

SUMMARY
The DNA-dependent pattern recognition receptor, cGAS (cyclic GMP-AMP synthase), mediates communication between the DNA damage and the immune responses. Mitotic chromosome missegregation stimulates cGAS activity; however, it is unclear whether progression through mitosis is required for cancercell-intrinsic activation of anti-tumor immune responses. Moreover, it is unknown whether cell cycle checkpoint disruption can restore responses in cancer cells that are recalcitrant to DNAdamage-induced inflammation. Here, we demonstrate that prolonged cell cycle arrest at the G2-mitosis boundary from either excessive DNA damage or CDK1 inhibition prevents inflammatory-stimulated gene expression and immune-mediated destruction of distal tumors. Remarkably, DNAdamage-induced inflammatory signaling is restored in a RIG-I-dependent manner upon concomitant disruption of p53 and the G2 checkpoint. These findings link aberrant cell progression and p53 loss to an expanded spectrum of damage-associated molecular pattern recognition and have implications for the design of rational approaches to augment anti-tumor immune responses.
In Brief
Chen et al. show that prolonged cell cycle arrest before mitosis prevents inflammatory signaling and anti-tumor immunity. Concomitant disruption of p53 and the G2 checkpoint restores DNAdamage-induced inflammatory signaling in a cGAS- and RIG-I-dependent manner.
Graphical Abstract
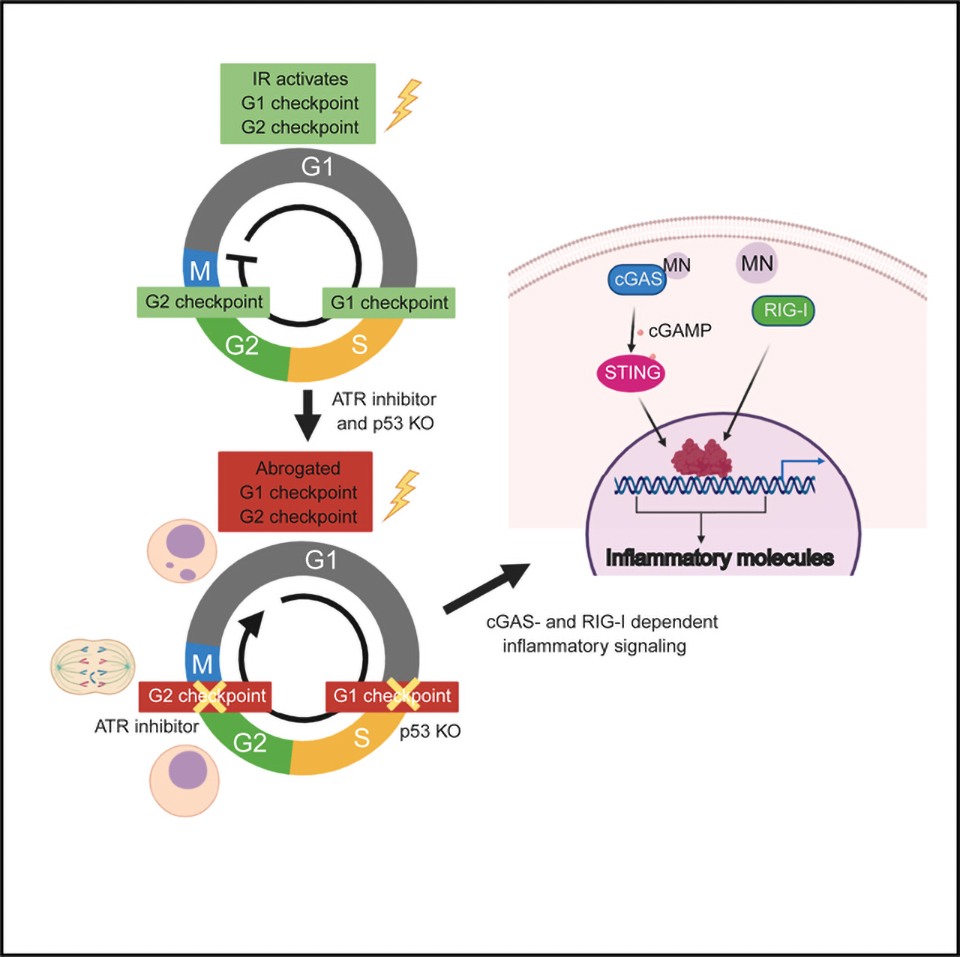
INTRODUCTION
Emerging evidence indicates that the efficacy of radio- and chemotherapies requires DNAdamage-induced activation of cytotoxic immune responses (Formenti et al., 2018; Lee et al., 2009; Liang et al., 2013; Postow et al., 2012). The underlying mechanism for how radiotherapy activates anti-tumor immune responses remains obscure but is thought to involve radiation-stimulated expression of type I interferon and other cytokines in cancer cells and surrounding stroma (Burnette et al., 2011; Deng et al., 2014; Woo et al., 2014). Studies from our laboratory and from others demonstrate mitotic progression after genotoxic stress is required to activate type-I interferon signaling that is associated with the pattern recognition receptor (PRR) cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS) localizing to cytosolic DNA within micronuclei (Bakhoum et al., 2018; Harding et al., 2017; Mackenzie et al., 2017; Santaguida et al., 2017; Yang et al., 2017). Whether cell cycle progression affects the efficacy of combined DNA damaging and immune therapies remains unknown. Similarly, it is unclear whether activation of such responses is feasible in cells that demonstrate persistent cell cycle arrest or loss of the cGAS-stimulator of interferon response cGAMP interactor 1 (STING) pathway.
