

Abstract
Prostate cancer is the second leading cause of male cancer death in the United States. The androgen receptor (AR) transcription factor is a master regulator of normal glandular homeostasis in the prostate, as well as growth and survival of prostate cancer cells. Therefore, AR-targeted therapies are effective for improving overall survival of patients with advanced prostate cancer that is incurable by surgery or radiation. However, prostate cancer will inevitably progress on AR-targeted therapies to a castration-resistant prostate cancer (CRPC) phenotype that accounts for virtually all prostate cancer-specific death. mRNA transcript variants of the AR gene are expressed in CRPC cells and can be translated to produce AR variant (AR-V) proteins that function as ligand-independent, constitutively active transcription factors. AR-Vs are able to support growth of CRPC cells by promoting expression of AR target genes that are normally suppressed by AR-targeted therapies. Knowledge of mechanisms that govern expression of AR-Vs is incomplete. Studies have shown genomic rearrangements of the AR gene underlie expression of diverse AR-Vs in certain CRPC tumors, but post-transcriptional processes represent a broader regulatory mechanism for expression of AR-Vs in CRPC. This review focuses on alternative splicing, 3′ end processing, miRNA-mediated mRNA repression, of AR and AR-V expression and the potential these mechanisms hold as therapeutic targets for CRPC.
Introduction
Prostate cancer is the most common non-cutaneous cancer in men. While localized disease can be cured by radiation or surgery, locally advanced or metastatic prostate cancer presents a clinical challenge. Advanced prostate cancer can initially be treated by endocrine therapies targeting the androgen receptor (AR), a ligand-dependent transcription factor that mediates the actions of androgens such as testosterone (1). AR-targeted therapies improve overall survival, but patients will inevitably progress to a lethal castration-resistant prostate cancer (CRPC) phenotype (2). Based on significant evidence that reactivation of AR transcriptional activity drives progression to CRPC (3,4),
potent second-generation AR-targeted drugs such as abiraterone and enzalutamide were developed and shown to extend overall survival in phase III clinical trials of post-chemotherapy CRPC patients (5,6). Subsequent phase III trials have tested abiraterone and enzalutamide in pre-chemotherapy CRPC patients as well as castration-sensitive prostate cancer patients and showed significantly better overall survival compared to standard of care (7–11). These trials have indicated that the earlier these second-generation AR-targeted drugs are used in advanced prostate cancer patients, the better the overall survival. Despite these clinical advances, all patients will eventually succumb to the disease, often with rising serum levels of prostate-specific antigen (encoded by the AR-regulated KLK3 gene). This emphasizes a continued need to understand mechanisms that drive AR expression and activity during CRPC progression.
AR is a master regulator transcription factor in cells of prostatic lineage as evidenced by AR being required for development of the prostate gland as well as homeostasis of the adult prostate (12). The AR gene contains eight canonical exons that encode four discrete functional domains in the AR protein (13). Exon 1 encodes an N-terminal transactivation domain (NTD), exons 2 and 3 encode a DNA-binding domain (DBD), exon 4 encodes a hinge region, and exons 5 through 8 encode a C-terminal ligand-binding domain (LBD) (Fig. 1A). Binding of androgen ligand to the LBD induces a conformational change in the AR protein that promotes nuclear localization, DNA binding and transcriptional activation of target genes (14). AR target genes regulate crucial cellular functions such as growth, proliferation and survival (15).
Figure 1.
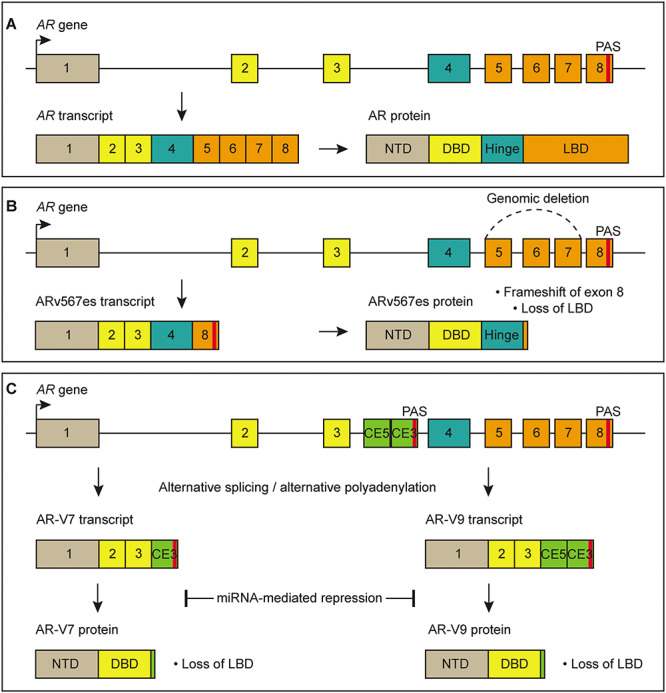
Genomic alterations and RNA-based mechanisms that regulate expression of AR and AR-Vs. (A) Canonical splicing of the AR gene to generate full-length AR mRNA and AR protein. (B) Example of a genomic alteration that causes expression of the AR-V ARv567es. Genomic deletion of exons 5 through 7 in the AR gene causes truncation of AR protein and loss of the LBD. (C) Examples of RNA-based mechanisms that regulate expression of AR-V7 and AR-V9. Alternative splicing and/or alternative polyadenylation can promote splicing of cryptic exons to generate mRNAs that encode truncated AR-Vs lacking the LBD. miRNAs can reduce stability or translation of AR-V mRNAs, causing reduced AR-V protein expression. NTD = N-terminal transcriptional domain, DBD = DNA-binding domain, LBD = C-terminal ligand-binding domain, CE5 = cryptic exon 5, CE3 = cryptic exon 3, PAS = polyadenylation site.
