

In cancer, active expression of the TERT gene paradoxically correlates with a hypermethylated CpG island. Here, we show that in 833 cancer cell lines representing 23 tissue types, the proximal promoter contains conserved hypomethylation. In lines with monoallelic TERT expression, decreased proximal promoter methylation associates with the active allele. Thus, the proximal TERT promoter has canonical DNA methylation.
Keywords: epigenetics, monoallelic gene expression, promoter CpG methylation, telomerase reverse transcriptase, TERT
Abstract
Telomerase reverse transcriptase (TERT) is pathologically expressed in the vast majority of human cancers, but the epigenetic regulation of its expression is only beginning to be understood. In particular, the active TERT gene in cancer cells has been characterized as having a hypermethylated CpG island, opposite to the general association of DNA methylation with gene repression. Here, we analyzed TERT promoter CpG methylation in 833 human cancer cell lines representing 23 different tissue types and found hypermethylation of the upstream portion of the CpG island and more conserved hypomethylation of a region including the proximal TERT promoter and exon 1. In cell lines with monoallelic expression of TERT, we found allelic methylation of the proximal TERT promoter. This included cell lines with the −124 or −146 activating promoter mutation as well as wild‐type TERT cancer lines. In these cell line types, decreased proximal promoter methylation is associated with the active allele. Compared to cells with monoallelic expression of TERT, lines with biallelic expression of TERT had even lower methylation in the proximal TERT promoter. Thus, in cell lines from cancers of many different tissues, the TERT proximal promoter has canonical DNA methylation, with low methylation correlating with increased TERT expression.
Abbreviations
- BAE
biallelic expression
- CCLE
Cancer Cell Line Encyclopedia
- CGI
CpG island
- ChIP‐Bis‐Seq
ChIP–bisulfite conversion sequencing
- CpG
cytosine–guanine sequence
- ETS
E‐twenty‐six
- FISH
fluorescence innonbreakingspacesitu hybridization
- GABP
GA‐binding protein
- gDNA
genomic DNA
- H3ac
histone H3 acetylation
- MAE
monoallelic expression
- SNP
single‐nucleotide polymorphism
- TERT
telomerase reverse transcriptase
- TF
transcription factor
- WT
wild‐type
1. Introduction
Serving as protective caps at the ends of eukaryotic chromosomes, telomeres maintain chromosomal and genome stability. Telomeric DNA can be lengthened by telomerase, a ribonucleoprotein enzyme [1, 2]. While telomerase is expressed during normal human development, it becomes inactive in most somatic cells. Telomeres then progressively shorten due to the ‘end replication problem’ until they reach a critical length, and the aged cells undergo senescence. However, in most malignant human cancers (≈ 80–90%), telomerase is pathologically active, allowing cellular immortalization [3, 4, 5]. The expression of the catalytic subunit of telomerase, TERT [6], is limiting for telomerase in most cells, because the RNA subunit (hTR) is constitutively present. Indeed, some human cells can be immortalized simply by ectopic expression of TERT [7, 8].
In ≈ 22% of cancer cell lines, TERT reactivation is clearly genetic, occurring through activating promoter mutations [9]. Two common TERT‐activating mutations are C>T transitions located −124 and −146 bp upstream of the TERT translation start codon (AUG) (chr5:1295228 and chr5:1295250, or C228T and C250T, respectively; hg19 genomic coordinates). These mutations create a new binding site for E‐twenty‐six (ETS) transcription factors and recruit the GA‐binding protein (GABP) [2, 10]. These mutations are heterozygous, and only the promoter‐mutant TERT allele is active, resulting in monoallelic expression (MAE) of TERT. An alternative explanation for MAE of TERT is that the promoter mutation abrogates silencing of that allele [11]. Although this pathway is distinct from the reactivation model, the end result is the same: The promoter‐mutant TERT allele is transcriptionally active, while the other alleles of TERT are epigenetically silenced.
TERT reactivation can also be entirely epigenetic, and this appears to be the case for the majority of cancers. Approximately 71% of these TERT WT cell lines show biallelic expression (BAE) of TERT [9]. Intriguingly, cancer cell lines with wild‐type (WT) TERT sequences, containing no known activating cis‐acting genetic alterations, sometimes show MAE [9, 12, 13]. These MAE vs. BAE line classifications are based on expression of exonic TERT single‐nucleotide polymorphisms (SNPs). However, using single‐cell fluorescence in situ hybridization (FISH) imaging, we recently found that there is actually considerable line‐to‐line (and cell‐to‐cell) heterogeneity in the number of TERT transcription sites and gene copies, and in the ratio of the two [14]. In other words, while MAE cell lines express only one version of the TERT gene and BAE lines express multiple versions, the ratio of active to inactive TERT gene copies is not simply 1 : 1 and 2 : 0 in MAE and BAE lines, respectively. Regardless of ‘MAE’ or ‘BAE’ classification, all lines have inactive copies as well as one or multiple active WT TERT gene copies. It is important to understand the epigenetic mechanisms reactivating these copies (or failing to silence them). Epigenetic activation involves histone modifications [15] and DNA methylation, described next in this section.
In mammalian genomes, DNA CpG methylation impacts gene transcription, though not always in a straightforward manner. CpG sites contain a methylated cytosine ≈ 80% of the time [16]. This methylation affects chromatin structure and binding of transcription‐associated factors. Genome‐wide demethylation is an early cancer hallmark [17]. This primarily manifests as demethylation of intergenic and highly repeated sequences, possibly causing activation of noncoding RNAs [16, 17, 18]. Hypomethylation of promoters is also apparent, which correlates with overexpression of oncogenes and other genes associated with tumor invasion or metastasis. Intriguingly, cancer‐associated hypermethylation of promoters in CpG islands (CGIs; GC‐rich regions typically spanning ≈ 1 kb) represses transcription of tumor suppressors [16, 17, 18, 19, 20]. In healthy adult somatic cells, promoters in CGIs are typically unmethylated and associated with active gene expression [16].
Because promoter hypermethylation canonically associates with repressed transcription, the TERT promoter has appeared noncanonical, though this may be due to a focus on the upstream promoter region. The relatively large TERT promoter CGI spans ≈ 4 kb, approximately −1800 to +2200 bp relative to the TERT AUG (chr5:1295228, hg19). It is extremely GC‐rich, possessing up to 70% GC content [16, 21]. Paradoxically, the TERT promoter CGI is primarily hypomethylated and inactive in healthy adult somatic cells [22], yet hypermethylated and active in cancer cells. In the upstream TERT promoter, hypermethylation of a CpG (cg11625995, −628 of the AUG) has been used as a reliable biomarker for TERT expression, tumor progression, and prognosis [23]. Methylation of repressor binding sites here may lead to TERT reactivation [16, 24]. However, because this region is hypomethylated in pluripotent cells that actively express TERT [22, 25], this explanation is insufficient. Additionally, decreased methylation in this upstream region actually associates with active transcription histone marks [26].
A limited hypomethylated region flanking the TERT transcription start site region may serve as a ‘minimal promoter’. This region roughly spans −200 to +100 bp of the transcription start site or −260 to +40 bp of the AUG [27]. This hypomethylation is consistent between both WT and TERT promoter‐mutant cancer cell lines [26]. Within it are multiple methylation‐sensitive transcription factor (TF) binding sites. These include two E‐Box sites (−236 and −28 bp of the AUG) that repressive Mad/Max or activating c‐Myc/Max TFs bind [16, 24, 28, 29] (Table S1) (for a detailed description of TERT promoter TF binding sites, see Ref. [24]). Active chromatin marks have been associated with unmethylated DNA in this region [16, 27].
Better understanding of TERT promoter methylation patterns may enable us to decipher the different mechanisms by which TERT is reactivated in different cancer types. For example, TERT‐activating mutations are more prevalent in some types of cancers (e.g., melanoma and medulloblastoma) [9]. These possess allelic methylation in the hypermethylated, upstream promoter region, where decreased methylation associates with the active allele [26]. Allelic methylation in the minimal promoter of mutant cells has only been reported in thyroid cancer cell lines [30], but its more general occurrence and its presence in WT lines, with either MAE or BAE of TERT, are unknown. Additionally, while WT MAE and BAE lines associate with different cancer types [9]—for example, pancreatic cancers are frequently WT MAE, while lung cancers are primarily WT BAE—it has been unknown whether there are associated TERT promoter methylation patterns.
Here, we investigated DNA methylation patterns in the TERT promoter across 23 different cancer tissue types and 833 different cancer cell lines. In all cancerous tissue types, we found hypermethylation in the upstream promoter region and hypomethylation of a proximal promoter region and exon 1. The proximal promoter hypomethylation appeared more conserved across different tissue types than the upstream hypermethylation, which varied significantly between different tissues. Within the hypomethylated proximal promoter, apparently BAE lines had significantly decreased methylation relative to MAE WT and mutant lines, suggesting decreased methylation here to be important for increased TERT transcription. This region also contained allelic methylation in MAE lines, with decreased methylation associating with active transcription. Overall, it appears that hypomethylation of the TERT proximal promoter is important for TERT expression in cancer cells.