Ionizing radiation (IR)-induced DNAdamage responses invoke double-strand break (DSB) repair and cell cycle checkpoints that delay entry into S phase or mitosis. Such events are thought to allow adequate time for DSB repair. The IR-induced G2/M cell cycle checkpoint requires the ataxia telangiectasia and Rad3-related (ATR)-checkpoint kinase 1(CHK1) pathway with additional contributions from the ataxia telangiectasia mutated (ATM) kinase (Abraham, 2001; Liu et al., 2000; Xu et al., 2002). The G1/S cell cycle checkpoint is dependent on ATM-mediated p53 induction and transcriptional activation of its target genes (Barlow et al., 1997; Canman et al., 1998; Kastan et al., 1992). Tumor cells with unstable genomes more frequently missegregate chromosomes during mitosis, leading to the formation of micronuclei. Nuclear envelope integrity is compromised in approximately 50% of micronuclei, allowing cGAS and other cytoplasmic proteins to recognize their double-stranded DNA (dsDNA) contents (Hatch et al., 2013, 2018; Liu et al., 2018). This localization correlates with cGAMP and subsequent activation of its signal transducer STING in cells that have experienced genotoxic stress (Coquel et al., 2018; Dou et al., 2017; Glück et al., 2017; Harding et al., 2017; Mackenzie et al., 2017). The detection of foreign cytosolic RNA is mediated largely by RIG-I-like receptors (RLRs), including RIG-I, MDA5, and LGP2 (Ablasser and Hur, 2020). RNA polymerase-III-dependent transcription on cytosolic DNA has also been reported to stimulate RIG-I-dependent inflammatory cytokine production (Ablasser et al., 2009; Chiu et al., 2009).
Recent findings illuminate several distinct possibilities to limit inflammatory responses to genotoxic agents. We reported that deficiency in canonical non-homologous end joining (c-NHEJ) abrogates micronuclei formation and renders cells unable to activate cGAS-STING-dependent inflammatory signaling in response to IR-induced DNA damage (Harding et al., 2017). Tumor cells can also escape immune surveillance by silencing the cytosolic DNA-sensing pathway (Kwon and Bakhoum, 2020), preventing signaling responses to inflammatory cytokines or suppression of antigenic peptide presentation (Benci et al., 2019; Ishizuka et al., 2019; Patel and Minn, 2018). cGAS or STING expression is reported to be reduced in many cancer cell lines, including melanoma, and in tumor cells that rely on alternative telomere maintenance or express oncogenic DNA tumor viruses (Chen et al., 2017; Lau et al., 2015; Xia et al., 2016). These clinically relevant obstacles necessitate alternative approaches that can abrogate persistent cell cycle checkpoint activation and promote inflammatory signaling, irrespective of canonical DNA sensing by cGAS-STING. Here, we delineate the importance of DNAdamage-induced cell cycle checkpoints in relation to anti-tumor immune responses and describe cooperation between ATR- and p53-dependent cell cycle checkpoints in limiting activation of DNA- and RNA-sensing pattern recognition receptors.
