

Abstract
Diffuse gastric cancer (DGC) is a distinct histopathologic and molecular disease, characterized by mutations in CDH1, RHOA, and others. In addition, DGC is associated with familial syndromes, including hereditary DGC and germline mutation in CDH1. Clinically, this subtype of gastric adenocarcinoma is associated with a poor prognosis and possible resistance to available systemic therapies. An understanding of the genetic and molecular underpinnings of DGC may help inform of its clinical behavior and aid in screening, diagnosis, and response to treatment. In this review, we will review the current histologic, molecular, and genetic landscape of DGC and its relevance to clinical practice.
Keywords: Diffuse, gastric, cancer
Gastric cancer (GC) is the fifth most common malignancy with over 1,000,000 cases diagnosed annually and is the third leading cause of cancer death globally (1). The incidence is highest in Asia (with over half of all GC cases globally diagnosed in East Asia), Eastern Europe, and South America with comparatively lower rates in Africa, North America, and Europe. Approximately 95% of gastric tumors are gastric adenocarcinomas (GAC) which can be further divided into intestinal type gastric cancer (IGC), diffuse type gastric cancer (DGC), and mixed histology based on the 1965 Lauren classification (2-4). Approximately 50% of GAC are IGC, 30% are DGC, and 15–20% are mixed or indeterminate. DGC tends to occur in younger patients and in females, while IGC typically presents in older patients and in men (5-7). In the United States, the incidence of DGC also appears to be higher in the Hispanic population compared to non-Hispanic whites (8). The overall incidence of GAC has been declining since 1973 which may be due to a decrease in chronic H. Pylori infection, decreased tobacco use, and changes in diet (9). However, during the same period of time the incidence of DGC and signet-ring cell carcinoma (SRCC) had been increasing in both Asian and Western cohorts before decreasing in recent years (10-12). Reasons for this trend, as well as differences in ethnic and racial subgroups require additional research.
The etiology of DGC is diverse. While intestinal metaplasia from chronic infectious etiologies (i.e., H. Pylori) is more commonly associated with IGC rather than DGC (13), Epstein-Barr virus (EBV) appears to be associated with DGC, though the strength of this association is not as clear (4,14). Tobacco use also appears to be related to the development of DGC (15,16). However, the role of other lifestyle factors such as diet and alcohol, along with the strength of these associations, merit further research (17-19). In addition to environmental etiologies, somatic and germline mutations in a number of genes can contribute to the development of DGC. These genes include CDH1, TP53, RHOA, CTNN1A, and CMTM2 (4,20-22). Approximately 1–3% of all GCs are due to hereditary diffuse gastric cancer (HDGC), with 40% of those associated with germline mutations in CDH1 (23,24). The risk of developing DGC in patients with germline CDH1 mutations may be as high as 70% in men and 56% in women by age 80 years, and can also be associated with the development of other cancers such as colon cancer and lobular breast cancer in women (25,26).
Histologically, DGC lack expression of adhesion molecules and typically show an infiltrative, poorly differentiated, poorly cohesive appearance (Figures 1 and 2). Tumor cells nestle within a rich fibrous stroma that extends into the submucosal layers and arise in the background of normal mucosa. This is in contrast to IGC, which typically appears as cells arranged in tubular or glandular structures that elevate from the mucosal surface and arise from a metaplastic background (2,20,23,27). SRCC, which contain nuclei that have been pushed to the periphery due to excess production of intracellular mucin, is a subtype of DGC based on the Lauren classification. However, even though SRCC are classified as DGC, not all DGC have signet ring cells (28,29). DGC and SRCC are frequently characterized by fibrous stroma, which has been associated with overexpression in TGF-β signaling and worse clinical outcomes (3,30,31). A genomic analysis of 428 GC tumor samples, including 118 diffuse-type patients, revealed that gene-expression profiles associated with increased stroma were found more commonly in DGC as compared to IGC (30). Those patients who had increased stromal gene-expression also had a poorer survival compared to patients who had lower levels of stromal gene-expression. All of these factors lead to a more aggressive tumor biology and later stage at diagnosis, contributing to poorer clinical outcomes.
Figure 1.
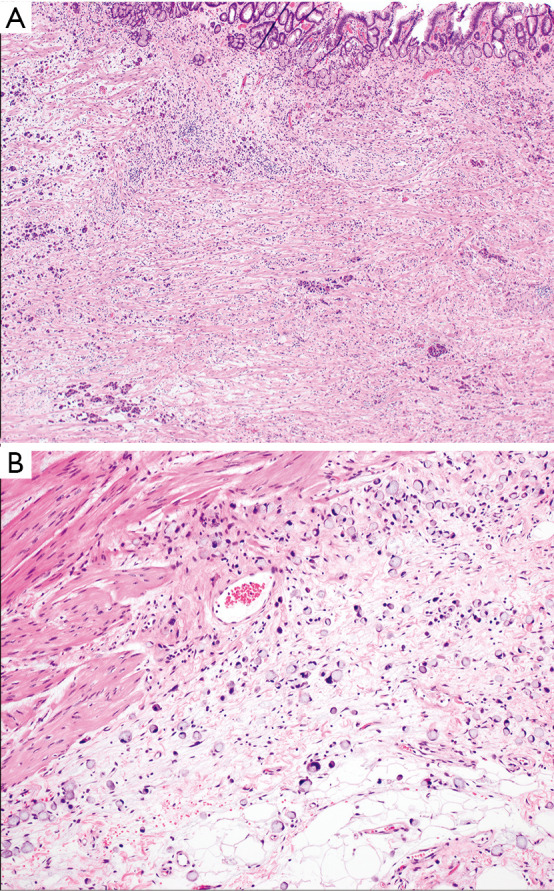
Intestinal type gastric adenocarcinoma. (A) Malignant glands of this intestinal (tubular) adenocarcinoma infiltrate the gastric wall, with benign oxyntic type mucosa at the top of the image. (B) The malignant glands are composed of dilated and slit-like branching tubules. Hematoxylin and eosin stained sections, original magnification, A: 4×, B: 10×.
Figure 2.
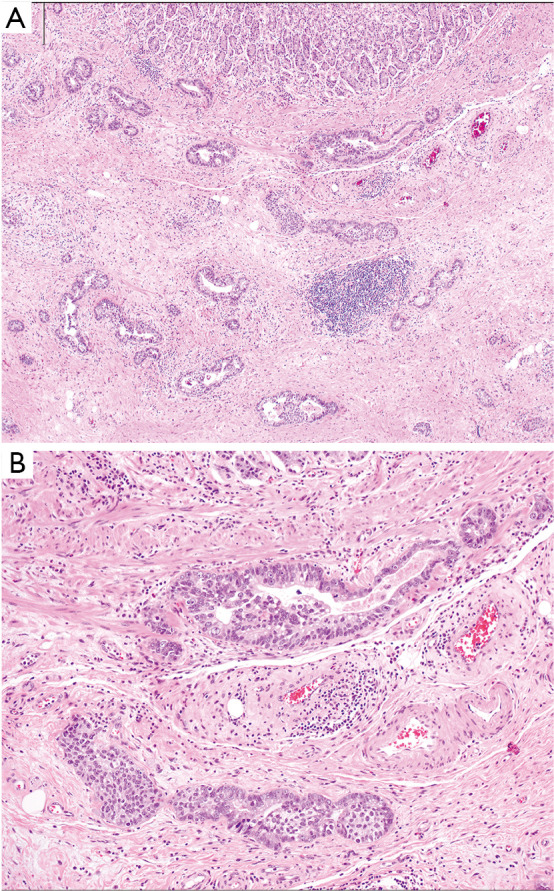
Diffuse type gastric adenocarcinoma. (A) At low power the malignant cells have a diffuse pattern of involvement, and may be difficult to detect. (B) At higher power, the tumor is easier to recognize as a poorly cohesive, signet-ring cell carcinoma, and is infiltrating through the muscularis propria into subserosal adipose tissue. Hematoxylin and eosin stained sections, original magnification, A: 4×, B: 10×.
Molecular profile of DGC
With the advent of next generation sequencing (NGS), RNA sequencing (RNA-seq), and whole genome sequencing (WES), the unique molecular characteristics of DGC have become more apparent, including their impact on clinical outcomes. One of the most robust assessments was performed using WES and RNA-seq as part of The Cancer Genome Atlas (TCGA) which sequenced 295 patients with early stage GC treated with surgery alone (4). Four molecular subtypes were identified in GC-EBV positive, microsatellite unstable tumors, genomically stable (GS) tumors, and tumors with chromosomal instability. DGC were predominantly found within the GS subgroup (40/55=73%, P=7.5×10−17), though DGC were also seen within all of the other subgroups in small numbers. In keeping with the known epidemiology of DGC, the GS subgroup tended to be diagnosed at a younger age. Within the TCGA study, the GS cluster also had high rate of somatic CDH1 mutations (37% of cases), RHOA mutations (15% of cases), and CLDN18-ARHGAP fusions (15% of cases), with RHOA and CLDN18-ARHGAP mutations being mutually exclusive. Further work by Sohn et al. created a prediction model based on the TCGA classification and tested it on two large cohorts (n=267 and 432) (32). They found the GS cohort was associated with a lack of significant benefit from adjuvant therapy (HR for recurrence after chemotherapy =0.83, P=0.66) and an overall worse prognosis (P=0.004).
