with GranzymeB+ CD8+ T-cell infiltration. Oncotarget. 2015;6:2981–2991. doi: 10.18632/oncotarget.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schumacher T.N., Schreiber R.D. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 48.Schumacher T.N., Hacohen N. Neoantigens encoded in the cancer genome. Curr. Opin. Immunol. 2016;41:98–103. doi: 10.1016/j.coi.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 49.Yarchoan M., Johnson B.A., 3rd, Lutz E.R., Laheru D.A., Jaffee E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer. 2017;17:209–222. doi: 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deniger D.C., Pasetto A., Tran E., Parkhurst M.R., Cohen C.J., Robbins P.F., Cooper L.J., Rosenberg S.A. Stable, Nonviral Expression of Mutated Tumor Neoantigen-specific T-cell Receptors Using the Sleeping Beauty Transposon/Transposase System. Mol. Ther. 2016;24:1078–1089. doi: 10.1038/mt.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bottcher J.P., Bonavita E., Chakravarty P., Blees H., Cabeza-Cabrerizo M., Sammicheli S., Rogers N.C., Sahai E., Zelenay S., Reis E Sousa C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell. 2018;172:1022–1037.e14. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barry K.C., Hsu J., Broz M.L., Cueto F.J., Binnewies M., Combes A.J., Nelson A.E., Loo K., Kumar R., Rosenblum M.D. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018;24:1178–1191. doi: 10.1038/s41591-018-0085-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salmon H., Idoyaga J., Rahman A., Leboeuf M., Remark R., Jordan S., Casanova-Acebes M., Khudoynazarova M., Agudo J., Tung N. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016;44:924–938. doi: 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dudziak D., Kamphorst A.O., Heidkamp G.F., Buchholz V.R., Trumpfheller C., Yamazaki S., Cheong C., Liu K., Lee H.W., Park C.G. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 55.Theisen D.J., Davidson J.T., 4th, Briseño C.G., Gargaro M., Lauron E.J., Wang Q., Desai P., Durai V., Bagadia P., Brickner J.R. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science. 2018;362:694–699. doi: 10.1126/science.aat5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.