

Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the preferential death of motor neurons. Approximately 10% of ALS cases are familial and 90% are sporadic. Fused in sarcoma (FUS) is a ubiquitously expressed RNA-binding protein implicated in familial ALS and frontotemporal dementia (FTD). The physiological function and pathological mechanism of FUS are not well understood, particularly whether post-translational modifications play a role in regulating FUS function. In this study, we discovered that FUS was acetylated at lysine-315/316 (K315/K316) and lysine-510 (K510) residues in two distinct domains. Located in the nuclear localization sequence, K510 acetylation disrupted the interaction between FUS and Transportin-1, resulting in the mislocalization of FUS in the cytoplasm and formation of stress granule-like inclusions. Located in the RNA recognition motif, K315/K316 acetylation reduced RNA binding to FUS and decreased the formation of cytoplasmic inclusions. Treatment with deacetylase inhibitors also significantly reduced the inclusion formation in cells expressing ALS mutation P525L. More interestingly, familial ALS patient fibroblasts showed higher levels of FUS K510 acetylation as compared with healthy controls. Lastly, CREB-binding protein/p300 acetylated FUS, whereas both sirtuins and histone deacetylases families of lysine deacetylases contributed to FUS deacetylation. These findings demonstrate that FUS acetylation regulates the RNA binding, subcellular localization and inclusion formation of FUS, implicating a potential role of acetylation in the pathophysiological process leading to FUS-mediated ALS/FTD.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurological disorder characterized by the gradual degeneration of motor neurons leading to progressive weakening of muscles, paralysis and death (1). About 90% of ALS cases are sporadic, whereas the remaining 10% of the cases are inherited (2,3). Several gene mutations have been identified to cause the familial form of ALS (fALS) (4). Mutations in fused in sarcoma (FUS, also called translocated in liposarcoma) have been found in the fALS (5). Moreover, FUS pathology is reported in ~10% cases of another clinically overlapping disease frontotemporal dementia (FTD–FUS) (6).
FUS is a ubiquitously expressed RNA-binding protein that plays a role in different cellular processes such as DNA repair (7–9), transcription (10–20), RNA splicing (19,21,22), nucleocytoplasmic RNA shuttling (23) and dendritic RNA transport (24–26). FUS contains an N-terminal prion-like domain, a glycine-rich region, an RNA recognition motif (RRM), a zinc finger domain flanked by two arginine–glycine–glycine (RGG)-rich domains, and a C-terminal nuclear localization sequence (NLS) (27). FUS is mainly localized in the nucleus, although it is also present in the cytoplasm of neuronal cells at lower levels (28). Many of the fALS-related FUS mutations are localized in the C-terminal NLS, causing mislocalization of FUS to the cytoplasm where it forms stress granule-like structures (29–32). A loss of FUS function in the nucleus and a gain of toxic function in the cytoplasm can both contribute to the disease mechanism concomitantly (33).
Protein post-translational modifications (PTMs) refer to covalent attachments of a functional group to a protein that can regulate its functions. Common eukaryotic PTMs include methylation, phosphorylation, acetylation, ubiquitination and sumoylation (34). Regarding FUS, various studies have shown that FUS is extensively methylated at arginine residues in the RGG-rich domains and this modification regulates the nuclear import of FUS (12,35,36). Lysine acetylation is a major PTM that modifies a great number of mammalian proteins and has also been implicated in neurodegenerative disorders (37–40). For instance, acetylation of misfolded Tau was reported as a feature of Alzheimer’s disease pathology (40). Acetylation of TDP-43 was found to impair its RNA binding and promote cytoplasmic aggregation that resembles the TDP-43 pathology in ALS patients (38). However, it is unknown whether FUS protein undergoes lysine acetylation or how acetylation may regulate FUS protein function.
In this study, we performed mass spectrometric analysis of 3× FLAG-tagged FUS immunoprecipitated from HEK293T cells and identified acetylated lysine residues K315/K316 and K510. Acetylation of K315 and K316 in the RRM decreased RNA-binding capability, whereas acetylation of K510 in the NLS affected the interaction of FUS with Transportin-1 and consequently its subcellular localization. Acetylation of K510 resulted in the formation of cytoplasmic inclusions that co-localized with stress granule marker G3BP1. However, additional acetylation of K315/K316 decreased the formation of inclusions, probably by decreasing the RNA binding to FUS. Moreover, deacetylase inhibitor (DACi) treatment and acetylation mimicking mutant K315Q/K316Q of FUS decreased the inclusion formation by the ALS disease-related P525L mutant. Fibroblast cells from fALS patients showed increased K510 acetylation as compared with healthy controls. Further studies demonstrated that FUS was acetylated by CREB-binding protein (CBP)/p300 and that both histone deacetylases (HDAC) and sirtuins (SIRT) played a role in FUS deacetylation. In summary, this study establishes that FUS acetylation affects RNA binding, cellular localization and formation of cytoplasmic inclusions, which may contribute to the pathophysiology of ALS and FTD.
Results
FUS is acetylated at specific lysine residues in two distinct functional domains
To evaluate whether FUS was acetylated, we immunoprecipitated endogenous FUS from N2A cells treated with a DACi cocktail [30 mM nicotinamide, 50 mM sodium butyrate and 3 μM trichostatin-A (TSA)], followed by immunoblotting with a pan-acetylated lysine antibody. A prominent acetylated FUS band was observed in the presence of DACi cocktail as compared with a significantly weaker band in the absence of DACi (Fig. 1A). The result suggests that FUS is acetylated and the acetylation is dynamic.
Figure 1.
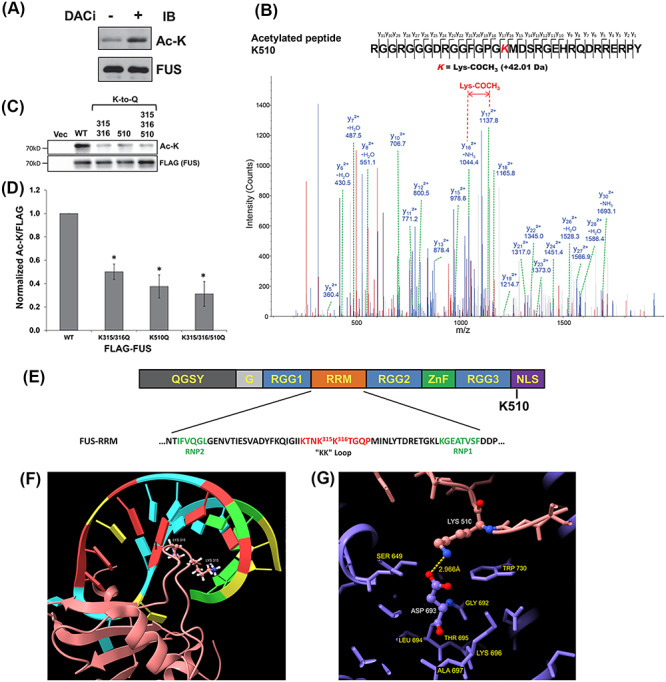
FUS is acetylated. (A) Endogenous FUS-IP from N2A cells treated with DACi cocktail (nicotinamide (30 mM), sodium butyrate (50 mM) and TSA (3 μM)) for 6 h. Immunoblotting was performed using the indicated antibodies. (B) Mass spectrometric identification of the acetylated FUS peptide RGGRGGGDRGGFGPGK510MDSRGEHRQDRRERPY. (C) 3× FLAG-tagged WT, K315Q/K316Q, K510Q, K315Q/K316Q/K510Q or FLAG vector control were transfected into HEK293T cells. After 24 h, cells were treated with DACi cocktail for 6 h, followed by FLAG-IP and immunoblotting with the indicated antibodies. (D) Quantification of FUS acetylation from three independent experiments ±standard deviation (SD). Student’s t-test was performed for individual comparisons against WT (*P ≤ 0.05). (E) The domain structure of FUS showing the RRM domain sequence and acetylation sites. (F) NMR solution structure of FUS RRM domain (pink) showing K315 and K316 in the KK-loop bound to stem–loop RNA (protein data bank entry 6GBM). (G) Crystal structure of TNPO1/FUS-NLS (protein data bank entry 4FQ3) illustrating the FUS-NLS domain (pink) K510 adjacent to TNPO1 (purple) D693. Molecular graphics of FUS RRM and NLS domains visualized using UCSF ChimeraX (79).
Next, we used liquid chromatography tandem mass spectrometry (LC–MS/MS) to identify the lysine acetylation sites. 3× FLAG-tagged FUS was transfected into HEK293T cells and treated with the DACi cocktail overnight. Subsequently, a FLAG immunoprecipitation (FLAG-IP) was performed, and the eluates were run on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by SYPRO Ruby staining. FUS bands at ~72 kDa were excised, subjected to in-gel chymotrypsin digestion and analyzed by LC–MS/MS as previously described (41). Figure 1B shows the mass spectrometric evidence of FUS acetylation at K510 and Supplementary Material, Figure S1A shows the di-acetylation at K315 and K316.