The Asian Cancer Research Group (ACRG) classified 300 GC cases into four different subtypes-microsatellite unstable tumors with the best overall prognosis and three microsatellite stable (MSS) groups: MSS/TP53-active, MSS/TP53-inactive, and MSS/EMT (epithelial-to-mesenchymal transition, the process through which epithelial cells lose their intercellular adhesion and increase motility) (33). Similar to the GS cohort in TCGA, MSS/EMT subgroup was defined by loss of CDH1, poorest prognosis, advanced disease stage at diagnosis, and highest rates of diffuse histology tumors (>80%, P<0.0001). This subtype also had the highest rates of recurrence and development of peritoneal carcinomatosis, with the lowest rate of mutational events. However, while there was an overall higher incidence of DGC in the entire ACRG cohort compared to the TCGA cohort (45% vs. 24%, respectively), there was less clustering of DGC to a single subtype in the ACRG cohort (27% in the MSS/EMT), suggesting more heterogeneity in this patient population (33). In addition, CDH1 mutations were less common in the ACRG MSS/EMT subtype and RHOA mutations were seen in both the MSS/TP53+ and MSS/TP53- groups, suggesting that the TCGA GS and ACRG MSS/EMT subtypes are not the same despite significant overlap in characteristics.
In order to better classify the heterogeneity seen within the TCGA and ACRG analyses of DG, Ge et al. conducted a proteomic analysis of 84 DGC cases (29). The study analyzed shared proteomic features between DGC and nearby normal tissue from the same patients, ultimately classifying DGC into three proteomic subgroups—PX1, PX2, and PX3. Cell cycle dysregulation was exhibited PX1 and PX2, EMT in PX2, and immune-response proteins in PX3. Notably, PX1 subtype had the best overall survival (OS) while PX3 exhibited no significant benefit from adjuvant chemotherapy (log rank P=0.831), expressed immune-response proteins IDO1 and ARG1, and had the worst prognosis (log-rank P=0.038). Other proposed classification systems have focused more on clinicopathologic characteristics, including one by Shah et al. which divided GCs into three clinical subtypes – proximal non-diffuse, distal non-diffuse, and diffuse – and found distinct gene expression profiles for each subgroup (34). Further analyses of DGC should provide insight into the heterogeneity of DGC and may eventually inform clinical decision making regarding optimal treatment strategies and combinations for this disease.
In addition to differences in genomic, proteomic, and clinical profiles between DGC and other types of GC, mutations in adhesion and motility proteins in DGC may contribute to its infiltrative and poorly cohesive histology. CDH1 is a tumor suppressor gene that codes for E-cadherin and is one of the most common mutations seen in DGC. E-cadherin is calcium-dependent transmembrane glycoprotein that interacts with the extracellular domain to form the adherens junctions while the cytoplasmic domains connect to the cytoskeleton and mediate cellular signaling (24,35). This protein controls embryogenesis, oversees cell growth, regulates cell maturation, and maintains epithelial integrity and tissue architecture (36,37). WES and RNA-seq studies have confirmed higher rates of somatic CDH1 mutations in DGC and SRCC, especially in younger patients and those with peritoneal carcinomatosis (21,23). The most common CDH1 mutations seen are truncating mutations or predicted to be deleterious missense mutations in the cadherin domain, occurring 2.5 times more frequently in those with signet ring cell phenotype and predicted to be associated with loss of function (23). In all cases, CDH1 mutations were clonal and co-occurred with mutations in TP53, another important tumor suppressor gene, suggesting that this mutation is acquired early in the pathogenesis of the disease.
RHOA is a gene that encodes a small GTPase, and is involved in cell migration via actin organization, cytokinesis, and the cell cycle; it is somatically mutated in up to 25.3% of DG cases (20,38). These mutations tend to occur in specific hotspots and sometimes co-occur with mutations in its regulatory molecules, although mutations in the regulatory molecules are not specific to DGC (20). In addition, only 4.5% of RHOA-mutant DGC stained positive for HER2, suggesting that these tumors represent a subtype with little overlap to the more common HER2-positive GC. In vitro models also suggest that increased RHOA activity appears to correlate with resistance to chemotherapy and may provide a growth advantage in disease progression (20,39).
In addition to somatic mutations in CDH1 and RHOA, a study of WES on 23 DGC samples identified mutations in CMTM2, a member of a chemokine-like factor superfamily, as a driver mutation in DGC with lymph node metastases (11). In this study, patients with lower CMTM2 expression had shorter OS. The study also found a difference in mutational patterns between N0 and N3 tumors, suggesting both molecular and clinical heterogeneity within DGC. N3 tumors showed more frequent DNA copy number alterations compared to N0 tumors, and although N0 and N3 tumors both had a large number of mutated genes, only 7% of the total mutations were shared between the two groups. N0 tumors contained more mutations related to cell adhesion and transport, while N3 tumors had more tumors related to cell development. N3-specific mutations included TP53, ARID1A, and RHOA, providing evidence that certain mutational profiles may be correlated with a more aggressive disease course.
CLDN18 is a cellular tight-junction protein only expressed in gastric mucosa and ARHGAP is a regulator of G proteins which may be involved in cell motility (4). CLDN18-ARHGAP fusions have been noted in ~15% of DGC and may be associated with larger tumors, greater rate of lymph node metastases, and more advanced disease presentation compared to fusion-negative DGC patients (38,40). The CLDN18-ARHGAP fusion may also be associated with resistance to oxaliplatin and fluoropyrimidine-based chemotherapy (41).
In addition to these common mutations, a recent study of GCs presenting with peritoneal carcinomatosis provided further clues as to genomic alterations most commonly seen in sporadic DGC (23). Mutations in TAF1 and KMT2C were exclusively seen in DGC, more commonly occurring in those with signet ring cell histology, with truncating or deleterious TAF1 mutations typically co-occurring with CDH1 or TP53 mutations. DGC specimens also had higher ploidy than intestinal type cancers, with frequent heterozygous deletion of chromosome 3p, an area encompassing multiple tumor suppressor genes, 9p24 involving JAK2 and CD274 (PD-L1), and duplication of 20q, which includes multiple oncogenes. DGC tumors also had higher rates of dysregulation of important cancer signaling pathways when compared to the intestinal type, including G2/M cell cycle checkpoint, mTOR, MYC, and mitotic spindle assembly. Finally, DGC samples had higher levels of CD4 memory T cells with lower levels of cytotoxic lymphocytes, monocytes, NK cells, myeloid dendritic cells, and normal peritoneal fibroblasts, suggesting a change in tumor microenvironment within this histologic subtype that facilitates development of peritoneal carcinomatosis.
With regard to the commonly tested biomarkers in GC, HER2 overexpression or amplification is seen less commonly in DGC (6.2% vs. 32% in intestinal type) and appears to be mutually exclusive with RHOA mutations (20,42). Further, DGC are not likely to be microsatellite unstable (MSI-H), with at least one study finding no MSI-high tumors amongst the DGC cohort (20). Other molecular biomarkers which have been associated with DGC include alterations in the PI3K/Akt/mTOR pathway (such as overexpression of phosphorylated mTOR and HER, and FGFR amplifications leading to PI3K/Akt activation) and c-MET overexpression or amplification (43). On the other hand, VEGF/VEGFR, which is associated with NF-kB activation by H. Pylori, is more commonly associated with IGC. These findings may have implications for currently available treatment, resulting in lower efficacy of HER2-targeted and immune checkpoint inhibitors in certain subsets of DGC and highlighting the need for alternative targeted therapies for this subtype.
HDGC
While most GCs are sporadic, 1–3% of cases occur in the setting of hereditary syndromes, which are more common in Western populations than in Asians (44-47). There are three main indications for genetic testing for CDH1 mutations or other germline genetic alterations (Table 1) according to the International Gastric Cancer Linkage Consortium (IGCLC) (26): (I) patient with a diagnosis of DGC before age 40 years; (II) two or more GC cases regardless of age, with at least one confirmed DGC, in first-degree or second-degree relatives; (III) personal or family history of DGC and lobular breast cancer, with at least one diagnosed before age 50 years. The presence of oral facial clefts in patients with DGC, bilateral lobular breast cancer diagnosed younger than 50 years of age, two family members with lobular breast cancer diagnosed younger than 50 years of age, and the pathological finding of signet-ring cells are all soft indications for genetic testing (24).
