

Abstract
Most anti-cancer agents and radiotherapy exert their therapeutic effects via the production of free radicals. Ferroptosis is a recently described cell death process that is accompanied by iron-dependent lipid peroxidation. Hydrogen peroxide (H2O2) has been reported to induce cell death. However, it remains controversial whether H2O2-induced cell death is ferroptosis. In the present study, we aimed to elucidate the involvement of mitochondria in H2O2-induced ferroptosis and examined the molecules that regulate ferroptosis. We found that one mechanism underlying H2O2-induced cell death is ferroptosis, which occurs soon after H2O2 treatment (within 3 h after H2O2 treatment). We also investigated the involvement of mitochondria in H2O2-induced ferroptosis using mitochondrial DNA-depleted ρ0 cells because ρ0 cells produce more lipid peroxidation, hydroxyl radicals (•OH), and are more sensitive to H2O2 treatment. We found that ρ0 cells contain high Fe2+ levels that lead to •OH production by H2O2. Further, we observed that aquaporin (AQP) 3, 5, and 8 bind nicotinamide-adenine dinucleotide phosphate oxidase 2 and regulate the permeability of extracellular H2O2, thereby contributing to ferroptosis. Additionally, the role of mitochondria in ferroptosis was investigated using mitochondrial transfer in ρ0 cells. When mitochondria were transferred into ρ0 cells, the cells exhibited no sensitivity to H2O2-induced cytotoxicity because of decreased Fe2+ levels. Moreover, mitochondrial transfer upregulated the mitochondrial quality control protein prohibitin 2 (PHB2), which contributes to reduced AQP expression. Our findings also revealed the involvement of AQP and PHB2 in ferroptosis. Our results indicate that H2O2 treatment enhances AQP expression, Fe2+ level, and lipid peroxidation, and decrease mitochondrial function by downregulating PHB2, and thus, is a promising modality for effective cancer treatment.
Keywords: Mitochondria, Ferroptosis, Aquaporin, Hydrogen peroxide, Fe2+
Graphical abstract
1. Introduction
There are numerous chemotherapeutic agents that exert their effects via production of free radicals and/or reactive oxygen species (ROS) [[1], [2], [3], [4], [5]]. Among broad sense ROS, hydrogen peroxide (H2O2) is used as a sensitizer in cancer treatment during radiation therapy. H2O2 treatment resolves the hypoxic state in tumor tissue by downregulating internal peroxidase activity and enables the generation of superoxide (O2 • -) for radiation therapy [6,7]. ROS are highly reactive and oxidize intracellular components such as DNA, proteins, and lipids, leading to cell death [8]. Intracellular ROS are generated by various enzymatic reactions such as nicotinamide-adenine dinucleotide phosphate oxidase (NOX) in the cytoplasm, but the mitochondrial electron transport chain (ETC) is thought to be the main source of intracellular ROS, especially hydroxyl radicals (•OH) [9,10].
Mitochondria have their own DNA (mtDNA) that encodes 13 proteins, which are components of the ETC. Damage to mtDNA produces a higher amount of ROS that, in turn, plays an important role in cancer initiation, promotion, and chemo/radio resistance [11,12]. We previously established mtDNA-depleted cells (ρ0 cells) from two cancer cell lines, i.e. cervical cancer (HeLa) and oral squamous cell carcinoma (SAS). We observed that the ρ0 cells exhibit sensitivity to ROS, particularly H2O2, because the ρ0 cell plasma membrane includes more lipid peroxides than their parental cells. In short, the membrane lipid components were changed by the influence of H2O2, and H2O2 more easily permeates the plasma membrane. Indeed, liposome membrane experiments showed that increased lipid peroxidation content leads to more H2O2 permeation, at least up to 5–10% lipid peroxidation [13,14]. Furthermore, the ρ0 cells showed higher aquaporin (AQP) gene expression [15]. Importantly, AQPs are involved in the diffusion of H2O2 as well as H2O [[16], [17], [18]].
Mitochondria are not only the main intracellular organelle of ROS production, but also the main metabolic site for iron regulation. The influx of cytoplasmic Fe2+ into mitochondria mainly uses a system of heme and iron-sulfur (Fe/S) clusters. Heme functions as an active center of hemoglobin, cytochrome p450, and cytochrome oxidase, while Fe/S clusters function in the ETC and in vitamin synthesis [19,20]. When Fe2+ is increased, •OH is produced through the Fenton reaction in the presence of Fe2+ and H2O2. •OH induces lipid peroxidation in the plasma membrane, which leads to cell death, including ferroptosis.
Ferroptosis is a new type of cell death where Fe2+, •OH, and lipid peroxidation play crucial role [[21], [22], [23]]. Recently, ferroptosis was implicated in several diseases such as neuronal degeneration, kidney injury, and cancer [21,24]. Ferroptosis is regulated by a number of genes/proteins. Glutathione peroxidase 4 (GPx4) was initially reported as a regulator of ferroptosis, however, other genes/proteins such as lipoxygenase, transferrin receptor, and frataxin were also reported as ferroptosis regulators [23,[25], [26], [27]]. Although mitochondrial by-products play an important role in ferroptosis, the involvement of mitochondria in ferroptosis is currently under debate [23,[27], [28], [29]]. For example, osteosarcoma ρ0 cells are not sensitive to erastin-induced cell death [28]. In addition, erastin and RSL3 induce cell death, even when mitochondria are depleted by parkin overexpression and carbonyl cyanide 3-chlorophenylhydrazone treatment [23]. Other reports describe a relationship among mitochondria, ferroptosis, and frataxin, a mitochondrial protein [27,29]. However, there are few reports that ferroptosis contributes to ρ0 cell sensitivity to H2O2
In the present in vitro study, we investigated the involvement of mitochondria in H2O2-induced ferroptosis and examined the molecules that regulate ferroptosis.
2. Materials and methods
2.1. Cell culture and mitochondrial isolation
The HeLa and SAS human cancer cell lines were obtained from the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University, Sendai, Japan. HeLa and SAS ρ0 cells were established by culturing cells with 50 ng/mL ethidium bromide as described previously [13]. Cells were cultured in RPMI 1640 (189–02025; Fujifilm Wako Pure Chemical Corporation, Osaka, Japan) with 10% FBS (Biological Industries, Cromwell, CT, USA), 110 μg/mL pyruvate (Sigma-Aldrich, St Louis, MO, USA), and 50 μg/mL uridine (TOKYO Chemical Industry Co. Ltd, Tokyo, Japan) in a humidified atmosphere at 37 °C with 5% CO2. Mitochondria were isolated from WI-38 cells (RIKEN BRC, Ibaraki Japan) using a mitochondrial isolation kit (ab110171, Abcam, Cambridge, UK) for 24 h, as described previously [30]. Then, transferred-mitochondria (Mito) cells were established by culture with 5 μg/mL isolated mitochondria. HeLa and SAS parental cells and Mito cells were cultured with RPMI 1640 with 10% FBS in a humidified atmosphere at 37 °C with 5% CO2. Exponentially growing cells were used in all experiments.