Research on the genomic landscape of CRPC patients has identified multiple alterations of the AR gene that lead to overexpression of AR and aberrant AR activity that can promote activation of downstream target genes. Amplification or copy number gain of the AR gene body or upstream enhancer in patient samples has been found to cause AR overexpression, which sensitizes CRPC cells to castrate levels of androgens (3,16). Somatic mutations in the AR ligand-binding domain (LBD) have also been identified, which can broaden the repertoire of ligands that activate AR transcriptional activity (3,16). Further, AR structural rearrangements such as deletions and inversions have been identified in CRPC tumors, which can promote high-level expression of diverse AR variant (AR-V) species such as ARv567es (Fig. 1B) (17,18).
There are many additional AR-Vs expressed in CRPC that do not appear to be expressed as a result of underlying AR gene rearrangements. The well-studied AR variant-7 (AR-V7) is generated through alternative splicing of AR pre-mRNA, which has high clinical relevance because detection of AR-V7 in circulating tumor cells (CTCs) from patients with CRPC has been found to predict resistance to enzalutamide and abiraterone (19–22). Because of this, AR-V7 in CTCs is being used as a treatment selection biomarker for CRPC patients (23). Accordingly, mechanisms that regulate expression of AR-V7 and other AR-Vs have become an area of significant interest. While genomic alterations of AR in CRPC patients are well-documented (3,4,18), post-transcriptional regulation of AR and AR-V mRNA has emerged as another level of regulation that is manipulated in advanced prostate cancer (Fig. 1C). This review will focus on the multiple RNA-based mechanisms that regulate AR and AR-V expression and the potential these mechanisms hold as therapeutic targets in CRPC.
Androgen receptor variants in castration-resistant prostate cancer
Several well-characterized AR-Vs, such as AR-V7 and AR variant-9 (AR-V9), arise from splicing of different cryptic exons (CEs) located within intron 3 of the AR gene. When these CEs are spliced downstream of AR exon 3, they contain in-frame translation stop codons that cause truncation of the protein (Fig. 1C). Because the translation stop codon occurs after exon 3, AR-Vs contain the same NTD and DBD as full-length AR but lack the LBD encoded by exons 5 through 8. For example, AR-V7 mRNA is encoded from spliced AR exons 1, 2, 3 and CE3, and AR-V9 mRNA arises from spliced AR exons 1, 2, 3 and CE5 (Fig. 1C). AR-V7, AR-V9 and many other related AR-Vs have been shown to function as ligand-independent, constitutively active transcription factors (24–32). Numerous AR-Vs have been discovered in CRPC patient samples (33), and research has focused on the function of AR-Vs in metastatic prostate cancer progression and treatment resistance.
AR-Vs have been shown to support growth of CRPC cells by targeting genes that are normally suppressed by endocrine therapies (24–32,34–40). AR-V7 and AR-V9 are broadly enriched in CRPC cell lines and patient samples (35,41,42), and expression of AR-Vs is proposed to confer resistance to second-generation AR inhibitors in patients because these isoforms lack the ligand-binding domain (LBD) that is targeted by these inhibitors (Fig. 1B and C) (19,20).
It should be noted that the clinical relevance of AR-Vs as drivers of CRPC resistance to AR-targeted therapies remains a controversial topic. Excellent recent reviews have covered this topic (43,44). This debate in the field illustrates the need for continued research on the molecular regulation of AR-V expression and biological effects of these regulatory mechanisms to better understand the role of AR-Vs in CRPC.
Alternative AR mRNA splicing
Alternative splicing of AR pre-mRNA is responsible for the generation of many AR-Vs such as AR-V7 and AR-V9, but factors that specifically regulate the inclusion and exclusion of exons and introns of the AR gene are still not well-understood. Virtually all studies on splicing regulation of AR-Vs have focused on splicing of full-length AR versus AR-V7, and many of these studies have employed an AR minigene reporter first described in 2014 (45). The AR minigene reporter contains exon CE3 of the human AR gene and its flanking ~400 bp nucleotide sequence in intron 3 (Fig. 2A). Using this AR minigene reporter, the authors found that a 5′ATGTCTCTCTTTCATAC intronic splicing enhancer (ISE) and a 5′CCCTGAAGAAAGGCTGAC exonic splicing enhancer (ESE) near the 3′ splice site of exon CE3 were necessary for optimal splicing of AR exon 3 to exon CE3 in hormone-sensitive VCaP and LNCaP prostate cancer cells (45). Using RNA oligo-nucleotides that mimicked the ISE and ESE sequences, they found that serine/arginine-rich splicing factor 1 (SRSF1) bound to the ESE site and that polypyrimidine tract-binding protein 1 (PTBP1) and 65 kDa U2 auxiliary factor (U2AF65) bound to the ISE site (Fig. 2A). They found depletion of U2AF65 and SRSF1 in VCaP cells significantly reduced expression of AR-V7 mRNA and protein but not full-length AR mRNA or protein. However, depletion of PTBP1 in VCaP cells reduced expression of both AR-V7 and full-length AR mRNA and protein, suggesting PTBP1 is a non-specific regulator of AR transcription and/or splicing of all AR isoforms.