Table 1. Criteria for genetic risk assessment in the setting of suspected hereditary diffuse gastric cancer.
| IGCLC 2015 criteria (26): including 1st or 2nd degree relatives |
| Established criteria |
| One case of DGC <40 years of age |
| Family history of 2 gastric cancer cases regardless of age, at least one confirmed DGC |
| Personal or family history of DGC and LBC, one diagnosed <50 |
| Families in whom testing can be considered |
| A personal or family history of cleft lip/palate in a patient with DGC |
| Individual with bilateral LBC or family history of 2 or more cases of LBC <50 years of age |
| In situ signet ring cells and/or pagetoid spread of signet ring cells |
IGCLC, International Gastric Cancer Linkage Consortium; DGC, diffuse gastric cancer.
In addition to the available multigene hereditary cancer panels, newer technologies have been developed including next-generation sequencing panels to cover all the 16 exons of the CDH1 gene and liquid biopsies to detect circulating-tumor DNA and test for CDH1 promoter methylation in patients with GC (44,45). These NGS panels may pick up mutations in many of the genes implicated in hereditary syndromes to which DGC may be linked. However, the IGCLC guidelines clearly do not capture all patients who should be screened for a CHD1 mutation. In one study examining 26,936 patients who underwent multigene panel testing for any reason, 65% of patients who were identified to have a pathogenic CDH1 variant would not have otherwise met IGCLC guidelines for testing (47).
In addition to CDH1 germline mutation on chromosome 16q22 (44,48,49), germline mutations in the PALB2 gene, which encodes a protein that binds to the breast cancer 2 (BRCA2) early onset protein, have been linked to HDGC (50). Other familial cancer syndromes that are associated with GC include: hereditary non-polyposis colorectal cancer syndrome or Lynch syndrome (mutations in MLH1, MSH2, MSH6, or PSM2) (51), Peutz-Jeghers syndrome (mutations in STK11) (52), familial adenomatous polyposis (APC gene mutation) (53), gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) (54), MUTYH gene-associated polyposis (MAP) (55), Cowden syndrome (PTEN gene mutation) (56), juvenile Polyposis syndrome (germline mutations in the BMPR1A and SMAD4/MADH4 genes) (57,58), Li-Fraumeni syndrome (germline TP53 mutations) (59), MAP3K6 germline mutations (60), and loss-of-function ATM gene mutations (21). CTNNA1 mutation has also been implicated in HDGC (22,61).
Since the initial report of CDH1 germline mutation associated HDGC in indigenous New Zealand Māori kindred by Guilford et al. in 1998 (48), 155 distinct germline CDH1 mutations across the gene have been identified with 126 mutations being pathogenic (24). Truncating mutations or deficient mRNA expression are the leading causes for pathogenic non-functional E-cadherin protein (22,26,62). Autosomal dominant inheritance has been described in HDGC syndrome with oncological process initiated after somatic inactivation of the second CDH1 allele (63). HDGC, hereditary lobular breast cancer, oral facial clefts and blepharocheilodontic syndrome constitute the pleiotropic constellation of manifestations in patients with CDH1 germline mutations (24,25,64,65). These patients are thought to have a cumulative risk of 56% for females and 70% for males to develop GC by age of 80 years (66). However, these numbers came from families who were tested based on IGCLC criteria. A recent review of the Surveillance, Epidemiology, and End Results (SEER) database suggested that incidence of GC among all patients with pathogenic CDH1 variants (not just those tested based on IGCLC guidelines) may be significantly lower than previously reported, closer to 42% for men and 33% for women (67). The average age of HDGC diagnosis is 37 years (68,69). Symptomatic patients are more often found to have metastatic disease and a cure rate as low as 10% (26,70).
In CDH1-negative index cases, 11.1% have been found to have pathogenic variants driving familial disease (66). Novel truncating mutations in CTNNA1, a gene encoding for alpha-catenin and whose mutation results in alpha-catenin expression loss, have been reported in 2 unrelated families. As CTNNA1 is involved in intracellular adhesion, this loss of function mutation likely mirrors that seen with CDH1 mutations and serves as a susceptibility gene for DGC. In families affected by CTNNA1 mutations, DGC arises at a later age and has incomplete penetrance (22,61). Novel truncating mutations in BRCA2, PRSS2, ATM, and PALB2 as well as missense variants in SDHB, STK11, and MSR1 were also identified, suggesting that hereditary DGC may be more heterogeneous than previously thought and may require more liberal referral to genetic counseling.
Management: presentation and treatment outcomes
DGC is associated with a poorer prognosis compared to IGC. In one meta-analysis of 60,000 GC patients across 73 studies with all stages of disease, there was an increased risk of death in DGC patients compared to IGC (HR 1.23, P<0.0001) (27). This adverse prognosis was found across multiple subgroups including loco-regional disease (HR 1.21, P<0.0001), advanced disease (HR 1.25, P=0.014), both Western and Asian patients, and regardless of chemotherapy exposure. A second large series of over 3000 patients (30% of which had DGC) demonstrated similar findings, with a poorer 5-year survival in patients with DGC compared to IGC (45% vs. 57.7%, P<0.001) (6).
The poor prognosis associated with DGC is due in part to its aggressive nature and in part to the difficulty in detecting the disease (71). In the early stages of disease, DGC can present with vague symptoms such as reflux or no symptoms at all as the tumor infiltrates the musculature of the gastric wall leading to a rigid, “leather-bottle” stomach (linitis plastica). In more advanced stages, DGC is more likely to present with direct extension into the nearby anatomic structures including; the peritoneum, omentum, and mesocolon, while IGC is more frequently associated with hematogenous dissemination and hepatic metastases (2,5,7,72). In addition, DGC is more likely to result in peritoneal carcinomatosis, which may not be measurable on standard imaging (23). Endoscopic detection is challenging due to the presence of isolated tumor cells or small tumor cell clusters which infiltrate in an unpredictable pattern (23,71). Therefore, the disease can be visually absent on endoscopy and even random sampling of gastric tissue might miss cells which are too deep or scattered. Computer tomography and positron emission tomography, routinely used to stage GC, are limited in detecting peritoneal or infiltrating DGC, due to low standard uptake values (SUV), perhaps due to a lack of expression of glycolytic pathways compared to the upregulation of these pathways in IGC (8,34). Given the predilection for peritoneal metastases in DGC, diagnostic laparoscopy may be a useful tool in evaluation of this histologic subtype (73).
Suspected or confirmed HDGC patients are best managed at a comprehensive cancer center, where a multi-disciplinary team including a gastroenterologist, genetic counsellor, surgical oncologist, medical oncologist, experienced pathologist, and nutritionist is available to provide optimal oncological outcomes. The growing arsenal of cost-effective genetic and genomic assessment tools, combined with the unique role of genetic counsellors, have an important role in the evaluation and diagnosis of HDGC (74). Early detection of CDH1 mutations using multigene hereditary cancer panels in otherwise unsuspected patients provides timely comprehensive oncological care for both patients and families, and may improve OS (70), while newly emerging multigene panels help with the detection of CDH1 mutations in young patients who are adopted or have unknown family history (70).
Genetic testing is followed by upper endoscopy using the Cambridge protocol to maximize the detection of focal aggregations of signet-ring cells and also evaluate gastric distensibility to assess for pagetoid spread and submucosal infiltration which would be concerning for linitis plastic (46,75). The Cambridge protocol includes a minimum of 30 biopsies divided into 6 groups, each containing five random biopsies from the pre-pyloric mucosa, antrum, transitional zone, body, fundus, and cardia (46). This should be done using a white light high-definition endoscope over at least 30 minutes to allow for careful inspection of the entire gastric mucosa (46). The utility of this extensive endoscopic protocol is to increase the yield of detecting cancerous foci that do not manifest as visible lesions, although missing minute foci is common (26,76). Screening for asymptomatic patient with positive CDH1 mutation should begin at age of 16–18 years (26).
Considerations in surgical approaches
Histologically, DGC as seen in the HDGC setting can be confined to the epithelium (in situ), spreading in pagetoid fashion, or invasive (44). Total gastrectomy and Roux-en-Y esophagojejunostomy is the curative surgical approach in asymptomatic patients but should only be undertaken after baseline endoscopy. The IGCLC recommends prophylactic total gastrectomy for CDH1 mutation carriers during their third decade regardless of endoscopic features (26,46,77). Frozen sections from the proximal and distal portions of the specimen should be sent intraoperatively to confirm the removal of the entire gastric mucosa and prevent future recurrence (78). These operations can be performed either laparoscopic or robotic techniques. Patients who are not willing to undergo surgery should be followed closely by endoscopic surveillance with recommendation for surgery once an early pathological abnormality is detected (26). For those with a missense mutation, the decision to undertake a prophylactic total gastrectomy is less clear, as the pathogenicity of these mutations is less understood (44). Pathological staging is commonly T1 N0 for patients who undergo prophylactic gastrectomy after incidental detection of CDH1 mutations (70), thought to be related to the low proliferative index of signet ring adenocarcinoma and early surgical intervention. There is no consensus on the level of lymph node dissection; however, D1 lymph node dissection is typically recommended in patients undergoing prophylactic gastrectomy, as missing cancerous focus in the setting of prophylactic gastrectomy is not uncommon (26,70). We do not currently advocate for a D2 dissection when performing prophylactic gastrectomy. Prophylactic bilateral mastectomy is not routinely performed and breast cancer surveillance with magnetic resonance imaging starting between the age 25–35 years is recommended (44).