2. Materials and methods
2.1. CCLE data analysis
Cancer cell line DNA CpG methylation data were downloaded from the Broad Institute's Cancer Cell Line Encyclopedia (CCLE) [31] (www.broadinstitute.org/ccle, June 14, 2018, release, TERT gene) for the TERT gene and 1 kb upstream of the translation start site. Data were visualized using python (Python Software Foundation, Beaverton, OR, USA) with the libraries plotly [32] and pandas (NumFOCUS, Austin, TX, USA). Full CCLE dataset includes data from 833 cancer cell lines with bisulfite conversion sequencing (Bis‐Seq) CpG methylation data at 224 genomic positions. For Fig. 2A, raw read Bis‐Seq DNA CpG methylation data from CCLE's Bis‐Seq dataset were obtained by direct request to the Broad Institute's CCLE and analyzed for allelic patterns using stringent cutoff criteria (reads analyzed contained 3–6 CpGs per read, and coverage of ≥ 5 reads per cell line). Dataset BAM files were analyzed using python with pysam [33] and scipy libraries [34], as well as the methylation analysis tool ‘quma’ (http://quma.cdb.riken.jp/) [35]. Graphs were prepared using prism 8 (GraphPad, San Diego, CA, USA, version 8.4.0).
Fig. 2.
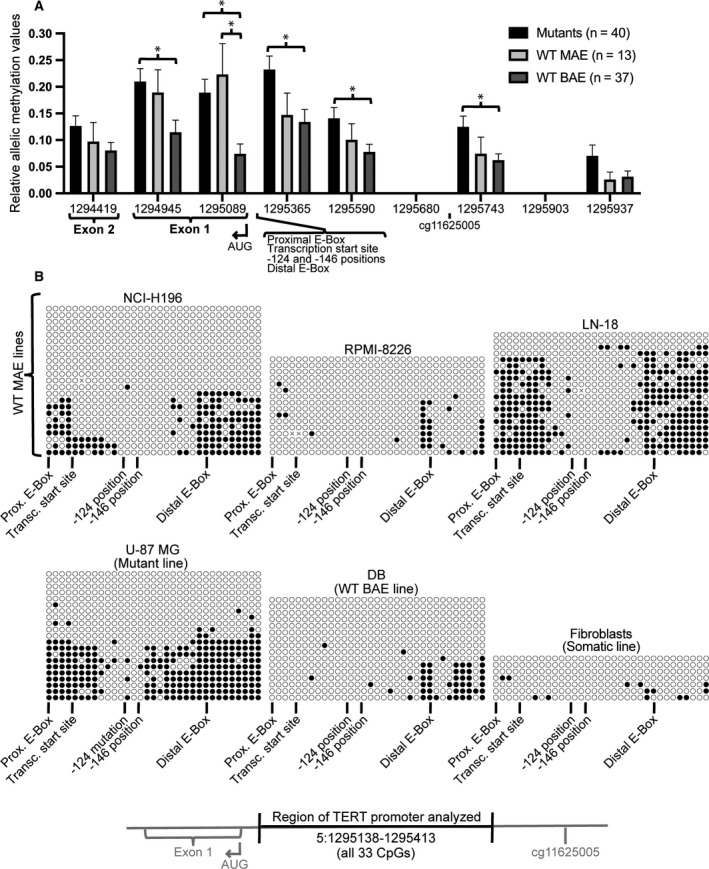
Cells with monoallelic expression of TERT have allelic methylation of the proximal TERT promoter. (A) Extent of allelic methylation across the TERT gene, which is transcribed from right to left. Relative allelic methylation was measured by calculating the difference between the mean and mode values of raw read Bis‐Seq CpG methylation data, where greater levels suggest greater allelic methylation behavior (see Fig. S3 for examples). Positions included contained 3–6 CpGs per read and coverage of ≥ 5 reads per cell line. Error bars represent standard error of the mean. n represents the number of cancer cell lines. *P ≤ 0.05, where statistical analysis was performed using 2‐tailed Student's t‐test with unequal variance. (B) Bisulfite conversion cloning data from genomic DNA of select CpGs flanking the TERT transcription start site (5:1295138–1295413, spanning 33 CpGs). Each row represents a different clone (or genome copy, or allele) and each circle represents a CpG, with black circles representing a methylated CpG and white circles representing an unmethylated CpG. For chromosomal positions of noted TERT features, see Table S1.
2.2. ENCODE UCSC Genome Browser data analysis
Cell line data for normal, cancer, and human embryonic stem cell (hESC) line DNA CpG methylation data (Fig. S2 only) were downloaded as BED files from the Encyclopedia of DNA Elements (ENCODE) at UC Santa Cruz (UCSC) Genome Browser for the TERT gene and 1 kb upstream of the translation start site [36]. Data were visualized using python (Python Software Foundation) with the libraries plotly [32] and pandas (NumFOCUS).
2.3. Cell lines and culture
Lines DB, NCI‐H196, and RPMI 8226 [American Type Culture Collection (ATCC), Manassas, VA, USA] were maintained in RPMI‐1640 medium (Gibco Thermo Fisher Scientific, Waltham, MA, USA). Lines U‐87 MG [University of Colorado Cancer Center, Protein Production/MoAB/Tissue Culture Shared Resource (PPSR)] and LN‐18 (ATCC) were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco Thermo Fisher Scientific). SK‐N‐SH (PPSR) and adult human foreskin fibroblasts were maintained in Eagle's minimum essential medium (EMEM) (Gibco Thermo Fisher Scientific). All media were supplemented with 100 μg·mL−1 penicillin and 100 μg·mL−1 streptomycin (Gibco Thermo Fisher Scientific) and 10% (Sigma‐Aldrich, St. Louis, MO, USA) or 5% (only line LN‐18) fetal bovine serum (FBS) (Peak Serum Inc., Wellington, Colorado, US). All lines were cultured according to recommended protocols.
2.4. DNA isolation, PCR, and sequencing
DNA isolation, PCR, and Sanger sequencing (GENEWIZ, South Plainfield, NJ, USA) were performed as previously described [14]. Briefly, gDNA was isolated from cells using Quick‐DNA Miniprep Kit (11‐317AC; Zymo Research, Irvine, CA, USA). PCRs (20 μL) were performed using 50 ng of gDNA and Phusion High‐Fidelity DNA Polymerase (F‐530; Thermo Fisher Scientific, Grand Island, NY, USA) supplemented with 7‐deaza‐2′‐deoxy‐guanosine‐5′‐triphosphate (7‐Deaza‐dGTP) (10988537001; Sigma‐Aldrich) to aid in amplifying GC‐rich regions. Sequences for primers (Integrated DNA Technologies, Coralville, IA, USA) are listed in Table S4. PCR products were purified using E.Z.N.A. Cycle Pure Kit (D6492; Omega Bio‐Tek).
2.5. RNA extraction, cDNA synthesis, and RT‐PCR
RNA extraction, cDNA synthesis, and RT‐PCR were performed as previously described [14]. Briefly, total RNA was isolated using the E.Z.N.A. Total RNA Kit I (R6834; Omega Bio‐Tek, Norcross, GA, USA) and RNase‐free DNase Set I (E1091‐02; Omega Bio‐Tek). RNA (1 μg) was used to synthesize cDNA with the SuperScript IV First‐Strand Synthesis System (Invitrogen Thermo Fisher Scientific, Waltham, MA, USA; 18091050). RT‐PCR was performed with SYBR Select Master Mix (4472908; Thermo Fisher Scientific) supplemented with 7‐Deaza‐dGTP using the lightcycler 480 software (Roche, Basel, Switzerland). Primers used were previously described except for TERT exon 2 primers (primer sequences and citations are listed in Table S4). RT‐PCRs (10 μL) were run in triplicate on a 96‐well plate and data normalized to the geometric mean of 3 ‘housekeeping’ genes [glucose phosphate isomerase (GPI), peptidylprolyl isomerase A (PPIA), and hydroxymethylbilane synthase (HMBS)] [14]. PCR products were purified using E.Z.N.A. Cycle Pure Kit (D6492; Omega Bio‐Tek) and underwent Sanger sequencing (GENEWIZ).