Figure 2.
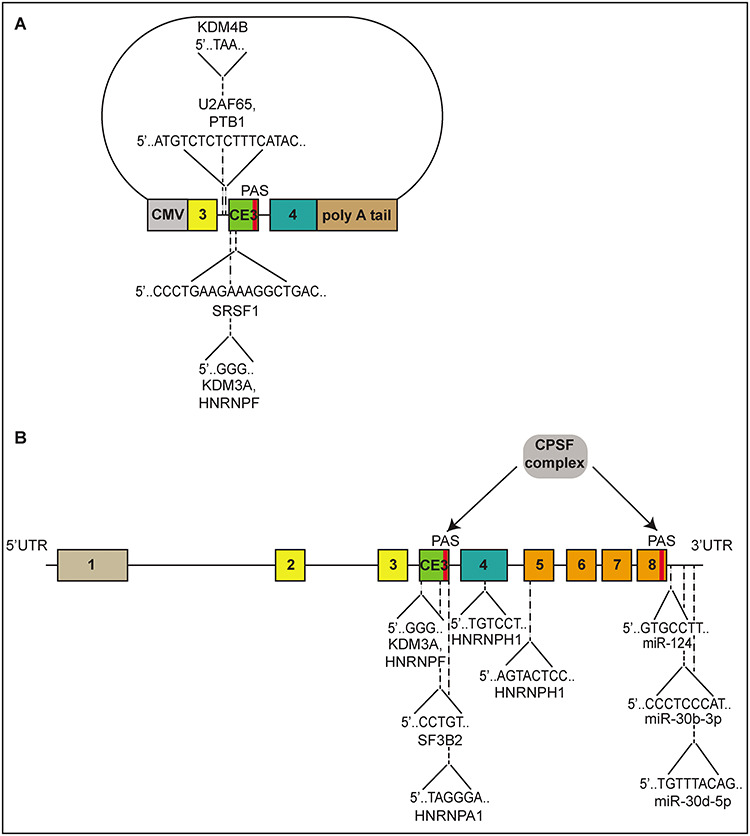
Cis-elements and trans-acting factors that regulate expression of AR and AR-Vs. (A) Diagram of a minigene splicing reporter to mimic AR and AR-V7 alternative splicing. The reporter contains AR exon and flanking ~400 bp nucleotide sequence from AR intron 3. Factors found to directly recognize and regulate splicing of the reporter are illustrated. (B) Schematic of the endogenous AR gene including the 5′UTR, 3′UTR and exon CE3. Factors found to directly recognize and regulate splicing of endogenous AR pre-mRNAs are illustrated. UTR = untranslated region, CE3 = cryptic exon 3, PAS = polyadenylation site, CMV = human cytomegalovirus promoter.
Additional studies employing this AR minigene reporter determined histone demethylases function in the splicing of AR-V7. One study found overexpression of a member of the Jumonji C (JmjC) KDM4 histone lysine demethylase (KDM) family, KDM4B, induced expression of AR-V7 mRNA and protein in LNCaP cells, whereas depletion of KDM4B reduced expression of AR-V7 mRNA and protein in VCaP and 22Rv1 cells (46). They found KDM4B bound to an intronic sequence located upstream of exon CE3 in the AR minigene reporter. Using RNA oligonucleotide binding assays, they found that a 5′TAA motif was necessary for this interaction (Fig. 2A). Another study found the histone demethylase KDM3A also promotes the alternative splicing of AR-V7 through the recruitment of heterogeneous nuclear ribonucleoprotein F (HNRNPF) (47). The authors showed knockdown of either KDM3A or HNRNPF reduced expression of AR-V7 mRNA and protein but not full-length AR mRNA or protein in 22Rv1 cells. In RNA oligonucleotide binding assays, they found HNRNPF bound to a G-tract sequence in exon CE3 (Fig. 2A), adjacent to the 3′ splice site. In the AR minigene reporter, binding of HNRNPF to this G-tract sequence was required for recruitment of KDM3A (47). The authors validated these interactions in cells, finding both HNRNPF and KDM3A directly bound the G-tract of endogenous AR pre-mRNA in 22Rv1 cells (Fig. 2B).
In addition to studies that have explored regulation of an AR minigene reporter, several studies have examined regulation of endogenous AR splicing. For instance, a recent study identified splicing factor 3b subunit 2 (SF3B2) as an important regulator of AR-V7 splicing (48). The authors used genome engineering in 22Rv1 cells to knock in a GFP reporter downstream of exon CE3 in the endogenous AR locus, which they used as a surrogate for AR-V7 expression. Using this reporter line, they showed knockdown of SF3B2 caused reduced AR-V7/GFP reporter expression. They performed photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP) analysis of SF3B2, which revealed that SF3B2 bound to AR exons 1 and CE3. Accordingly, disruption of the SF3B2 recognition motif in exon CE3 (Fig. 2B) significantly increased the number of 22Rv1 cells that were GFP-negative for the AR-V7 reporter, suggesting this motif functions in AR-V7 splicing (48). Noteworthy, high SF3B2 levels were correlated with adverse pathology as determined by Gleason scoring as well as shortened progression-free survival in prostate cancer patients (48).
