

Abstract
Alcoholic liver disease (ALD) and nonalcoholic fatty liver disease (NAFLD) account for the majority of hepatic morbidity and deaths due to cirrhosis in the United States. ALD is an umbrella term for a number of conditions linked to excessive alcohol consumption including simple steatosis, cirrhosis, acute alcoholic hepatitis (AH) with or without cirrhosis, and hepatocellular carcinoma (HCC) as a complication of cirrhosis. Although it presents with histological features resembling alcohol-induced liver injury, NAFLD occurs in patients with little or no history of alcohol consumption. NAFLD is a broad-spectrum term used to describe anything from fat accumulation in hepatocytes without inflammation or fibrosis (simple hepatic steatosis) to hepatic steatosis with a necroinflammatory component (steatohepatitis) with or without associated fibrosis. The pathogenesis is not fully understood for either disease. Development of severe liver disease is highly variable amongst chronic abusers of alcohol. Sex, age, genetics, host microbiome, and behavior are all factors linked to the development of ALD. These factors also contribute to NAFLD, but by contrast, insulin resistance is widely believed to be the main driver of nonalcoholic hepatic steatosis. The mechanism behind the transition from nonalcoholic steatosis to steatohepatitis remains a matter of debate with insulin resistance, oxidative injury, hepatic iron, gut hormones, antioxidant deficiency, and host microbiome all suspected to play part of the role.
Keywords: Alcohol, alcoholic hepatitis (AH), liver, fibrosis, portal hypertension
Pathogenesis of ethanol-mediated liver injury (Figure 1)
Figure 1.
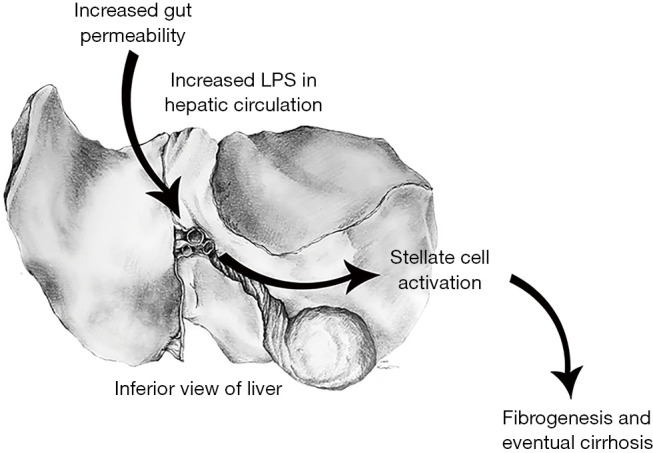
Increased gut permeability: alcohol damages tight junction integrity in the distal epithelium leading to increased gut permeability (1). Increased LPS in hepatic circulation: increased gut permeability leads to increased levels of bacterial lipopolysaccharide in intestinal venous circulation. This blood eventually enters hepatic circulation through the portal vein (2). Stellate cell activation: lipopolysaccharide binds to LPS-binding protein on liver macrophages known as hepatic stellate cells, leading to downstream proinflammatory cytokine and neutrophil recruiting chemokine activation. Activated hepatic stellate cells proliferate and take on a pro-fibrotic role (3). Fibrogenesis & cirrhosis: activated hepatic stellate cells lay down a fibrotic extracellular matrix composed of collagens type I and III, glycoproteins, and proteoglycans (4). LPS, lipopolysaccharide.
Biochemical injury
Ethanol is primarily metabolized to acetaldehyde by alcohol dehydrogenase (ADH) located in the cytosol of hepatocytes. Acetaldehyde is further metabolized to acetate in mitochondria by aldehyde dehydrogenase (ALDH). Acetaldehyde is toxic due to its high reactivity and may go on to form DNA or protein adducts (2). A genetically linked deficiency in the enzyme ALDH2 may lead to the accumulation of acetaldehyde. Interestingly, this deficiency has not been shown to increase the risk of ALD, as these variants experience higher rates of flushing and disulfiram like reactions upon ingestion of alcohol. These unwanted effects tend to cause an aversion to alcohol and significantly lower rates of alcoholism (5,6).