2.2. Flow cytometry analysis
To investigate H2O2-induced cell death, a BD Accuri C6 Flow Cytometer (BD Biosciences, San Jose, CA, USA) was used. Briefly, 2 × 105 HeLa and SAS ρ0 cells were cultured in 60 mm dishes for 24 h and treated with 75 μM (for HeLa ρ0 cells) or 50 μM (for SAS ρ0 cells) H2O2 (Nacalai Tesque, Kyoto, Japan) for 3 h. After H2O2 treatment, the cells were trypsinized and resuspend with 1x binding buffer (10 mM HEPES pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2). After filtration through a 40 μm cell strainer (352,235; BD Biosciences), 1 × 105 cells/100 μL solutions were mixed with 4 μg/mL propidium iodide (PI; Sigma-Aldrich) and 20 μM Liperfluo (DOJINDO Laboratories, Kumamoto, Japan) or 5 μL Annexin V-FITC (4700-100; MEDICAL & BIOLOGICAL LABORATORIES CO. LTD., Aichi, Japan) at room temperature for 20 min. Then, 400 μL 1x binding buffer were added and fluorescence images were obtained.
2.3. Annexin V and Liperfluo detection by fluorescence microscopy
HeLa and SAS ρ0 cells were cultured in glass-bottom dishes (Matsunami Glass Ind., Ltd., Osaka, Japan) with 20 μM Liperfluo or 5 μL Annexin V-FITC following H2O2 treatment as described above. Then, cells were washed three times with 1x binding buffer. Fluorescence images were obtained using a BZ-8000 fluorescence microscope (KEYENCE Corporation, Osaka, Japan) with a GFP-BP filter (excitation and absorption wavelengths: 470/40 nm). No autofluorescence was detected under the conditions of this experiment (Fig. S1). ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997–2012) was used to measure fluorescence intensity.
2.4. Intracellular and mitochondrial Fe2+ detection
FerroOrange (Goryo Chemical Inc., Hokkaido, Japan) and Mito-FerroGreen (Dojindo) were used to detect intracellular and mitochondrial Fe2+. HeLa and SAS ρ0 cells were cultured overnight in glass-bottom dishes (Matsunami Glass). Then, the cells were washed twice with Hank's Balanced Salt Solution (HBSS) (Fujifilm Wako Pure Chemical Corporation) to remove residual FBS. The cells were treated with 1 μM FerroOrange or 5 μM Mito-FerroGreen in HBSS for 30 min at 37 °C. After incubation, FerroOrange and Mito-FerroGreen were removed by washing three times with HBSS. Fluorescence images were obtained using a BZ-8000 fluorescence microscope with GFP-BP and TRITC filters (excitation and absorption wavelengths: 540/25 and 605/55 nm). ImageJ software was used to measure fluorescence intensity.
2.5. The role of iron in H2O2 cytotoxicity using WST assay
Phenanthroline (Phe: Nacalai Tesque), deferoxamine (DFO: Sigma-Aldrich) and deferasirox (DFX: Cayman Chemical, Ann Arbor, MI, USA) were used to investigate the involvement of iron during H2O2 treatment. HeLa and SAS ρ0 cells were cultured in 48 well plates. Then, 20 μM Phe, DFO, and DFX were mixed with the cultured cells for 30 min, followed by 50 μM H2O2 for 1 h. The cell survival ratio was analyzed using the water-soluble tetrazolium (WST) assay using a CCK-8 assay kit (Dojindo), as previously described [14].
2.6. Immunostaining
HeLa and SAS ρ0 cells were cultured in glass-bottom dishes. Cells were fixed with 4% formaldehyde in PBS for 30 min and rinsed three times with PBS. Plasma membranes were permeabilized by incubation in 95% ethanol with 5% acetic acid for 10 min. After washing five times with PBS, the cells were incubated for 30 min in blocking solution (5% skim milk in PBS-T; PBS with 0.05% Tween 20). Rabbit anti-AQP3 antibody (PA5-36552; Thermo Fisher Scientific, Waltham, MA, USA; dilution factor: 1:500), rabbit anti-AQP5 antibody (AQP-005; Alomone Labs, Jerusalem, Israel; dilution factor: 1:200), mouse anti-AQP8 antibody (SAB1403559; Sigma-Aldrich; dilution factor: 1:200), rabbit anti-gp91-phox (NOX2) antibody (07–024; EMD Millipore; dilution factor: 1:500) and rabbit anti-PHB antibody (GTX32812; GeneTex, Inc. Irvine, CA, USA; dilution factor: 1:1000) were used as primary antibodies. Cells were incubated at 4 °C overnight. Then, the cells were incubated with Alexa Fluor 488 goat anti-mouse IgG, Alexa Fluor 488 goat anti-rabbit IgG, or Alexa Fluor 568 goat anti-rabbit IgG (Thermo Fisher Scientific; A11001, A11008, and A11011) secondary antibodies (dilution factor: 1:200, for 1 h at room temperature. A BZ-8000 fluorescence microscope was used to obtain fluorescence images with GFP-BP and Texas Red filters (excitation and absorption wavelengths: 560/40 and 630/60 nm) and ImageJ software was used to measure fluorescence intensity.
2.7. Western blotting
Cells were extracted in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.1% sodium deoxycholate, 1 mM sodium fluoride, 1 mM sodium vanadate, and 1 mM phenylmethylsulfonyl fluoride: PMSF). A bicinchoninic acid (BCA) Protein Assay Kit (Thermo Fisher Scientific) was used to estimate the protein concentration. Proteins (10 μg per lane) were analyzed by SDS-PAGE using a 15% polyacrylamide gel. SDS-PAGE was performed under reducing conditions. Proteins were subsequently blotted on a PVDF membrane. After blocking with 5% skim milk in PBS-T, the membranes were incubated with primary antibodies in blocking solution [rabbit anti-AQP3, 5, NOX2, prohibitin 2 (PHB2), or mouse anti-AQP8]. After washing five times with PBS-T, the membranes were incubated with peroxidase-conjugated anti-rabbit IgG antibody or anti-mouse IgG antibodies (#7074, #7076; Cell Signaling Technology, Danvers, MA, USA) at room temperature for 2 h. Immunoreactive proteins were visualized with ImmunoStar Zeta (Fujifilm Wako) using a ChemiDoc XRS Plus instrument (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Anti-β-actin antibody (NB100-56874; Novus Biologicals LLC, Centennial, CO, USA; dilution factor: 1:1000) was used as loading control. All antibody dilution factors except for β-actin antibody were same as immunofluorescence assays. All Western blot analyses were performed using an identical sample amount in each well and were blotted under the same conditions.
2.8. Immunoprecipitation
Cells were suspended and homogenized with ten times volume of Homogenize solution (HS; 20 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 100 μg/mL DNase, 50 μg/mL RNaseA, 1 mM PMSF, and protease inhibitor cocktail). Homogenized samples were pre-incubated with Protein A-Sepharose 4B beads (Sigma-Aldrich) that were previously incubated with NOX2 antibody or normal rabbit IgG. An equal volume of sample (1 mg) and NOX2 or normal rabbit IgG-bound beads were incubated at 4 °C for 4 h. After the incubation, beads were washed three times with HS containing 1 mg/mL BSA. The washed beads were mixed with sample buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 6% 2-mercaptoethanol, and 20% glycerol) to extract NOX2-bound proteins. Extracted samples were analyzed by SDS-PAGE and western blotting as described above.