Another study examining endogenous AR-V7 splicing determined that overexpression of the splicing factor heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1) in 22Rv1 cells resulted in increased AR-V7 mRNA and protein expression (49). Additionally, knockdown of HNRNPA1 reduced expression of AR-V7 mRNA and protein and re-sensitized 22Rv1 cells to enzalutamide (49). Using immunohistochemistry, the authors found that HNRNPA1 was overexpressed in prostate tumors compared with benign prostate tissue. Further, enzalutamide-resistant CRPC cells were found to exhibit higher levels of HNRNPA1, suggesting this splicing factor may contribute to treatment resistance (49). Several HNRNPA1 binding sites (5′TAGGGA) were identified in AR, and through RNA immunoprecipitation experiments in enzalutamide-resistant 22Rv1 cells, HNRNPA1 was found to be enriched near the poly(A) signal in AR exon CE3 (Fig. 2B) (49). Interestingly, knockdown of HNRNPA1 also reduced full-length AR mRNA and protein expression in VCaP cells (49), suggesting this factor may regulate AR expression through non-specific mechanisms such as decreased transcription of the AR gene or inhibition of global splicing.
Lastly, a study examining global expression of splicing factors in prostate cancer identified dysregulation of numerous splicing factors compared to benign tumor adjacent regions (50). They found high expression of splicing factors such as small nuclear ribonucleoprotein U5 subunit 200 (SNRNP200), serine/arginine-rich splicing factor 3 (SRSF3), and serine/arginine repetitive matrix protein 1 (SRRM1) in prostatectomy specimens was associated with high Gleason score, metastasis, biochemical recurrence, and high AR-V7 expression, suggesting these splicing factors may also contribute to AR-V production. Together, these studies illustrate trans-acting splicing factors can function specifically to regulate AR-V expression. These factors represent new drug targets for future studies to examine as an alternative or to complement traditional CRPC therapies.
AR mRNA 3′ end processing
mRNA cleavage and polyadenylation, which defines the 3′ end of mRNA transcripts, is another post-transcriptional mechanism that governs gene expression. The process of mRNA polyadenylation was discovered nearly 50 years ago (51–53), but recent research has found dysregulation of this mechanism in numerous cancers including prostate cancer (54–57). mRNA species undergo cleavage and polyadenylation at a canonical 5′AAUAAA polyadenylation (poly(A)) sequence that is typically located within the 3′UTR encoded by the last exon of the transcript. Alternative polyadenylation is the usage of a poly(A) site that is proximal or distal to the predominant poly(A) and provides a mechanism to modify the 3′ end of mRNA transcripts, which can also modify the amino acid sequence and/or length of protein C-termini. Four core complexes recognize and bind the poly(A) site to promote endonucleolytic cleavage: the cleavage and polyadenylation specificity factor (CPSF) complex, the cleavage stimulation factor (CstF) complex, the cleavage factor I (CFIm) complex, and the CFIIm complex (58).
Our group showed that a poly(A) site in AR exon CE3 is utilized in CRPC cells to generate an abundance of AR-Vs, including AR-V7 and AR-V9 (55). Further, the CPSF complex component CPSF1 regulated selection of this alternative poly(A) site based on the finding that knockdown of CPSF1 in 22Rv1 cells reduced expression of AR-Vs and increased expression of full-length AR (Fig. 2B) (55). Thus, similar to alternative splicing, alternative polyadenylation is an additional RNA-based mechanism that controls levels of AR and AR-Vs and highlights targetable cis-elements and trans-acting factors that function in the selection of the poly(A) site in exon CE3 of AR.
miRNA-mediated repression of AR
Another well-studied mechanism of post-transcriptional gene regulation is miRNA-mediated repression. miRNAs regulate the expression of genes by directly binding mRNAs through sequence elements typically located within the 3′UTR and reducing stability or translation of the target mRNA (59). The full-length AR mRNA contains numerous miRNA binding sites (60,61), and studies have explored the role of miRNA-mediated repression as a mechanism of regulating AR and AR-V expression in prostate cancer.
miR-124 binding sites are found in the 3′UTRs of both full-length AR and AR-V7 mRNAs (Fig. 2B), as well as the 3′UTR of other AR-Vs (62,63). Administering miR-124 to 22Rv1 cells grown in vitro or as mouse xenografts in vivo reduced AR-V7 protein expression and tumor growth (63). Full-length AR protein or mRNA was not examined in this study, and it remains unknown whether miR-124 regulates AR-V7 specifically. Importantly, the authors found expression of miR-124 also promoted sensitivity of 22Rv1 cells to enzalutamide in vitro and in vivo, suggesting this miRNA has potential to overcome enzalutamide resistance.
Another study found significantly lower levels of miR-212 in prostate tumors relative to normal prostate tissue and showed ectopic expression of miR-212 mimics in C4-2B CRPC cells reduced expression of full-length AR and AR-V7 mRNA and protein (64). Interestingly, this study found ectopic expression of miR-212 also reduced levels of the splicing regulator HNRNPH1 and showed HNRNPH1 directly interacted with AR pre-mRNA (Fig. 2B). The authors suggested that lower levels of HNRNPH1 could reduce transcription and/or splicing of AR and AR-Vs, suggesting miRNAs may also indirectly regulate AR and AR-V expression.
miR-145 is another miRNA shown to target AR, and ectopic expression of miR-145 in 22Rv1 cells reduced expression of both full-length AR and AR-V protein (65). The authors found that miR-145 expression inversely correlated with the occurrence of metastases and androgen deprivation therapy response in a prostate cancer cohort, suggesting this miRNA may also function in treatment resistance. However, whether miR-145 regulates full-length AR and AR-V expression through direct or indirect mechanisms is unclear.