For those without a known hereditary syndrome and histologically confirmed DGC, surgical resection to achieve negative surgical margins remains the mainstay of curative therapy. While endoscopic resection modalities such as endoscopic submucosal dissection (ESD) and endoscopic mucosal resection (EMR) (79,80) can be used for early non-ulcerated GC (T1 tumors) with low risk of lymph node metastasis (80,81), the majority of patients will require minimally invasive or open approaches. D1 dissection is commonly performed in patients with cT1aN0 and well-differentiated ≤1.5 cm cT1bN0 tumors who do not have access to endoscopic resection at experienced centers (82). Gastrectomy with D2 dissection is the standard surgical treatment for cT1N+ and resectable T2–T4 tumors in East Asia since its introduction in 1960’s, whereas D2 dissection did not show survival benefits in Western surgical oncology practices (82-85). Therefore, the D2 dissection is a recommended but not a mandatory procedure in Western countries and should be performed by experienced surgeons in high-volume comprehensive cancer centers. If GC invades the duodenum, D2 dissection is extended to involve retro-pancreatic lymph nodes and this dissection is defined as D2+ (82). There is no role for D3 dissection, as it confers a higher morbidity and worse survival (86). It is recommended that ≥15 lymph nodes to be harvested and examined for accurate staging, and is used as a quality metric for surgery (87). Roux-en-Y gastrojejunostomy is the ideal reconstruction after total gastrectomy and has been used more frequently after distal gastrectomy due to reduced postoperative complications and improved quality of life (88). Cytoreductive surgery (CRS) and hyperthermic intraperitoneal chemotherapy (HIPEC) have been offered to patients with peritoneal metastases from GC. Many studies have argued the benefit and morbidity of CRS/HIPEC with many showing no survival advantage and others demonstrating few extra months of survival; nonetheless, this is still a topic of debate and controversy and should only be performed on clinical trial (89,90).
Considerations in systemic therapy
From a therapeutic perspective, DGC is associated with decreased responsiveness to chemotherapy and chemoradiation (23,39,91-93). In the non-metastatic setting, perioperative therapy has become a standard of care since the publication of the MAGIC trial, a landmark randomized phase III trial that established the survival benefit of this approach when compared to surgery alone in resectable gastric, GEJ, or lower esophageal adenocarcinoma (HR 0.75, P=0.009). However, this study did not analyze therapeutic outcome differences by histologic subtype or Lauren classification, and multiple studies since then have suggested a resistance of SRCC to chemotherapy (91,92). Given this observed resistance in SRCC, the phase II PRODIGE 19 was designed to examine upfront surgery followed by adjuvant ECF for 6 cycles versus the MAGIC regimen of 3 cycles before surgery and 3 cycles after (94). The study met its endpoint of OS at 2 years, with a median OS (mOS) favoring perioperative chemotherapy rather than upfront surgery [39 vs. 28 months, exploratory hazard ratio 0.71 (95% confidence ratio 0.40−2.64)]; final results are pending (95).
More recently, the FLOT4-AIO demonstrated the advantage of using perioperative FLOT (infusional 5-fluorouracil, leucovorin, oxaliplatin, and docetaxel) over the prior MAGIC regimen (epirubicin, cisplatin, infusional 5-fluorouracil/capecitabine) (96). In the study, approximately 27% of patients had DGC, 45% with IGC, 20% unevaluable, and 8% unknown. An improvement in OS favoring the FLOT cohort (mOS 50 vs. 35 months, P=0.012) was found, with benefit for both DGC and non-diffuse cohorts. However, the non-diffuse cohort had a numerically favorable hazard ratio for survival with FLOT compared to the diffuse cohort (0.74 vs. 0.85, respectively). In a pooled subgroup analysis evaluating the rate of complete pathologic response (pCR) across both arms of chemotherapy, pCR was seen more frequently in IGC compared to DGC (16% vs. 3%, P=0.004) (97). Therefore, while resectable DGC appears to respond to chemotherapy in the perioperative setting, this effect may not be as robust as in IGC tumors. Further research can help clarify this degree of response and help identify which patients will benefit from adjuvant chemotherapy.
DGC may also be less responsive to chemoradiotherapy in the adjuvant setting. INT0116 was a phase III trial of adjuvant chemoradiation versus observation after resection for GC and found benefit to chemoradiation for IGC but not DGC (P=0.077) (98). Although not statistically significant, these findings were replicated in the ARTIST trial, which randomized 458 GC (of which 60% had DGC) patients to adjuvant chemotherapy or chemotherapy plus chemoradiotherapy. Across the entire cohort, there was no benefit in disease-free-survival (DFS) or OS with the addition of chemoradiotherapy to chemotherapy (P=0.086 and 0.52, respectively). However, subgroup analysis found a significant improvement in DFS for the IGC cohort but not DGC, and the effect of radiotherapy differed significantly between IGC and DGC for both DFS and OS (P<0.01 and P<0.01, respectively) (99), suggesting that DGC may not be as sensitive to adjuvant chemoradiotherapy as IGC.
Landmark studies of systemic therapy in advanced disease have typically incorporated subgroup analyses examining treatment effects in DGC and IGC subgroups (Table 2). However, even though DGC and SRCC have been associated with a poorer response to chemotherapy in some studies (91,92), many of the landmark studies of cytotoxic chemotherapy in GC/GEJC have not found a statistically significant difference in treatment effect between diffuse and intestinal histology possibly due to small numbers of patients or the study not being powered to detect such a difference. While early platinum/fluoropyrimidine studies did not include these analyses, the WJOG 4007 trial, a phase 3 trial comparing irinotecan to paclitaxel in second-line, found no significant difference in OS between arms (100); intestinal and diffuse histology had similar HR for OS (1.15 vs. 1.11, respectively). The TAGS study, which evaluated trifluridine/tipiracil versus best supportive care in treatment refractory GC/GEJ, showed an improvement in OS (5.7 vs. 3.6 months, P<0.001) with hazard ratios for OS between diffuse and intestinal histology appearing relatively similar (0.69 vs. 0.58) and subgroup analysis not detecting a significant difference in survival (101). Although these studies did not detect a difference between histological subtypes, other studies have begun to specifically look at the use of chemotherapy in DGC and SRCC. For example, TEFOX (docetaxel, 5-fluorouracil, and oxaliplatin) has shown efficacy in a single arm study of 65 patients with advanced signet-ring carcinoma (ORR 66%, DCR 87%) (108).
Table 2. Hazard ratios of systemic therapy trials where an analysis by histological subtype was available.
| Study | Intervention vs. control | Hazard ratio for overall survival for diffuse cohort (95% CI, if given) | Hazard ratio for overall survival in non-diffuse cohort (95% CI, if given) | P value |
|---|---|---|---|---|
| Cytotoxic chemotherapy | ||||
| FLOT4-AIO (96) | Perioperative FLOT vs. ECF | 0.85 | 0.74 | 0.58 |
| WJOG 4007 (100) | Irinotecan vs. paclitaxel in 2nd line | 1.11 (0.75–1.63) favoring paclitaxel | 1.15 (0.77–1.71) | N/A |
| TAGS (101) | Trifluridine/tipiracil (TAS-102) vs. best supportive care | 0.69 (0.36 vs. 1.31) favoring TAS-102 | 0.58 (0.39 vs. 0.87) | N/A |
| Targeted therapy | ||||
| REGARD (102) | Ramucirumab vs. best supportive care | 0.56 (0.37 vs. 0.86) favoring ramucirumab | 1.01 (0.58–1.74) favoring placebo | N/A |
| RAINBOW (103) | Ramucirumab + paclitaxel vs. paclitaxel alone in 2nd line | 0.86 (0.64–1.14) favoring ramucirumab | 0.71 (0.53–0.93) | N/A |
| TOGA (42) | Trastuzumab + chemotherapy versus chemotherapy alone in 1st line HER2+ cancer | 1.07 (0.56–2.05) favoring chemotherapy alone | 0.69 (0.54–0.88) favoring trastuzumab + chemotherapy | N/A |
| JACOB (104) | Trastuzumab/chemotherapy + pertuzumab vs. trastuzumab/chemotherapy alone in 1st line HER2+ cancer | 0.82 (0.40–1.69) favoring pertuzumab | 0.85 (0.71–1.02) | N/A |
| Immunotherapy | ||||
| Keynote 062 (105) | Pembrolizumab vs. chemotherapy PD-L1 CPS >1 in the 1st line | 0.73 (0.53–0.99) favoring pembrolizumab | 0.92 (0.67–1.25) | N/A |
| Keynote 061 (106) | Pembrolizumab vs. paclitaxel in PD-L1 CPS >1 in the 2nd line | 0.88 (0.54–1.45) favoring pembrolizumab | 0.66 (0.40–1.11) | N/A |
| ATTRACTION 2 (107) | Nivolumab vs. placebo in 3rd line | 0.82 (0.57–1.17) favoring nivolumab | 0.59 (0.41–0.85) | 0.377 |
PD-L1, programmed death ligand-1; CPS, combined positive score.