2.9. siRNA gene silencing
HeLa and SAS cells were transfected with synthetic miRNA corresponding to AQP3 (360-1-B, 360-2B; Bioneer, Daejeon, Korea) and AQP5, AQP8, or PHB2 (sc-2917, sc-42369, sc-45849; Santa Cruz Biotechnology, Dallas, TX, USA) using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific). AccuTarget Negative Control siRNA (SN-1003: Bioneer) was used as a control. Cell viability was measured using CCK-8 assay, as described above.
2.10. Measurement of intracellular H2O2
Intracellular H2O2 was visualized using HYDROP (Goryo Chemical Inc.) as described previously [13]. Briefly, cells in glass-bottom dishes (Matsunami Glass) were cultured in RPMI 1640 with 50 μM H2O2 for 1 h. After washing out the H2O2 twice with RPMI 1640, the cells were treated with 2.5 μM HYDROP in RPMI 1640 at 37 °C for 20 min. Then, the cells were washed twice with RPMI 1640. Fluorescence images were obtained using a BZ-8000 fluorescence microscope (KEYENCE) with a GFP-BP filter. ImageJ software was used to measure fluorescence intensity.
2.11. Quantitative PCR
Total RNA was extracted using ISOGEN reagent (Nippon Gene Toyama, Japan). The quality of RNA was checked by absorbance and electrophoresis. All cDNAs were prepared by reverse transcription of 1 μg total RNA using oligo dT (20) primer (0.4 μM/50 μl final volume) and ReverTra Ace (TOYOBO CO Ltd., Osaka, Japan). After 10x dilution with Tris-EDTA buffer (TE: 10 mM Tris-HCl pH 8.0, 1 mM EDTA), 0.5 μL cDNA (equivalent to 1 ng total RNA) was used for quantitative polymerase chain reaction (qPCR). The qPCR reactions were performed using an Applied Biosystems 7300 instrument (Applied Biosystems; Foster City, CA, USA) using TUNDERBIRD qPCR Mix (TOYOBO). β-actin was used as the loading control. cDNA was amplified as follows: one cycle at 95 °C for 10 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 60 s. Each experiment was performed in triplicate. Table 1 shows the primer sequences used in this experiment.
Table 1.
Primer sequences used in this study.
| Primer name | Primer sequence |
|---|---|
| AQP3-F | 5′-TTTTTACAGCCCTTGCGGGCTGGG-3′ |
| AQP3-R | 5′-ATCATCAGCTGGTACACGAAGACACC-3′ |
| AQP5-F | 5′-ATGAACCCAGCCCGCTCTTTTGGC-3′ |
| AQP5-R | 5′-ACGCTCACTCAGGCTCAGGGAGTT-3′ |
| AQP8-F | 5′-AACCACTGGAACTTCCACTGGATCTACT-3′ |
| AQP8-R | 5′-ATCTCCAATGAAGCACCTAATGAGCAGTC-3′ |
| PHB2–F | 5′-AAGATGCTTGGAGAAGCACTGAGCAAGAA-3′ |
| PHB2-R | 5′-AGCACAAGGTTGTCAGCTGTGAGATAGATA-3′ |
| β actin-F | 5′-AGAGCTACGAGCTGCCTGAC-3′ |
| β actin-R | 5′-AGCACTGTGTTGGCGTACAG-3′ |
2.12. Data analysis
Relative fluorescence intensities were obtained by measuring the fluorescence intensity of each cell using all the cells from three independent dishes. Fluorescence was normalized by subtracting the background fluorescence intensity of each dish from the fluorescence intensity of each cell. One-way ANOVA with Scheffe's F test was performed for the WST assay. All other statistical analyses were performed using Student's t-test. p < 0.05 was considered statistically significant. The results are expressed as means ± standard error.
3. Results
3.1. Induction of ferroptosis by H2O2 treatment in ρ0 cells
To determine whether H2O2-mediated cell death occurs via apoptosis or ferroptosis, the cells were treated with Liperfluo or Annexin V and PI followed by flow cytometry analysis. Liperfluo is a ferroptosis marker [31] and Annexin V is an apoptosis marker. Our results showed that Liperfluo increased more than Annexin V in both HeLa and SAS ρ0 cells after 3-h H2O2 treatment (1.55 vs. 1.15-fold in HeLa ρ0 cells and 3.79 vs 1.63-fold in SAS ρ0 cells, Fig. 1 A). Moreover, similar results were detected using fluorescence microscopy (Fig. 1B). Indeed, Liperfluo labeling intensity increased significantly after 3 h of H2O2 treatment in both HeLa and SAS ρ0 cells. In contrast, the intensity of Annexin V labeling increased slightly, but it was not significant (Fig. 1C). These results strongly suggest that cell death after H2O2 treatment occurs via ferroptosis, and that cell death occurs relatively quickly.
Fig. 1.

Detection of H2O2-induced ferroptosis in ρ0 cells.
To investigate H2O2-induced cell death, cells were stained with Liperfluo (a ferroptosis marker) or Annexin V (an apoptosis marker) and analyzed by flow cytometry. A: Liperfluo expression increased after 3-h H2O2 treatment. However, Annexin V did not increase. The concentration of H2O2 was 75 µM (for HeLa ρ0 cells) or 50 µM (for SAS ρ0 cells). B: Apoptosis and ferroptosis detected by fluorescence microscopy. Liperfluo or Annexin V was used to detect ferroptosis or apoptosis after H2O2 treatment. The conditions for H2O2 treatment were the same as in A. C: Relative intensity of Liperfluo or Annexin V. **: p < 0.01 using Student’s t-test (vs. negative control: N.C.).
3.2. Fe2+ amount is involved in H2O2-induced cell death in ρ0 cells
Intracellular and mitochondrial Fe2+ levels and the effect of iron chelators were examined to investigate the involvement of Fe2+ during H2O2 sensitivity in ρ0 cells. Intracellular Fe2+ was measured using FerroOrange (Fig. 2 A and B) and mitochondrial Fe2+ was measured using Mito-FerroGreen (Fig. 2C and D). Both intracellular and mitochondrial Fe2+ in ρ0 cells were significantly higher than in parental cells. We confirmed that the Mito-FerroGreen signal originated from mitochondria using Mito-Tracker red CMXRos (Fig. S2). No significant differences were detected in the number of mitochondria in each cell between parental cells and ρ0 cells (see details in discussion).
Fig. 2.

Effect of Fe2+ on H2O2 treatment in ρ0 cells.
To investigate the involvement of Fe2+ during H2O2 treatment in ρ0 cells, intracellular and mitochondrial Fe2+ and the effect of iron chelators were examined. A: Detection of intracellular Fe2+ levels by FerroOrange. B: Relative intensity of FerroOrange. C: Detection of mitochondrial Fe2+ by Mito-FerroGreen. D: Relative intensity of Mito-FerroGreen. The FerroOrange and Mito-FerroGreen signals in ρ0 cells were significantly higher than in parental cells. **: p < 0.01 using Student’s t test (vs. parent). E and F: Effect of iron chelators to H2O2 treatment in HeLa (E) and SAS (F) ρ0 cells. Iron chelating suppressed H2O2-induced cell death. Phe: Phenanthroline, DFO: Deferoxamine, DFX: Deferasirox. *: p < 0.05, **: p < 0.01 using Scheffe’s F test (vs. H2O2).
We examined whether iron chelators could recover H2O2 sensitivity. The typical iron chelators, Phe, DFO, and DFX, were used. Phe and DFX treatment significantly reduced cell death caused by H2O2 treatment (Fig. 2E).