2.6. Bisulfite conversion cloning
For Fig. 2B, gDNA was isolated from cells using the Quick‐DNA Miniprep Kit (11‐317AC; Zymo Research) and 300 ng underwent bisulfite conversion using the EZ DNA Methylation‐Gold Kit (D5005; Zymo Research). Twenty nanogram of bisulfite‐converted DNA was used in 25 μL PCR amplification reactions with primers flanking the TERT proximal promoter (5:1295138–1295413; 33 CpGs included; 331 bp PCR product), 1.25 units of EpiMark Hot Start Taq DNA Polymerase [M0490; New England BioLabs (NEB), Ipswich, MA, USA], and the following thermocycling conditions: 95 °C for 30 s, then 40 cycles of 95 °C for 30 s, 55 °C for 60 s, and 68 °C for 30 s, followed by a final extension of 68 °C for 5 min. Primers used were modified from a previous publication [22] which had designed primers to complement and amplify methylated CpGs in hypomethylated cell types. Here, in initial experiments we found that these primers led to unrepresentative overamplification of methylated CpGs in cancer cells. Hence, we modified these primers to complement unmethylated CpGs; sequences for primers (Integrated DNA Technologies) are listed in Table S4. To prepare PCR products for blunt‐end cloning, 5′‐end phosphorylation was performed using T4 polynucleotide kinase (M0201; NEB) and 3′ overhang removal using T4 DNA polymerase (M0203; NEB) and purified using E.Z.N.A. Cycle Pure Kit (D6492; Omega Bio‐Tek). To prepare the vector, 1 μg pUC19 DNA was digested using SmaI restriction enzyme (R01415; NEB), treated with alkaline phosphatase (M0290; NEB), and purified using E.Z.N.A. Cycle Pure Kit. Prepared PCR products were ligated into the vector using Quick Ligation (M2200; NEB). Supercompetent cells were transformed and incubated on Carb LB agar plates overnight at 37 °C, colonies were picked and incubated in 5 mL LB overnight at 37 °C while shaking, plasmids were purified using E.Z.N.A. Plasmid Mini Kit I (D6942; Omega Bio‐Tek), and inserts were sequenced using Sanger sequencing. All cloning details were according to recommended manufacturer's protocols. Methylation sites were visualized, and quality control was performed using the online tool ‘quma’ (http://quma.cdb.riken.jp/) [35].
2.7. Long‐range bisulfite conversion PCR
For Fig. 3B,C, long‐range bisulfite conversion PCR was optimized following previously published guidelines for generating large bisulfite‐converted PCR products [37]. gDNA was isolated from cells using Quick‐DNA EZ DNA Methylation‐Gold Kit and underwent bisulfite conversion using the Methylamp DNA Modification Kit (P‐1001; EpiGentek, Farmingdale, NY, USA). A two‐step PCR amplification was used: For the first step, 50 ng of bisulfite‐converted DNA was used in 20 μL reactions with 3.5 units of EpiMark Hot Start Taq DNA Polymerase and the following thermocycling conditions: 94 °C for 2 min, then 35 cycles of 94 °C for 20 s, 62 °C for 45 s, and 65 °C for 1 min and 55 s, followed by a final extension of 65 °C for 5 min; for the second step, 1 μL of a 1 : 50 dilution of the first‐step PCR product was used in 25 μL reactions with 3.5 units of EpiMark Hot Start Taq DNA Polymerase and the same thermocycling conditions as the first step except the 62 °C annealing temperature was increased to 67 °C. PCR products were validated on an agarose gel, gel‐purified using the MinElute Gel Extraction Kit (28606; Qiagen, Hilden, Germany), and sequenced using Sanger sequencing. Unmethylated‐ and methylated‐specific primers in the TERT promoter upstream of the TERT translation start site contained three CpGs to amplify unmethylated or methylated DNA by containing a C (to complement methylated DNA) or T (to complement unmethylated DNA) within the primers at all three CpGs. For both of these promoter primers, the same reverse primer was used, which was downstream of the exon 2 SNP in a methylated region. The bisulfite conversion PCR generated a relatively large (1448 bp) PCR product, which was sequenced using Sanger sequencing. Sequences for primers (Integrated DNA Technologies) are listed in Table S4.
Fig. 3.
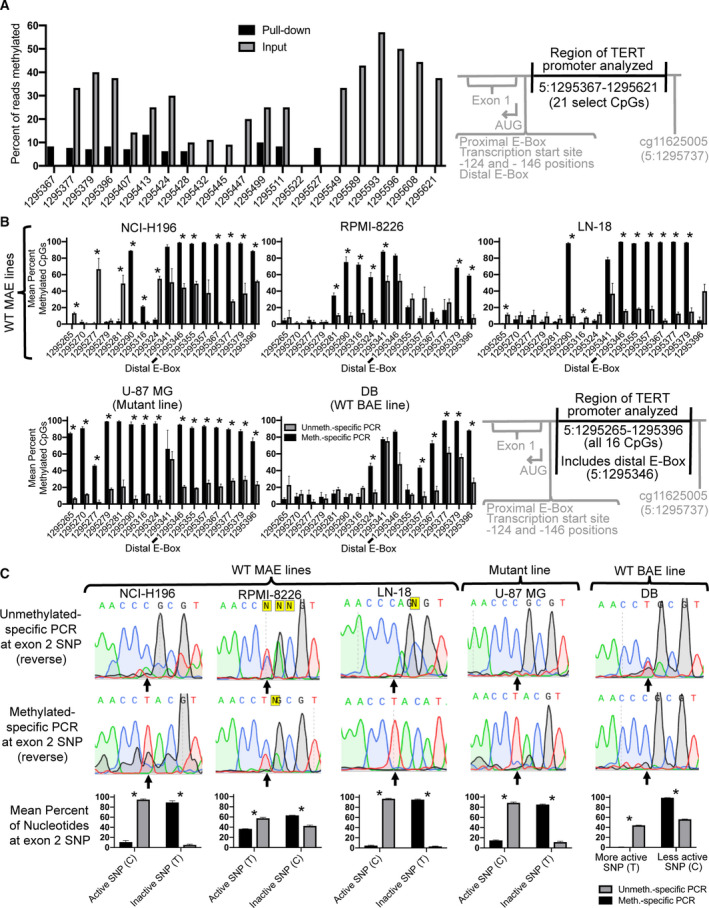
Decreased TERT promoter methylation associates with histone marks of active transcription and an active exonic SNP. (A) ChIP‐Bis‐Seq of the TERT promoter using an H3ac antibody shows enrichment of unmethylated DNA in the pulled‐down samples (black) relative to the input (gray) in LN‐18 cells. The absence of any bars indicates zero percent methylation. Inclusion criteria for read positions were a greater number of reads in the pull‐down relative to the input and ≥ 10 reads in the pull‐down (mean input coverage was 9 reads; mean pull‐down coverage was 13 reads; P = 0.01 for pull‐down efficiency). (B) Confirmation of long‐range bisulfite conversion PCR enriching for unmethylated or methylated CpGs at the TERT proximal promoter (16 CpGs spanning 5:1295265–1295396; region overlaps with some of the CpGs analyzed in 3A) using unmethylated (gray)‐ or methylated (black)‐specific bisulfite conversion PCR, respectively. PCR products generated a 1448‐bp product including the proximal promoter and the exon 2 SNP analyzed in Panel C. *P ≤ 0.05 (C) Long‐range bisulfite conversion PCR (same PCRs as shown in Panel B) showing representative Sanger sequencing results (upward arrow indicates position of the exon 2 SNP) and graphs of the sequencing results (n = 2–3 sequenced reactions). ‘Active SNP’ means that the nucleotide at the position of the SNP is the one found in the TERT mRNA transcribed in that cell line. The active SNP was either previously identified in all cell lines [14] or was identified here (Fig. S6). Error bars represent standard error of the mean. *P ≤ 0.01, where statistical analysis was performed using 2‐tailed Student's t‐test with unequal variance.
2.8. ChIP‐Bis‐Seq library construction and data analysis
ChIP was performed as previously described [15, 26] with modifications. LN‐18 cells were cultured until approximately 80% confluent, rinsed with PBS, fixed with freshly prepared 1% (v/v) formaldehyde (BP531500; Fisher Scientific) in PBS at room temperature (RT) for 10 min, and inactivated using 1.25 m glycine solution for 2 min at RT. Following solution aspiration, cells were scraped and collected, resuspended in ice‐cold PBS, and centrifuged at 1000 g at 4 °C for 5 min. The resultant pellet was frozen at −80 °C for at least 60 min and then lysed for 10 min on ice in 300 μL lysis buffer (50 mm Tris/Cl pH 8.1, 10 mm EDTA, 0.5% SDS) with 6 μL 50× protease inhibitor (A32965; Thermo Fisher Pierce, Waltham, MA, USA) added immediately prior to use. Chromatin was sonicated using a BioRuptor for 4 × 10 min on ‘high’, for 30 s ‘on’ and 30 s ‘off’, in ice water, resulting in fragmented pieces of approximately 200–500 bp, as confirmed using a purified sample via gel electrophoresis on a 1% agarose gel. Following sonication, chromatin was initially cleared via centrifugation at 16 000 g for 15 min at 4 °C and supernatant transferred to a new tube and quantified via NanoDrop. Next, 20 μg of cleared chromatin (15–50 μL per sample) was further cleared to reduce background via nutation for 2 h at 4 °C in 1–2 mL immunoprecipitation (IP) buffer (16.7 mm Tris/Cl pH 8.1, 1.2 mm EDTA, 167 mm NaCl, 1% Triton X‐100) with 50–100 μL A/G magnetic beads (88803; Pierce Protein, Waltham, MA, USA) that had been washed twice in IP buffer. Using a magnetic rack, cleared chromatin was recovered in 500 μL IP buffer and incubated with 5 μg anti‐H3ac antibody (06‐599; Millipore, Burlington, MA, USA) or 5 μg rbt IgG (12‐370; Millipore) overnight with nutation at 4 °C. Immunoprecipitation was performed using 25 μL washed A/G magnetic beads for 1 h with nutation at RT. Following immunoprecipitation, IP buffer was removed and nonspecific binding reduced by washing the beads in 1 mL low‐salt buffer (20 mm Tris/Cl pH 8.0, 2 mm EDTA, 150 mm NaCl, 0.1% SDS, 1% Triton X‐100), high‐salt buffer (20 mm Tris/Cl pH 8.0, 2 mm EDTA, 500 mm NaCl, 0.1% SDS, 1% Triton X‐100), and lithium chloride (LiCl) buffer (10 mm Tris/Cl pH 8.0, 1 mm EDTA, 250 mm LiCl, 1% sodium deoxycholate, 1% IGEPAL), and then washed in Tris/EDTA (TE) (20 mm Tris pH 8, 2 mm EDTA) to remove salts. The antibody:IP complex was eluted in 120 μL elution buffer (100 mm NaHCO3, 1% SDS, 200 mm NaCl) at RT with mixing for 20 min. During elution, an input sample was included that had been frozen immediately following the first clearing step. For crosslink reversal, supernatant was incubated at 65 °C for > 6 h. Protein and RNA were removed through incubation with 100 mm Tris pH 6.5, 11 mm EDTA, 60 μg Proteinase K (AM2544; Life Technologies, Carlsbad, CA, USA), and 1 μg DNase‐free RNase (EN0531; Thermo Scientific) for 60 min at 37 °C. DNA was purified using E.Z.N.A. Cycle Pure Kit (D6492; Omega Bio‐Tek), resuspended in TE, and underwent RT‐PCR as described in Section 2.5 above.