Vascular endothelial growth factor (VEGF) and VEGF-receptor (VEGFR) expression have been associated with GC, though the degree VEGF/VEGFR expression between diffuse versus intestinal histology remains controversial (109,110). Some studies have found an association between VEGF/VEGFR expression and intestinal type histology, while others have found no correlation. Studies on anti-angiogenic therapy have also failed to show a strong connection between Lauren classification and response to therapy. The REGARD trial, which evaluated single-agent ramucirumab versus best supportive care in the second-line setting, demonstrated an OS benefit to ramucirumab, with diffuse type tumors appearing to benefit from ramucirumab similarly versus intestinal type tumors in terms of PFS (hazard ratio 0.49 vs. 0.47), with a numerically better OS (hazard ratio 0.56 vs. 1.01, respectively) (102). The RAINBOW trial, however, in which ramucirumab was combined with paclitaxel in the second-line setting, appeared to show a greater benefit for the combination therapy in intestinal type tumors compared to diffuse-type tumors (hazard ratio for OS 0.71 vs. 0.86, respectively) (103).
HER2 amplification is seen less frequently DGC compared to IGC (42,43,111), and large trials have suggested a possible larger benefit to HER2-directed therapy in the intestinal subgroup. The ToGA trial, demonstrating benefit to adding trastuzumab to platinum/fluoropyrimidine therapy in the front-line setting, demonstrated a numerically greater benefit in the in the intestinal group compared to the diffuse cohort (HR for OS 0.69 vs. 1.07), though treatment effect could not be excluded in either cohort (42). The JACOB trial, a negative trial of platinum/fluoropyrimidine/trastuzumab-based therapy with or without pertuzumab, showed no global difference in OS (17.5 vs. 14.2 months, P=0.057), and no difference between diffuse or intestinal histology (HR 0.82 vs. 0.85, respectively) (104). However, numbers for diffuse histology in the study were small (n=39).
Immunotherapy in advanced GC has become an area of active investigation but may have a decreased response in DGC compared to IGC. One study showed that the genome-stable TCGA subtype (of which DGC are enriched) demonstrated significantly lower responses to pembrolizumab compared to other subtypes (112). The larger clinical trials have been mixed with respect to efficacy of immunotherapy in DGC, however. In KEYNOTE 062, a front-line trial of pembrolizumab alone versus pembrolizumab plus chemotherapy versus chemotherapy alone showed that single agent pembrolizumab was noninferior to chemotherapy in the intention to treat population, and the hazard ratios for survival were relatively similar between diffuse and intestinal subtypes (0.73 vs. 0.92 favoring pembrolizumab, respectively) (105). KEYNOTE 061 was a randomized phase 3 trial which compared patients with programmed death ligand-1 (PD-L1) combined positive score (CPS) >1 to pembrolizumab versus paclitaxel in the second line setting for advanced GC/GEJC (106). While the overall study was negative for pembrolizumab benefit, the hazard ratio for survival was numerically better in intestinal type tumors compared to diffuse type (0.66 vs. 0.88, respectively). Similar results were seen in ATTRACTION 2, which randomized refractory GC patients in the third-line setting to nivolumab versus placebo and found an OS benefit (5.26 vs. 4.14 months P<0.0001) (107); intestinal histology appeared to have a numerically better hazard ratio for OS (HR 0.59 vs. 0.82, P=0.377) versus DGC, though this was not statistically significant. Additional post-hoc analyses of banked tumor specimens and pre-planned analysis by histologic subtype in large phase 3 trials will be necessary before definitive conclusions regarding optimal treatment strategies for DGC can be made.
While previous targeted therapy trials have focused on pathways more common in IGC, a novel agent targeting CLDN18 may have some impact in DGC. Zolbetuximab is a chimeric IgG1 monoclonal antibody which binds to CLDN18.2 on the surface of tumor cells and mediates killing of CLDN18-positive cancer cells via antibody-depended cellular cytotoxicity. In a single arm phase II trial of single agent zolbetuximab in refractory GC (MONO trial), zolbetuximab demonstrated an objective response rate of 9% and a clinical benefit rate of 23%, with 90% of those achieving clinical benefit having moderate or high expression of CLDN18.2 in at least 70% of tumor cells (113). Of note, 41% of the patients enrolled on this trial had DGC. The FAST trial, a phase II trial of front-line epirubicin, oxaliplatin, and capecitabine with or without zolbetuximab in patients with CLDN18-positive advanced GC, found an improvement in its primary endpoint of PFS (5.7 vs. 7.9 months, P=0.001) as well as a numerically better OS (median 8.7 vs. 12.5 months) with increased efficacy in very high expressing tumors (114,115). Similar to the MONO trial, 45% of patients enrolled had DGC, likely secondary to the fact that both studies required centralized testing of CLDN18.2 membrane expression and only enrolled those patients with staining intensity of at least 50% of tumor cells. This strategy likely selects for DGC patients, and we await the results of a large phase III trial of infusional 5-fluorouracil/oxaliplatin (FOLFOX) with or without zolbetuximab, currently underway, to potentially provide additional insight in the management of this subtype (NCT03504397).
Conclusions and future directions
From its initial classification in 1965, DGC has demonstrated unique histological characteristics which may explain its clinical behavior. In addition, emerging molecular sequencing technologies and the recognition of hereditary syndromes have added insight into risk profiles and potential therapeutic targets. However, the poorly cohesive and infiltrating nature of tumor cells often leads to locally invasive disease, lower success rates in early detection, and peritoneal carcinomatosis. This difficulty with screening and surveillance especially affects members with suspected or known familial predisposition to DGC, resulting in recommendations for highly morbid procedures such as prophylactic gastrectomy. Ultimately, patients with DGC, especially those with hereditary syndromes, are best managed at high-volume comprehensive cancer centers which can provide multidisciplinary care.
In terms of treatment, DGC, including signet ring cell cancer subtypes, appear to demonstrate resistance to standard cytotoxic chemotherapy and chemoradiotherapy in early stage disease, with less apparent trends in the metastatic setting. However, there appears to be a heterogeneity in response which goes beyond just histologic classification. More recently, molecular subtyping has allowed us to better characterize the disease and understand its behavior. Analyses from the TCGA and ACRG have provided insight into molecular profiles which can help classify DGC into more predictive and prognostic subgroups. As novel agents are investigated, including immunotherapy and targeted therapy, understanding how the various subtypes of DGC may respond to treatment will be essential. The majority of prospective studies in GC have historically included subgroup analyses of DGC versus IGC in terms of histology. In the future, prospective trials which study the molecular subtypes of GC (including DGC) will allow us to more precisely tailor treatments for patients based on their disease characteristics.
Acknowledgments
We thank Dr. Nicholas Mackrides for his contribution of the photomicrographs included in this paper.
Funding: This publication was supported by grant number P30 CA006927 from the National Cancer Institute, NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Conflicts of Interest: Dr. Crystal Denlinger has received advisory board honoraria from Astellas, Exelixis, Bayer, Merck, Bristol Myer Squibb, Eli Lilly & Co, EMD Serono, and BeiGene. Dr. Denlinger has received institutional grant support from Array BioPharma, Amgen, Advaxis, Bristol Myer Squibb, Astra Zeneca, Sanofi, BeiGene, Merrimack Pharmaceuticals, Roche/Genentech, Macrogenics, Lycera, Zymeworks, and Agios Pharmaceuticals. Dr. Jeffrey Farma is on the data safety monitoring board for Delcath. The other authors have no conflicts of interest to declare.