3.3. Upregulation of AQPs in ρ0 cells
The spatial distribution of AQPs in ρ0 cells was investigated because some AQPs allow H2O2 flux. In both HeLa and SAS ρ0 cells, the expression of AQP 3, 5, and 8, which were reported to pass H2O2, was higher than in parental cells. The expression of AQPs in ρ0 cells was strongly observed at the cell margin, i.e. the plasma membrane (Fig. 3 ). We further investigated the amount of AQP protein by Western blot. AQP3, 5, and 8 expression was upregulated in both HeLa and SAS ρ0 cells (Fig. 4 A).
Fig. 3.

Spatial distribution of AQPs that function as H2O2 channels.
Immunostaining of AQPs was performed to investigate the contribution of AQPs to H2O2 permeability. A: Immunostaining of AQP3 in HeLa and SAS ρ0 cells. B: Relative fluorescence intensity of AQP3 in HeLa and SAS ρ0 cells. C: Immunostaining of AQP5. D: Relative intensity of AQP5. E: Immunostaining of AQP8. F: Relative fluorescence intensity of AQP8. In HeLa and SAS ρ0 cells, AQPs were strongly expressed in the plasma membrane, and average expression intensities were significantly higher than in parental cells. **: p < 0.01 using Student’s t-test (vs. parent).
Fig. 4.

AQP3, 5, and 8 directly bind to NOX2, which produces H2O2 in the cell.
Western blot analysis of AQPs was performed to investigate protein expression, and immunoprecipitation was performed to confirm if AQP and NOX2 directly interact. A: Western blot and immunoprecipitation of AQPs and NOX2. AQP3, 5, and 8 directly bound with NOX2. To investigate the spatial distribution of NOX2, immunostaining was also performed. B: Immunostaining of NOX2 in HeLa and SAS ρ0 cells. C: Relative fluorescence intensity of NOX2 in HeLa and SAS ρ0 cells. NOX2 expression was significantly higher than in parental cells. **: p < 0.01 using Student’s t-test (vs. parent).
3.4. Interaction between AQPs and NOX2
To investigate whether AQPs directly bind to NOX2, immunoprecipitation experiments were performed. We observed that AQP3, 5, and 8 bind to NOX2 (Fig. 4A). Next, we investigated the spatial distribution of NOX2 by fluorescence microscopy. NOX2 was detected in nuclei and in the plasma membrane (Fig. 4B). Stronger intensity of NOX2 was detected in both HeLa and SAS ρ0 cells compared with parental cells (Fig. 4C).
3.5. AQP knockdown abolishes H2O2-induced ferroptosis
To investigate whether AQP3, 5, and 8 are involved in H2O2 sensitivity, we knocked down these genes with siRNA. After AQP3, 5, and 8 knockdown with specific siRNA, the cells were treated with H2O2 for 1 h. Cell viability was measured using CCK-8 assays. The results revealed that cell viability was improved by knocking down AQP3, 5, and 8 compared with negative control siRNA transfection. Internal H2O2 amount was also measured by HYDROP after H2O2 treatment. Our results show that the internal H2O2 amount was significantly decreased after siAQP treatment (Fig. 5 C and D).
Fig. 5.

AQP knockdown rescues H2O2 sensitivity by reducing internal H2O2.
To investigate the involvement of AQPs in H2O2 sensitivity, AQPs were knocked down by siRNA. A: Changes in H2O2 sensitivity after siAQP treatment in HeLa ρ0 cells. The cell viability results for Negative Control (N.C.) vs. siAQP are summarized in Table 2. B: Changes in H2O2 sensitivity after siAQP treatment in SAS ρ0 cells. C: Internal H2O2 amount visualized by HYDROP after 50 µM H2O2 treatment for 1 h. D: Relative intensity of HYDROP in HeLa and SAS ρ0 cells. Significantly lower internal H2O2 levels were observed by knockdown of AQPs. **: p < 0.01 using Student’s t-test (vs. N.C.).
3.6. Transfer of normal mitochondria reduces H2O2 sensitivity in ρ0 cells
To clarify the relationship between mitochondrial function and AQP expression, isolated normal mitochondria were transferred into ρ0 cells (Mito cells). After confirming that normal mitochondria were transferred into ρ0 cells, AQP expression, H2O2 sensitivity, and Fe2+ levels were investigated. In the Mito cells, AQP3, 5, and 8 expression (Fig. 6 A- C), H2O2 sensitivity (Fig. 6. D, E), and Fe2+ levels (Fig. 6 F-I) were all significantly decreased. Overall, these findings suggest the importance of mitochondria for H2O2-induced ferroptosis.
Fig. 6.

Mitochondrial transfer rescues H2O2 sensitivity by decreasing the expression of AQPs and reducing Fe2+ levels.
To clarify the relationship between mitochondrial function and AQP expression, mitochondrial transfer experiments were performed. A-C: AQP expression after mitochondrial transfer. A: AQP3. B: AQP5. C: AQP8. The expression of AQPs was significantly lower after mitochondrial transfer. D and E: Cell viability after H2O2 treatment. D: HeLa ρ0 cells vs. HeLa Mito cells. E: SAS ρ0 cells vs. SAS Mito cells. Significant H2O2 resistance was observed after mitochondrial transfer. F: Detection of intracellular Fe2+ by FerroOrange. G: Detection of mitochondrial Fe2+ by Mito-FerroGreen. H: Relative intensity of FerroOrange. I: Relative intensity of Mito-FerroGreen. The FerroOrange and Mito-FerroGreen signals in Mito cells were significantly lower after mitochondria transfer. *: p < 0.05, **: p < 0.01 using Student’s t-test (vs. ρ0 cells).
3.7. Mitochondrial PHB2 regulates AQP expression
Since PHB2 plays an important role in mitochondrial functions such as membrane potential and mitochondrial morphology, PHB2 expression was examined at the mRNA and protein levels in ρ0 cells. PHB2 gene expression was significantly downregulated in ρ0 cells and was rescued in Mito cells (Fig. 7 A). Furthermore, significantly weaker PHB2 expression was observed in ρ0 cells compared to parental and Mito cells using immunofluorescence microscopy (Fig. 7B and C). Western blot analysis confirmed that PHB2 expression was decreased in ρ0 cells in comparison with parental and Mito cells (Fig. 7D).
Fig. 7.

Prohibitin 2 (PHB2) expression is upregulated by mitochondrial transfer.
PHB2 expression was examined to investigate whether mitochondrial function was rescued after mitochondrial transfer. A: PHB2 gene expression after mitochondrial transfer. B: Immunostaining of PHB2. C: Relative intensity of PHB2. D: Western blot of PHB2. PHB2 expression was lower in ρ0 cells than in parental cells. In contrast, PHB2 expression increased after mitochondrial transfer. *: p < 0.05, **: p < 0.01 using Scheffe’s F test.
Finally, to investigate whether PHB2 regulates AQP expression, PHB2 knockdown was performed. PHB2 knockdown upregulated AQP3, 5, and 8 gene expression (Fig. 8 ), indicating that PHB2 negatively regulates AQP expression.
Fig. 8.

Knockdown of PHB2 upregulates AQP expression in parental cells.