We next validated these three acetylation sites using a mutagenesis approach, in which acetylation mimicking mutants (Lys–Gln or K/Q) were generated for each site. Mutating lysine to glutamine is a method widely used in the field to study protein acetylation (38,41) In our data, replacing lysine with glutamine at K315 or K316 alone did not result in significant loss of FUS acetylation (Supplementary Material, Fig. S1B and C). However, when these two sites were mutated together, FUS acetylation was decreased significantly (Fig. 1C and D). This suggests that acetylation at both of these sites may occur interchangeably, which is consistent with the detection of di-acetylation of K315 and K316 in the LC–MS/MS analysis. Similarly, K510Q mutant decreased FUS acetylation significantly, and mutating the three sites also showed acetylation decrease (Fig. 1C and D). FUS lysine acetylation was not completely abrogated in the triple K315/316/510Q mutant, suggesting that additional acetylation sites might be present beyond the detection limit in our study.
Lysine residues 315 and 316 are localized in the RRM, specifically in the very positively charged KK-loop that facilitates the electrostatic interaction between FUS and DNA/RNA (Fig. 1E). NMR structure of the RRM domain found that K315 and K316 are inserted into the major groove of the stem–loop RNA and in contact with the ribose-phosphate backbone of both strands as shown in Figure 1F (42). Previous studies have shown that replacing lysine residues by alanine in the KK-loop disrupted nucleic acid binding (43). Thus, we hypothesized that acetylation of K315/K316 could regulate the RNA binding of FUS by a similar mechanism. In contrast, lysine 510 is localized in the C-terminal NLS (Fig. 1E). The NLS of FUS interacts with the nuclear transport receptor transportin-1 (TNPO1) at multiple positively charged residues including K510, and this interaction is crucial for nuclear localization and function of FUS (32,44). Our hypothesis was that acetylation of K510 disrupts the interaction between FUS and D693 of TNPO1 (Fig. 1G) by removing the positive charge of lysine and imposing steric hindrance. The net effect was predicted to be that K510 acetylation affects the interaction with TNPO1 and thereby regulates the subcellular localization of FUS.
Acetylation of FUS at K315/K316 regulates its RNA-binding activity
To test the hypothesis that acetylation of K315/K316 regulates RNA binding to FUS, we first tested FUS binding to its own pre-mRNA surrounding exon 7 as previously reported (45). The 3× FLAG-tagged FUS [wild-type (WT) and K/Q acetylation mimicking mutants] was immunoprecipitated and the quantity of FUS pre-mRNA was measured by reverse transcription and quantitative polymerase chain reaction (qPCR). The K315/316Q acetylation mimicking mutant co-precipitated significantly less FUS mRNA as compared with WT FUS (Fig. 2A). In contrast, K510Q did not change the RNA binding. As a control, we verified that the total FUS pre-mRNA levels were comparable in all cell extracts (Fig. 2A).
Figure 2.
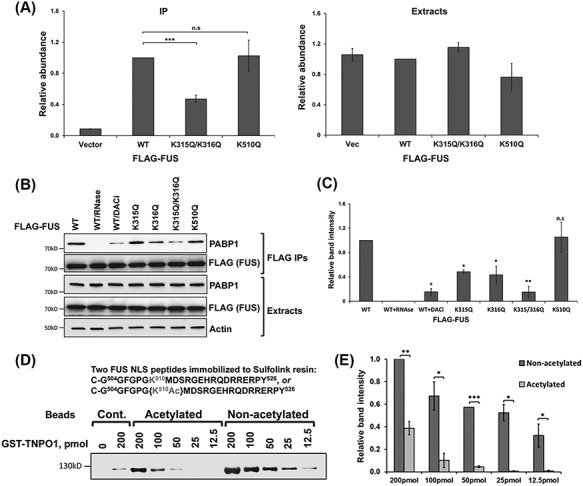
Acetylation of FUS impairs RNA binding and the FUS-NLS-TNPO1 interaction. (A) 3× FLAG-tagged FUS constructs were transfected into N2A cells. After 48 h, cells were lysed and FLAG-IP followed by reverse transcription and qPCR against FUS pre-mRNA surrounding exon 7 was performed. FUS mRNA was normalized with the FLAG levels in the FLAG-FUS-IPs and with the Rpl13a mRNA levels in the total extracts. Averages of three independent experiments are shown, ±SD. Student’s t-test was performed for individual comparisons against WT. (B) The indicated 3× FLAG-tagged FUS constructs were transfected into N2A cells and treated with DACi cocktail where indicated. FLAG-IP was performed 48 h after transfection with the inclusion of RNase cocktail as indicated, followed by immunoblotting with the indicated antibodies. (C) Quantification of (B) from three independent experiments. Student’s t-test was performed for individual comparisons against WT. (D) In vitro TNPO1 pulldown with Sulfolink-immobilized acetylated or non-acetylated FUS-NLS peptides. Different amounts of GST–TNPO1 were incubated with FUS-NLS peptide immobilized on the beads at 4°C for 3 h. The amount of TNPO1 pulled down with FUS-NLS peptides were evaluated by western blot. (E) Quantification of (D) from three independent experiments, ±SD. Student’s t-test was performed comparing the band intensities of non-acetylated and acetylated peptide pulldowns at the same concentration (*P ≤ 0.05; **P ≤ 0.005; ***P ≤ 0.001; NS, not significant).
A second assay was used to test FUS RNA binding by examining the interaction between FUS and poly-A binding protein (PABP1), which was reported to be RNA dependent (30). We performed a FLAG-IP from N2A cells transfected with 3× FLAG-tagged WT or K/Q mutant FUS to examine the FUS–PABP1 interaction. As expected, in the presence of RNase cocktail containing a mixture of RNase A and RNase T1, the WT FUS–PABP1 interaction was completely abrogated (Fig. 2B and C). When the cells were treated with DACi cocktail, the WT FUS–PABP1 interaction decreased significantly, suggesting that acetylation affected RNA binding. We also found that the PABP1–FUS interaction was affected even by single K/Q mutations in the KK-loop, and when both sites were mutated to glutamine, the interaction was impaired at similar levels as the DACi treatment itself (Fig. 2B and C). In contrast, K510Q mutant did not affect the FUS–PABP1 interaction. Overall, these results support the hypothesis that lysine acetylation of K315/K316 impairs FUS binding to RNA.
The effect of FUS K510 acetylation on the FUS-NLS–TNPO1 interaction
To test the hypothesis that acetylation of K510 affects the interaction between FUS and TNPO1 and influences the subcellular localization of FUS, we employed chemically synthesized FUS-NLS containing the acetylated K510 in an in vitro interaction study with TNPO1. The acetylated and non-acetylated FUS-NLS peptides were immobilized to Sulfolink resin and incubated with different amounts of purified glutathione S-transferase (GST) tagged TNPO1. The pulldown of TNPO1 by the non-acetylated FUS peptide was detectable with the lowest amount of 12.5 pmol TNPO1 (Fig. 2D). However, the K510-acetylated FUS-NLS peptide pulled down significantly less TNPO1 noting that the interaction was almost undetectable with 25 pmol of TNPO1 (Fig. 2D and E). As a control, Sulfolink beads without FUS peptide were subjected to the same immobilization protocol and incubated with 0 or 200 pmol of TNPO1. No signal was detected at 0 pmol of TNPO1 but a weak band was detected when 200 pmol of TNPO1 was incubated with the blank beads. Thus, acetylation of K510 affected the interaction between FUS and TNPO1, suggesting that this PTM in the NLS reduces the nuclear import of FUS and hence may alter cytoplasmic localization.
Acetylation of FUS at distinct sites differentially alter cellular localization and stress granule formation
Since the acetylation at K315/K316 and K510 residues affects the FUS–RNA and the FUS–TNPO1 interaction, respectively, we used acetylation mimic K/Q mutants to determine whether acetylation of these lysine residues differentially affect its subcellular localization and inclusion formation by fluorescence microscopy. In Figure 3A, enhanced green fluorescent protein (EGFP)-tagged WT FUS was localized to the nuclei of N2A cells. Similarly, the K315Q/K316Q mutant was localized to the nuclei of the cells, suggesting that acetylation of K315/K316 residues did not affect FUS nuclear localization. In contrast, ~ 30% of the K510Q mutant showed cytoplasmic localization, whereas ~ 40% of the triple K/Q mutant FUS was in the cytoplasm (Fig. 3A and B). More interestingly, ~ 59% of cells expressing the K510Q mutant FUS showed cytoplasmic inclusions, whereas only ~ 22% of cells expressing the triple mutant K315Q/K316Q/K510Q showed cytoplasmic inclusions (Fig. 3C, P < 0.001). The reduced formation of cytoplasmic inclusions by the triple K/Q mutant suggests that that RNA binding is important for the formation of FUS cytoplasmic inclusions, which is consistent with our previous report (19). Next, we assessed whether the cytoplasmic inclusions formed by acetylation mimicking mutants were co-localized with the stress granule marker G3BP1 in N2A cells using fluorescence microscopy. As expected, EGFP–FUS-WT and K315Q/316Q neither formed inclusions nor co-localized with G3BP1. In contrast, EGFP–FUS K510Q and K315Q/K316Q/K510Q cytoplasmic inclusions were co-localized with G3BP1 (Supplementary Material, Fig. S2). These results are consistent with previous studies that ALS mutant FUS forms cytoplasmic inclusions co-localized with stress granule markers (30,46). Taken together, we conclude that the acetylation of K510 affects the interaction between FUS and TNPO1, causing the mislocalization of FUS to the cytoplasm and the formation of stress granule-like inclusions. In addition, the acetylation of K315/K316 reduces the RNA binding and suppresses FUS-positive stress granules.