For ChIP‐Bis‐Seq library construction, ChIP was performed using biological duplicates. Duplicate pull‐down and input samples were used for library construction. Fifty nanogram of pulled‐down ChIP DNA was used with the NuGEN Ovation Ultralow Methyl‐Seq DR Multiplex System (0335‐32; NuGEN, Tecan Genomics, Inc., Redwood City, CA, USA), according to the manufacturer's instructions. DNA was assessed using a Qubit Fluorometer 3.0 (Thermo Fisher Scientific). Sonicated, pulled‐down gDNA fragments underwent end repair, ligation of kit‐provided methylated adaptors, and final repair, following the manufacturer's instructions. Eluted library samples underwent qPCR to determine the number (N) of PCR cycles required for library amplification (QuantStudio 6 Real‐Time PCR; Thermo Fisher Scientific). Amplified libraries were purified with Agencourt beads and eluted in low‐EDTA TE buffer. Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA) was used to validate and quantify libraries. Amplified libraries were normalized and pooled, denatured, and diluted for sequencing on MiSeq (Illumina, San Diego, CA, USA) according to manufacturer's guidelines.
For ChIP‐Bis‐Seq data analysis, prior to alignment, single‐end reads were filtered using FastQC (Babraham Institute, Cambridge, UK) and adaptor‐trimmed using cutadapt [38]. Only reads with a Q score ≥ 20 and matching length criteria were used for mapping. Alignment of trimmed bisulfite‐converted sequences was carried out using Bismark (Babraham Institute, Cambridge, UK) [39] against the human reference genome for chromosome 5 (GRCh38, release 93), yielding methylation call percentages for each CpG and non‐CpG site within the chromosome. Duplicate reads arising from artifacts in library preparation and sequencing were deduplicated using Bismark, and Bismark methylation extractor was used to generate postalignment methylation counts. Aligned reads and methylation were visualized using Integrated Genomics Viewer (Broad Institute and UC San Diego, La Jolla, CA, USA). Graphs were prepared using prism 8 (GraphPad, version 8.4.0).
3. Results
3.1. Upstream TERT promoter hypermethylation and proximal promoter hypomethylation conserved across cancer tissue types
Using publicly available bisulfite conversion sequencing (Bis‐Seq) data (from the Broad Institute's Cancer Cell Line Encyclopedia [CCLE]: www.broadinstitute.org/ccle), we compared TERT promoter CpG methylation across 23 different cancer tissue types and 833 cancer cell lines. The comparison spanned a 2788‐bp region containing the upstream promoter through exon 2 (5:1296377–1293589; hg19 genomic coordinates) (Table S1). The mean methylation across this region varied for different tissue types from 46.4% to 78.9%, with 91% of tissues (21/23) being overall hypermethylated (> 50% methylated) (Fig. 1A). Values for each tissue are as shown in Table 1 (for individual cell line data, see Fig. S1 and Table S2).
Fig. 1.
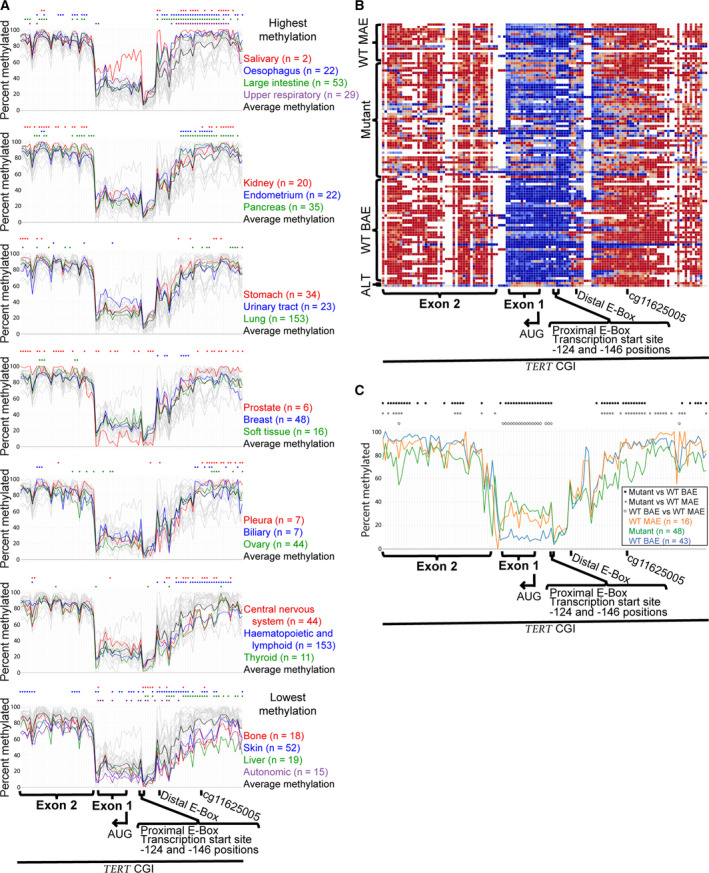
TERT promoter is characterized by conserved upstream hypermethylation and proximal hypomethylation across different cancer tissue types. (A) Bisulfite conversion sequencing (Bis‐Seq) DNA CpG methylation data for 95 positions across the TERT promoter for 23 different cancerous tissues, showing mean values from 833 cancer cell lines (n represents the number of cell lines per tissue). Colored circles indicate individual CpG sites with statistically significant (P ≤ 0.005) differences between the tissue and all other tissues. Each graph groups tissues by total mean percent methylated, from most to least methylated (top to bottom, respectively). Each chromosomal position includes data from at least two cell lines for all 23 tissues. (B) Bis‐Seq DNA CpG methylation data for 129 positions across the TERT promoter for 109 cell lines with known allelic expression and activating mutation classifications. Lines had been classified as having wild‐type (WT) monoallelic expression (MAE) of TERT (‘WT MAE’), −124 or −146 C>T activating promoter mutations (‘mutant’), biallelic expression (BAE) of TERT (‘WT BAE’), or alternative lengthening of telomeres (ALT). Each row represents a different cell line. Colors range from red to blue for more to less methylated CpGs, respectively. White represents unavailable data. Each chromosomal position includes data from at least 10 cell lines. (C) Bis‐Seq DNA CpG methylation data for 122 positions across the TERT promoter for 107 cell lines shown in 1B (n represents the number of cell lines per tissue). Colored circles indicate statistically significant (P ≤ 0.05) differences in the listed pairwise comparisons, where statistical analysis was performed using 2‐tailed Student's t‐test with unequal variance. Each chromosomal position includes data from at least two cell lines for all three cell types. See Table S1 for chromosomal positions and Table S2 for cell line data.
Table 1.