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424. 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 2.Ajani JA, Lee J, Sano T, et al. Gastric adenocarcinoma. Nat Rev Dis Primers 2017;3:17036. 10.1038/nrdp.2017.36 [DOI] [PubMed] [Google Scholar]
- 3.Lauren P. The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma. An Attempt at a Histo-Clinical Classification. Acta Pathol Microbiol Scand 1965;64:31-49. 10.1111/apm.1965.64.1.31 [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202-9. 10.1038/nature13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korivi BR, Faria S, Aly A, et al. Intestinal and diffuse gastric cancer: a retrospective study comparing primary sites. Clin Imaging 2019;56:33-40. 10.1016/j.clinimag.2019.03.002 [DOI] [PubMed] [Google Scholar]
- 6.Chen YC, Fang WL, Wang RF, et al. Clinicopathological Variation of Lauren Classification in Gastric Cancer. Pathol Oncol Res 2016;22:197-202. 10.1007/s12253-015-9996-6 [DOI] [PubMed] [Google Scholar]
- 7.Nie RC, Yuan SQ, Li YF, et al. Clinicopathological Characteristics and Prognostic Value of Signet Ring Cells in Gastric Carcinoma: A Meta-Analysis. J Cancer 2017;8:3396-404. 10.7150/jca.21017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanjeevaiah A, Cheedella N, Hester C, et al. Gastric Cancer: Recent Molecular Classification Advances, Racial Disparity, and Management Implications. J Oncol Pract 2018;14:217-24. 10.1200/JOP.17.00025 [DOI] [PubMed] [Google Scholar]
- 9.Balakrishnan M, George R, Sharma A, et al. Changing Trends in Stomach Cancer Throughout the World. Curr Gastroenterol Rep 2017;19:36. 10.1007/s11894-017-0575-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henson DE, Dittus C, Younes M, et al. Differential trends in the intestinal and diffuse types of gastric carcinoma in the United States, 1973-2000: increase in the signet ring cell type. Arch Pathol Lab Med 2004;128:765-70. [DOI] [PubMed] [Google Scholar]
- 11.Choi JH, Kim YB, Ahn JM, et al. Identification of genomic aberrations associated with lymph node metastasis in diffuse-type gastric cancer. Exp Mol Med 2018;50:6. 10.1038/s12276-017-0009-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu H, Rusiecki JA, Zhu K, et al. Stomach carcinoma incidence patterns in the United States by histologic type and anatomic site. Cancer Epidemiol Biomarkers Prev 2009;18:1945-52. 10.1158/1055-9965.EPI-09-0250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsonnet J, Vandersteen D, Goates J, et al. Helicobacter pylori infection in intestinal- and diffuse-type gastric adenocarcinomas. J Natl Cancer Inst 1991;83:640-3. 10.1093/jnci/83.9.640 [DOI] [PubMed] [Google Scholar]
- 14.Lee JH, Kim SH, Han SH, et al. Clinicopathological and molecular characteristics of Epstein-Barr virus-associated gastric carcinoma: a meta-analysis. J Gastroenterol Hepatol 2009;24:354-65. 10.1111/j.1440-1746.2009.05775.x [DOI] [PubMed] [Google Scholar]
- 15.Cormedi MCV, Guindalini RSC, Branas PA, et al. Fraction of diffuse gastric cancer attributable to dietary risk factors. J Clin Oncol 2018;36:e16042-e.
- 16.Praud D, Rota M, Pelucchi C, et al. Cigarette smoking and gastric cancer in the Stomach Cancer Pooling (StoP) Project. Eur J Cancer Prev 2018;27:124-33. 10.1097/CEJ.0000000000000290 [DOI] [PubMed] [Google Scholar]
- 17.Duell EJ, Travier N, Lujan-Barroso L, et al. Alcohol consumption and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort. Am J Clin Nutr 2011;94:1266-75. 10.3945/ajcn.111.012351 [DOI] [PubMed] [Google Scholar]
- 18.Rota M, Pelucchi C, Bertuccio P, et al. Alcohol consumption and gastric cancer risk-A pooled analysis within the StoP project consortium. Int J Cancer 2017;141:1950-62. 10.1002/ijc.30891 [DOI] [PubMed] [Google Scholar]
- 19.Peleteiro B, Lopes C, Figueiredo C, et al. Salt intake and gastric cancer risk according to Helicobacter pylori infection, smoking, tumour site and histological type. Br J Cancer 2011;104:198-207. 10.1038/sj.bjc.6605993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kakiuchi M, Nishizawa T, Ueda H, et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet 2014;46:583-7. 10.1038/ng.2984 [DOI] [PubMed] [Google Scholar]
- 21.Cho J, Ahn S, Son DS, et al. Bridging genomics and phenomics of gastric carcinoma. Int J Cancer 2019;145:2407-17. 10.1002/ijc.32228 [DOI] [PubMed] [Google Scholar]
- 22.Oliveira C, Pinheiro H, Figueiredo J, et al. Familial gastric cancer: genetic susceptibility, pathology, and implications for management. Lancet Oncol 2015;16:e60-70. 10.1016/S1470-2045(14)71016-2 [DOI] [PubMed] [Google Scholar]
- 23.Wang R, Song S, Harada K, et al. Multiplex profiling of peritoneal metastases from gastric adenocarcinoma identified novel targets and molecular subtypes that predict treatment response. Gut 2020;69:18-31. 10.1136/gutjnl-2018-318070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Figueiredo J, Melo S, Carneiro P, et al. Clinical spectrum and pleiotropic nature of CDH1 germline mutations. J Med Genet 2019;56:199-208. 10.1136/jmedgenet-2018-105807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corso G, Figueiredo J, La Vecchia C, et al. Hereditary lobular breast cancer with an emphasis on E-cadherin genetic defect. J Med Genet 2018;55:431-41. 10.1136/jmedgenet-2018-105337 [DOI] [PubMed] [Google Scholar]
- 26.van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361-74. 10.1136/jmedgenet-2015-103094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petrelli F, Berenato R, Turati L, et al. Prognostic value of diffuse versus intestinal histotype in patients with gastric cancer: a systematic review and meta-analysis. J Gastrointest Oncol 2017;8:148-63. 10.21037/jgo.2017.01.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu B, El Hajj N, Sittler S, et al. Gastric cancer: Classification, histology and application of molecular pathology. J Gastrointest Oncol 2012;3:251-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge S, Xia X, Ding C, et al. A proteomic landscape of diffuse-type gastric cancer. Nat Commun 2018;9:1012. 10.1038/s41467-018-03121-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Y, Grabsch H, Ivanova T, et al. Comprehensive genomic meta-analysis identifies intra-tumoural stroma as a predictor of survival in patients with gastric cancer. Gut 2013;62:1100-11. 10.1136/gutjnl-2011-301373 [DOI] [PubMed] [Google Scholar]
- 31.Komuro A, Yashiro M, Iwata C, et al. Diffuse-type gastric carcinoma: progression, angiogenesis, and transforming growth factor beta signaling. J Natl Cancer Inst 2009;101:592-604. 10.1093/jnci/djp058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sohn BH, Hwang JE, Jang HJ, et al. Clinical Significance of Four Molecular Subtypes of Gastric Cancer Identified by The Cancer Genome Atlas Project. Clin Cancer Res 2017;23:4441-9. 10.1158/1078-0432.CCR-16-2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cristescu R, Lee J, Nebozhyn M, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 2015;21:449-56. 10.1038/nm.3850 [DOI] [PubMed] [Google Scholar]
- 34.Shah MA, Khanin R, Tang L, et al. Molecular classification of gastric cancer: a new paradigm. Clin Cancer Res 2011;17:2693-701. 10.1158/1078-0432.CCR-10-2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci 2008;65:3756-88. 10.1007/s00018-008-8281-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onitilo AA, Aryal G, Engel JM. Hereditary diffuse gastric cancer: a family diagnosis and treatment. Clin Med Res 2013;11:36-41. 10.3121/cmr.2012.1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paredes J, Figueiredo J, Albergaria A, et al. Epithelial E- and P-cadherins: role and clinical significance in cancer. Biochim Biophys Acta 2012;1826:297-311. [DOI] [PubMed]
- 38.Tanaka A, Ishikawa S, Ushiku T, et al. Frequent CLDN18-ARHGAP fusion in highly metastatic diffuse-type gastric cancer with relatively early onset. Oncotarget 2018;9:29336-50. 10.18632/oncotarget.25464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoon C, Cho SJ, Aksoy BA, et al. Chemotherapy Resistance in Diffuse-Type Gastric Adenocarcinoma Is Mediated by RhoA Activation in Cancer Stem-Like Cells. Clin Cancer Res 2016;22:971-83. 10.1158/1078-0432.CCR-15-1356 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Nakayama I, Shinozaki E, Sakata S, et al. Enrichment of CLDN18-ARHGAP fusion gene in gastric cancers in young adults. Cancer Sci 2019;110:1352-63. 10.1111/cas.13967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shu Y, Zhang W, Hou Q, et al. Prognostic significance of frequent CLDN18-ARHGAP26/6 fusion in gastric signet-ring cell cancer. Nat Commun 2018;9:2447. 10.1038/s41467-018-04907-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687-97. 10.1016/S0140-6736(10)61121-X [DOI] [PubMed] [Google Scholar]