A minor proportion of ethanol is alternatively metabolized by the microsomal ethanol oxidizing system (MEOS) located within the smooth endoplasmic reticulum of hepatocytes. MEOS converts ethanol to acetaldehyde through a redox reaction that depletes NADPH, generates NADP+, and dissipates heat. MEOS activity increases with chronic alcohol consumption. MEOS also produces increased levels of reactive oxygen species which subsequently leads to lipid peroxidation, mitochondrial glutathione depletion, and S-adenosylmethionine depletion. The combination of these effects produces increased oxidative stress and liver injury (7-9).
Immunologic injury
Once hepatocytes have been damaged by the effects of alcohol metabolism, they release endogenous damage-associated molecular patterns (DAMPs). DAMPs activate cellular pattern recognition receptors which trigger production of proinflammatory cytokines through the actions of a cytosolic protein complex known as the inflammasome. The proinflammatory cytokines produced lead to the localization of immune cells to the site of hepatocyte injury (7). Of these immune cells, neutrophils serve as a highly characteristic pathologic sign of AH, as they are typically absent in most other forms of inflammatory liver disease. Patients with AH demonstrate increased plasma IL-8 levels, which may explain the presence of neutrophils (10). In addition to increased IL-8 levels, in vitro hepatocyte ethanol metabolism leads to the production of a leukotriene-B4-related arachidonic acid metabolite which serves as a further neutrophil chemoattractant in human, but not mouse models (11).
Steatosis
Alcoholic steatosis is marked by reduced oxidation of hepatic fatty acids and increased lipogenesis contributing to the accumulation of triglycerides, phospholipids, and cholesterol esters in hepatocytes. Ethanol may exert these effects through changes in the NADH/NAD+ ratio and the associated downstream effects on β-oxidation or through changes in the regulation of lipid metabolism transcription factors. Ethanol upregulates sterol regulatory element-binding transcription factor 1 (SREBP-1), which stimulates lipogenesis through its effects on target genes, such as fatty acid synthase, acetyl-coenzyme A carboxylase, and steroyl-CoA desaturase. This upregulation of SREBP-1 occurs directly from the actions of the ethanol metabolite, acetaldehyde and indirectly from other inflammatory effects of ethanol metabolism. In addition, ethanol downregulates the negative regulators of SREBP-1, thereby further contributing to lipogenesis (12). Ethanol inhibits DNA-binding and transcriptional activity of peroxisome proliferator-activated receptor α (PPAR-α), with resulting downregulation of fatty acid oxidation (13). This inhibition occurs directly through the effects of acetaldehyde or indirectly from CYP2E1-generated oxidative stress, adenosine, downregulation of adiponectin, and zinc deficiency (7).
By the time alcoholic steatosis is present but before the onset of alcoholic steatohepatitis, innate immune system signaling is already present through the action of interferon regulatory factor 3 (IRF3). IRF3 activation is a necessary step in hepatocyte specific mitochondrial apoptosis. A single exposure to ethanol phosphorylates (activates) IRF3 as a result of endoplasmic reticulum stress. The regulatory actions of IRF3 appear to have both pro-inflammatory and anti-inflammatory effects, as whole-body knock-out IRF3 mice are protected from ethanol induced liver injury, but IRF3 deficiency in parenchymal cells aggravates ethanol-induced liver injury in mice (14).
Microbiome disruption
Ethanol is believed to contribute to bacterial overload in the gut, in part due to small intestinal dysmotility and changes in bile. Ethanol also contributes to increased gut permeability by disruption of intestinal tight junction integrity. ALD-associated zinc deficiency contributes to this disruption in tight-junction integrity (15). The effects of bacterial overload and increased gut permeability lead to an elevated level of bacterial lipopolysaccharide (LPS) within the portal circulation and the liver. LPS binds to LPS binding protein, which activates CD14 on the surface of liver macrophages known as Kupffer cells. CD14 then activates TLR4 leading to a downstream cytokine activation. In addition, ethanol activates the complement system, and the interaction between the complement system and Kupffer cells leads to further production of proinflammatory cytokines. The effects of proinflammatory cytokines (TNF-α, IL-17) on hepatocytes and hepatic stellate cells results in the production of neutrophil recruiting chemokines such as IL-8, CXCL1, and IL-17 (7,16). Evidence of the contribution of TLR4 activation and proinflammatory cytokines to ALD has been demonstrated in mouse models. Mouse models in which LPS-binding protein, CD14, or TLR4 has been knocked out demonstrated decreased risk of developing hepatic injury when fed ethanol (1,3,17). In addition, hepatic inflammation and necrosis were significantly decreased in ethanol fed rats who had been administered neutralizing antibodies to TNF-α or who lacked the TNF receptor 1 (but not 2) gene (18).