Figure 3.
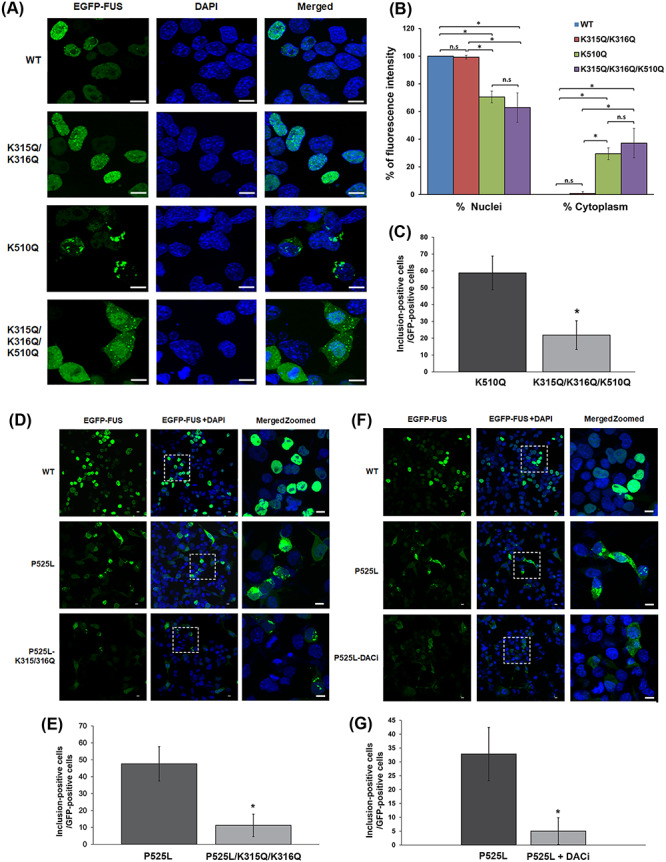
The effect of FUS acetylation on cellular localization and stress granule formation. (A) N2A cells were transfected with EGFP-tagged WT, K315Q/K316Q, K510Q and K315Q/K316Q/K510Q FUS. The nuclei were visualized with DAPI. Samples were examined by confocal microscopy. Scale bars, 10 μm. (B) Quantification of nuclear and cytoplasmic EGFP-tagged FUS intensity ± SD (n > 200 cells) using ImageJ scripts. One-way ANOVA was performed to determine statistical significance (*P ≤ 0.05; **P ≤ 0.005; ***P ≤ 0.001). (C) The percentage of EGFP-positive cells with cytoplasmic inclusions (n > 100 cells; *P ≤ 0.001). (D) N2A cells were transfected with EGFP-tagged WT, P525L and P525L/K315Q/K316Q FUS. The nuclei were visualized with DAPI. Samples were examined by confocal microscopy. Scale bars, 10 μm. (E) The percentage of EGFP-positive cells with cytoplasmic inclusions (n > 100 cells; *P ≤ 0.001). (F) N2A cells were transfected with EGFP-tagged WT or P525L FUS. One set of P525L-transfected cells were treated with DACi cocktail (30 mM nicotinamide, 50 mM sodium butyrate and 3 μM TSA). The nuclei were visualized with DAPI. Samples were examined by confocal microscopy; scale bars, 10 μm. (G) The percentage of EGFP-positive cells with cytoplasmic inclusions (n > 100 cells; *P ≤ 0.001).
FUS acetylation in the RRM domain decreases the formation of ALS mutant inclusions
To further test whether acetylation of K315/K316 in the KK-loop of the RRM domain affects the formation of FUS cytoplasmic inclusions by disrupting RNA binding, we transfected N2A cells with the EGFP-tagged FUS ALS mutant P525L with or without harboring the acetylation mimicking mutation K315Q/K316Q. The P525L mutation itself caused mislocalization to the cytoplasm as previously reported (47) and ~48% of the P525L mutant-expressing cells showed cytoplasmic inclusions (Fig. 3D and E). However, only ~ 11% of cells expressing the P525L/K315Q/K316Q mutant FUS showed cytoplasmic inclusions (Fig. 3D and E, P < 0.001). Furthermore, cells expressing EGFP–P525L FUS were treated with the DACi cocktail and the percentage of cells with cytoplasmic inclusions decreased from ~ 33% in the absence of DACi to ~ 5% in the presence of DACi (Fig. 3F and G, P < 0.001). Thus, the results support that the acetylation of the K315/K316 residues reduces the ability of ALS mutants of FUS to form cytoplasmic inclusions.
K510 acetylation is increased in FUS ALS patients
Since the K510 acetylation mimicking mutant significantly increased cytoplasmic mislocalization and inclusion formation (Fig. 3A–C), we examined the status of K510 acetylation in ALS patients. We first generated a site-specific antibody against acetylated K510. The rabbit polyclonal antibody specifically detected hyperacetylated FUS in human HEK293T cells and mouse N2A cells in the presence of DACi, but did not detect any signal in FUS-null N2A cells (Supplementary Material, Fig. S3A). Additionally, we transfected FUS-null N2A cells with either empty vector or WT, K510Q or K510R FUS with or without DACi cocktail. The antibody detected a strong band in extracts from WT FUS-expressing cells treated with DACi, whereas it did not detect any signal in K510R mutant extract treated with DACi (Supplementary Material, Fig. S3B). The acetylation mimicking mutant K510Q produced a very faint band; thus, the antibody is specific for FUS acetylation of K510. We used this acetyl-K510 specific antibody to analyze human skin fibroblast samples from FUS ALS patients and healthy controls. Levels of K510 acetylation were normalized against total FUS. We found ∼50% increased levels of acetylated FUS K510 in ALS patients (P = 0.015) (Fig. 4A and B). These results suggest that acetylation of FUS could be associated with pathological characteristics of FUS ALS.
Figure 4.
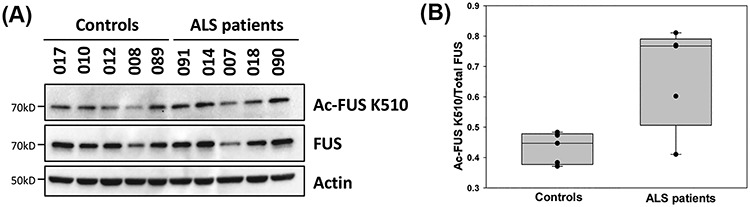
K510 acetylation is increased in familiar FUS ALS. (A) FUS-K510 acetylation levels in ALS patients with R521G or P525R FUS mutations versus control subjects. Immunoblotting was performed using the indicated antibodies. (B) Quantification of FUS-K510 acetylation normalized against total FUS levels. Error bars represent SD between individuals. One-way ANOVA was performed to determine statistical significance (*P ≤ 0.05).
FUS is acetylated by CBP/p300
The CBP and p300 form a select family of lysine acetyltransferases due to their structural and functional similarities (48,49). These proteins are responsible for a large portion of the acetylation of histone and non-histone proteins in mammalian cells (49). FUS has been shown to interact with CBP/p300 (10), and to verify that CBP/p300 acetylates FUS we co-transfected HA-tagged CBP and FLAG-tagged FUS into N2A cells. After FLAG-IP, a pan-acetylated lysine antibody showed a strong signal when HA–CBP was present compared to HA–empty vector (Fig. 5A), indicating that FUS was acetylated by CBP/p300.
Figure 5.