To investigate whether PHB2 regulates AQP expression, PHB knockdown experiments were performed. A: Relative PHB2 expression. B: Relative AQP3 expression. C: Relative AQP5 expression. D: Relative AQP8 expression. PHB2 knockdown led to upregulated AQP expression. **: p < 0.01 using Student’s t-test (vs. N.C.).
4. Discussion
It has previously been reported that cell death induced by H2O2 treatment occurs via apoptosis or necroptosis [32]. However, in our present study, ferroptosis occurred in ρ0 cells at a relatively early stage after H2O2 treatment. Notably, H2O2-induced ferroptosis was recently reported in rat glioma cells [33]. The induction of apoptosis by H2O2 treatment was confirmed by costaining with Annexin V and PI (early apoptosis is stained by only Annexin V and late apoptosis is stained with Annexin V and PI). The induction of ferroptosis was confirmed with Liperfluo and PI. As a result, more Liperfluo-positive cells were observed than Annexin V-positive cells 3 h after H2O2 treatment, confirming the induction of ferroptosis after H2O2 (Fig. 1, Fig. S3). Interestingly, treating ρ0 cells with H2O2 for 2 h downregulated the key apoptotic genes Caspase 8 and 9 (Fig. S4). Furthermore, the GPx4 gene, which acts as a suppressor of lipid peroxidation and ferroptosis [21,34], was not upregulated in ρ0 cells 2 h after H2O2 treatment. However, in parental cells, GPx4 expression was upregulated 2 h after H2O2 treatment (Fig. S4). These results highlight the involvement of mitochondria in the ferroptosis process. Furthermore, nuclear factor erythroid 2-related factor 2 (Nrf2) contributes in regulation of GPx4 gene expression [35], however, its gene expression was suppressed in ρ0 cells (Fig. S5). The nuclear factor erythroid 2–related factor 2 (Nrf2)-Kelch-like ECH-associated protein 1 (keap1) pathway enables the upregulation of antioxidant enzymes such as GPx4, but does not work in ρ0 cells. It seems that the promotion of ferroptosis occurs differently than apoptosis during the early stage of H2O2 treatment, at least in ρ0 cells. However, more studies are necessary to develop our understanding about the mechanism of ferroptosis induction after H2O2 treatment.
Ferroptosis is cell death from iron-dependent lipid peroxidation. ρ0 cells are sensitive to H2O2-mediated cell death because ρ0 cells are susceptible lipid peroxidation compared to parental cells [14]. However, the importance of the intracellular Fe2+ content has not yet been addressed. Our findings reveal that both intracellular and mitochondrial Fe2+ were significantly increased in ρ0 cells. Interestingly, when endogenous Fe2+ was suppressed by iron chelators, H2O2 sensitivity was ameliorated (Fig. 2E and F). The effect of DFO was limited, likely because it is water-soluble and does not penetrate the plasma membrane. Collectively, our results indicate that H2O2 sensitivity in ρ0 cells is due to increased ferroptosis.
It has previously been reported that ferroptosis occurs by lipid peroxidation of the plasma membrane. The lipid peroxidation of the plasma membrane occurs by •OH that results from the “Fenton reaction,” where H2O2 reacts with Fe2+. The amount of •OH and lipid peroxidation is initially higher in ρ0 cells than in parental cells [14]. H2O2 enters ρ0 cells more readily when treated with H2O2 compared to parental cells [13]. It has also been reported that AQP3, 5, and 8 expressed on the plasma membrane also regulate the permeability of the extracellular H2O2 via H2O2 channel activity [[16], [17], [18]]. Therefore, we examined the spatial and quantitative expression of AQP3, 5, and 8 in the present study. Indeed, AQP3, 5, and 8 expression was enhanced in ρ0 cells according to both immunostaining and Western blot analysis (Fig. 3, Fig. 4A). AQP8 and NOX2 directly interact, and H2O2 produced by NOX2 enters cells via AQP8 [36]. Therefore, an immunoprecipitation experiment was performed to investigate whether AQPs bind to NOX2 directly. Our results indicate that NOX2 expression is upregulated in ρ0 cells, and that NOX2 binds to AQP3, 5, and 8 in both HeLa and SAS cells (Fig. 4). Furthermore, knockdown of AQP3, 5, and 8 increased cell viability after H2O2 treatment and decreased the amount of endogenous H2O2 (Fig. 5, Fig. S6). When H2O2 is administered to ρ0 cells, lipid peroxidation in the plasma membrane is enhanced, leading to increased ferroptosis because intracellular H2O2, AQP and NOX expression, and Fe2+levels are higher in ρ0 cells than in parental cells. Together, these factors would produce more •OH. These results indicate that drugs that enhance AQP expression may be effective in cancer treatment. Candidates that enhance AQP expression are vasopressin, epidermal growth factor (EGF), the Chinese herb “Keigai”, and nuclear receptor estrogen receptor α (ERα). Vasopressin, an antidiuretic hormone, enhances AQP2 expression in the kidney [37], EGF increases AQP3 expression in MPC-83 pancreatic cancer [38], and the Chinese herb “Keigai” enhances AQP3 expression [39]. Furthermore, ERα up-regulates AQP7 expression [40]. However, further investigations will be needed to address some questions, including whether vasopressin or ERα activate AQP3, 5, and 8 and promote H2O2 permeability in the plasma membrane. The combination of these candidate molecules with anti-cancer agents or radiation might lead to more effective cancer treatment.
To verify whether enhanced AQP expression and H2O2 sensitivity in ρ0 cells are due to mitochondrial dysfunction, mitochondria transfer experiments were performed. As a result, mitochondrial transfer reduced the expression of AQP3, 5, and 8, and rescued cellular sensitivity to H2O2. In addition, mitochondrial transfer decreased intracellular and mitochondrial Fe2+ levels (Fig. 6). We speculate that mitochondrial dysfunction causes enhanced mitochondrial membrane permeability by AQPs, produces more ROS by the Fenton reaction, and induces leak of Fe2+ from mitochondrial interior, leading to cell death via ferroptosis. Therefore, it may be possible to extract mitochondria after establishing ρ0 cells from the patient's own tissue and introduce them into cancer cells that have normal mitochondria, which could offer a new treatment to increase cellular sensitivity to ROS and drugs. We believe that mitochondria transfer might be an effective therapeutic strategy in the near future. However, mitochondria transfer is only in the initial development stage, so further investigation is needed to clarify technical and ethical issues.
PHB2 is an important protein for maintaining mitochondrial function. Indeed, PHB2 is expressed in mitochondria, and is also present in the cytoplasm, nucleus, and plasma membrane, and controls various functions [41,42]. For example, PHB2 maintains mitochondrial morphology and controls mitophagy [43]. Further, PHB2 regulates the cell cycle and cytoplasmic signaling pathways [44,45]. PHB2 is also involved in transcriptional regulation with ERα in the nucleus [46]. On the plasma membrane, PHB2 controls insulin signaling by binding to the insulin receptor, and protects against viral infections such as coronavirus [47]. Our results indicate that the expression of PHB2 in the parental, ρ0, and Mito cells is different and is downregulated in ρ0 cells. Furthermore, knocking down PHB2 with siRNA in the parental cells enhances AQP expression (Fig. 7, Fig. 8). Since the PHB2 gene was not rescued by AQP knockdown (Fig. S7), it is likely that PHB2 downregulates AQP gene expression. PHB2 translocates to the nucleus with ERα in HeLa and MCF-7 cells and represses ERα-dependent transcription [46,48]. Moreover, ERα up-regulates AQP expression, as mentioned in the Results section [41]. From these results, we propose that mitochondrial PHB2 plays an important role in the regulation of ROS sensitivity by downregulating AQP expression, probably through nuclear receptors such as ERα.