Immune exhaustion
The products of ethanol metabolism, including lipid peroxidation products, acetaldehyde, and hydroxyethyl radicals can all form protein adducts with proteins located within or on the surface of hepatocytes. These protein-adducts can lead to cellular dysfunction or the creation of neoantigens which may elicit a T-lymphocyte mediated immune reaction while in the presence of monocytes. This immune reaction may result in the death of hepatocytes (13).
Despite the increased inflammation and immune activity observed in ALD, the leading cause of death in AH is sepsis-induced multiorgan failure due to overwhelming bacterial infection. Chronic inflammation may lead to immune exhaustion through the action of inhibitory receptors on T-lymphocytes such as programmed cell death protein 1 (PD-1) and T-cell immunoglobulin & mucin protein 3 (TIM-3). The downstream effects of T-lymphocyte inhibition elicited by these receptors is associated with dysfunction in neutrophil phagocytosis and oxidative burst reaction. This dysfunction may be reversed by the use of antibodies against PD-1 and TIM-3 (19).
Cirrhosis and carcinoma
Ethanol induced chronic liver injury may lead to fibrosis and eventual cirrhosis through the action of hepatic stellate cells. Hepatic stellate cells are perisinusoidal cells present throughout the liver which store the majority of hepatic vitamin A. Upon activation, hepatic stellate cells lose vitamin A, proliferate, and assume a profibrotic role. Evidence has shown that acetaldehyde and ethanol are unlikely to directly contribute to hepatic stellate cell activation. However, MEOS produced free radicals contribute to the production of lipid aldehydes, which are believed to play a role. Activated hepatic stellate cells lay down a fibrotic extracellular matrix composed of collagens type I and III, glycoproteins, and proteoglycans (4).
HCC may develop in the setting of alcoholic cirrhosis. Ethanol has immunosuppressive effects and its metabolite, acetaldehyde, acts as a direct carcinogen due to its mutagenic effects on DNA. The replicative environment created by chronic liver injury and cirrhosis contributes to the development of HCC. High hepatocyte turnover leads to the accumulation of errors of replication. In addition, cells with normal checkpoint inhibition function and telomere length eventually stop division and reduce the competition for resources experienced by abnormal cells. Ultimately, there is an increase in the proportion of hepatocytes with the greatest replicative potential, and the highest risk of tumorigenesis (20).
Pathogenesis of NAFLD (Figure 2)
Figure 2.
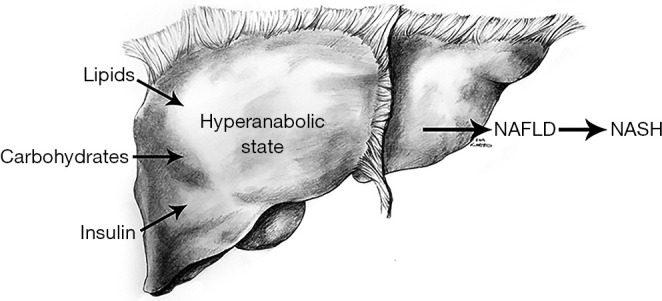
Lipids: insulin resistance causes decreased glucose delivery to tissues leading to peripheral lipolysis and increased free fatty acids in the bloodstream. Carbohydrates: excess carbohydrate consumption and decreased glucose uptake by insulin resistant muscle and adipose tissue leads to increased hepatocyte exposure to blood glucose. Insulin: as muscle and adipose tissue develop decreased insulin sensitivity, pancreatic beta cells secrete increasing levels of insulin to maintain blood glucose level homeostasis. Hyper-anabolic state: the liver maintains greater insulin sensitivity than adipose or skeletal-muscle tissue. The liver is exposed to increased blood glucose, free fatty acid, and insulin levels. NAFLD: insulin drives hepatic uptake of free fatty acids and carbohydrates. Both are converted to triglycerides and stored by hepatocytes of the liver (21). NASH: the mechanism behind the transition from nonalcoholic steatosis to steatohepatitis remains a matter of debate with insulin resistance, oxidative injury, hepatic iron, gut hormones, antioxidant deficiency, and host microbiome all suspected to play part of the role (22). NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis.