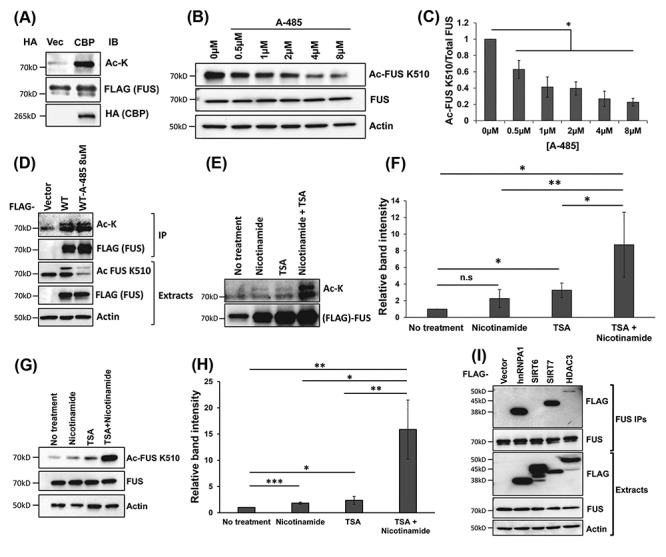
The regulators of FUS acetylation. (A) 3× FLAG-FUS or 3× FLAG-vector was co-transfected with HA-CBP or HA-vector into N2A cells. FLAG-IP was performed, followed by immunoblotting using a pan-acetylated lysine antibody and other indicated antibodies. (B) N2A cells were treated with different concentrations of the CBP/p300 inhibitor A-485. Immunoblotting was performed using the FUS K510 acetylation antibody and other antibodies as indicated. (C) Quantification of (B) from three independent experiments ±SD. Student’s t-test was performed for individual comparisons against no treatment (*P ≤ 0.05). (D) N2A cells were transfected with 3× FLAG-vector or 3× FLAG-FUS and treated with 8 μM CBP/p300 inhibitor A-485 for 16 h in the presence of DACi cocktail. FLAG-IP was performed followed by immunoblotting with a pan-acetylated lysine antibody and a FLAG antibody. The cell lysate was examined using the FUS K510 acetylation antibody and other indicated antibodies. (E). N2A cells were treated with 30 mM nicotinamide and/or 3 μM TSA for 6 h. FLAG-IP was performed followed by immunoblotting with a pan-acetylated lysine antibody and a FLAG antibody. (F) Quantification of (E) from three independent experiments, ±SD. One-way ANOVA was performed to determine statistical significance (*P ≤ 0.05; **P ≤ 0.005). (G). N2A cells were treated with 30 mM nicotinamide and/or 3 μM TSA for 6 h. Cells were harvested and lysed after treatment and immunoblotting was performed using the FUS K510 acetylation antibody and other indicated antibodies. (H) Quantification of (G) from three independent experiments ±SD. One-way ANOVA was performed to determine statistical significance (*P ≤ 0.05; **P ≤ 0.005; ***P ≤ 0.001). (I) HEK293T cells were transfected with 3× FLAG-empty vector, 3× FLAG-SIRTs 6–7, 3× FLAG-HDAC3 and 3× FLAG-ROA1 (hnRNPA1) used as a positive control. After 48 h, endogenous FUS-IP was performed, followed by immunoblotting using the indicated antibodies.
We then treated N2A cells with DACi cocktail plus compound A-485, a highly selective CBP/p300 inhibitor (50), at different concentrations for 24 h. We detected a significant signal decrease in acetylated-K510 FUS with increasing concentrations of A-485 (Fig. 5B and C). Interestingly, treatment of 8 μM A-485 did not change the total acetylation as detected by the pan-acetylated lysine antibody, whereas K510 acetylation significantly decreased (Fig. 5D). The results suggest that endogenous CBP/p300 is a major acetyltransferase at the K510 site of FUS.
FUS is deacetylated by HDACs and SIRTs lysine deacetylases
Lysine deacetylases are enzymes that play an important role in regulating epigenetic changes that are critical for gene expression. They are classified into different families: HDACs are Zn2+-dependent lysine deacetylases, whereas SIRTs require nicotinamide adenine dinucleotide (NAD+) (51). To identify which family of lysine deacetylases deacetylates FUS, we treated N2A cells with two DACis for 24 h: nicotinamide that inhibits SIRTs (52), and TSA that inhibits a broad range of HDACs (53). We first examined the total acetylation by FLAG-IP followed by western blot using a pan-acetylated lysine antibody (Fig. 5E and F). Cells treated with nicotinamide showed a trend of increase without statistical significance. Cells treated with TSA showed a significant increase of total FUS acetylation. Cells with both nicotinamide and TSA showed ~8× increase of total FUS acetylation as compared with the control (Fig. 5E and F). Next, we evaluated the effect of these inhibitors on the K510 site. In cells treated with nicotinamide or TSA alone, we observed a significant increase in FUS-K510 acetylation compared with the vehicle control. In the same fashion, treating with both nicotinamide and TSA together, we detected a ~15× increase in FUS-K510 acetylation (Fig. 5G and H). This suggests that both HDACs and SIRTs are able to deacetylate FUS.
We next used co-precipitation assays to identify candidate FUS lysine deacetylases. A set of FLAG-tagged expression constructs for all known human lysine deacetylases (HDACs 1–11 and SIRTs 1–7) were transfected into HEK293T cells, followed by endogenous FUS-IP. Among the 11 HDACs and 7 SIRTs (Supplementary Material, Fig. S4A and B) tested, only HDAC3 and SIRT7 co-precipitated with the endogenous FUS (Fig. 5I), suggesting that these lysine deacetylases might be involved in FUS deacetylation. SIRT6 and hnRNPA1 were included as a negative and positive control in the co-precipitation study, respectively (Fig. 5I). The results in Figure 5I are consistent with the results in Figure 5E and H that the FUS acetylation level increased in the presence of either HDAC or SIRT inhibitors.
Discussion
FUS is a DNA/RNA-binding protein that belongs to the FET (FUS/EWS/TLS) or TET (TAF15/EWS/TLS) family of proteins including TATA-Box Binding Protein Associated Factor 15 (TAF15) and Ewing sarcoma (EWS). FUS mutations have been linked to fALS (5) and FUS pathology is also found in a subset of FTD (6). FUS is a ubiquitously expressed protein that has many molecular functions in the cell. In this study, we found that the biochemical properties of FUS are modulated by acetylation. First, using a mass spectrometry approach, we identified lysine acetylation sites in the RRM domain and C-terminal NLS of FUS. Lysine residues 315 and 316 are located in the KK-loop, which is a structural component of the RRM domain and is involved directly in DNA and RNA binding (43). The lysine residue 510 is located in the NLS of FUS, a domain that is primarily responsible for importing FUS into the nucleus. Remarkably, mutations in this site (K510E, K510R and K510M) have been reported in fALS cases (54–56). These findings led us to hypothesize that acetylation of lysine at these sites could influence the biochemical function of FUS in the cell.
FUS-NLS domain harbors most of the reported ALS mutations, resulting in aberrant FUS protein mislocalization and aggregation in the cytoplasm (29–32). The interaction between FUS-NLS and TNPO1 is critical for importing FUS into the nucleus. Structural analysis showed that the positively charged K510 residue interacts with the negatively charged D693 residue of TNPO1 (Fig. 1G) (44). It is conceivable that the addition of an acetyl group to K510 could disrupt the interaction between K510 and D693. In addition, the acetyl group can also cause steric hindrance between the K510 and D693 residues. Thus, K510 acetylation would reduce FUS binding affinity to TNPO1 and increase the cytoplasmic localization of FUS. It was reported that fALS patients with the K510R mutation manifest a mild phenotype and long survival after disease onset (56). Although both arginine and lysine contain positively charged side chains, the arginine side chain is spatially larger than lysine, consistent with the notion that the K510R mutation may disrupt the FUS-NLS–TNPO1 interaction to a lesser degree, accounting for mild disease phenotypes. In contrast, FUS K510E ALS patients showed early onset and rapid progression of the disease supporting the concept that a negative charge at position 510 is particularly deleterious (57). Our previous data showed that K510E has only 1% binding affinity to TNPO1 (44), and it has been shown that a severe disruption of nuclear import could be correlated with a rapid progression of the disease (58). These results suggest that FUS-NLS is sensitive to perturbations at the K510 residue, including mutations or PTMs like acetylation discovered in this study. Indeed, our results showed that FUS acetylation at K510 disrupted the interaction with TNPO1 (Fig. 2D and E), leading to the cytoplasmic accumulation of FUS and the formation of G3BP1-positive stress granule-like inclusions in the cytoplasm (Fig. 3A–C, Supplementary Material, Fig. S2). Several transgenic mice models have shown that cytoplasmic aggregation and inclusion formation contribute to neurodegeneration (59–61), supporting the gain-of-toxic function hypothesis.
FUS-RRM domain has been reported to bind various nucleic acids, ranging from DNA to G-quadruplex RNA (43). The K315/K316 motif in FUS-RRM domain exhibits tandem positive charges at the protruding tip of the KK-loop and contributes significantly to the electrostatic interaction with the negatively charged nucleic acids. Point mutations K315A/K316A abolished FUS nucleic acid binding (43). Similarly, our results show that acetylation mimicking mutations in the KK-loop significantly reduce the RNA-binding capability of FUS (Fig. 2A–C), most likely by neutralizing the positive charge of lysine, leading to a decreased affinity to the negatively charged backbone of the nucleic acids (62). Finally, the attachment of an acetyl group to lysine could cause a steric clash with its interaction partners.
An interesting observation from our immunofluorescence experiments was that transfecting the cells with EGFP-K315Q/K316Q/K510Q caused FUS to mislocalize to the cytoplasm, but the percentage of cells with cytoplasmic inclusions decreased significantly when compared with EGFP-K510Q (Fig. 3C). These results are in agreement with our previous study that FUS–RNA binding is required for the formation of cytoplasmic inclusions (46). That study demonstrated that when FUS–RNA binding domains were truncated, the formation of inclusions significantly decreased as compared with the R495X FUS ALS mutant. Other work showed that mutating K315 and K316 to alanine residues prevented the formation of cytoplasmic inclusions by the R495X mutant (43). These data are consistent with the results obtained in this study that introducing the acetylation mimics at the RNA-binding sites into the ALS mutant P525L (P525L/K315Q/K316Q) significantly reduced the percentage of cells with cytoplasmic inclusions as compared with the P525L mutation alone (Fig. 3D and E). Consistent with these findings, we showed that treating cells expressing the P525L ALS mutant with DACi cocktail significantly reduced the percentage of cells with cytoplasmic inclusions (Fig. 3F and G). Our results are relevant to a recent study showing that dynamic multivalent interaction between FUS and RNA is critical to maintaining the fluidity and function of FUS (63). FUS mutations that constrain the FUS–RNA interactions lead to higher order complex of ribonucleoprotein aggregates (63). Finally, RNA binding was also shown to play a critical role in the formation of stress granules and pathological inclusion of TDP-43 (64). These studies support our conclusion that RNA binding is critical for self-assembly of FUS into cytoplasmic inclusions and that lysine acetylation in the RRM domain can effectively modulate RNA binding and inclusion formation of mutant FUS.