TERT promoter methylation values for different tissue types from Fig. 1A. n, number of cell lines analyzed
| Tissue type | Mean percent methylated |
|---|---|
| Salivary | 78.9 (n = 2) |
| Esophagus | 73.3 (n = 22) |
| Large intestine | 71.5 (n = 53) |
| Upper respiratory | 69.8 (n = 29) |
| Kidney | 69.6 (n = 20) |
| Endometrium | 69.1 (n = 22) |
| Pancreas | 68.6 (n = 35) |
| Stomach | 68.5 (n = 34) |
| Urinary tract | 65.4 (n = 23) |
| Lung | 65.2 (n = 153) |
| Prostate | 64.9 (n = 6) |
| Breast | 64.6 (n = 48) |
| Soft tissue | 64.6 (n = 16) |
| Pleura | 63.7 (n = 7) |
| Biliary | 63.2 (n = 7) |
| Ovary | 59.8 (n = 44) |
| Central nervous system | 59.7 (n = 44) |
| Haematopoietic and lymphoid | 57.4 (n = 153) |
| Thyroid | 55.0 (n = 11) |
| Bone | 51.7 (n = 18) |
| Skin | 50.5 (n = 52) |
| Liver | 46.4 (n = 19) |
| Autonomic | 46.4 (n = 15) |
While significant variation was apparent between different tissue types, some conserved patterns of TERT methylation were observed (Fig. 1A). We describe these from right to left on the genome browser traces, because this is the direction in which TERT transcription occurs. A region of the upstream promoter (≈ 1296377–1295699) was highly methylated (52.4–94.5% methylated; mean = 82.5% for all tissues). This included a previously identified hypermethylated CpG biomarker (cg11625005; 1295737). A hyper‐ to hypomethylation transition occurred over a span of ≈ 347 bp, from ≈ 581 to 234 bp upstream of the translation start site (AUG) (≈ 1295685–1295338) (Fig. 1A). The distal E‐Box was within this region. The proximal promoter, which includes the promoter mutation sites, the somewhat heterogeneous transcription start site region, and the proximal E‐Box, contained the point of lowest methylation for nearly all tissue types (1295247, ≈ 143 bp upstream of AUG, between the −124 and −146 mutations). This point ranged from 0.4% to 11.2% methylated, with a mean value of 6.5% across all tissue types. Levels increased but remained hypomethylated through exon 1 (until 1294448) for nearly all tissue types (96%; 22/23); greater methylation levels in exon 1 were observed in some cell line types compared to others (see Section 3.2, below) (Fig. 1A). The transition from hypo‐ to hypermethylation occurred in intron 1, which contained data from relatively few CpGs (Fig. 1A); if data were available from additional CpG sites, this transition might appear more gradual. Exon 2 (≈ 1294441–1293589) was hypermethylated in all tissue types. This overall pattern is consistent with previous studies on more limited tissue types [16, 24, 26, 27]. Interestingly, in most tissues the hypermethylated upstream region showed significant variation between different tissue types, while the hypomethylated proximal promoter region was more constant.
To compare CpG methylation in normal adult cells and a human embryonic stem cell (hESC) line with some of the cancer cell lines, a limited analysis across the same region was performed using data from the Encyclopedia of DNA Elements (ENCODE) (Fig. S2, Table S1, Table S3). In contrast to the cancer cell lines, the cg11625005 biomarker and surrounding distal promoter region were hypomethylated in normal adult cells and hESCs (Fig. S2), in agreement with previous work [22]. Because hESCs express TERT, it seems unlikely that methylation and inactivation of repressor binding sites in this distal promoter region by themselves lead to TERT reactivation in cancer cells, as has been suggested [16, 24]. Hypomethylation of the proximal promoter–exon 1 region was similar between cancer and normal cells (Fig. S2). Because normal adult cells do not express TERT, hypomethylation of the proximal promoter is clearly insufficient, though potentially necessary, for gene expression.
3.2. TERT promoter methylation patterns associate with allelic expression classifications
TERT expression was analyzed in 107 cancer cell lines that were previously classified as follows: WT promoter, MAE (n = 16); −124 and −146 promoter mutations, which cause MAE (n = 48); and WT, BAE (n = 43) [9, 13]. Patterns of TERT methylation showed general similarity among cell lines in these categories (Fig. 1B,C). Mean methylation values were 68.2% for WT MAE, 66.7% for WT BAE, and 59.8% for mutants. Interestingly, some regions of the CGI did show methylation differences that associated with allelic expression and activating mutation classifications. Mutant lines displayed significantly decreased methylation compared to WT BAE and WT MAE lines (P < 0.05) across most of the upstream hypermethylated promoter region and part of the adjacent transition region (≈ 1296377–1295458), and at multiple positions in exon 2. In addition, WT BAE lines showed significantly decreased methylation relative to both WT and mutant lines across most of the hypomethylated proximal promoter region (≈ 1295139–1294873) (Fig. 1C). Specifically, this region of increased methylation in WT and mutant lines relative to WT BAE lines included exon 1, the AUG, and the proximal E‐Box. However, it did not include the more upstream elements of the transcription start site region and −124 and −146 sites, where low methylation levels were observed in all cell line types. This finding supports the conclusion that hypomethylation associates with transcriptional activity, because the WT BAE lines only have active TERT alleles, whereas the methylation data in MAE lines are an average of active and inactive alleles.
In addition, two lines analyzed were telomerase‐negative and used the alternative lengthening of telomeres (ALT) mechanism [40] (Table S2). ALT lines displayed relatively higher mean methylation values (70.2%), particularly across the transition region and adjacent hypomethylated proximal promoter. This is consistent with the conclusion that increased methylation of the proximal promoter associates with transcriptional inactivity.
3.3. Monoallelic expression of TERT correlates with allelic proximal promoter methylation
Using publicly available Bis‐Seq data (from CCLE), raw read analysis revealed allelic methylation behavior within the TERT proximal promoter and exon 1. Relative allelic methylation values were calculated based on the difference between the mean and mode of the percent methylation of reads at a given read position (Fig. S3). Using this calculation, the greater the difference between mean and mode, the more suggestive it is of allelic behavior. Averaged over all cell lines, the highest allelic values were in the hypomethylated proximal promoter region (1294945–1295363; Fig. 2A), including exon 1, the AUG, the transcription start site region, the −124 and −146 mutations, and both E‐Boxes. In this region and elsewhere, WT BAE lines displayed significantly lower allelic values compared to both WT MAE and mutant cell lines. This is consistent with the expectation that WT MAE and mutant cell lines have more allelic methylation than WT BAE lines.
To validate the apparent allelic methylation behavior, we performed bisulfite conversion cloning within the proximal TERT promoter (5:1295138–1295413; 33 CpGs total) (Fig. 2B). Bisulfite conversion cloning gives the methylation status of successive CpGs along single cloned copies of a gene, where each sequence must represent a single allele [41]. Each sequence is shown as a row of circles (with a white circle for an unmethylated CpG and black for a methylated CpG). The results aligned well with our Bis‐Seq analysis of this region (Fig. 1 and Fig. S4). Bis‐Seq analysis of WT MAE lines found the lowest methylation point to be the 5:1295247 read position (Fig. 1), and bisulfite conversion cloning showed the lowest methylation to be in the same region, ≈ 5:1295195–1295324. Also supporting the Bis‐Seq analysis, one WT MAE line (RPMI‐8226) had much lower overall methylation levels. The mutant line analyzed (U‐87 MG) contained a narrower hypomethylated area (≈ 5:1295189–1295260), also agreeing with the Bis‐Seq analysis for this cell line (Fig. S4). Furthermore, a WT BAE line (DB) had lower overall methylation, as we had seen for BAE lines in general, as well as for this specific line (Fig. 1B,C, and Fig. S4). Somatic cells (fibroblasts) had low levels of methylation throughout the region [0% (0/33) – 9% (3/33) of CpGs methylated per clone], consistent with earlier reports [22, 42].
In MAE lines, the CpGs flanking the hypomethylated region displayed allelic methylation behavior. Specifically, in this flanking region a given sequence was either entirely or mostly unmethylated (white circles in Fig. 2B), or the sequence was heavily methylated (black circles). Two distinct patterns of methylation are the expectation for allelic methylation. This was clearly seen in the U‐87 MG mutant line, where 43% of clones (9/21) had ≤ 6% of CpGs methylated and 48% (10/21) had 49% (16/33) – 79% (26/33) methylated CpGs. In the DB WT BAE line, methylation clustered on some alleles within an upstream region (≈ 5:1295341–1295413), potentially demonstrating allelic methylation here [56% (9/16) of clones contained no methylated CpGs, while 38% (6/16) contained 60% (6/10) – 80% (8/10) methylated CpGs]. Overall, allelic TERT methylation patterns were evident in most MAE lines analyzed, both TERT promoter mutant and WT.
3.4. Decreased methylation of the TERT promoter associates with histone marks of active transcription
Using chromatin immunoprecipitation Bis‐Seq (ChIP‐Bis‐Seq), we found decreased TERT proximal promoter methylation to associate with increased histone marks of active transcription. The H3 acetylation (H3ac) antibody used binds acetylated lysine 9 (H3K9ac) and acetylated lysine 14 (H3K14ac) present together. This is tightly associated with active transcription start site regions and active genes [43]. ChIP using the H3ac antibody yielded efficient pull‐down and significant selection for the −124 mutant, active allele in the TERT promoter (Fig. S5). H3ac ChIP‐Bis‐Seq at the proximal TERT promoter had pull‐down of 21 CpGs spanning 5:1295250–1295504 (Fig. 3A). The majority of CpGs (18/21, or 86%) had decreased methylation in the H3ac‐pull‐down compared to the input (Fig. 3A). This indicates an association between decreased methylation and increased histone marks of active transcription in the TERT proximal promoter.