NAFLD has both environmental and genetic components. Its prevalence in the United States has more than doubled over the past 30 years as other components of metabolic syndrome have correspondingly increased in the population (23). In addition, bariatric surgery has been shown to ameliorate NASH in 85% of patients within the first year and to reduce fibrosis in 30% (24,25). Twin studies have shown a roughly 50% genetic contribution to hepatic fat content and hepatic fibrosis (26). Genetic variations in hepatic lipid metabolism and insulin signaling have been demonstrated to contribute to development and progression of NAFLD (27-29).
Steatosis
Nonalcoholic hepatic steatosis is the result of excessive lipid accumulation within the liver. This largely results from the excessive importation, diminished exportation, and/or diminished breakdown of free fatty acid in the liver. During periods of excess macronutrient intake, the liver will convert unused carbohydrates and proteins to triglycerides and store them. During later periods of diminished macronutrient intake or heightened caloric utilization these lipids may be broken down and released. Complications of obesity may include hyperlipidemia leading to increased levels of circulating triglycerides and increased delivery and storage of triglycerides in the liver (30).
Insulin resistance
Insulin resistance correlates strongly with the development of nonalcoholic hepatic steatosis and causes profound changes to normal systemic lipid metabolism. Insulin regulates blood glucose levels by driving nutrients from the bloodstream into cells postprandially. The development of insulin resistance occurs chiefly in muscle and adipose tissue, whereas hepatic tissue remains largely spared. As muscle and adipose tissue develop diminished sensitivity to the effects of insulin, less glucose is delivered to these tissues, and a catabolic state occurs where peripheral adipose tissue is broken down and free fatty acids are released to systemic circulation. To compensate for elevated blood glucose levels, pancreatic beta cells secrete increasing levels of insulin resulting in hyperinsulinemia. The liver, having remained largely insulin sensitive, and being exposed to increasing levels of blood glucose, serum triglycerides, and insulin enters a hyper-anabolic state where it continues to both synthesize and store lipids (21). Therapies focused on increasing insulin sensitivity such as rosiglitazone, pioglitazone and liraglutide show promise as potential treatments for NAFLD. Liraglutide, in particular, has shown additional benefits of satiety and weight loss in treated patients. A recent study of 68 patients treated with liraglutide for 6 months showed a 31% reduction in liver fat content as well as significant reductions in body weight, BMI, and ALT levels (31).
Biochemical injury
How the buildup of lipids within the liver leads to the hepatocellular injury seen in nonalcoholic steatohepatitis remains a matter of debate. Roughly one in ten patients who develop NAFLD will go on to develop NASH (32). This disparity has led to the development of a second hit hypothesis, with some believing the effects of insulin resistance alone insufficient to explain the development of NASH. Free fatty acids may directly lead to damage through the induction of oxygen radical forming cytochrome p-450 microsomal lipoxygenases (22). Increased lipid metabolism may lead to increased lipid peroxidation in which chain reactions between oxygen free radicals and lipids can damage mitochondrial DNA and mitochondrial membrane structures necessary for beta-oxidation (33,34). Patients with NASH have been shown to have significant mitochondrial structural abnormalities within their hepatocytes. Whether lipid peroxidation alone is sufficient to produce these abnormalities is unknown. In the setting of mitochondrial abnormalities, it theorized that the shift to free fatty acid beta-oxidation seen in NAFLD & NASH produces further increased free radical production resulting in hepatocellular injury and eventual fibrosis (35).