PHB2 functions as a putative membrane scaffold in mitochondria and stabilizes phospholipids such as cardiolipin in the inner mitochondrial membrane [49]. Knockdown of PHB2 produces more intracellular ROS, reduces adipogenesis, and reduces lipid accumulation in 3T3-L1 cells [50]. Furthermore, the depletion of PHB2 promotes fatty acid oxidation and decreases fatty acid uptake in cardiomyocytes [51]. We previously reported that ROS generation and lipid peroxidation in ρ0 cells is higher than in parental cells. The expression of lipoxygenase, an enzyme that oxidizes fatty acids, is also higher than in parental cells [14]. In this study, we showed low PHB2 expression and high Fe2+ content in ρ0 cells, and showed that mitochondrial transfer rescues this condition. Oxidative stress such as selenite treatment leads to iron-sulfur cluster degradation and increases Fe2+ levels in mitochondria followed by lipid peroxidation [52]. These damaged mitochondria are degraded and the mitochondrial contents, including Fe2+, are released into the cytoplasm for degradation in lysosomes [53]. It has been reported that mitochondria morphology is different between parental and ρ0 cells, but the total mitochondrial volume is similar [54,55]. We confirmed that the volume of mitochondria was not significantly different among parent, ρ0, and Mito cells (Fig. S8). When the morphology of mitochondria in ρ0 cells was observed by confocal microscopy and transmission electron microscopy, the network structure appeared disrupted, the mitochondrial appeared swollen, the matrix appeared to be electron-empty, and structure of cristae was destroyed [54]. Taken together, these results indicate that the downregulation of PHB2 by mitochondrial dysfunction leads to decreased fatty acid turnover and increased Fe2+ contents, failing to rescue the lipid peroxidation that leads to cell death. Therefore, downregulating PHB2 expression could create a ROS-sensitive condition, which may enable more effective cancer treatment.
In this study, we showed that H2O2 mediates ferroptosis in ρ0 cells. Mitochondrial dysfunction, such as mtDNA depletion and conditions such as decreased PHB2, leads to more ferroptosis because mitochondrial dysfunction, like PHB2 reduction, increases intracellular H2O2, AQP, NOX, and Fe2+ levels, and could result in increased •OH production, resulting in lipid peroxidation (summarized in Fig. 9 ). Some anti-cancer agents kill cancer cells through the production of ROS. Furthermore, H2O2 is used as a sensitizer in cancer treatment. Therefore, amplifying AQP expression before sensitizer treatment will likely enhance the therapeutic effect. Further progress in this field will likely facilitate improved cancer treatment.
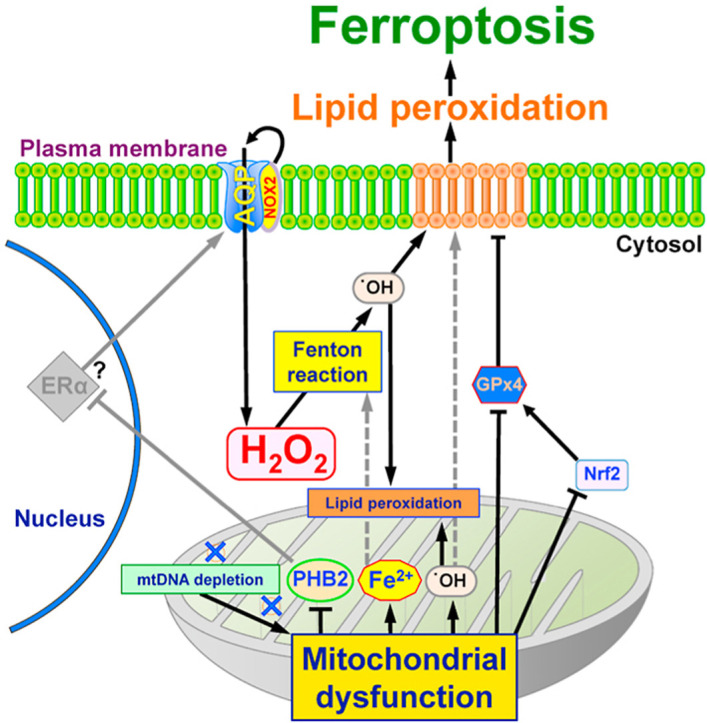
Fig. 9.

Schematic diagram of mitochondria-mediated ferroptosis by H2O2.
H2O2 permeability is regulated by cell surface AQPs. Intracellular H2O2 becomes •OH by the Fenton reaction. The peroxidized phospholipids induced by •OH in the plasma and mitochondrial membrane suggest the high probability of ferroptosis. We propose that internal H2O2 levels also increase via NOX2, which is bound to AQPs and produces H2O2 at the plasma membrane. In mitochondria, oxidative phosphorylation produces O2• -, which is converted to •OH. ρ0 cells could produce more •OH than parental cells because of lacking mtDNA and mitochondrial dysfunction, such as enhancement of mitochondrial membrane permeability and PHB2 reduction. PHB2 may negatively regulates AQP expression via ERa, and inhibits enhanced H2O2 permeability through the plasma and mitochondrial membranes. In other words, mitochondrial dysfunction, which is present in ρ0 cells, enhance mitochondrial leak of Fe2+, which further promotes mitochondrial and cytoplasmic Fenton reactions, leading to ferroptosis via enhanced •OH production and lipid peroxidation. Reduction of GPx4 via Nrf2 would be caused by mitochondrial dysfunction and accelerate plasma membrane lipid peroxidation. See the detail for discussion section.
Declaration of competing interest
The authors declare no conflicts of interest.
Acknowledgments
This work was supported by a Grant from the Kodama Memorial Fund for Medical Research to K.T. and JSPS KAKENHI (Grant-in Aid for Scientific Research C: No. 18K09772 to Y.T.; 19K10318 to K.T.).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.freeradbiomed.2020.09.027.
Table 2.