3.5. Decreased methylation of the TERT promoter associates with actively transcribed exon SNP
As an independent test of the relationship between TERT methylation and transcription, we performed long‐range PCR on bisulfite‐converted genomic DNA using unmethylated‐ or methylated‐specific primers. The resulting 1448 bp PCR product spanned the TERT proximal promoter and an exon 2 SNP (rs2736098, G/A) (see Table S4 for primer sequences). In genomic DNA containing a mixture of methylated and unmethylated alleles, unmethylated‐specific primers should amplify primarily unmethylated DNA, and methylated‐specific primers should amplify primarily methylated DNA. The exon 2 SNP was included to determine whether a primarily unmethylated or methylated product was associated with the active allele. We analyzed genomic DNA from cancer cell lines that were heterozygous for the TERT exon 2 SNP. The sequence of the active allele at this SNP was identified in all lines using RT‐PCR of RNA followed by Sanger sequencing of the PCR product [14] (Fig. S6). Additionally, in this way, lines with expression of only one allele were confirmed to have MAE; this included all WT MAE and mutant lines tested here. Lines with expression of both alleles were confirmed to have BAE of TERT; this included the WT BAE line tested here (Table S2 and Fig. S6) [14]. As a validation of the method, in the proximal TERT promoter all WT MAE and mutant lines had more methylated DNA in the methylated‐specific PCR products compared to the unmethylated‐specific products (Fig. 3B) [56.1 ± 1.0% vs. 30.9 ± 5.6% for NCI‐H196 (P = 0.07); 38.1 ± 3.4% vs. 16.6 ± 4.3% for RPMI‐8226 (P = 0.03); 50.0 ± 1.2% vs. 14.9 ± 3.9% for LN‐18 (P = 0.01); and 87.3 ± 2.8% vs. 19.1 ± 3.0% (P = 1 × 10−14) for U87].
This long‐range bisulfite conversion PCR approach confirmed associations between decreased TERT proximal promoter methylation and active TERT expression. In the same bisulfite‐converted PCR products, DNA sequencing was performed across exon 2, covering the position of the SNP. DNA sequencing of the exon 2 SNP showed significantly higher levels of the transcription‐associated SNP sequence in unmethylated‐specific products compared to methylated‐specific products (Fig. 3C; P ≤ 0.01). In agreement, in a WT BAE line that expressed one SNP nucleotide at relatively greater levels, the unmethylated‐specific PCR also significantly correlated with the more active nucleotide at the position of the SNP (P = 1 × 10−5). Overall, these long‐range bisulfite conversion PCR findings demonstrate an association between decreased TERT proximal promoter methylation and increased TERT expression.
4. Discussion
While the TERT promoter contains a strikingly hypermethylated distal promoter region in human cancer cells, here we found that hypomethylation at the proximal promoter, flanking the transcription start site region, is also strongly associated with active TERT transcription in cancer cells. This methylation pattern, and the proximal promoter hypomethylation in particular, was observed across 23 different cancerous tissue types, using Bis‐Seq data from 833 different cancer cell lines (Fig. 1A). In the hypomethylated proximal promoter region, cancer cell lines with apparent biallelic expression (BAE) of TERT had significantly lower methylation compared to lines with monoallelic expression (MAE) of TERT, consistent with decreased methylation here to be associated with TERT expression (Fig. 1B,C). In this proximal promoter region, methylation was allele‐specific (Fig. 2) and decreased methylation associated with marks of active TERT transcription (Fig. 3). Thus, TERT expression in cancer lines may be canonical after all, in terms of its association with low DNA CpG methylation.
The overall pattern of TERT promoter methylation is relatively conserved across a range of cancer tissue types, particularly in a proximal hypomethylated region, in alignment with previous studies [26]. While a TERT ‘minimal promoter’ has been previously described, those studies typically focused on one or a few tissues and/or a handful of cancer cell lines per study, making it challenging to determine tissue‐specific differences and similarities. In particular, a ≈ 300 bp region of hypomethylation flanking the transcription start site region was previously labeled the ‘minimal promoter’, ranging roughly −260 to +40 bp of the translation start site (AUG) (5:1295364–1295064)[27]. Here, we provide resolution at the level of individual CpG sites in this hypomethylated region in TERT, which appears to be larger and slightly more downstream than previously described, spanning ≈ −220 to +231 bp of the AUG (or 5:1295321–1294873) (Fig. 1A). This includes all of TERT exon 1 and does not include the distal E‐Box site [16, 24]. This suggests that, in cancer cells, the more proximal E‐Box (5:1295138) may be more important for binding of methylation‐sensitive activating factors, such as c‐Myc/Max transcription factors [44, 45].
Hypomethylation of this TERT proximal promoter region may be a universal correlation and possibly a necessity for TERT expression in cancer cells, regardless of allelic expression status. We found this region to have significantly decreased methylation in TERT BAE cancer cells compared to MAE cells (Fig. 1B,C). Because WT BAE lines have only active TERT alleles, whereas MAE lines have both active and inactive alleles, this finding supports the idea that hypomethylation in this region is important for transcriptional activity. In this region, we also found decreased methylation to correlate with transcription in MAE cancer cells (Fig. 3). This agrees with previous findings showing active chromatin marks associated with unmethylated DNA in this region [16, 27]. Surprisingly, we saw similar, though less striking, allelic methylation correlations in apparently BAE cells, suggesting that even in BAE cells some allelic expression may occur and may depend upon decreased methylation of the more active alleles. Our group previously reported that in −124 mutant cells, active transcriptional histone marks (specifically H3K4me2/3) associate with decreased promoter methylation in the upstream hypermethylated region (1295619–1295738) [26]. It is also worth noting that human pluripotent stem cells, which express TERT, are largely hypomethylated across the TERT promoter [22]. Taken together, TERT activation appears canonically associated with decreased methylation in the proximal promoter. Ultimately, increasing methylation of the proximal TERT promoter to decrease TERT expression may be a route to explore for cancer therapeutics.
While we found the hypomethylated proximal TERT promoter region to contain apparently allelic methylation (Fig. 2), this should not be interpreted as one allele very lowly methylated and one allele with more methylation. Cancer lines often contain more than two TERT gene copies, with considerable line‐to‐line and cell‐to‐cell heterogeneity [14]. We previously performed TERT DNA FISH on several of the lines used here and found mean values of TERT DNA FISH spots per nucleus as 10.98 for NCI‐H196, 3.07 for LN‐18, 2.17 for U‐87 MG, and 3.88 for DB (RPMI‐8226 was not investigated). Hence, because in these cells there are multiple TERT gene copies, one or more of which may be inactive, drawing precise conclusions about allelic methylation patterns is challenging.
Considering the hypermethylation of the distal TERT promoter around cg11625005, which serves as a biomarker for identification and prognosis of cancer cells, it could contribute positively to gene expression [46]. If so, then the hESCs, which have a methylation pattern resembling telomerase‐minus normal human cells, would be an exception. Alternatively, the presence of this hypermethylation may be associated with, but not causal of, the cancer cell state, possibly due to larger‐scale chromatin structure remodeling events that occur in cancer cells.
A limitation of our findings here is that our data are correlative and not causal. Previous studies have altered the DNA CpG methylation of the TERT promoter with varying and apparently contradictory effects on TERT expression (reviewed by [47]). These disparate results may be due to using approaches that altered methylation in a nontargeted manner or using exogenous TERT promoter constructs. Specifically, most of these studies used 5‐azacytidine or 5‐aza‐2′‐deoxycytidine (decitabine; DAC) treatment, which results in global genomic demethylation. In some studies using cancer cells, these treatments caused decreased TERT expression, which may actually be due to demethylation of a downstream CTCF repressor binding site [28, 42, 48]. However, senescing fibroblasts, in which the TERT promoter had become hypermethylated, gained increased TERT expression upon promoter demethylation via DAC treatment [49]. An area of future investigation would be to alter DNA methylation in the TERT promoter in a targeted manner. For example, dCas9 techniques that tether DNMT3A to increase DNA methylation [50] could be targeted to the hypomethylated proximal promoter region; decreased TERT expression would then be predicted based on our findings here. Using a dCas9 technique tethering TET1 to locally decrease DNA methylation [51] in this region may not significantly increase TERT expression since this region is already hypomethylated.
Overall, our findings that hypomethylation of the proximal TERT promoter is shared across a range of cancer tissue types and that decreased methylation is associated with increased transcription regardless of cancer cell line type suggest that this hypomethylated area may be necessary for active TERT expression. However, because the proximal promoter region is also hypomethylated in somatic cells that do not express TERT, it is clearly not sufficient for TERT expression. Yet, the possibility of an underlying TERT activation mechanism that is necessary, specific, and universal to cancer cells is noteworthy. While −124 mutants are known to have a mutation that recruits ETS transcription factors such as GABP to activate TERT expression [10], still approximately 78% of cancer cell lines have wild‐type TERT promoters and do not contain known activating cis‐acting genetic alterations [9]. This makes it unclear how the majority of cancer lines have active TERT expression. It may be that TERT expression is activated by aberrant upregulation of activating transcription factors in cancer cells that recognize binding motifs in this hypomethylated region, such as c‐Myc/Max binding of the proximal TERT promoter E‐Box. Future studies may investigate whether targeted methylation or other inhibition of the proximal E‐Box results in decreased TERT expression in cancer cells. If this is found to be the case, such targeted approaches may be useful to explore for potential cancer therapeutics.
5. Conclusion
Across 23 cancerous tissue types, we found DNA hypermethylation in the upstream TERT promoter region and relatively more conserved hypomethylation of a proximal promoter region and exon 1. This region showed allelic methylation in cancer lines with MAE of TERT. Decreased methylation of the proximal TERT promoter is associated with, and may be important for, active TERT expression in cancer cells.