Antioxidants protect hepatic tissue from the damaging effects of oxygen radicals and lipid peroxidation by donating electrons and neutralizing these molecules before they react with DNA or proteins. However, antioxidants may be depleted in the setting of continued oxidative stress or insufficient dietary intake. Glutathione, vitamin E, vitamin C, and beta-carotene may all become depleted in the setting of NASH (36,37). Studies have shown improvements to hepatic inflammation and fibrosis in NASH patients with long term vitamin C and E supplementation (38,39). In addition, another study has shown decreased dietary intake of vitamins C and E in NASH patients (40).
Hepatic iron content may also contribute to the oxygen free radical damage seen in NASH. Hepatic iron levels correlate with insulin resistance and severity of fibrosis (41,42). However, homozygosity for the HFE gene, which leads to the development of hereditary hemochromatosis, has not been shown to increase the risk of NAFLD (43). In addition, no improvements in NAFLD measures were observed in a study of 74 patients, where active participants underwent 6 months of phlebotomy to lower serum ferritin levels. Even those patients with hyperferritinemia at baseline showed no significant difference in the endpoints of hepatic steatosis, ALT levels, or insulin sensitivity (44).
Microbiome disruption
The gut microbiome may contribute to the pathogenesis of NAFLD in a similar manner to its contribution in ALD. A study showed significantly increased intestinal permeability and small intestinal bacterial overgrowth in patients with NAFLD (45,46). These patients were shown to have compromised intestinal tight junction integrity. Colonic bacteria may damage intestinal tight junctions through endogenous production of alcohol and acetaldehyde (47). Increased intestinal permeability allows endotoxins produced by intestinal bacteria to enter portal circulation and activate TLR-4 signaling in Kupffer cells, leading to downstream increases in proinflammatory cytokines (48,49). TLR-4 may trigger MyD88 dependent or MyD88 independent pathways, with MyD88-dependent signaling playing a more critical role in the pathogenesis of NASH than in that of ALD (50-52). Both pathways lead to activation of NF-κB, with additional activation of proinflammatory cytokines being characteristic of MyD88-dependent signaling and induction of type I IFNs being characteristic of MyD88 independent signaling (53). Antibiotics and probiotics have been shown to decrease hepatic steatosis in mouse models and in human patients (54-56). Probiotic administration has been shown to decrease NF-κB signaling in mouse models of NAFLD (56).
Conclusions
ALD and NAFLD are complex multifactorial disorders, and the understanding of their pathogenesis remains incomplete. Genetics, nutrition, immune activation, reactive oxygen damage, and hepatic fat accumulation are all probable contributors to their development. The major drivers of ALD and NAFLD are the damaging effects of excess alcohol consumption and insulin resistance secondary to metabolic syndrome and obesity respectively. While potential therapies may target any of the above pathways, the most effective therapies remain abstinence from alcohol for ALD and weight loss for NAFLD. Any attempt to understand the pathogenesis, meaning the manner of development, of ALD and NAFLD would be remiss to ignore the psychosocial and environmental factors at play and their importance in patient choice. Behavior and environment remain the largest contributors to these disorders. For example, admission to alcohol rehabilitation within 30 days of hospitalization for AH reduces mortality as much as 80% (9) and low socioeconomic status increases the risk of alcohol related mortality by 66% for men and 78% for women (57). The combination of obesity and smoking is a greater risk factor for the progression of alcoholic steatosis to alcoholic cirrhosis than any allele thus far studied (58-60). This is all to say that in the pathogenesis and the treatment of ALD and NAFLD, there are as many intrahepatic factors as extrahepatic factors and psychology plays as strong of a role as biology. As the understanding of the pathogenesis of these disorders grows so too will our options for medical intervention, but lifestyle modification is likely to remain the cornerstone of treatment for the foreseeable future.
Acknowledgments
The authors thank Terri J. Johnson for administrative support and Karina Laenartowicz for the liver drawings she provided to the paper.
Funding: This work was supported by the National Institutes of Health [AA21171, AA21788, AA26974, AA26886, AA26887 to VH Shah].