Lastly, a library of over 800 miRNA mimics was screened to identify regulators of AR activity, leading to the finding that miR-30 family members functioned as direct AR inhibitors (Fig. 2B) (60). This study identified reductions in expression of miR-30c-5p and miR-30d-5p in metastatic CRPC compared to normal prostate tissue and reduction of miR-30d-5p in primary prostate cancer compared to normal prostate tissue. They found that ectopic expression of miR-30d-5p and miR-30b-3p reduced expression of full-length AR protein in LNCaP cells and AR-V7 protein in VCaP cells. The authors also found inhibition of miR-30d-5p and miR-30b-3p promoted androgen-independent growth of LNCaP cells, suggesting these miRNAs may be important for sustaining normal androgen-dependent growth of prostate cancer cells.
Together these studies emphasize the importance of miRNAs in regulating AR and AR-V expression, but it still remains unclear whether miRNAs are capable of specifically targeting AR-Vs. Interestingly, an additional study identified circular RNAs to be depleted in enzalutamide-resistant CRPC cells compared to enzalutamide-sensitive cells, suggesting circular RNAs may regulate prostate cancer therapeutic responses (66). The authors identified numerous miRNA binding sites in the circular RNA sequences, including miR-124 sites. This finding suggests that circular RNAs may function as sponges for miRNAs, effectively decreasing the availability of these miRNAs to regulate endogenous targets. Future work is warranted to understand the coordinate regulation of miRNAs such as miR-124 and miR-212, as well as interplay with circRNAs, and their resultant effects on AR and AR-V expression. These factors could hold potential as therapeutic targets for CRPC.
Post-transcriptional drug targets of AR-Vs and future directions
While second-generation AR-targeted therapies such as enzalutamide improve overall survival of prostate cancer patients, these therapies are not curative. This highlights the need for development of new therapies for CRPC. AR-Vs are insensitive to ligands such as enzalutamide because AR-Vs lack the LBD targeted by these drugs. The mechanisms of RNA-based regulation of AR and AR-Vs described in this review highlight new opportunities for drug development in CRPC, which could serve as alternatives or complements to current therapies.
Antisense nucleic-acid based therapies have been investigated as potential therapeutics in CRPC. For instance, inhibition of AR-V7 splicing with antisense oligo-nucleotides was shown to be effective in reducing androgen-independent growth of CRPC-derived 22Rv1, DuCaP, and VCaP cell lines (67). Our group developed an antisense morpholino targeted to the alternative poly(A) sequence in AR exon CE3, and found this morpholino reduced growth of 22Rv1 cells in androgen-deficient medium but not androgen-proficient medium (55). These studies highlight the potential of antisense nucleic acid-based strategies for inhibiting AR-Vs in CRPC.
miRNA mimics are another promising option and have shown success in other cancer models including liver cancer (68) and non-small cell lung cancer (69). miR-124 mimics have already shown effectiveness in targeting and repressing AR-V7 in animal models (63), and represent another therapy option to inhibit AR-V7 expression in CRPC.
Pladienolide B is a macrolide compound with antitumor activity that has been shown to inhibit mRNA splicing (70). Pladienolide B binds to SF3B3 and inhibits interactions with other splicing factors such as SF3B1 (71,72) to disrupt downstream splicing. Treatment of 22Rv1 cells with pladienolide B reduced expression of AR-V7 and full-length AR mRNA, but the effect was more pronounced for AR-V7 (48). Additionally, Pladienolide B inhibited the growth of 22Rv1 xenografts in mice (48). Pladienolide B as well as other splicing inhibitors could provide yet another opportunity for alternative treatment for CRPC that does not respond to traditional treatments such as enzalutamide.
The RNA-based mechanisms presented in this review are important levels of regulation manipulated by prostate cancer and provide an exciting avenue for druggable targets outside of the traditional therapies that target the AR. Continued research on the trans-acting factors and cis-elements that regulate these mechanisms will bring us closer to the reality of manipulating these pathways in CRPC patients for better long-term survival.
Conflict of Interest statement: The authors declare no conflicts or financial interests.
Funding
National Institutes of Health (grant R01CA174777 to S.M.D.); Kiel Tietz was supported by T32 CA009138.