Conflict of interest
TRC is on the board of directors of Merck and Co., Inc., and an advisor to Storm Therapeutics and Eikon Therapeutics.
Author contributions
TJR and TRC conceived the project and designed the research. TJR designed the experiments. TJR, AJB, and TRC analyzed the data. TJR and TRC wrote the manuscript.
Supporting information
Fig. S1. TERT promoter consistently has upstream hypermethylation and proximal hypomethylation across different cancer tissue types. Bis‐Seq DNA CpG methylation data for 129 positions across the TERT promoter (same positions as shown in Fig. 1B) for all 23 tissues and 833 cell lines (also analyzed in Fig. 1A). 109 cell lines had been classified as having wildtype (WT) monoallelic expression (MAE) of TERT (“WT MAE”), ‐124 or ‐146 C>T activating promoter mutations (“mutant”), biallelic expression (BAE) of TERT (“WT BAE”), or alternative lengthening of telomeres (ALT). All other cell lines either do not belong to one of these categories or were unknown to be classified for this analysis (“Unclassified”). Each row represents a different cell line. Colors range from red to blue for more to less methylated CpGs, respectively. White represents unavailable data. See Table S1 for chromosomal positions and Table S2 for cell line data.
Fig. S2. TERT promoter CpG methylation data in select cancer cell lines, normal adult cells, and hESCs from the ENCODE UCSC Genome Browser. DNA CpG methylation data for 151 positions across the TERT promoter (spanning the same region shown in Fig. 1 and Fig. S1) for 12 cancer cell lines (all cancer lines are also present in Fig. S1), 6 normal adult cell lines, and 1 hESC line. Each row represents a different cell line. Colors range from red to blue for more to less methylated CpGs, respectively. White represents unavailable data. See Table S1 for chromosomal positions and Table S3 for cell line data.
Fig. S3. Examples of bisulfite conversion sequencing (Bis‐Seq) raw read analysis (CCLE Bis‐Seq dataset). Each graph shows an example analysis of Bis‐Seq reads for a single cancer cell line, at a single CpG position, to determine potentially different degrees of allelic methylation (used in Figure 2A). Graphs are shown in order of increasing likelihood of possessing allelic methylation (S5A‐S5D). Read positions included for analysis contained 3 ‐ 6 CpGs per read and coverage of ≥5 reads per cell line. For calculations, methylated CpGs were assigned a value of 1 and unmethylated CpGs a value of 0. The greater the difference between the mean and mode calculations, the more suggestive it is of allelic methylation behavior.
Fig. S4. Select cell line data from Figure 1A showing bisulfite conversion sequencing (Bis‐Seq) data (CCLE Bis‐Seq dataset) for cell lines used in Fig. 2B, 3B, and 3C. CCLE Bis‐Seq data was unavailable for line LN‐18.
Fig. S5. Chromatin immunoprecipitation (ChIP) validation of H3ac antibody that was used in ChIP bisulfite sequencing (ChIP‐Bis‐Seq). (A) ChIP using H3ac antibody showed effective pull‐down at the proximal TERT promoter, with 2.6‐4.0% of input pulled down (for primers used, see Table S4). (B) ChIP pull‐down in this same region demonstrated significant enrichment for the active, mutant ‐124 allele in two different ‐124 mutant cell lines (U‐87 MG and SK‐N‐SH). Error bars represent standard error of the mean (SEM). *p≤0.02, where statistical analysis was performed using 2‐tailed Student's t‐test with unequal variance.
Fig. S6. Identification of the active TERT SNP in Exon 2. Genomic DNA (gDNA) and Reverse‐Transcription PCR (RT‐PCR) sequencing of TERT exon 2 SNP in RPMI‐8226 cells (WT MAE for TERT). All other cell lines shown in Fig. 3B and 3C had been previously analyzed in this manner to identify the active exon 2 TERT SNP [14].
Table S1. TERT promoter features and positions, including specific CpGs, interrogated in Figures 1A, 1B, and 2B, and Supplemental Figures 1 and 2.
Table S2. Cell line and tissue Bis‐Seq DNA CpG methylation values from 833 cell lines and 23 different cancerous tissue types shown in Figure 1 and Supplemental Figure 1, where methylated CpGs receive a value of 1 and unmethylated CpGs receive a 0.
Table S3. Cell line DNA CpG methylation values from 12 cancer cell lines, 6 normal adult cell lines, and 1 hESC line from the ENCODE UCSC Genome Browser shown in Fig. S2, where methylated CpGs receive a value of 1 and unmethylated CpGs receive a 0.
Table S4. Primer sequences for Figures 2B, 3B, 3C and Figures S5, S6.
Acknowledgements
We thank TRC laboratory members Dan Youmans, Yicheng Long, Josh Stern, Ci Ji Lim, Anne Gooding, and Arthur Zaug for useful discussions. We thank Theresa Nahreini and Nicole Kethley for use of the Cell Culture Facility (University of Colorado Boulder). We thank Amber Scott for assistance with the ChIP‐Bis‐Seq library construction at the BioFrontiers Next‐Generation Sequencing Facility (University of Colorado Boulder). Bis‐Seq and RNA expression datasets were obtained from CCLE, either from their website or by direct request, as described in the Materials and Methods section. This work was funded by National Institutes of Health Grant R01 GM099705 to TRC. TRC is an investigator of the Howard Hughes Medical Institute.
References
- 1. Greider CW & Blackburn EH (1985) Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43(2), 405–413. [DOI] [PubMed] [Google Scholar]
- 2. Roake CM & Artandi SE (2020) Regulation of human telomerase in homeostasis and disease. Nat Rev Mol Cell Biol 21, 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL & Shay JW (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 226, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 4. Shay JW & Bacchetti S (1997) A survey of telomerase activity in human cancer. Eur J Cancer 33, 787–791. [DOI] [PubMed] [Google Scholar]
- 5. Umbricht CB, Sherman ME, Dome J, Carey LA, Marks J, Kim N & Sukumar S (1999) Telomerase activity in ductal carcinoma in situ and invasive breast cancer. Oncogene 18, 3407–3414. [DOI] [PubMed] [Google Scholar]
- 6. Nakamura TM, Gregg BM, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB & Cech TR (1997) Telomerase catalytic subunit homologs from fission yeast and human. Science 277, 955–959. [DOI] [PubMed] [Google Scholar]
- 7. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu C, Morin GB, Harley CB, Shay JW, Lichtsteiner S & Wright WE (1998) Extension of life‐span by introduction of telomerase into normal human cells. Science 279, 349–352. [DOI] [PubMed] [Google Scholar]
- 8. Vaziri H & Benchimol S (1998) Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol 8, 279–282. [DOI] [PubMed] [Google Scholar]
- 9. Huang FW, Bielski CM, Rinne ML, Hahn WC, Sellers WR, Stegmeier F, Garraway LA & Kryukov GV (2015) TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis 4, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bell RJA, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, Choi S, Hong C, He D, Pekmezci M et al (2015) The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 348, 1036–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM & Hockemeyer D (2015) Cancer‐associated TERT promoter mutations abrogate telomerase silencing. Elife 4, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K et al (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339, 959–961. [DOI] [PubMed] [Google Scholar]
- 13. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L & Garraway LA (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rowland TJ, Dumbovic G, Hass EP, Rinn JL & Cech TR (2019) Single‐cell imaging reveals unexpected heterogeneity of telomerase reverse transcriptase expression across human cancer cell lines. Proc Natl Acad Sci USA 116, 18488–18497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stern JL, Theodorescu D, Vogelstein B, Papadopoulos N & Cech TR (2015) Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev 29, 2219–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Avin B, Umbricht C & Zeiger M (2016) Human telomerase reverse transcriptase regulation by DNA methylation, transcription factor binding and alternative splicing. Int J Oncol 49, 2199–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ehrlich M (2010) DNA hypomethylation in cancer cells. Epigenomics 1, 239–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Łuczak MW & Jagodzinski PP (2006) The role of DNA methylation in cancer development. Folia Histochem Cytobiol 44, 143–154. [PubMed] [Google Scholar]
- 19. Cheung H‐H, Lee T, Rennet OM & Chan W‐Y (2009) DNA methylation of cancer genome birth defects. Res Part C Embryo Today Rev 87, 335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pfeifer GP (2018) Defining driver DNA methylation changes in human cancer. Int J Mol Sci 19, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dessain SK, Yu H, Reddel RR, Beijersbergen RL & Weinberg RA(2000) Advances in brief methylation of the human telomerase gene CpG island. Cancer Res 60, 537–541. [PubMed] [Google Scholar]
- 22. Takasawa K, Arai Y, Yamazaki‐Inoue M, Toyoda M, Akutsu H, Umezawa A & Nishino K (2018) DNA hypermethylation enhanced telomerase reverse transcriptase expression in human‐induced pluripotent stem cells. Hum Cell 31, 78–86. [DOI] [PubMed] [Google Scholar]
- 23. Castelo‐Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, Zhukova N, Walker EJ, Martin D, Merino D et al (2013) Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol 14, 534–542. [DOI] [PubMed] [Google Scholar]