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Conflicts of Interest: KE Robinson has no conflicts of interest to declare. VH Shah, MD is a consultant for Afimmune, Ltd.; Durect Corporation; Enterome SAB; Merck Research Laboratories; Novartis Pharmaceuticals; and Vital Therapies.
References
- 1.Uesugi T, Froh M, Arteel GE, et al. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol 2002;168:2963-9. 10.4049/jimmunol.168.6.2963 [DOI] [PubMed] [Google Scholar]
- 2.Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longev 2010;3:178-85. 10.4161/oxim.3.3.12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin M, Bradford BU, Wheeler MD, et al. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol 2001;166:4737-42. 10.4049/jimmunol.166.7.4737 [DOI] [PubMed] [Google Scholar]
- 4.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;134:1655-69. 10.1053/j.gastro.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosron WF, Ehrig T, Li TK. Genetic factors in alcohol metabolism and alcoholism. Semin Liver Dis 1993;13:126-35. 10.1055/s-2007-1007344 [DOI] [PubMed] [Google Scholar]
- 6.Enomoto N, Takase S, Yasuhara M, et al. Acetaldehyde metabolism in different aldehyde dehydrogenase-2 genotypes. Alcohol Clin Exp Res 1991;15:141-4. 10.1111/j.1530-0277.1991.tb00532.x [DOI] [PubMed] [Google Scholar]
- 7.Dunn W, Shah VH. Pathogenesis of Alcoholic Liver Disease. Clin Liver Dis 2016;20:445-56. 10.1016/j.cld.2016.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zima T, Kalousová M. Oxidative stress and signal transduction pathways in alcoholic liver disease. Alcohol Clin Exp Res 2005;29:110S-115S. 10.1097/01.alc.0000189288.30358.4b [DOI] [PubMed] [Google Scholar]
- 9.Peeraphatdit TB, Kamath PS, Karpyak VM, et al. Alcohol Rehabilitation Within 30 Days of Hospital Discharge is Associated With Reduced Readmission, Relapse, and Death in Patients With Alcoholic Hepatitis. Clin Gastroenterol Hepatol 2019. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheron N, Bird G, Koskinas J, et al. Circulating and tissue levels of the neutrophil chemotaxin interleukin-8 are elevated in severe acute alcoholic hepatitis, and tissue levels correlate with neutrophil infiltration. Hepatology 1993;18:41-6. [PubMed] [Google Scholar]
- 11.Roll FJ, Perez HD, Serhan CN. Characterization of a novel arachidonic acid-derived neutrophil chemoattractant. Biochem Biophys Res Commun 1992;186:269-76. 10.1016/S0006-291X(05)80803-4 [DOI] [PubMed] [Google Scholar]
- 12.Ji C, Chan C, Kaplowitz N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J Hepatol 2006;45:717-24. 10.1016/j.jhep.2006.05.009 [DOI] [PubMed] [Google Scholar]
- 13.Li HH, Tyburski JB, Wang YW, et al. Modulation of fatty acid and bile acid metabolism by peroxisome proliferator-activated receptor alpha protects against alcoholic liver disease. Alcohol Clin Exp Res 2014;38:1520-31. 10.1111/acer.12424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrasek J, Iracheta-Vellve A, Csak T, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci U S A 2013;110:16544-9. 10.1073/pnas.1308331110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong W, McClain CJ, Cave M, et al. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am J Physiol Gastrointest Liver Physiol 2010;298:G625-633. 10.1152/ajpgi.00350.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedman, SL. Pathogenesis of alcoholic liver disease. In: Post TW. Editor. UpToDate. Waltham, MA, 2019. [Google Scholar]
- 17.Uesugi T, Froh M, Arteel GE, et al. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001;34:101-8. 10.1053/jhep.2001.25350 [DOI] [PubMed] [Google Scholar]
- 18.Yin M, Wheeler MD, Kono H, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology 1999;117:942-52. 10.1016/S0016-5085(99)70354-9 [DOI] [PubMed] [Google Scholar]