| HeLa ρ0 | 12.5 μM | 25 μM | 50 μM | 100 μM | 200 μM |
|---|---|---|---|---|---|
| siAQP3 | * | * | ** | * | |
| siAQP5 | * | ** | |||
| siAQP8 | * | ** | ** | ** | ** |
|
| |||||
| SAS ρ0 |
12.5 μM |
25 μM |
50 μM |
100 μM |
200 μM |
| siAQP3 | ** | ** | ** | ** | |
| siAQP5 | ** | ** | ** | ** | |
| siAQP8 | ** | * | ** | ** | |
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Tada-Oikawa S., Oikawa S., Kawanishi M., Yamada M., Kawanishi S. Generation of hydrogen peroxide precedes loss of mitochondrial membrane potential during DNA alkylation-induced apoptosis. FEBS Lett. 1999;442:65–69. doi: 10.1016/s0014-5793(98)01618-4. [DOI] [PubMed] [Google Scholar]
- 2.Varbiro G., Veres B., Gallyas F., Jr., Sumegi B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic. Biol. Med. 2001;31:548–558. doi: 10.1016/s0891-5849(01)00616-5. [DOI] [PubMed] [Google Scholar]
- 3.Murata M., Suzuki T., Midorikawa K., Oikawa S., Kawanishi S. Oxidative DNA damage induced by a hydroperoxide derivative of cyclophosphamide. Free Radic. Biol. Med. 2004;37:793–802. doi: 10.1016/j.freeradbiomed.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Magda D., Miller R.A. Motexafin gadolinium: a novel redox active drug for cancer therapy. Semin. Cancer Biol. 2006;16:466–476. doi: 10.1016/j.semcancer.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Alexandre J., Nicco C., Chéreau C., Laurent A., Weill B., Goldwasser F., Batteux F. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J. Natl. Cancer Inst. 2006;98:236–244. doi: 10.1093/jnci/djj049. [DOI] [PubMed] [Google Scholar]
- 6.Fang Y., Moore B.J., Bai Q., Cook K.M., Herrick E.J., Nicholl M.B. Hydrogen peroxide enhances radiation-induced apoptosis and inhibition of melanoma cell proliferation. Anticancer Res. 2013;33:1799–1807. [PubMed] [Google Scholar]
- 7.Kariya S., Sawada K., Kobayashi T., Karashima T., Shuin T., Nishioka A., Ogawa Y. Combination treatment of hydrogen peroxide and X-rays induces apoptosis in human prostate cancer PC-3 cells. Int. J. Radiat. Oncol. Biol. Phys. 2009;75:449–454. doi: 10.1016/j.ijrobp.2009.04.092. [DOI] [PubMed] [Google Scholar]
- 8.Halliwell B., Gutterridge J.M.C. Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Arch. Biochem. Biophys. 1986;246:501–514. doi: 10.1016/0003-9861(86)90305-x. [DOI] [PubMed] [Google Scholar]
- 9.Indo H.P., Davidson M., Yen H.-C., Suenaga S., Tomita K., Nishii T., Higuchi M., Koga Y., Ozawa T., Majima H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007;7:106–118. doi: 10.1016/j.mito.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 10.Ogura A., Oowada S., Kon Y., Hirayama A., Yasui H., Meike S., Kobayashi S., Kuwabara M., Inanami O. Redox regulation in radiation-induced cytochrome c release from mitochondria of human lung carcinoma A549 cells. Cancer Lett. 2009;277:64e71. doi: 10.1016/j.canlet.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 11.Chatterjee A., Mambo E., Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 12.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2003;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 13.Tomita K., Kuwahara Y., Takashi Y., Tsukahara T., Kurimasa A., Fukumoto M., Nishitani Y., Sato T. Sensitivity of mitochondrial DNA depleted ρ0 cells to H2O2 depends on the plasma membrane status. Biochem. Biophys. Res. Commun. 2017;490:330–335. doi: 10.1016/j.bbrc.2017.06.044. [DOI] [PubMed] [Google Scholar]
- 14.Tomita K., Takashi Y., Ouchi Y., Kuwahara Y., Igarashi K., Nagasawa T., Nabika H., Kurimasa A., Fukumoto M., Nishitani Y., Sato T. Lipid peroxidation increases hydrogen peroxide permeability leading to cell death in cancer cell lines that lack mtDNA. Canc. Sci. 2019;110:2856. doi: 10.1111/cas.14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takashi Y., Tomita K., Kuwahara Y., Nabika H., Igarashi K., Nagasawa T., Kurimasa A., Fukumoto M., Nishitani Y., Sato T. Data on the aquaporin gene expression differences among ρ0, clinically relevant radioresistant, and the parental cells of human cervical cancer and human tongue squamous cell carcinoma. Data Brief. 2018;20:402–410. doi: 10.1016/j.dib.2018.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller E.W., Dickinson B.C., Chang C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. U.S.A. 2010;107:15681–15686. doi: 10.1073/pnas.1005776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodrigues C., Pimpão C., Mósca A.F., Coxixo A.S., Lopes D., da Silva I.V., Pedersen P.A., Antunes F., Soveral G. Human aquaporin-5 facilitates hydrogen peroxide permeation affecting adaption to oxidative stress and cancer cell migration. Cancers. 2019;11:932. doi: 10.3390/cancers11070932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bienert G.P., Moller A.L., Kristiansen K.A., Schulz A., Moller I.M., Schjoerring J.K., Jahn T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 19.Beri R., Chandra R. Chemistry and biology of heme. Effect of metal salts, organometals, and metalloporphyrins on heme synthesis and catabolism, with special reference to clinical implications and interactions with cytochrome P-450. Drug Metab. Rev. 1993;25:49–152. doi: 10.3109/03602539308993973. [DOI] [PubMed] [Google Scholar]
- 20.Johnson D.C., Dean D.R., Smith A.D., Johnson M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- 21.Stockwell B.R., F Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J., Fulda S., Gascón S., Hatzios S.K., Kagan V.E., Noel K., Jiang X., Linkermann A., Murphy M.E., Overholtzer M., Oyagi A., Pagnussat G.C., Park J., Ran Q., Rosenfeld C.S., Salnikow K., Tang D., Torti F.M., Torti S.V., Toyokuni S., Woerpel K.A., Zhang D.D. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang L., Kon N., Li T., Wang S.J., Su T., Hibshoosh H., Baer R., Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., 3rd Morrison B., Stockwell B.R. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bebber C.M., Müller F., Clemente L.P., Weber J., von Karstedt S. Ferroptosis in cancer cell biology. Cancers. 2020;12:164. doi: 10.3390/cancers12010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wenzel S.E., Tyurina Y.Y., Zhao J., St Croix C.M., Dar H.H., Mao G., Tyurin V.A., Anthonymuthu T.S., Kapralov A.A., Amoscato A.A., Mikulska-Ruminska K., Shrivastava I.H., Kenny E.M., Yang Q., Rosenbaum J.C., Sparvero L.J., Emlet D.R., Wen X., Minami Y., Qu F., Watkins S.C., Holman T.R., VanDemark A.P., Kellum J.A., Bahar I., Bayır H., Kagan V.E. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171:628–641. doi: 10.1016/j.cell.2017.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng H., Schorpp K., Jin J., Yozwiak C.E., Hoffstrom B.G., Decker A.M., Rajbhandari P., Stokes M.E., Bender H.G., Csuka J.M., Upadhyayula P.S., Canoll P., Uchida K., Soni R.K., Hadian K., Stockwell B.R. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30:3411–3423. doi: 10.1016/j.celrep.2020.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du J., Zhou Y., Li Y., Xia J., Chen Y., Chen S., Wang X., Sun W., Wang T., Ren X., Wang X., An Y., Lu K., Hu W., Huang S., Li J., Tong X., Wang Y. Identification of frataxin as a regulator of ferroptosis. Redox Biol. 2020;32:101483. doi: 10.1016/j.redox.2020.101483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaschler M.M., Hu F., Feng H., Linkermann A., Min W., Stockwell B.R. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem. Biol. 2018;13:1013–1020. doi: 10.1021/acschembio.8b00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H., Liu C., Zhao Y., Gao G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020;99:151058. doi: 10.1016/j.ejcb.2019.151058. [DOI] [PubMed] [Google Scholar]