- 43.Wadhwa R, Song S, Lee JS, et al. Gastric cancer-molecular and clinical dimensions. Nat Rev Clin Oncol 2013;10:643-55. 10.1038/nrclinonc.2013.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo W, Fedda F, Lynch P, et al. CDH1 Gene and Hereditary Diffuse Gastric Cancer Syndrome: Molecular and Histological Alterations and Implications for Diagnosis And Treatment. Front Pharmacol 2018;9:1421. 10.3389/fphar.2018.01421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsujiura M, Ichikawa D, Konishi H, et al. Liquid biopsy of gastric cancer patients: circulating tumor cells and cell-free nucleic acids. World J Gastroenterol 2014;20:3265-86. 10.3748/wjg.v20.i12.3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fitzgerald RC, Hardwick R, Huntsman D, et al. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet 2010;47:436-44. 10.1136/jmg.2009.074237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lowstuter K, Espenschied CR, Sturgeon D, et al. Unexpected CDH1 Mutations Identified on Multigene Panels Pose Clinical Management Challenges. JCO Prec Oncol 2017;(1):1-12. [DOI] [PubMed]
- 48.Guilford P, Hopkins J, Harraway J, et al. E-cadherin germline mutations in familial gastric cancer. Nature 1998;392:402-5. 10.1038/32918 [DOI] [PubMed] [Google Scholar]
- 49.Berx G, Staes K, van Hengel J, et al. Cloning and characterization of the human invasion suppressor gene E-cadherin (CDH1). Genomics 1995;26:281-9. 10.1016/0888-7543(95)80212-5 [DOI] [PubMed] [Google Scholar]
- 50.Fewings E, Larionov A, Redman J, et al. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: a whole-exome sequencing study. Lancet Gastroenterol Hepatol 2018;3:489-98. 10.1016/S2468-1253(18)30079-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aarnio M, Salovaara R, Aaltonen LA, et al. Features of gastric cancer in hereditary non-polyposis colorectal cancer syndrome. Int J Cancer 1997;74:551-5. [DOI] [PubMed] [Google Scholar]
- 52.Boardman LA, Thibodeau SN, Schaid DJ, et al. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Annals of internal medicine 1998;128:896-9. 10.7326/0003-4819-128-11-199806010-00004 [DOI] [PubMed] [Google Scholar]
- 53.Kashiwagi H, Spigelman AD. Gastroduodenal lesions in familial adenomatous polyposis. Surg Today 2000;30:675-82. 10.1007/s005950070077 [DOI] [PubMed] [Google Scholar]
- 54.Worthley DL, Phillips KD, Wayte N, et al. Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS): a new autosomal dominant syndrome. Gut 2012;61:774-9. 10.1136/gutjnl-2011-300348 [DOI] [PubMed] [Google Scholar]
- 55.Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009;137:1976-85.e1-10. [DOI] [PubMed]
- 56.Hamby LS, Lee EY, Schwartz RW. Parathyroid adenoma and gastric carcinoma as manifestations of Cowden's disease. Surgery 1995;118:115-7. 10.1016/S0039-6060(05)80018-2 [DOI] [PubMed] [Google Scholar]
- 57.Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol 1998;5:751-6. 10.1007/BF02303487 [DOI] [PubMed] [Google Scholar]
- 58.Brosens LA, van Hattem A, Hylind LM, et al. Risk of colorectal cancer in juvenile polyposis. Gut 2007;56:965-7. 10.1136/gut.2006.116913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oliveira C, Ferreira P, Nabais S, et al. E-Cadherin (CDH1) and p53 rather than SMAD4 and Caspase-10 germline mutations contribute to genetic predisposition in Portuguese gastric cancer patients. Eur J Cancer 2004;40:1897-903. 10.1016/j.ejca.2004.04.027 [DOI] [PubMed] [Google Scholar]
- 60.Gaston D, Hansford S, Oliveira C, et al. Germline mutations in MAP3K6 are associated with familial gastric cancer. PLoS genetics 2014;10:e1004669. 10.1371/journal.pgen.1004669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Majewski IJ, Kluijt I, Cats A, et al. An alpha-E-catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. J Pathol 2013;229:621-9. 10.1002/path.4152 [DOI] [PubMed] [Google Scholar]
- 62.Norton JA, Ham CM, Van Dam J, et al. CDH1 truncating mutations in the E-cadherin gene: an indication for total gastrectomy to treat hereditary diffuse gastric cancer. Ann Surg 2007;245:873-9. 10.1097/01.sla.0000254370.29893.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grady WM, Willis J, Guilford PJ, et al. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet 2000;26:16-7. 10.1038/79120 [DOI] [PubMed] [Google Scholar]
- 64.Frebourg T, Oliveira C, Hochain P, et al. Cleft lip/palate and CDH1/E-cadherin mutations in families with hereditary diffuse gastric cancer. J Med Genet 2006;43:138-42. 10.1136/jmg.2005.031385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghoumid J, Stichelbout M, Jourdain AS, et al. Blepharocheilodontic syndrome is a CDH1 pathway-related disorder due to mutations in CDH1 and CTNND1. Genet Med 2017;19:1013-21. 10.1038/gim.2017.11 [DOI] [PubMed] [Google Scholar]
- 66.Hansford S, Kaurah P, Li-Chang H, et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol 2015;1:23-32. 10.1001/jamaoncol.2014.168 [DOI] [PubMed] [Google Scholar]
- 67.Roberts ME, Ranola JMO, Marshall ML, et al. Comparison of CDH1 Penetrance Estimates in Clinically Ascertained Families vs. Families Ascertained for Multiple Gastric Cancers. JAMA Oncol 2019;5:1325-31. 10.1001/jamaoncol.2019.1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pharoah PD, Guilford P, Caldas C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001;121:1348-53. 10.1053/gast.2001.29611 [DOI] [PubMed] [Google Scholar]
- 69.Shepard B, Yoder L, Holmes C. Prophylactic Total Gastrectomy for Hereditary Diffuse Gastric Cancer. ACG Case Rep J 2016;3:e179. 10.14309/crj.2016.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moslim MA, Heald B, Tu C, et al. Early genetic counseling and detection of CDH1 mutation in asymptomatic carriers improves survival in hereditary diffuse gastric cancer. Surgery 2018;164:754-9. 10.1016/j.surg.2018.05.059 [DOI] [PubMed] [Google Scholar]
- 71.Kumar S, Long JM, Ginsberg GG, et al. The role of endoscopy in the management of hereditary diffuse gastric cancer syndrome. World J Gastroenterol 2019;25:2878-86. 10.3748/wjg.v25.i23.2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marrelli D, Roviello F, de Manzoni G, et al. Different patterns of recurrence in gastric cancer depending on Lauren's histological type: longitudinal study. World J Surg 2002;26:1160-5. 10.1007/s00268-002-6344-2 [DOI] [PubMed] [Google Scholar]
- 73.Rawicz-Pruszyński K, Mielko J, Pudlo K, et al. Yield of staging laparoscopy in gastric cancer is influenced by Lauren histologic subtype. J Surg Oncol 2019;120:1148-53. 10.1002/jso.25711 [DOI] [PubMed] [Google Scholar]
- 74.Slavin TP, Niell-Swiller M, Solomon I, et al. Clinical Application of Multigene Panels: Challenges of Next-Generation Counseling and Cancer Risk Management. Front Oncol 2015;5:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barber M, Murrell A, Ito Y, et al. Mechanisms and sequelae of E-cadherin silencing in hereditary diffuse gastric cancer. J Pathol 2008;216:295-306. 10.1002/path.2426 [DOI] [PubMed] [Google Scholar]
- 76.Mi EZ, Mi EZ, di Pietro M, et al. Comparative study of endoscopic surveillance in hereditary diffuse gastric cancer according to CDH1 mutation status. Gastrointest Endosc 2018;87:408-18. 10.1016/j.gie.2017.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blair V, Martin I, Shaw D, et al. Hereditary diffuse gastric cancer: diagnosis and management. Clin Gastroenterol Hepatol 2006;4:262-75. 10.1016/j.cgh.2005.12.003 [DOI] [PubMed] [Google Scholar]
- 78.Network NCC. Gastric Cancer (version 3.2019) 2019. Available online: https://www.nccn.org/professionals/physician_gls/pdf/gastric.pdf
- 79.Asano M. Endoscopic submucosal dissection and surgical treatment for gastrointestinal cancer. World J Gastrointest Endosc 2012;4:438-47. 10.4253/wjge.v4.i10.438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ahn JY, Jung HY, Choi KD, et al. Endoscopic and oncologic outcomes after endoscopic resection for early gastric cancer: 1370 cases of absolute and extended indications. Gastrointest Endosc 2011;74:485-93. 10.1016/j.gie.2011.04.038 [DOI] [PubMed] [Google Scholar]
- 81.Wang J, Yu JC, Kang WM, et al. Treatment strategy for early gastric cancer. Surg Oncol 2012;21:119-23. 10.1016/j.suronc.2010.12.004 [DOI] [PubMed] [Google Scholar]
- 82.Mihmanli M, Ilhan E, Idiz UO, et al. Recent developments and innovations in gastric cancer. World J Gastroenterol 2016;22:4307-20. 10.3748/wjg.v22.i17.4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hartgrink HH, van de Velde CJ, Putter H, et al. Extended lymph node dissection for gastric cancer: who may benefit? Final results of the randomized Dutch gastric cancer group trial. J Clin Oncol 2004;22:2069-77. 10.1200/JCO.2004.08.026 [DOI] [PubMed] [Google Scholar]