- 19.Markwick LJ, Riva A, Ryan JM, et al. Blockade of PD1 and TIM3 restores innate and adaptive immunity in patients with acute alcoholic hepatitis. Gastroenterology 2015;148:590-602.e10. 10.1053/j.gastro.2014.11.041 [DOI] [PubMed] [Google Scholar]
- 20.McKillop IH, Schrum LW. Role of alcohol in liver carcinogenesis. Semin Liver Dis 2009;29:222-32. 10.1055/s-0029-1214377 [DOI] [PubMed] [Google Scholar]
- 21.Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343-51. 10.1172/JCI23621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002;346:1221-31. 10.1056/NEJMra011775 [DOI] [PubMed] [Google Scholar]
- 23.Younossi ZM, Stepanova M, Afendy M, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol 2011;9:524-530.e1; quiz e60. [DOI] [PubMed]
- 24.Murphy SK, Yang H, Moylan CA, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013;145:1076-87. 10.1053/j.gastro.2013.07.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahrens M, Ammerpohl O, von Schönfels W, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab 2013;18:296-302. 10.1016/j.cmet.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 26.Loomba R, Schork N, Chen CH, et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 2015;149:1784-93. 10.1053/j.gastro.2015.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J Hepatol 2018;68:268-79. 10.1016/j.jhep.2017.09.003 [DOI] [PubMed] [Google Scholar]
- 28.Dongiovanni P, Romeo S, Valenti L. Genetic Factors in the Pathogenesis of Nonalcoholic Fatty Liver and Steatohepatitis. Biomed Res Int 2015;2015:460190. [DOI] [PMC free article] [PubMed]
- 29.Dongiovanni P, Valenti L, Rametta R, et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010;59:267-73. 10.1136/gut.2009.190801 [DOI] [PubMed] [Google Scholar]
- 30.Noureddin M, Mato JM, Lu SC. Nonalcoholic fatty liver disease: update on pathogenesis, diagnosis, treatment and the role of S-adenosylmethionine. Exp Biol Med (Maywood) 2015;240:809-20. 10.1177/1535370215579161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petit JM, Cercueil JP, Loffroy R, et al. Effect of Liraglutide Therapy on Liver Fat Content in Patients With Inadequately Controlled Type 2 Diabetes: The Lira-NAFLD Study, J Clin Endocrinol Metab 2017;102:407-15. [DOI] [PubMed] [Google Scholar]
- 32.Tesfay M, Goldkamp WJ, Neuschwander-Tetri BA. NASH: The Emerging Most Common Form of Chronic Liver Disease. Mo Med 2018;115:225-9. [PMC free article] [PubMed] [Google Scholar]
- 33.Hruszkewycz AM. Evidence for mitochondrial DNA damage by lipid peroxidation. Biochem Biophys Res Commun 1988;153:191-7. 10.1016/S0006-291X(88)81207-5 [DOI] [PubMed] [Google Scholar]
- 34.Chen J, Schenker S, Frosto TA, et al. Inhibition of cytochrome c oxidase activity by 4-hydroxynonenal (HNE). Role of HNE adduct formation with the enzyme subunits. Biochim Biophys Acta 1998;1380:336-44. 10.1016/S0304-4165(98)00002-6 [DOI] [PubMed] [Google Scholar]
- 35.Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001;120:1183-92. 10.1053/gast.2001.23256 [DOI] [PubMed] [Google Scholar]
- 36.Sastre J, Pallardó FV, Llopis J, et al. Glutathione depletion by hyperphagia-induced obesity. Life Sci 1989;45:183-7. 10.1016/0024-3205(89)90293-2 [DOI] [PubMed] [Google Scholar]
- 37.Strauss RS, Barlow SE, Dietz WH. Prevalence of abnormal serum aminotransferase values in overweight and obese adolescents. J Pediatr 2000;136:727-33. [PubMed] [Google Scholar]
- 38.Lavine JE. Vitamin E treatment of nonalcoholic steatohepatitis in children: a pilot study. J Pediatr 2000;136:734-8. 10.1016/S0022-3476(00)05040-X [DOI] [PubMed] [Google Scholar]
- 39.Harrison SA, Torgerson S, Hayashi P, et al. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am J Gastroenterol 2003;98:2485-90. 10.1111/j.1572-0241.2003.08699.x [DOI] [PubMed] [Google Scholar]
- 40.Musso G, Gambino R, De Michieli F, et al. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003;37:909-16. 10.1053/jhep.2003.50132 [DOI] [PubMed] [Google Scholar]