To identify the lysine deacetylases that might regulate FUS acetylation, we first used nicotinamide and TSA to test whether FUS acetylation is susceptible to SIRTs or HDACs, respectively. Our results showed that total FUS acetylation as well as FUS K510 acetylation can be mediated by both SIRTs and HDACs (Fig. 5E–H). It is noted that the effect of nicotinamide alone on the total acetylation level was not statistically significant. It has been reported that the expected inhibitory effect of nicotinamide could be unreliable; in particular, nicotinamide was reported to stimulate the SIRT1 activity (65). We next examined which specific SIRT or HDAC interacted with FUS using IP. We found that SIRT7 and HDAC3 interact with FUS. The FUS–SIRT7 interaction was previously reported in a proteomics study (66) but was not experimentally validated. SIRT7 is a NAD+-dependent deacetylase that acts on histone and non-histone proteins (67) and it is localized in the nucleus, but primarily in the nucleolus (68). Moreover, we identified a novel interaction between HDAC3 and FUS. HDAC3 is a Zn2+-dependent class-I HDAC that has been reported to shuttle between the nucleus and the cytoplasm (69). An interaction between FUS and HDAC1 was reported previously (9); however, we could not confirm this interaction under our experimental conditions. Future studies of genetically reducing HDAC3 and SIRT7 are needed to confirm that they indeed deacetylate FUS.
We also demonstrated that endogenous CBP/p300 is a major acetyltransferase for FUS, particularly the K510 site (Fig. 5A–D). The CBP/p300 inhibitor A-485 (50) significantly decreased FUS K510 acetylation in a dose-dependent manner in N2A cells (Fig. 5B and C); however, the total FUS acetylation did not show significant change, suggesting that the acetylation of the specific K510 site is more sensitive to CBP/p300 inhibitor. Reports show that CBP/p300 is localized to the nucleus (70) and is responsible for acetylation of over two-thirds of the lysine acetylation sites. Overall, the process of FUS acetylation/deacetylation appears to be very dynamic, and it is likely that other acetyltransferase enzymes play a role in FUS acetylation at other sites such as K315/K316. The functional consequence of inhibiting the specific acetyltransferases (e.g. CBP/p300) and deacetylases (e.g. SIRT7 and HDAC3) on the subcellular localization and inclusion formation of FUS remains to be determined in future studies.
To our knowledge, lysine acetylation of human FUS has not been reported previously. A proteomics study reported the acetylation of K502 in rat FUS and K640 in rat EWS, the equivalent of K510 in human FUS. The acetylation of the K263 residue in rat TAF15 was also reported, which corresponds to K316 in human FUS (71). Although the acetylation of those residues was not further validated and their functional relevance was not studied, the proteomic identification of acetylation of these conserved residues in the FET family proteins across species is supportive of the importance of our findings demonstrating acetylation of K315, K316 and K510 residues. Our mutagenesis and pharmacological studies demonstrate that acetylation of these residues bestowed a profound impact on FUS subcellular localization as well as the formation of stress granule-like cytoplasmic inclusions.
More interestingly, a higher K510 acetylation level was observed in familial FUS ALS patients as compared with healthy controls (Fig. 4A and B), suggesting that acetylation could play a role in disease pathogenesis and/or serve as a molecular hallmark of the disease. Particularly, K510 acetylation increased the cytoplasmic inclusions (Fig. 3A–C). Based on all results in this study, a working model is proposed to illustrate the role of FUS acetylation (Fig. 6). Acetylation of K315/K316 disrupts the RNA binding but does not interfere with the TNPO1-mediated nuclear import of FUS. In contrast, acetylation of K510 disrupts the interaction with TNPO1 and promotes the formation of cytoplasmic inclusions. When all three lysine residues are acetylated, FUS accumulates in the cytoplasm but forms less inclusions due to impaired RNA binding. We propose this process to be dynamic and the three forms of FUS acetylation can be present simultaneously. For ALS-linked mutations, mutant FUS shows reduced TNPO1 binding, impaired nuclear import and cytoplasmic accumulation. Moreover, mutant FUS shows a higher level of K510 acetylation in ALS patients, facilitating the formation of cytoplasmic inclusions. Future studies are needed to test whether CBP/p300 inhibitors can mitigate mutant FUS-mediated disease pathology by suppressing the K510 acetylation.
Figure 6.
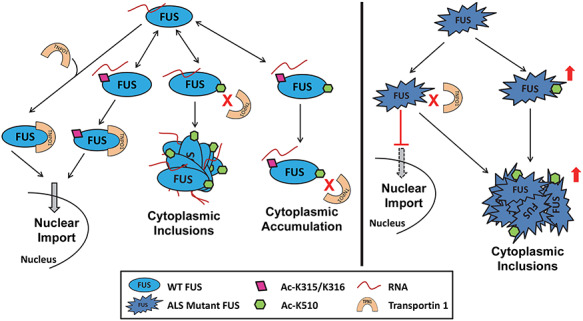
Proposed model of the role of FUS acetylation in the modulation of FUS subcellular localization and inclusion formation.
The pathological inclusions are a clinical characteristic of mutant FUS-mediated ALS/FTD. Studies show that different types of stress granules can form in a G3BP1-dependent (72) or independent (73) fashion, and can generate different functional outcomes (74). The role of acetylation of FUS in stress granules and the functional consequence of such acetylation-dependent granules remains to be determined in future studies. In addition, a recent study reported that nuclear-to-cytoplasmic mislocalization of FUS without aggregation in the cytoplasm occurred in different models of FUS ALS (75). We propose that pharmacologically modulating acetylation of FUS can prevent protein mislocalization or formation of pathological inclusions, providing new targets for therapeutic treatments against FUS-mediated ALS/FTD and other neurodegenerative diseases.
Materials and Methods
Cell culture and transfection
HEK293T and N2A cells were cultured in Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich, D5796) with 10% fetal bovine serum (FBS), penicillin–streptomycin, and amphotericin B at 37°C in 5% CO2/95% air with humidification. The pCMV10–3× FLAG-FUS-WT and pEGFP–C3 FUS plasmids were generated as reported (30). FUS mutations were introduced using the QuikChange II site-directed mutagenesis kit (Agilent). HEK293T and N2A cells were transfected with polyethylenimine ‘Max’ (Polysciences, Inc. 24 765) and lipofectamine 2000 (Thermo Fisher Scientific, 11 668), respectively.
Immunoprecipitation
Cells were lysed with 1× RIPA buffer (Millipore Sigma, 20-188) supplemented with protease inhibitor cocktail (Millipore Sigma, P8340, 1:500), sodium orthovanadate (1 mM), and DACi cocktail (nicotinamide (20 mM), sodium butyrate (20 mM) and TSA (1.5 μM)). Lysates were homogenized by sonication and centrifuged at 1000 g for 15 min at 4°C. IPs were performed overnight at 4°C using EZview™ Red ANTI-FLAG® M2 Affinity Gel (Millipore Sigma, F2426) or 4 μg/ml mouse monoclonal ANTI-FLAG® M2 antibody (Millipore Sigma, F3165) and Protein G UltraLink Resin (Thermo Fisher Scientific, 53 126). Beads were washed three times with lysis buffer and bound proteins were eluted with 0.15 μg/μl 3× FLAG peptide (Millipore Sigma, F4799) in 1× RIPA buffer at 4°C for 1 h. Samples were resolved by SDS-PAGE followed by immunoblotting. Where indicated, a cocktail of RNase A and RNase T1 (Life Technologies, AM2286) was added to the lysates at 1:100 dilution before the overnight IP.
Immunoblotting
Proteins resolved by SDS-PAGE were transferred to nitrocellulose membranes (Pall, 66 485). The membranes were blocked with 5% non-fat dry milk in TBST (100 mM Tris–HCl, pH 7.5, 0.9% NaCl, 0.1% Tween-20) or bovine serum albumin (BSA) for 1 h. The antibodies used include: mouse anti-FUS (Santa Cruz, sc-47 711), mouse monoclonal ANTI-FLAG® M2-peroxidase (horseradish peroxidase) (Millipore Sigma, A8592), rabbit anti-acetylated-lysine (Cell Signaling Technology, 9441), rabbit anti-PABP1 (Abcam, ab21060), mouse anti-β-actin (Santa Cruz Biotechnology, sc-81 178), rabbit anti-β-actin (Cell Signaling 8457) and a custom-made rabbit anti-K510-acetylated FUS antibody (see below).