References
- 1. Watson P.A., Arora V.K. and Sawyers C.L. (2015) Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer, 15, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Logothetis C.J., Gallick G.E., Maity S.N., Kim J., Aparicio A., Efstathiou E. and Lin S.H. (2013) Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov., 3, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robinson D., Van Allen E.M., Wu Y.M., Schultz N., Lonigro R.J., Mosquera J.M., Montgomery B., Taplin M.E., Pritchard C.C., Attard G. et al. (2015) Integrative clinical genomics of advanced prostate cancer. Cell, 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chandrasekar T., Yang J.C., Gao A.C. and Evans C.P. (2015) Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol., 4, 365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Bono J.S., Logothetis C.J., Molina A., Fizazi K., North S., Chu L., Chi K.N., Jones R.J., Goodman O.B. Jr., Saad F. et al. (2011) Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med., 364, 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scher H.I., Fizazi K., Saad F., Taplin M.E., Sternberg C.N., Miller K., de Wit R., Mulders P., Chi K.N., Shore N.D. et al. (2012) Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med., 367, 1187–1197. [DOI] [PubMed] [Google Scholar]
- 7. Ryan C.J., Smith M.R., de Bono J.S., Molina A., Logothetis C.J., de Souza P., Fizazi K., Mainwaring P., Piulats J.M., Ng S. et al. (2013) Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med., 368, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beer T.M., Armstrong A.J., Rathkopf D.E., Loriot Y., Sternberg C.N., Higano C.S., Iversen P., Bhattacharya S., Carles J., Chowdhury S. et al. (2014) Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med., 371, 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. James N.D., de Bono J.S., Spears M.R., Clarke N.W., Mason M.D., Dearnaley D.P., Ritchie A.W.S., Amos C.L., Gilson C., Jones R.J. et al. (2017) Abiraterone for prostate cancer not previously treated with hormone therapy. N. Engl. J. Med., 377, 338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fizazi K., Tran N., Fein L., Matsubara N., Rodriguez-Antolin A., Alekseev B.Y., Ozguroglu M., Ye D., Feyerabend S., Protheroe A. et al. (2017) Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N. Engl. J. Med., 377, 352–360. [DOI] [PubMed] [Google Scholar]
- 11. Davis I.D., Martin A.J., Stockler M.R., Begbie S., Chi K.N., Chowdhury S., Coskinas X., Frydenberg M., Hague W.E., Horvath L.G. et al. (2019) Enzalutamide with standard first-line therapy in metastatic prostate cancer. N. Engl. J. Med., 381, 121–131. [DOI] [PubMed] [Google Scholar]
- 12. Brinkmann A.O. (2001) Molecular basis of androgen insensitivity. Mol. Cell. Endocrinol., 179, 105–109. [DOI] [PubMed] [Google Scholar]
- 13. Gao W., Bohl C.E. and Dalton J.T. (2005) Chemistry and structural biology of androgen receptor. Chem. Rev., 105, 3352–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davey R.A. and Grossmann M. (2016) Androgen receptor structure, function and biology: from bench to bedside. Clin. Biochem. Rev., 37, 3–15. [PMC free article] [PubMed] [Google Scholar]
- 15. Bolton E.C., So A.Y., Chaivorapol C., Haqq C.M., Li H. and Yamamoto K.R. (2007) Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev., 21, 2005–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quigley D.A., Dang H.X., Zhao S.G., Lloyd P., Aggarwal R., Alumkal J.J., Foye A., Kothari V., Perry M.D., Bailey A.M. et al. (2018) Genomic hallmarks and structural variation in metastatic prostate cancer. Cell, 174, 758–769.e759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y., Yang R., Henzler C.M., Ho Y., Passow C., Auch B., Carreira S., Nava Rodrigues D., Bertan C., Hwang T.H. et al. (2020) Diverse AR gene rearrangements mediate resistance to androgen receptor inhibitors in metastatic prostate cancer. Clin. Cancer Res., 26, 1965–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Henzler C., Li Y., Yang R., McBride T., Ho Y., Sprenger C., Liu G., Coleman I., Lakely B., Li R. et al. (2016) Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat. Commun., 7, 13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Antonarakis E.S., Lu C., Wang H., Luber B., Nakazawa M., Roeser J.C., Chen Y., Mohammad T.A., Chen Y., Fedor H.L. et al. (2014) AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med., 371, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Antonarakis E.S., Lu C., Luber B., Wang H., Chen Y., Nakazawa M., Nadal R., Paller C.J., Denmeade S.R., Carducci M.A. et al. (2015) Androgen receptor splice variant 7 and efficacy of Taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol., 1, 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sharp A., Coleman I., Yuan W., Sprenger C., Dolling D., Rodrigues D.N., Russo J.W., Figueiredo I., Bertan C., Seed G. et al. (2019) Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Invest., 129, 192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scher H.I., Graf R.P., Schreiber N.A., Jayaram A., Winquist E., McLaughlin B., Lu D., Fleisher M., Orr S., Lowes L. et al. (2018) Assessment of the validity of nuclear-localized androgen receptor splice variant 7 in circulating tumor cells as a predictive biomarker for castration-resistant prostate cancer. JAMA Oncol., 4, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Graf R.P., Hullings M., Barnett E.S., Carbone E., Dittamore R. and Scher H.I. (2020) Clinical utility of the nuclear-localized AR-V7 biomarker in circulating tumor cells in improving physician treatment choice in castration-resistant prostate cancer. Eur. Urol., 77, 170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Z., Wu D., Thomas-Ahner J.M., Lu C., Zhao P., Zhang Q., Geraghty C., Yan P.S., Hankey W., Sunkel B. et al. (2018) Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl. Acad. Sci. U. S. A., 115, 6810–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chan S.C., Li Y. and Dehm S.M. (2012) Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J. Biol. Chem., 287, 