- 41.Valenti L, Fracanzani AL, Bugianesi E, et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010;138:905-12. 10.1053/j.gastro.2009.11.013 [DOI] [PubMed] [Google Scholar]
- 42.George DK, Goldwurm S, MacDonald GA, et al. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 1998;114:311-8. 10.1016/S0016-5085(98)70482-2 [DOI] [PubMed] [Google Scholar]
- 43.Chitturi S, Weltman M, Farrell GC, et al. HFE mutations, hepatic iron, and fibrosis: ethnic-specific association of NASH with C282Y but not with fibrotic severity. Hepatology 2002;36:142-9. 10.1053/jhep.2002.33892 [DOI] [PubMed] [Google Scholar]
- 44.Adams LA, Crawford DH, Stuart K, et al. The impact of phlebotomy in nonalcoholic fatty liver disease: A prospective, randomized, controlled trial. Hepatology 2015;61:1555-64. 10.1002/hep.27662 [DOI] [PubMed] [Google Scholar]
- 45.Wigg AJ, Roberts-Thomson IC, Dymock RB, et al. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001;48:206-11. 10.1136/gut.48.2.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009;49:1877-87. 10.1002/hep.22848 [DOI] [PubMed] [Google Scholar]
- 47.Cope K, Risby T, Diehl AM. Increased gastrointestinal ethanol production in obese mice: implications for fatty liver disease pathogenesis. Gastroenterology 2000;119:1340-7. 10.1053/gast.2000.19267 [DOI] [PubMed] [Google Scholar]
- 48.Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol 2013;28:38-42. 10.1111/jgh.12019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology 2009;136:65-80. 10.1053/j.gastro.2008.10.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology 2010;139:323-34.e7. 10.1053/j.gastro.2010.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petrasek J, Dolganiuc A, Csak T, et al. Type I interferons protect from Toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology 2011;140:697-708.e4. 10.1053/j.gastro.2010.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao XJ, Dong Q, Bindas J, et al. TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol 2008;181:3049-56. 10.4049/jimmunol.181.5.3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petrasek J, Csak T, Ganz M, et al. Differences in innate immune signaling between alcoholic and non-alcoholic steatohepatitis. J Gastroenterol Hepatol 2013;28:93-8. 10.1111/jgh.12020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drenick EJ, Fisler J, Johnson D. Hepatic steatosis after intestinal bypass--prevention and reversal by metronidazole, irrespective of protein-calorie malnutrition. Gastroenterology 1982;82:535. 10.1016/S0016-5085(82)80403-4 [DOI] [PubMed] [Google Scholar]
- 55.Alisi A, Bedogni G, Baviera G, et al. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 2014;39:1276. 10.1111/apt.12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Z, Yang S, Lin H, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003;37:343. 10.1053/jhep.2003.50048 [DOI] [PubMed] [Google Scholar]
- 57.Probst C, Roerecke M, Behrendt S, et al. Socioeconomic differences in alcohol-attributable mortality compared with all-cause mortality: a systematic review and meta-analysis. Int J Epidemiol 2014;43:1314-27. 10.1093/ije/dyu043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corrao G, Lepore AR, Torchio P, et al. The effect of drinking coffee and smoking cigarettes on the risk of cirrhosis associated with alcohol consumption. A case-control study. Provincial Group for the Study of Chronic Liver Disease. Eur J Epidemiol 1994;10:657-64. 10.1007/BF01719277 [DOI] [PubMed] [Google Scholar]
- 59.Klatsky AL, Armstrong MA. Alcohol, smoking, coffee, and cirrhosis. Am J Epidemiol 1992;136:1248-57. 10.1093/oxfordjournals.aje.a116433 [DOI] [PubMed] [Google Scholar]
- 60.Naveau S, Giraud V, Borotto E, et al. Excess weight risk factor for alcoholic liver disease. Hepatology 1997;25:108-11. 10.1002/hep.510250120 [DOI] [PubMed] [Google Scholar]