- 30.Roushandeh A.M., Tomita K., Kuwahara Y., Jahanian-Najafabadi A., Igarashi K., Roudkenar M.H., Sato T. Transfer of healthy fibroblast-derived mitochondria to HeLa ρ0 and SAS ρ0 cells recovers the proliferation capabilities of these cancer cells under conventional culture medium, but increases their sensitivity to cisplatin-induced apoptotic death. Mol. Biol. Rep. 2020;47(6):4401–4411. doi: 10.1007/s11033-020-05493-5. [DOI] [PubMed] [Google Scholar]
- 31.Stockwell B.R., Jiang X. The chemistry and biology of ferroptosis. Cell Chem. Biol. 2020;27:365–375. doi: 10.1016/j.chembiol.2020.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiang J., Wan C., Guo R., Guo D. Is hydrogen peroxide a suitable apoptosis inducer for all cell types? BioMed Res. Int. 2016:7343965. doi: 10.1155/2016/7343965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang T., Chu J., Chen H., Cheng H., Su J., Wang X., Cao Y., Tian S., Li Q. Gastrodin inhibits H2O2-induced ferroptosis through its antioxidative effect in rat glioma cell line C6. Biol. Pharm. Bull. 2020;43:480–487. doi: 10.1248/bpb.b19-00824. [DOI] [PubMed] [Google Scholar]
- 34.Imai H., Matsuoka M., Kumagai T., Sakamoto T., Koumura T. In: Nagata S., Nakano H., editors. vol. 403. Springer; Cham: 2017. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis; pp. 143–170. (Apoptotic and Non-apoptotic Cell Death. Curr. Top. Microbiol. Immunol). [DOI] [PubMed] [Google Scholar]
- 35.Dodson M., Castro-Portuguez R., Zhang D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi: 10.1016/j.redox.2019.101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bertolotti M., Farinelli G., Galli M., Aiuti A., Sitia R. AQP8 transports NOX2-generated H2O2 across the plasma membrane to promote signaling in B cells. J. Leukoc. Biol. 2016;100:1071–1079. doi: 10.1189/jlb.2AB0116-045R. [DOI] [PubMed] [Google Scholar]
- 37.Eto K., Noda Y., Horikawa S., Uchida S., Sasaki S. Phosphorylation of aquaporin-2 regulates its water permeability. J. Biol. Chem. 2010;285:40777–40784. doi: 10.1074/jbc.M110.151928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weijun L., Wang K., Gong K., Li X., Luo K. Epidermal growth factor enhances MPC-83 pancreatic cancer cell migration through the upregulation of aquaporin 3. Mol. Med. Rep. 2012;6:607–610. doi: 10.3892/mmr.2012.966. [DOI] [PubMed] [Google Scholar]
- 39.Isohama Y. Increase in aquaporin 3 expression in keratinocytes by Schizonepeta tenuifolia (in Japanese) Folia Pharmacol. Jpn. 2014;143:115–119. doi: 10.1254/fpj.143.115. [DOI] [PubMed] [Google Scholar]
- 40.Fu X., Zhu J., Zhang L., Shu J. Long non-coding RNA NEAT1 promotes steatosis via enhancement of estrogen receptor alpha-mediated AQP7 expression in HepG2 cells, Artif. Cells Nanomed. Biotechnol. 2019;47:1782–1787. doi: 10.1080/21691401.2019.1604536. [DOI] [PubMed] [Google Scholar]
- 41.Thuaud F., Ribeiro N., Nebigil C.G., Désaubry L. Prohibitin ligands in cell death and survival: mode of action and therapeutic potential. Chem. Biol. 2013;20:316–331. doi: 10.1016/j.chembiol.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bavelloni A., Piazzi M., Raffini M., Faenza I., Blalock W.L. Prohibitin 2: at a communications crossroads. IUBMB Life. 2015;67:239–254. doi: 10.1002/iub.1366. [DOI] [PubMed] [Google Scholar]
- 43.Wei Y., Chiang W.-C., Sumpter R., Jr., Mishra P., Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–238. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nuell M.J., Stewart D.A., Walker L., Friedman V., Wood C.M., Owens G.A., Smith J.R., Schneider E.L., Dell' Orco R., Lumpkin C.K., Danner D.B., McClung J.K. Prohibitin, an evolutionarily conserved intracellular protein that blocks DNA synthesis in normal fibroblasts and HeLa cells. Mol. Cell Biol. 1991;11:1372–1381. doi: 10.1128/mcb.11.3.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J.W., Akiyama M., Park J.-H., Lin M.-L., Shimo A., Ueki T., Daigo Y., Tsunoda T., Nishidate T., Nakamura Y., Katagiri T. Activation of estrogen/estrogen receptor signaling by BIG3 through its inhibitory effect on nuclear transport of PHB2/REA in breast cancer. Canc. Sci. 2009;100:1468–1478. doi: 10.1111/j.1349-7006.2009.01209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kasashima K., Ohta E., Kagawa Y., Endo H. Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 2006;281:36401–36410. doi: 10.1074/jbc.M605260200. [DOI] [PubMed] [Google Scholar]
- 47.Cornillez-Ty C.T., Liao L., Yates J.R., 3rd, Kuhn P., Buchmeier M.J. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J. Virol. 2009;83:10314–10318. doi: 10.1128/JVI.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim N.-H., Yoshimaru T., Chen Y.-A., Matsuo T., Komatsu M., Miyoshi Y., Tanaka E., Sasa M., Mizuguchi K., Katagir T. BIG3 inhibits the estrogen-dependent nuclear translocation of PHB2 via multiple karyopherin-alpha proteins in breast cancer cells. PLoS One. 2015;10 doi: 10.1371/journal.pone.0127707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Osman C., Haag M., Potting C., Rodenfels J., Dip P.V., Wieland F.T., Brügger B., Westermann B., Langercorresponding T. The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J. Cell Biol. 2009;184:583–596. doi: 10.1083/jcb.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu D., Lin Y., Kang T., Huang B., Xu W., Garcia-Barrio M., Olatinwo M., Matthews R., Chen Y.E., Thompson W.E. Mitochondrial dysfunction and adipogenic reduction by prohibitin silencing in 3T3-L1 cells. PLoS One. 2012;7 doi: 10.1371/journal.pone.0034315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu D., Jian C., Peng Q., Hou T., Wu K., Shang B., Zhao M., Wang Y., Zheng W., Ma Q., Li C.-Y., Cheng H., Wang X., Zhao L. Prohibitin 2 deficiency impairs cardiac fatty acid oxidation and causes heart failure. Cell Death Dis. 2020;11:181. doi: 10.1038/s41419-020-2374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scalcon V., Tonolo F., Folda A., Bindoli A., Rigobello M.P. Dimers of glutaredoxin 2 as mitochondrial redox sensors in selenite-induced oxidative stress. Metallomics. 2019;11:1241–1251. doi: 10.1039/c9mt00090a. [DOI] [PubMed] [Google Scholar]
- 53.Kim I., Rodriguez-Enriquez S., Lemasters J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuka A., Kukat C., Brocher J., Schäfer I., Krohne G., Trounce I.A., Villani G., Seibel P. Generation of rho0 cells utilizing a mitochondrially targeted restriction endonuclease and comparative analyses. Nucleic Acids Res. 2008;36:e44. doi: 10.1093/nar/gkn124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gilkerson R.W., Margineantu D.H., Capaldi R.A., Selker J.M. Mitochondrial DNA depletion causes morphological changes in the mitochondrial reticulum of cultured human cells. FEBS Lett. 2000;474:1–4. doi: 10.1016/s0014-5793(00)01527-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.