- 84.Cuschieri A, Weeden S, Fielding J, et al. Patient survival after D1 and D2 resections for gastric cancer: long-term results of the MRC randomized surgical trial. Surgical Co-operative Group. Br J Cancer 1999;79:1522-30. 10.1038/sj.bjc.6690243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439-49. 10.1016/S1470-2045(10)70070-X [DOI] [PubMed] [Google Scholar]
- 86.Sasako M, Sano T, Yamamoto S, et al. D2 lymphadenectomy alone or with para-aortic nodal dissection for gastric cancer. N Engl J Med 2008;359:453-62. 10.1056/NEJMoa0707035 [DOI] [PubMed] [Google Scholar]
- 87.Schwarz RE, Smith DD. Clinical impact of lymphadenectomy extent in resectable gastric cancer of advanced stage. Ann Surg Oncol 2007;14:317-28. 10.1245/s10434-006-9218-2 [DOI] [PubMed] [Google Scholar]
- 88.Fukuhara K, Osugi H, Takada N, et al. Reconstructive procedure after distal gastrectomy for gastric cancer that best prevents duodenogastroesophageal reflux. World J Surg 2002;26:1452-7. 10.1007/s00268-002-6363-z [DOI] [PubMed] [Google Scholar]
- 89.Ikeguchi M, Kondou A, Oka A, et al. Effects of continuous hyperthermic peritoneal perfusion on prognosis of gastric cancer with serosal invasion. Eur J Surg 1995;161:581-6. [PubMed] [Google Scholar]
- 90.Yang XJ, Huang CQ, Suo T, et al. Cytoreductive surgery and hyperthermic intraperitoneal chemotherapy improves survival of patients with peritoneal carcinomatosis from gastric cancer: final results of a phase III randomized clinical trial. Ann Surg Oncol 2011;18:1575-81. 10.1245/s10434-011-1631-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chirieac LR, Swisher SG, Correa AM, et al. Signet-ring cell or mucinous histology after preoperative chemoradiation and survival in patients with esophageal or esophagogastric junction adenocarcinoma. Clin Cancer Res 2005;11:2229-36. 10.1158/1078-0432.CCR-04-1840 [DOI] [PubMed] [Google Scholar]
- 92.Charalampakis N, Nogueras Gonzalez GM, Elimova E, et al. The Proportion of Signet Ring Cell Component in Patients with Localized Gastric Adenocarcinoma Correlates with the Degree of Response to Pre-Operative Chemoradiation. Oncology 2016;90:239-47. 10.1159/000443506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pattison S, Mitchell C, Lade S, et al. Early relapses after adjuvant chemotherapy suggests primary chemoresistance in diffuse gastric cancer. PLoS One 2017;12:e0183891. 10.1371/journal.pone.0183891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Piessen G, Messager M, Le Malicot K, et al. Phase II/III multicentre randomised controlled trial evaluating a strategy of primary surgery and adjuvant chemotherapy versus peri-operative chemotherapy for resectable gastric signet ring cell adenocarcinomas - PRODIGE 19 - FFCD1103 - ADCI002. BMC Cancer 2013;13:281. 10.1186/1471-2407-13-281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eveno C, Adenis A, Bouche O, et al. Adjuvant chemotherapy versus perioperative chemotherapy (CTx) for resectable gastric signet ring cell (SRC) gastric cancer: A multicenter, randomized phase II study (PRODIGE 19). J Clin Oncol 2019;37:4019 10.1200/JCO.2019.37.15_suppl.4019 [DOI] [Google Scholar]
- 96.Al-Batran SE, Homann N, Pauligk C, et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, phase 2/3 trial. Lancet 2019;393:1948-57. 10.1016/S0140-6736(18)32557-1 [DOI] [PubMed] [Google Scholar]
- 97.Al-Batran SE, Hofheinz RD, Pauligk C, et al. Histopathological regression after neoadjuvant docetaxel, oxaliplatin, fluorouracil, and leucovorin versus epirubicin, cisplatin, and fluorouracil or capecitabine in patients with resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4-AIO): results from the phase 2 part of a multicentre, open-label, randomised phase 2/3 trial. Lancet Oncol 2016;17:1697-708. 10.1016/S1470-2045(16)30531-9 [DOI] [PubMed] [Google Scholar]
- 98.Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327-33. 10.1200/JCO.2011.36.7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Park SH, Sohn TS, Lee J, et al. Phase III Trial to Compare Adjuvant Chemotherapy With Capecitabine and Cisplatin Versus Concurrent Chemoradiotherapy in Gastric Cancer: Final Report of the Adjuvant Chemoradiotherapy in Stomach Tumors Trial, Including Survival and Subset Analyses. J Clin Oncol 2015;33:3130-6. 10.1200/JCO.2014.58.3930 [DOI] [PubMed] [Google Scholar]
- 100.Hironaka S, Ueda S, Yasui H, et al. Randomized, open-label, phase III study comparing irinotecan with paclitaxel in patients with advanced gastric cancer without severe peritoneal metastasis after failure of prior combination chemotherapy using fluoropyrimidine plus platinum: WJOG 4007 trial. J Clin Oncol 2013;31:4438-44. 10.1200/JCO.2012.48.5805 [DOI] [PubMed] [Google Scholar]
- 101.Shitara K, Doi T, Dvorkin M, et al. Trifluridine/tipiracil versus placebo in patients with heavily pretreated metastatic gastric cancer (TAGS): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2018;19:1437-48. 10.1016/S1470-2045(18)30739-3 [DOI] [PubMed] [Google Scholar]
- 102.Fuchs CS, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014;383:31-9. 10.1016/S0140-6736(13)61719-5 [DOI] [PubMed] [Google Scholar]
- 103.Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol 2014;15:1224-35. 10.1016/S1470-2045(14)70420-6 [DOI] [PubMed] [Google Scholar]
- 104.Tabernero J, Hoff PM, Shen L, et al. Pertuzumab plus trastuzumab and chemotherapy for HER2-positive metastatic gastric or gastro-oesophageal junction cancer (JACOB): final analysis of a double-blind, randomised, placebo-controlled phase 3 study. Lancet Oncol 2018;19:1372-84. 10.1016/S1470-2045(18)30481-9 [DOI] [PubMed] [Google Scholar]
- 105.Tabernero J, Van Cutsem E, Bang YJ, et al. Pembrolizumab with or without chemotherapy versus chemotherapy for advanced gastric or gastroesophageal junction (G/GEJ) adenocarcinoma: The phase III KEYNOTE-062 study. J Clin Oncol 2019;37:LBA4007 10.1200/JCO.2019.37.18_suppl.LBA4007 [DOI] [Google Scholar]
- 106.Shitara K, Ozguroglu M, Bang YJ, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet 2018;392:123-33. 10.1016/S0140-6736(18)31257-1 [DOI] [PubMed] [Google Scholar]
- 107.Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:2461-71. 10.1016/S0140-6736(17)31827-5 [DOI] [PubMed] [Google Scholar]
- 108.Pernot S, Dubreuil O, Aparicio T, et al. Efficacy of a docetaxel-5FU-oxaliplatin regimen (TEFOX) in first-line treatment of advanced gastric signet ring cell carcinoma: an AGEO multicentre study. Br J Cancer 2018;119:424-8. 10.1038/s41416-018-0133-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li X, Zhu X, Wang Y, et al. Prognostic value and association of Lauren classification with VEGF and VEGFR-2 expression in gastric cancer. Oncol Lett 2019;18:4891-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Suzuki S, Dobashi Y, Hatakeyama Y, et al. Clinicopathological significance of platelet-derived growth factor (PDGF)-B and vascular endothelial growth factor-A expression, PDGF receptor-beta phosphorylation, and microvessel density in gastric cancer. BMC Cancer 2010;10:659. 10.1186/1471-2407-10-659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tanner M, Hollmen M, Junttila TT, et al. Amplification of HER-2 in gastric carcinoma: association with Topoisomerase IIalpha gene amplification, intestinal type, poor prognosis and sensitivity to trastuzumab. Ann Oncol 2005;16:273-8. 10.1093/annonc/mdi064 [DOI] [PubMed] [Google Scholar]
- 112.Kim ST, Cristescu R, Bass AJ, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 2018;24:1449-58. 10.1038/s41591-018-0101-z [DOI] [PubMed] [Google Scholar]
- 113.Türeci O, Sahin U, Schulze-Bergkamen H, et al. A multicentre, phase IIa study of zolbetuximab as a single agent in patients with recurrent or refractory advanced adenocarcinoma of the stomach or lower oesophagus: the MONO study. Ann Oncol 2019;30:1487-95. 10.1093/annonc/mdz199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Al-Batran SE, Schuler MH, Zvirbule Z, et al. FAST: An international, multicenter, randomized, phase II trial of epirubicin, oxaliplatin, and capecitabine (EOX) with or without IMAB362, a first-in-class anti-CLDN18.2 antibody, as first-line therapy in patients with advanced CLDN18.2+ gastric and gastroesophageal junction (GEJ) adenocarcinoma. J Clin Oncol 2016;34:LBA4001 10.1200/JCO.2016.34.18_suppl.LBA4001 [DOI] [Google Scholar]
- 115.Sahin U, Tureci Ö, Manikhas GM, et al. Zolbetuximab combined with EOX as first-line therapy in advanced CLDN18.2+ gastric (G) and gastroesophageal junction (GEJ) adenocarcinoma: Updated results from the FAST trial. J Clin Oncol 2019;37:16 10.1200/JCO.2019.37.4_suppl.16 [DOI] [Google Scholar]