19736–19749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chan S.C., Selth L.A., Li Y., Nyquist M.D., Miao L., Bradner J.E., Raj G.V., Tilley W.D. and Dehm S.M. (2015) Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res., 43, 5880–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dehm S.M., Schmidt L.J., Heemers H.V., Vessella R.L. and Tindall D.J. (2008) Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res., 68, 5469–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He Y., Lu J., Ye Z., Hao S., Wang L., Kohli M., Tindall D.J., Li B., Zhu R., Wang L. et al. (2018) Androgen receptor splice variants bind to constitutively open chromatin and promote abiraterone-resistant growth of prostate cancer. Nucleic Acids Res., 46, 1895–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu R., Dunn T.A., Wei S., Isharwal S., Veltri R.W., Humphreys E., Han M., Partin A.W., Vessella R.L., Isaacs W.B. et al. (2009) Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res., 69, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu R., Lu C., Mostaghel E.A., Yegnasubramanian S., Gurel M., Tannahill C., Edwards J., Isaacs W.B., Nelson P.S., Bluemn E. et al. (2012) Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res., 72, 3457–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu D., Zhan Y., Qi Y., Cao B., Bai S., Xu W., Gambhir S.S., Lee P., Sartor O., Flemington E.K. et al. (2015) Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res., 75, 3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu Z., Chen S., Sowalsky A.G., Voznesensky O.S., Mostaghel E.A., Nelson P.S., Cai C. and Balk S.P. (2014) Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin. Cancer Res., 20, 1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu C. and Luo J. (2013) Decoding the androgen receptor splice variants. Transl. Androl. Urol., 2, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo Z., Yang X., Sun F., Jiang R., Linn D.E., Chen H., Chen H., Kong X., Melamed J., Tepper C.G. et al. (2009) A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res., 69, 2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kohli M., Ho Y., Hillman D.W., Van Etten J.L., Henzler C., Yang R., Sperger J.M., Li Y., Tseng E., Hon T. et al. (2017) Androgen receptor variant AR-V9 is coexpressed with AR-V7 in prostate cancer metastases and predicts abiraterone resistance. Clin. Cancer Res., 23, 4704–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Y., Alsagabi M., Fan D., Bova G.S., Tewfik A.H. and Dehm S.M. (2011) Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res., 71, 2108–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li Y., Chan S.C., Brand L.J., Hwang T.H., Silverstein K.A. and Dehm S.M. (2013) Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res., 73, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Y., Hwang T.H., Oseth L.A., Hauge A., Vessella R.L., Schmechel S.C., Hirsch B., Beckman K.B., Silverstein K.A. and Dehm S.M. (2012) AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene, 31, 4759–4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun S., Sprenger C.C., Vessella R.L., Haugk K., Soriano K., Mostaghel E.A., Page S.T., Coleman I.M., Nguyen H.M., Sun H. et al. (2010) Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest., 120, 2715–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Watson P.A., Chen Y.F., Balbas M.D., Wongvipat J., Socci N.D., Viale A., Kim K. and Sawyers C.L. (2010) Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. U. S. A., 107, 16759–16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kallio H.M.L., Hieta R., Latonen L., Brofeldt A., Annala M., Kivinummi K., Tammela T.L., Nykter M., Isaacs W.B., Lilja H.G. et al. (2018) Constitutively active androgen receptor splice variants AR-V3, AR-V7 and AR-V9 are co-expressed in castration-resistant prostate cancer metastases. Br. J. Cancer, 119, 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tagawa S.T., Antonarakis E.S., Gjyrezi A., Galletti G., Kim S., Worroll D., Stewart J., Zaher A., Szatrowski T.P., Ballman K.V. et al. (2019) Expression of AR-V7 and ARv(567es) in circulating tumor cells correlates with outcomes to Taxane therapy in men with metastatic prostate cancer treated in TAXYNERGY. Clin. Cancer Res., 25, 1880–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uo T., Plymate S.R. and Sprenger C.C. (2018) The potential of AR-V7 as a therapeutic target. Expert Opin. Ther. Targets, 22, 201–216. [DOI] [PubMed] [Google Scholar]
- 44. Maughan B.L. and Antonarakis E.S. (2015) Clinical relevance of androgen receptor splice variants in castration-resistant prostate cancer. Curr. Treat. Options in Oncol., 16, 57. [DOI] [PubMed] [Google Scholar]
- 45. Liu L.L., Xie N., Sun S., Plymate S., Mostaghel E. and Dong X. (2014) Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene, 33, 3140–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Duan L., Chen Z., Lu J., Liang Y., Wang M., Roggero C.M., Zhang Q.J., Gao J., Fang Y., Cao J. et al. (2019) Histone lysine demethylase KDM4B regulates the alternative splicing of the androgen receptor in response to androgen deprivation. Nucleic Acids Res., 47, 11623–11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fan L., Zhang F., Xu S., Cui X., Hussain A., Fazli L., Gleave M., Dong X. and Qi J. (2018) Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc. Natl. Acad. Sci. U. S. A., 115, E4584–e4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kawamura N., Nimura K., Saga K., Ishibashi A., Kitamura K., Nagano H., Yoshikawa Y., Ishida K., Nonomura N., Arisawa M. et al. (2019) SF3B2-mediated RNA splicing drives human prostate cancer progression. Cancer Res., 79, 5204–5217. [DOI] [PubMed] [Google Scholar]