Identification of FUS acetylation by mass spectrometry
FLAG-tagged FUS was transfected into HEK293T cells. Transfected cells were treated overnight with a cocktail of lysine DACi (30 mM nicotinamide, 50 mM sodium butyrate and 3 μM TSA) 1 day post-transfection in regular medium. Anti-FLAG-IP was performed immediately after DACi treatment. Eluates were run on SDS-PAGE and the gel was stained with SYPRO Ruby protein gel stain (Thermo Fisher Scientific, S12000). The bands that corresponded to FLAG-FUS (electrophoretic mobility of ~72 kDa) were excised and subjected to dithiothreitol (DTT) reduction, iodoacetamide alkylation and in-gel chymotrypsin digestion. Peptides were extracted, concentrated and subjected to LC–MS/MS analysis at the University of Kentucky Proteomics Core Facility as previously reported (47). Briefly, LC–MS/MS analysis was performed using an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) coupled with an Eksigent Nanoflex cHiPLC™ system (Eksigent, Dublin, CA) through a nano-electrospray ionization source. The peptide samples were separated with a reversed phase cHiPLC column (75 μm × 150 mm) at a flow rate of 300 nl/min. Mobile phase A was water with 0.1% (v/v) formic acid, whereas B was acetonitrile with 0.1% (v/v) formic acid. The data-dependent acquisition method consisted of an Orbitrap MS scan (300–1800 m/z) with 60 000 resolution for parent ions followed by MS/MS for fragmentation of the seven most intense multiple charged ions. The LC–MS/MS data were submitted to a local MASCOT server for MS/MS protein identification via Proteome Discoverer (version 1.3, Thermo Fisher Scientific, Waltham, MA) against a custom database containing only RNA-binding protein FUS (FUS_HUMAN) downloaded from Uniprot. Typical parameters used in the MASCOT MS/MS ion search include: chymotrypsin digestion with a maximum of three missed cleavages; 10 ppm precursor ion and 1.2 Da fragment ion mass tolerances; lysine acetylation; cysteine carbamidomethylation; methionine oxidation.
Generation of FUS KO (∆FUS) N2A cells
The FUS knockout cells were generated by employing CRISPR technology. N2A cells were transfected with FUS double nickase CRISPR plasmid (Santa Cruz Biotechnology, sc-433 326-NIC) following the manufacturer’s instructions. Clonal cell lines were isolated with serial dilution, and the FUS status of the clones was determined with immunoblotting.
Generation of the anti-acetylated-K510 FUS antibody
All peptides below were synthesized with an added extra N-terminal cysteine residue to facilitate conjugation to Sulfolink resin or KLH (Genscript, Piscataway, NJ). The rabbit anti-acetylated-K510 FUS antibody was raised against the peptide antigen N-G504GFGPG(K510Ac)MDSRG515-C. The peptide was synthesized with C-terminal amidation and conjugated to Keyhole Limpet Hemocyanin (KLH) for immunization (Pocono Rabbit Farm and Laboratory, Canadensis, PA). The serum was first depleted with the non-acetylated FUS peptide N-G504GFGPGK510MDSRGEHRQDRRERPY526-C, then affinity purified with the acetylated FUS peptide N-G504GFGPG(K510Ac) MDSRGEHRQDRRERPY526-C conjugated to Sulfolink resin (Thermo Fisher Scientific, 44 999). The antibodies were eluted with 0.1 M glycine–HCl buffer, pH 2.5, neutralized by the addition of Tris–HCl, 1 M, pH 8.5 and dialyzed against storage buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 0.04% [w/v] sodium azide).
GST–TNPO1 purification
The human TNPO1 coding region was PCR amplified with primers 5′-CGTCGGATCCATGGAGTATGAGTGGAAACCT-3′ and 5′-CGTCGTCGACTTAAACACCATAAAAAGCTGCAAGA-3′ from MGC clone 4 178 989 as template, and inserted between the BamHI and SalI sites of pGEX-6P-3 (GE Healthcare 28-9546-51). The GST–TNPO1 fusion protein was expressed in Rosetta 2 (DE3) pLysS Escherichia coli cells (Millipore 71 401) at 28°C incubation temperature, purified using glutathione sepharose 4B resin (GE Life Sciences, 17-0756-01) following the manufacturer’s recommendations, and eluted with 15 mM reduced L-glutathione (Gold Biotechnology, G-155-25) in 100 mM Tris–HCl, pH 8.0 and 2 mM DTT. Aliquots were flash frozen in liquid nitrogen, and stored at −80°C until use.
GST–TNPO1 pulldown with Sulfolink-immobilized FUS peptides
The acetylated and non-acetylated FUS peptides synthesized with an added extra N-terminal cysteine residue were immobilized to Sulfolink resin following the manufacturer’s instructions (Thermo Fisher Scientific, 44 999). The beads were blocked with 0.5 mg/ml BSA in TNPO1 buffer (20 mM Na-PO4, pH 7.4, 150 mM KCl, 0.5 mM ethylenediaminetetraacetic acid, 5 mM MgCl2, 10% glycerol, 1 mM DTT, 20 mM nicotinamide) for 1 h at 4°C. The indicated amounts of GST–TNPO1 were added to the blocked beads with the immobilized FUS peptides, and incubated at 4°C for 3 h with mild rotation. The beads were washed with TNPO1 buffer three times and eluted by the addition of 2× Laemmli sample buffer with β-mercaptoethanol. After incubation at 94°C for 5 min, the eluted samples were subjected to SDS-PAGE and western blot.
IP followed by qPCR
IP was carried out as described above, with the difference that SUPERase-In RNase Inhibitor (Thermo Fisher Scientific, AM2696, 0.2 U/μl) was added to the lysis and elution buffers, and incubation with the antibody was performed for 2 h. In the last step of the IP, the eluted sample was divided into two aliquots. One aliquot was used for immunoblotting and the other was used for RNA isolation using TRIzol Reagent (Thermo Fisher Scientific, 15 596), following the manufacturer’s instructions. Equal aliquots of the isolated RNA were reverse transcribed using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, 18 080). The resulting cDNA was subjected to quantitative real-time PCR using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, 4 367 659). The qPCR primers and annealing temperatures were: mouse FUS pre-mRNA flanking exon 7, 5′-CAACCCTTTTGTAGCCGTTGG-3′ and 5′-CAGCAGGAGGCATTCTACCC-3′, 59°C; and mouse RPL13A, 5′-CTGTGAAGGCATCAACATTTCTG-3′and 5′-GACCACCATCCGCTT TTTCTT-3′ (76), 55°C. The qPCR results were analyzed using the ΔΔCT method.
Immunofluorescence and fluorescence microscopy
N2A cells were seeded on gelatin-treated coverglass and transfected with EGFP–FUS constructs. Cells were treated with the lysine DACi cocktail for 6 h when indicated. In total, 48 h after transfection, cells were fixed with 4% formaldehyde in 1× phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) and permeabilized with 1× PBS supplemented with 0.25% (v/v) Triton X-100. The coverslips were blocked with 10% [w/v] BSA in 1× PBS for 1 h, followed by incubation with primary antibody at room temperature overnight. Coverslips were rinsed with 1× PBS and incubated with secondary antibodies for 1 h at 37°C. The primary antibody was rabbit anti-G3BP1 (Proteintech, 13 057-2-AP) and the secondary antibody was Alexa Fluor 568 donkey anti-rabbit (Thermo Fisher Scientific, A10042). All the samples were stained with 4′,6-diamidino-2-phenylindole (DAPI) and mounted with Vectashield Mounting Medium (Vector Laboratories, H-1000-10). Confocal images were acquired using a Nikon A1 confocal microscope with a 40× objective. Z-stack images were obtained from random view fields using identical parameters for all the samples. Maximum intensity projections of the Z-stacks were analyzed using ImageJ (http://imagej.nih.gov/ij) as reported previously (77). Inclusion-positive cells were defined as any cell with one or more inclusions >5 pixels. The number of inclusion-positive cells was normalized with the total number of GFP-positive cells in each view field.
Patient skin fibroblast isolation and culture
Human skin fibroblasts were prepared and maintained as described (78). ALS patients and healthy family members consented to donate the samples. Protocol was reviewed and approved by the institutional review board of the University of Kentucky. A skin biopsy of 3 mm diameter was obtained from the subjects and the tissue was washed in PBS, minced and incubated in minimum essential medium (Sigma-Aldrich, M5650) supplemented with 20% FBS, 2 mM L-glutamine, 100 unit/ml penicillin and 100 μg/ml streptomycin) at 37°C under 5%CO2/95% air. Information on the subjects examined in this study is shown in Table 1.
Table 1.
Familial ALS patients and healthy controls examined in this study.
| ID | Group | FUS genotype | Gender | Age of sample collection |
|---|---|---|---|---|
| 017 | Control | WT/WT | Female | 43 |
| 010 | Control | WT/WT | Female | 36 |
| 012 | Control | WT/WT | Female | 63 |
| 008 | Control | WT/WT | Female | 24 |
| 089 | Control | WT/WT | Male | 20 |
| 091 | ALS patient | WT/R521G | Female | 31 |
| 014 | ALS patient | WT/R521G | Male | 42 |
| 007 | ALS patient | WT/R521G | Male | 58 |
| 018 | ALS patient | WT/R521G | Female | 40 |
| 090 | ALS patient | WT/P525R | Female | 26 |
Statistical analysis
The quantification of western blot bands was performed using Image Lab software by BioRad. Statistical analysis was performed with SigmaPlot 14.0 software. Band intensities were calculated and comparison between groups was performed using analysis of variance (ANOVA) with post hoc Tukey honestly significant difference (HSD) test. Student’s t test was used to determine statistical significance between two groups. Chi square was used to compare difference between proportions. Experiments were not blinded. Fluorescence microscopy was quantified from >10 different view fields and all experiments were done in triplicates.