- 24. Ramlee MK, Wang J, Toh WX & Li S (2016) Transcription regulation of the human telomerase reverse transcriptase (hTERT) gene. Genes 7, 1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu L, Saldanha SN, Pate MS, Andrews LG & Tollefsbol TO (2004) Epigenetic regulation of human telomerase reverse transcriptase promoter activity during cellular differentiation. Genes Chromosomes Cancer 41, 26–37. [DOI] [PubMed] [Google Scholar]
- 26. Stern JL, Paucek RD, Huang FW, Ghandi M, Nwumeh R, Costello JC & Cech TR (2017) Allele‐specific DNA methylation and its interplay with repressive histone marks at promoter‐mutant TERT. Cell Rep 21, 3700–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zinn RL, Pruitt K, Eguchi S, Baylin SB & Herman JG (2007) hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Am Assoc Cancer Res 67, 194–201. [DOI] [PubMed] [Google Scholar]
- 28. Kumari A, Srinivasan R & Wig JD (2009) Effect of c‐Myc and EcF1 gene silencing and of 5‐azacytidine treatment on telomerase activity in pancreatic cancer‐derived cell lines. Pancreatology 9, 360–368. [DOI] [PubMed] [Google Scholar]
- 29. Xu D, Popov N, Hou M, Wang Q, Björkholm M, Gruber A, Menkel AR & Henriksson M(2001) Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc Natl Acad Sci USA 98, 3826–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Avin B, Wang Y, Gilpatrick T, Workman RE, Lee I, Timp W, Umbricht CB & Zeiger M (2019) Characterization of human telomerase reverse transcriptase promoter methylation and transcription factor binding in differentiated thyroid cancer cell lines. Genes Chromosomes Cancer 58, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ghandi M, Huang FW, Jané‐Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H et al (2019) Next‐generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Plotly Technologies Inc . (2015) Collaborative data science. Montreal, QC: Retrieved from https://plot.ly [Google Scholar]
- 33. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G & Durbin R (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J et al (2020) SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumaki Y, Oda M & Okano M (2008) QUMA: quantification tool for methylation analysis. Nucleic Acids Res 36, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG et al (2013) ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 41 (Database issue), D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang Y, Sebra R, Pullman BS, Qiao W, Peter I & Desnick RJ (2015) Quantitative and multiplexed DNA methylation analysis using long‐read single‐molecule real‐time bisulfite sequencing (SMRT‐BS). BMC Genom 16, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J 17, 5–7. [Google Scholar]
- 39. Krueger F & Andrews SR (2011) Bismark: a flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bryan TM, Englezou A, Oalla‐pozza L, Ounham MA & Reddel RR (1997) Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor‐derived cell lines. Nat Med 3, 1271–1274. [DOI] [PubMed] [Google Scholar]
- 41. Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL & Paul CL (1992) A genomic sequencing protocol that yields a positive display of 5‐ methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Renaud S, Loukinov D, Abdullaev Z, Guilleret I & Bosman FT (2007) Dual role of DNA methylation inside and outside of CTCF‐binding regions in the transcriptional regulation of the telomerase hTERT gene. Nuc Acids Res 35, 1245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koch CM, Andrews RM, Flicek P, Dillon SC, Karaöz U, Clelland GK, Wilcox S, Beare DM, Fowler JC, Couttet P et al (2007) The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res 17, 691–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund‐Sayeed S, Das PK, Kivioja T, Dave K, Zhong F et al (2017) Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu H, Wang G & Qian J (2016) Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet 17, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee DD, Komosa M, Nunes NM & Tabori U (2020) DNA methylation of the TERT promoter and its impact on human cancer. Curr Opin Genet Dev 60, 17–24. [DOI] [PubMed] [Google Scholar]
- 47. Sui X, Kong NA, Wang Z & Pan H (2013) Epigenetic regulation of the human telomerase reverse transcriptase gene: a potential therapeutic target for the treatment of leukemia. Oncol Lett 6, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pettigrew KA, Armstrong RN, Colyer HA, Zhang S‐D, Rea IM, Jones RE, Baird DM & Mills KI (2012) Differential TERT promoter methylation and response to 5‐aza‐20‐deoxycytidine in acute myeloid leukemia cell lines: TERT expression, telomerase activity, telomere length, and cell death. Genes Chromosomes Cancer 51, 768–780. [DOI] [PubMed] [Google Scholar]
- 49. Shin K, Kang MK, Dicterow E & Park N (2003) Hypermethylation of the hTERT promoter inhibits the expression of telomerase activity in normal oral fibroblasts and senescent normal oral keratinocytes. Br J Cancer 89, 1473–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vojta A, Dobrinic P, Tadic V, Bockor L, Korac P, Julg B, Klasić M & Zoldos V (2018) Repurposing the CRISPR‐Cas9 system for targeted DNA methylation. Nuc Acids Res 44, 5615–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu X, Tao Y, Gao X, Zhang L, Li X & Zou W (2016) A CRISPR‐based approach for targeted DNA demethylation. Cell Discov 2, 16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. TERT promoter consistently has upstream hypermethylation and proximal hypomethylation across different cancer tissue types. Bis‐Seq DNA CpG methylation data for 129 positions across the TERT promoter (same positions as shown in Fig. 1B) for all 23 tissues and 833 cell lines (also analyzed in Fig. 1A). 109 cell lines had been classified as having wildtype (WT) monoallelic expression (MAE) of TERT (“WT MAE”), ‐124 or ‐146 C>T activating promoter mutations (“mutant”), biallelic expression (BAE) of TERT (“WT BAE”), or alternative lengthening of telomeres (ALT). All other cell lines either do not belong to one of these categories or were unknown to be classified for this analysis (“Unclassified”). Each row represents a different cell line. Colors range from red to blue for more to less methylated CpGs, respectively. White represents unavailable data. See Table S1 for chromosomal positions and Table S2 for cell line data.
Fig. S2. TERT promoter CpG methylation data in select cancer cell lines, normal adult cells, and hESCs from the ENCODE UCSC Genome Browser. DNA CpG methylation data for 151 positions across the TERT promoter (spanning the same region shown in Fig. 1 and Fig. S1) for 12 cancer cell lines (all cancer lines are also present in Fig. S1), 6 normal adult cell lines, and 1 hESC line. Each row represents a different cell line. Colors range from red to blue for more to less methylated CpGs, respectively. White represents unavailable data. See Table S1 for chromosomal positions and Table S3 for cell line data.
Fig. S3. Examples of bisulfite conversion sequencing (Bis‐Seq) raw read analysis (CCLE Bis‐Seq dataset). Each graph shows an example analysis of Bis‐Seq reads for a single cancer cell line, at a single CpG position, to determine potentially different degrees of allelic methylation (used in Figure 2A). Graphs are shown in order of increasing likelihood of possessing allelic methylation (S5A‐S5D). Read positions included for analysis contained 3 ‐ 6 CpGs per read and coverage of ≥5 reads per cell line. For calculations, methylated CpGs were assigned a value of 1 and unmethylated CpGs a value of 0. The greater the difference between the mean and mode calculations, the more suggestive it is of allelic methylation behavior.
Fig. S4. Select cell line data from Figure 1A showing bisulfite conversion sequencing (Bis‐Seq) data (CCLE Bis‐Seq dataset) for cell lines used in Fig. 2B, 3B, and 3C. CCLE Bis‐Seq data was unavailable for line LN‐18.
Fig. S5. Chromatin immunoprecipitation (ChIP) validation of H3ac antibody that was used in ChIP bisulfite sequencing (ChIP‐Bis‐Seq). (A) ChIP using H3ac antibody showed effective pull‐down at the proximal TERT promoter, with 2.6‐4.0% of input pulled down (for primers used, see Table S4). (B) ChIP pull‐down in this same region demonstrated significant enrichment for the active, mutant ‐124 allele in two different ‐124 mutant cell lines (U‐87 MG and SK‐N‐SH). Error bars represent standard error of the mean (SEM). *p≤0.02, where statistical analysis was performed using 2‐tailed Student's t‐test with unequal variance.
Fig. S6. Identification of the active TERT SNP in Exon 2. Genomic DNA (gDNA) and Reverse‐Transcription PCR (RT‐PCR) sequencing of TERT exon 2 SNP in RPMI‐8226 cells (WT MAE for TERT). All other cell lines shown in Fig. 3B and 3C had been previously analyzed in this manner to identify the active exon 2 TERT SNP [14].
Table S1. TERT promoter features and positions, including specific CpGs, interrogated in Figures 1A, 1B, and 2B, and Supplemental Figures 1 and 2.
Table S2. Cell line and tissue Bis‐Seq DNA CpG methylation values from 833 cell lines and 23 different cancerous tissue types shown in Figure 1 and Supplemental Figure 1, where methylated CpGs receive a value of 1 and unmethylated CpGs receive a 0.
Table S3. Cell line DNA CpG methylation values from 12 cancer cell lines, 6 normal adult cell lines, and 1 hESC line from the ENCODE UCSC Genome Browser shown in Fig. S2, where methylated CpGs receive a value of 1 and unmethylated CpGs receive a 0.
Table S4. Primer sequences for Figures 2B, 3B, 3C and Figures S5, S6.