- 49. Nadiminty N., Tummala R., Liu C., Lou W., Evans C.P. and Gao A.C. (2015) NF-kappaB2/p52:c-Myc:hnRNPA1 pathway regulates expression of androgen receptor splice variants and enzalutamide sensitivity in prostate cancer. Mol. Cancer Ther., 14, 1884–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jimenez-Vacas J.M., Herrero-Aguayo V., Montero-Hidalgo A.J., Gomez-Gomez E., Fuentes-Fayos A.C., Leon-Gonzalez A.J., Saez-Martinez P., Alors-Perez E., Pedraza-Arevalo S., Gonzalez-Serrano T. et al. (2020) Dysregulation of the splicing machinery is directly associated to aggressiveness of prostate cancer. EBioMedicine, 51, 102547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee S.Y., Mendecki J. and Brawerman G. (1971) A polynucleotide segment rich in adenylic acid in the rapidly-labeled polyribosomal RNA component of mouse sarcoma 180 ascites cells. Proc. Natl. Acad. Sci. U. S. A., 68, 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Edmonds M., Vaughan M.H. Jr. and Nakazato H. (1971) Polyadenylic acid sequences in the heterogeneous nuclear RNA and rapidly-labeled polyribosomal RNA of HeLa cells: possible evidence for a precursor relationship. Proc. Natl. Acad. Sci. U. S. A., 68, 1336–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Darnell J.E., Wall R. and Tushinski R.J. (1971) An adenylic acid-rich sequence in messenger RNA of HeLa cells and its possible relationship to reiterated sites in DNA. Proc. Natl. Acad. Sci. U. S. A., 68, 1321–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee S.H., Singh I., Tisdale S., Abdel-Wahab O., Leslie C.S. and Mayr C. (2018) Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature, 561, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Van Etten J.L., Nyquist M., Li Y., Yang R., Ho Y., Johnson R., Ondigi O., Voytas D.F., Henzler C. and Dehm S.M. (2017) Targeting a single alternative Polyadenylation site coordinately blocks expression of androgen receptor mRNA splice variants in prostate cancer. Cancer Res., 77, 5228–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xia Z., Donehower L.A., Cooper T.A., Neilson J.R., Wheeler D.A., Wagner E.J. and Li W. (2014) Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3'-UTR landscape across seven tumour types. Nat. Commun., 5, 5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xue Z., Warren R.L., Gibb E.A., MacMillan D., Wong J., Chiu R., Hammond S.A., Yang C., Nip K.M., Ennis C.A. et al. (2018) Recurrent tumor-specific regulation of alternative polyadenylation of cancer-related genes. BMC Genomics, 19, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gruber A.J. and Zavolan M. (2019) Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet., 20, 599–614. [DOI] [PubMed] [Google Scholar]
- 59. O'Brien J., Hayder H., Zayed Y. and Peng C. (2018) Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. (Lausanne), 9, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kumar B., Khaleghzadegan S., Mears B., Hatano K., Kudrolli T.A., Chowdhury W.H., Yeater D.B., Ewing C.M., Luo J., Isaacs W.B. et al. (2016) Identification of miR-30b-3p and miR-30d-5p as direct regulators of androgen receptor signaling in prostate cancer by complementary functional microRNA library screening. Oncotarget, 7, 72593–72607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ostling P., Leivonen S.K., Aakula A., Kohonen P., Makela R., Hagman Z., Edsjo A., Kangaspeska S., Edgren H., Nicorici D. et al. (2011) Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res., 71, 1956–1967. [DOI] [PubMed] [Google Scholar]
- 62. Shi X.B., Xue L., Ma A.H., Tepper C.G., Gandour-Edwards R., Kung H.J. and deVere White R.W. (2013) Tumor suppressive miR-124 targets androgen receptor and inhibits proliferation of prostate cancer cells. Oncogene, 32, 4130–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shi X.B., Ma A.H., Xue L., Li M., Nguyen H.G., Yang J.C., Tepper C.G., Gandour-Edwards R., Evans C.P., Kung H.J. et al. (2015) miR-124 and androgen receptor signaling inhibitors repress prostate cancer growth by downregulating androgen receptor splice variants, EZH2, and Src. Cancer Res., 75, 5309–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yang Y., Jia D., Kim H., Abd Elmageed Z.Y., Datta A., Davis R., Srivastav S., Moroz K., Crawford B.E., Moparty K. et al. (2016) Dysregulation of miR-212 promotes castration resistance through hnRNPH1-mediated regulation of AR and AR-V7: implications for racial disparity of prostate cancer. Clin. Cancer Res., 22, 1744–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Larne O., Hagman Z., Lilja H., Bjartell A., Edsjo A. and Ceder Y. (2015) miR-145 suppress the androgen receptor in prostate cancer cells and correlates to prostate cancer prognosis. Carcinogenesis, 36, 858–866. [DOI] [PubMed] [Google Scholar]
- 66. Greene J., Baird A.M., Casey O., Brady L., Blackshields G., Lim M., O'Brien O., Gray S.G., McDermott R. and Finn S.P. (2019) Circular RNAs are differentially expressed in prostate cancer and are potentially associated with resistance to enzalutamide. Sci. Rep., 9, 10739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Luna Velez M.V., Verhaegh G.W., Smit F., Sedelaar J.P.M. and Schalken J.A. (2019) Suppression of prostate tumor cell survival by antisense oligonucleotide-mediated inhibition of AR-V7 mRNA synthesis. Oncogene, 38, 3696–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Daige C.L., Wiggins J.F., Priddy L., Nelligan-Davis T., Zhao J. and Brown D. (2014) Systemic delivery of a miR34a mimic as a potential therapeutic for liver cancer. Mol. Cancer Ther., 13, 2352–2360. [DOI] [PubMed] [Google Scholar]
- 69. Jiang Q., Yuan Y., Gong Y., Luo X., Su X., Hu X. and Zhu W. (2019) Therapeutic delivery of microRNA-143 by cationic lipoplexes for non-small cell lung cancer treatment in vivo. J. Cancer Res. Clin. Oncol., 145, 2951–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bonnal S., Vigevani L. and Valcarcel J. (2012) The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov., 11, 847–859. [DOI] [PubMed] [Google Scholar]
- 71. Effenberger K.A., Urabe V.K., Prichard B.E., Ghosh A.K. and Jurica M.S. (2016) Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA, 22, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kotake Y., Sagane K., Owa T., Mimori-Kiyosue Y., Shimizu H., Uesugi M., Ishihama Y., Iwata M. and Mizui Y. (2007) Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol., 3, 570–575. [DOI] [PubMed] [Google Scholar]