Conflict of Interest statement. None declared.
Funding
National Institute of Neurological Disorder and Stroke (R01NS077284 to H.Z.); Muscular Dystrophy Association (MDA352743 to H.Z.); Department of Veteran Affairs Merit Review (I01 BX002149 to H.Z.); National Institute of Neurological Disorders and Stroke (R21 NS095299 to J.G.). National Institute of Environmental Health Sciences Training (T32ES07266); University of Kentucky College of Medicine Fellowship for Excellence in Graduate Research (to A.A). Molecular graphics program ChimeraX is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health (R01-GM129325) and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
Supplementary Material
Contributor Information
Alexandra Arenas, Department of Toxicology and Cancer Biology.
Jing Chen, Department of Molecular and Cellular Biochemistry.
Lisha Kuang, Department of Molecular and Cellular Biochemistry.
Kelly R Barnett, Department of Molecular and Cellular Biochemistry.
Edward J Kasarskis, Department of Neurology, College of Medicine, University of Kentucky, Lexington, KY 40536, USA.
Jozsef Gal, Department of Molecular and Cellular Biochemistry.
Haining Zhu, Department of Toxicology and Cancer Biology; Department of Molecular and Cellular Biochemistry; Lexington VA Medical Center, Research and Development, Lexington, KY 40502, USA.
References
- 1. Wijesekera L.C. and Leigh P.N. (2009) Amyotrophic lateral sclerosis. Orphanet. J. Rare Dis., 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maruyama H., Morino H., Ito H., Izumi Y., Kato H., Watanabe Y., Kinoshita Y., Kamada M., Nodera H., Suzuki H. et al. (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature, 465, 223–226. [DOI] [PubMed] [Google Scholar]
- 3. Turner M.R., Hardiman O., Benatar M., Brooks B.R., Chio A., de Carvalho M., Ince P.G., Lin C., Miller R.G., Mitsumoto H. et al. (2013) Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol., 12, 310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen S., Sayana P., Zhang X. and Le W. (2013) Genetics of amyotrophic lateral sclerosis: an update. Mol. Neurodegener., 8, 28–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kwiatkowski T.J. Jr., Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T. et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science, 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- 6. Mackenzie I.R.A., Neumann M., Bigio E.H., Cairns N.J., Alafuzoff I., Kril J., Kovacs G.G., Ghetti B., Halliday G., Holm I.E. et al. (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol., 119, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baechtold H., Kuroda M., Sok J., Ron D., Lopez B.S. and Akhmedov A.T. (1999) Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J. Biol. Chem., 274, 34337–34342. [DOI] [PubMed] [Google Scholar]
- 8. Mastrocola A.S., Kim S.H., Trinh A.T., Rodenkirch L.A. and Tibbetts R.S. (2013) The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem., 288, 24731–24741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang W.Y., Pan L., Su S.C., Quinn E.J., Sasaki M., Jimenez J.C., Mackenzie I.R., Huang E.J. and Tsai L.H. (2013) Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci., 16, 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang X., Arai S., Song X., Reichart D., Du K., Pascual G., Tempst P., Rosenfeld M.G., Glass C.K. and Kurokawa R. (2008) Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature, 454, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tan A.Y. and Manley J.L. (2010) TLS inhibits RNA polymerase III transcription. Mol. Cell. Biol., 30, 186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Du K., Arai S., Kawamura T., Matsushita A. and Kurokawa R. (2011) TLS and PRMT1 synergistically coactivate transcription at the survivin promoter through TLS arginine methylation. Biochem. Biophys. Res. Commun., 404, 991–996. [DOI] [PubMed] [Google Scholar]
- 13. Haile S., Lal A., Myung J.K. and Sadar M.D. (2011) FUS/TLS is a co-activator of androgen receptor in prostate cancer cells. PLoS One, 6, e24197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tan A.Y., Riley T.R., Coady T., Bussemaker H.J. and Manley J.L. (2012) TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl. Acad. Sci. U. S. A., 109, 6030–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwartz J.C., Ebmeier C.C., Podell E.R., Heimiller J., Taatjes D.J. and Cech T.R. (2012) FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev., 26, 2690–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fujioka Y., Ishigaki S., Masuda A., Iguchi Y., Udagawa T., Watanabe H., Katsuno M., Ohno K. and Sobue G. (2013) FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Sci. Rep., 3, 2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bronisz A., Carey H.A., Godlewski J., Sif S., Ostrowski M.C. and Sharma S.M. (2014) The multifunctional protein fused in sarcoma (FUS) is a coactivator of microphthalmia-associated transcription factor (MITF). J. Biol. Chem., 289, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dhar S.K., Zhang J., Gal J., Xu Y., Miao L., Lynn B.C., Zhu H., Kasarskis E.J. and St Clair D.K. (2014) FUsed in sarcoma is a novel regulator of manganese superoxide dismutase gene transcription. Antioxid. Redox Signal., 20, 1550–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang L., Gal J., Chen J. and Zhu H. (2014) Self-assembled FUS binds active chromatin and regulates gene transcription. Proc. Natl. Acad. Sci. U. S. A., 111, 17809–17814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Uranishi H., Tetsuka T., Yamashita M., Asamitsu K., Shimizu M., Itoh M. and Okamoto T. (2001) Involvement of the pro-oncoprotein TLS (translocated in liposarcoma) in nuclear factor-kappa B p65-mediated transcription as a coactivator. J. Biol. Chem., 276, 13395–13401. [DOI] [PubMed] [Google Scholar]
- 21. Yang L., Embree L.J., Tsai S. and Hickstein D.D. (1998) Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J. Biol. Chem., 273, 27761–27764. [DOI] [PubMed] [Google Scholar]
- 22. Dichmann D.S. and Harland R.M. (2012) Fus/TLS orchestrates splicing of developmental regulators during gastrulation. Genes Dev., 26, 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zinszner H., Sok J., Immanuel D., Yin Y. and Ron D. (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci., 110, 1741–1750. [DOI] [PubMed] [Google Scholar]
- 24. Fujii R. and Takumi T. (2005) TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell. Sci., 118, 5755–5765. [DOI] [PubMed] [Google Scholar]
- 25. Fujii R., Okabe S., Urushido T., Inoue K., Yoshimura A., Tachibana T., Nishikawa T., Hicks G.G. and Takumi T. (2005) The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr. Biol., 15, 587–593. [DOI] [PubMed] [Google Scholar]
- 26. Sephton C.F., Tang A.A., Kulkarni A., West J., Brooks M., Stubblefield J.J., Liu Y., Zhang M.Q., Green C.B., Huber K.M. et al. (2014) Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proc. Natl. Acad. Sci. U. S. A., 111, E4769–E4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sama R.R.K., Ward C.L. and Bosco D.A. (2014) Functions of FUS/TLS from DNA repair to stress response: implications for ALS. ASN Neuro, 6, 1759091414544472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andersson M.K., Stahlberg A., Arvidsson Y., Olofsson A., Semb H., Stenman G., Nilsson O. and Aman P. (2008) The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol., 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bosco D.A., Lemay N., Ko H.K., Zhou H., Burke C., Kwiatkowski T.J. Jr., Sapp P., McKenna-Yasek D., Brown R.H. Jr. and Hayward L.J. (2010) Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet., 19, 4160–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gal J., Zhang J., Kwinter D.M., Zhai J., Jia H., Jia J. and Zhu H. (2011) Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol. Aging, 32, 2323 e2327–2323 e2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ito D., Seki M., Tsunoda Y., Uchiyama H. and Suzuki N. (2011) Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol., 69, 152–162. [DOI] [PubMed] [Google Scholar]
- 32. Dormann D., Rodde R., Edbauer D., Bentmann E., Fischer I., Hruscha A., Than M.E., Mackenzie I.R., Capell A., Schmid B. et al. (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J., 29, 2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dormann D. and Haass C. (2013) Fused in sarcoma (FUS): an oncogene goes awry in neurodegeneration. Mol. Cell. Neurosci., 56, 475–486. [DOI] [PubMed] [Google Scholar]
- 34. Bürkle A. (2001) In Brenner S. and Miller J.H. (eds), Encyclopedia of Genetics. Academic Press, New York, p. 1533. [Google Scholar]
- 35. Guo L., Kim H.J., Wang H., Monaghan J., Freyermuth F., Sung J.C., O’Donovan K., Fare C.M., Diaz Z., Singh N. et al. (2018) Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell, 173, 677–692.e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hofweber M., Hutten S., Bourgeois B., Spreitzer E., Niedner-Boblenz A., Schifferer M., Ruepp M.-D., Simons M., Niessing D., Madl T. and Dormann D. (2018) Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell, 173, 706–719.e713. [DOI] [PubMed] [Google Scholar]
- 37. Cohen T.J., Guo J.L., Hurtado D.E., Kwong L.K., Mills I.P., Trojanowski J.Q. and Lee V.M. (2011) The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun., 2, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cohen T.J., Hwang A.W., Restrepo C.R., Yuan C.X., Trojanowski J.Q. and Lee V.M. (2015) An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun., 6, 5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Irwin D.J., Cohen T.J., Grossman M., Arnold S.E., McCarty-Wood E., Van Deerlin V.M., Lee V.M. and Trojanowski J.Q. (2013) Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am. J. Pathol., 183, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Irwin D.J., Cohen T.J., Grossman M., Arnold S.E., Xie S.X., Lee V.M. and Trojanowski J.Q. (2012) Acetylated tau, a novel pathological signature in Alzheimer's disease and other tauopathies. Brain J. Neurol., 135, 807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gal J., Chen J., Na D.Y., Tichacek L., Barnett K.R. and Zhu H. (2019) The acetylation of lysine-376 of G3BP1 regulates RNA binding and stress granule dynamics. Mol. Cell. Biol., 39, e00052-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Loughlin F.E., Lukavsky P.J., Kazeeva T., Reber S., Hock E.-M., Colombo M., Von Schroetter C., Pauli P., Cléry A., Mühlemann O. et al. (2019) The solution structure of FUS bound to RNA reveals a bipartite mode of RNA recognition with both sequence and shape specificity. Mol. Cell., 73, 490–504.e496. [DOI] [PubMed] [Google Scholar]
- 43. Liu X., Niu C., Ren J., Zhang J., Xie X., Zhu H., Feng W. and Gong W. (2013) The RRM domain of human fused in sarcoma protein reveals a non-canonical nucleic acid binding site. Biochim. Biophys. Acta, 1832, 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Niu C., Zhang J., Gao F., Yang L., Jia M., Zhu H. and Gong W. (2012) FUS-NLS/Transportin 1 complex structure provides insights into the nuclear targeting mechanism of FUS and the implications in ALS. PLoS One, 7, e47056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou Y., Liu S., Liu G., Ozturk A. and Hicks G.G. (2013) ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet., 9, e1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang L., Zhang J., Kamelgarn M., Niu C., Gal J., Gong W. and Zhu H. (2015) Subcellular localization and RNAs determine FUS architecture in different cellular compartments. Hum. Mol. Genet., 24, 5174–5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kamelgarn M., Chen J., Kuang L., Arenas A., Zhai J., Zhu H. and Gal J. (2016) Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Biophys. Acta, 1862, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ogryzko V.V., Schiltz R.L., Russanova V., Howard B.H. and Nakatani Y. (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- 49. Weinert B.T., Narita T., Satpathy S., Srinivasan B., Hansen B.K., Scholz C., Hamilton W.B., Zucconi B.E., Wang W.W., Liu W.R. et al. (2018) Time-resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell, 174, 231–244.e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lasko L.M., Jakob C.G., Edalji R.P., Qiu W., Montgomery D., Digiammarino E.L., Hansen T.M., Risi R.M., Frey R., Manaves V. et al. (2017) Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature, 550, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Van Dyke M.W. (2014) Lysine deacetylase (KDAC) regulatory pathways: an alternative approach to selective modulation. Chem. Med. Chem., 9, 511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bitterman K.J., Anderson R.M., Cohen H.Y., Latorre-Esteves M. and Sinclair D.A. (2002) Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem., 277, 45099–45107. [DOI] [PubMed] [Google Scholar]
- 53. Yoshida M., Kijima M., Akita M. and Beppu T. (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem., 265, 17174–17179. [PubMed] [Google Scholar]
- 54. Mochizuki Y., Kawata A., Maruyama H., Homma T., Watabe K., Kawakami H., Komori T., Mizutani T. and Matsubara S. (2014) A Japanese patient with familial ALS and a p.K510M mutation in the gene for FUS (FUS) resulting in the totally locked-in state. Neuropathology, 34, 504–509. [DOI] [PubMed] [Google Scholar]
- 55. Syriani E., Morales M. and Gamez J. (2011) FUS/TLS gene mutations are the second most frequent cause of familial ALS in the Spanish population. Amyotroph Lateral Scler, 12, 118–123. [DOI] [PubMed] [Google Scholar]
- 56. Waibel S., Neumann M., Rabe M., Meyer T. and Ludolph A.C. (2010) Novel missense and truncating mutations in FUS/TLS in familial ALS. Neurology, 75, 815–817. [DOI] [PubMed] [Google Scholar]
- 57. Suzuki N., Aoki M., Warita H., Kato M., Mizuno H., Shimakura N., Akiyama T., Furuya H., Hokonohara T., Iwaki A. et al. (2010) FALS with FUS mutation in Japan, with early onset, rapid progress and basophilic inclusion. J. Hum. Genet., 55, 252–254. [DOI] [PubMed] [Google Scholar]
- 58. Nolan M., Talbot K. and Ansorge O. (2016) Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol. Commun., 4, 99–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shelkovnikova T.A., Peters O.M., Deykin A.V., Connor-Robson N., Robinson H., Ustyugov A.A., Bachurin S.O., Ermolkevich T.G., Goldman I.L., Sadchikova E.R. et al. (2013) Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J. Biol. Chem., 288, 25266–25274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Scekic-Zahirovic J., Sendscheid O., El Oussini H., Jambeau M., Sun Y., Mersmann S., Wagner M., Dieterle S., Sinniger J., Dirrig-Grosch S. et al. (2016) Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J., 35, 1077–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sharma A., Lyashchenko A.K., Lu L., Nasrabady S.E., Elmaleh M., Mendelsohn M., Nemes A., Tapia J.C., Mentis G.Z. and Shneider N.A. (2016) ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun., 7, 10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mutskov V., Gerber D., Angelov D., Ausio J., Workman J. and Dimitrov S. (1998) Persistent interactions of core histone tails with nucleosomal DNA following acetylation and transcription factor binding. Mol. Cell. Biol., 18, 6293–6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Niaki A.G., Sarkar J., Cai X., Rhine K., Vidaurre V., Guy B., Hurst M., Lee J.C., Koh H.R., Guo L. et al. (2020) Loss of dynamic RNA interaction and aberrant phase separation induced by two distinct types of ALS/FTD-linked FUS mutations. Mol. Cell, 77, 82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mann J.R., Gleixner A.M., Mauna J.C., Gomes E., DeChellis-Marks M.R., Needham P.G., Copley K.E., Hurtle B., Portz B., Pyles N.J. et al. (2019) RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron, 102, 321–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hwang E.S. and Song S.B. (2017) Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell. Mol. Life Sci., 74, 3347–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tsai Y.C., Greco T.M., Boonmee A., Miteva Y. and Cristea I.M. (2012) Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol. Cell. Proteomics, 11, 60–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wu D., Li Y., Zhu K.S., Wang H. and Zhu W.-G. (2018) Advances in cellular characterization of the Sirtuin isoform, SIRT7. Front Endocrinol. (Lausanne), 9, 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Michishita E., Park J.Y., Burneskis J.M., Barrett J.C. and Horikawa I. (2005) Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell, 16, 4623–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Longworth M.S. and Laimins L.A. (2006) Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene, 25, 4495–4500. [DOI] [PubMed] [Google Scholar]
- 70. McManus K.J. and Hendzel M.J. (2001) CBP, a transcriptional coactivator and acetyltransferase. Biochem. Cell. Biol., 79, 253–266. [PubMed] [Google Scholar]
- 71. Lundby A., Lage K., Weinert B.T., Bekker-Jensen D.B., Secher A., Skovgaard T., Kelstrup C.D., Dmytriyev A., Choudhary C., Lundby C. and Olsen J.V. (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep., 2, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sahoo P.K., Lee S.J., Jaiswal P.B., Alber S., Kar A.N., Miller-Randolph S., Taylor E.E., Smith T., Singh B., Ho T.S.-Y. et al. (2018) Axonal G3BP1 stress granule protein limits axonal mRNA translation and nerve regeneration. Nat. Commun., 9, 3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Andrusiak M.G., Sharifnia P., Lyu X., Wang Z., Dickey A.M., Wu Z., Chisholm A.D. and Jin Y. (2019) Inhibition of axon regeneration by liquid-like TIAR-2 granules. Neuron, 104, 290–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Boeynaems S. and Gitler A.D. (2019) Axons gonna ride ‘til they can’t no more. Neuron, 104, 179–181. [DOI] [PubMed] [Google Scholar]
- 75. Tyzack G.E., Luisier R., Taha D.M., Neeves J., Modic M., Mitchell J.S., Meyer I., Greensmith L., Newcombe J., Ule J. et al. (2019) Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain, 142, 2572–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gibson C.J., Hossain M.M., Richardson J.R. and Aleksunes L.M. (2012) Inflammatory regulation of ATP binding cassette efflux transporter expression and function in microglia. J. Pharmacol. Exp. Ther., 343, 650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gal J., Kuang L., Barnett K.R., Zhu B.Z., Shissler S.C., Korotkov K.V., Hayward L.J., Kasarskis E.J. and Zhu H. (2016) ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol., 132, 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kuang L., Kamelgarn M., Arenas A., Gal J., Taylor D., Gong W., Brown M., St. Clair D., Kasarskis E.J. and Zhu H. (2017) Clinical and experimental studies of a novel P525R FUS mutation in amyotrophic lateral sclerosis. Neurol. Genet., 3, e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H. and Ferrin T.E. (2018) UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci., 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.