RESULTS
CDK1 or c-NHEJ Inhibition Suppresses DNADamage-Induced Inflammatory Signaling and Anti-Tumor Immune Responses
We performed a kinetic analysis of gene expression to decipher the complexity of inflammatory signaling pathways that are activated in response to ionizing radiation (IR)-induced DNA damage. MCF10A cells were irradiated with 10 Gy and collected for RNA sequencing (RNA-seq) analysis at 3 and 5 days, respectively (Figure 1A). Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) revealed that interferon-α, interferon-γ, and interleukin-6 (IL-6)-JAK-STAT3 signaling were the top activated inflammatory pathways following IR treatment (Figure 1B). Interestingly, interferon-α and interferon-γ signaling further increased at 5 days compared with 3 days after IR; however, IL6-JAK-STAT3 signaling plateaued from day 3 onward, suggesting these pathways might be regulated differently. Next, we asked whether each inflammatory signature would be affected if cell cycle arrest was achieved to prevent entry to mitosis. Indeed, the CDK1-specific inhibitor RO-3306 (CDK1i) prevented the activation of all three signaling pathways (Figure 1C). Interestingly, c-NHEJ deficiency by loss of XRCC4 (Rooney et al., 2004) also significantly compromised inflammatory signaling (Figure 1D). XRCC4 is not required for Ku70/Ku80 DSB end recognition or activation of DNA-PKcs, but rather for the final ligation step of c-NHEJ (Graham et al., 2016). We, therefore, postulated that reductions in interferon-stimulated gene expression are secondary to cell cycle arrest before mitosis because of excessive unrepaired DNA damage and not because of a direct role in inflammatory signaling. Indeed, we observed persistent DNA damage, indicated by γH2AX in XRCC4 knockout (KO) cells, even 3 days after 10-Gy IR (Figure S1A). In contrast to wild-type (WT) cells, which initially arrested at G2 and began to progress through mitosis 16 h after IR, XRCC4-deficient cells remained persistently arrested at the G2-mitosis boundary (Figure S1B). In contrast, a low dose of 2Gy irradiation, which induced less DNA damage and restored micronuclei formation and inflammatory signaling in MCF10A XRCC4 KO cells (Figures S1C and S1D). This observation is consistent with a model in which c-NHEJ deficiency impairs cell cycle progression as a consequence of excessive DNA damage after 10 Gy, rather than through the putative role of c-NHEJ factors (i.e., DNA-PK) in pattern recognition (Burleigh et al., 2020; Ferguson et al., 2012; Zhang et al., 2011). We further analyzed inflammatory gene expression in genetically defined MCF10A cells. IR treatment stimulated 81 differentially expressed genes, most of which were upregulated (Figure 1E). Loss of pattern recognition receptor cGAS or STING compromised that increase, confirming their roles in mediating IR-induced inflammatory signaling. Notably, CDK1i-treated cells suppressed these inflammatory signatures to a greater extent, suggesting that progression through mitosis is the more critical determinant of IR-induced inflammatory signatures.
Figure 1. G2/M Cell Cycle Arrest or c-NHEJ Deficiency Suppresses IR-Induced Inflammatory Signaling.

(A) Scheme showing the library preparation workflow for RNA-seq. Created with BioRender.
(B–D) Gene set enrichment analysis (GSEA) of RNA-seq data in MCF10A cells to identify enriched biological pathways at 3 days after 10 Gy versus untreated cells, 5 days after 10 Gy versus untreated cells, and 5 days versus 3 days after 10 Gy (B); 5 days after 10 Gy with CDK1i versus 5 days after 10 Gy with DMSO (C); and 5 days after 10 Gy in XRCC4 KO versus 5 days after 10 Gy in WT (D), respectively. Significant GSEA enrichment score curves were noted for interferon-α response, interferon-γ response, and IL6-JAK-STAT3 signaling. The green curve in the displayed GSEA thumbnails represents the enrichment score curve. Genes on the far left (red) correlated with treatment condition, and genes on the far right (blue) correlated with the control condition. The vertical black lines indicate the position of each gene in the studied gene set. The normalized enrichment score (NES) and false discovery rate (FDRq) are shown for each pathway.
(E) Heatmap showing the Z score of FPKM expressions in control, CDK1i, cGAS KO, STING KO, and XRCC4 KO MCF10A cells for genes differentially expressed at 5 days after 10-Gy treatment (fold change > 2; p value < 1e10).
See also Figure S1.
Given the necessity of mitotic progression in DNAdamage-activated inflammatory signaling, we speculated that progression through mitosis might also have a critical role in anti-tumor immunity. To that end, we employed the B16F10 murine melanoma model to determine whether cell cycle arrest or c-NHEJ deficiency affected anti-tumor immunity (Figure 2A). Untreated B16 WT cells were subcutaneously injected in one flank of C57BL/ 6J mice 2 days before injection of ex vivo-treated B16 cells in the opposite flank. This model provides a method to track anti-tumor immunity without the confounding effect of systemic administration of drugs to animals (e.g., CDK1i), which may have unanticipated consequences, including on the immune system. Importantly, we have previously shown that ex vivo irradiation and subsequent implantation of tumor cells produced equivalent responses to in vivo radiation, as is classically done in abscopal models (Harding et al., 2017). Moreover, anti-CTLA-4 alone had limited effect on the abscopal tumors, but addition of radiotherapy markedly increased that response (Harding et al., 2017; Twyman-Saint Victor et al., 2015). The different treatments of the primary tumor included 10-Gy IR and CDK1i- or CRISPR-Cas9-mediated deletion of the c-NHEJ gene Ku70 (Ku70 KO). Immune checkpoint blockade was achieved using an anti-CTLA-4 (clone 9H10) antibody administered by intraperitoneal injection every 3 days for a total of three doses, as previously described (Harding et al., 2017). Growth rates of untreated tumors were then monitored after the final anti-CTLA-4 antibody injection. Consistent with suppression of inflammatory signatures in RNA-seq data in Figure 1E, cell cycle arrest by CDK1i abolished the IR-induced anti-tumor effect (Figures 2B and 2D). These data agree with the requirement for mitotic progression in irradiated tumor cells to activate systemic immune responses. We considered an alternative possibility that an extended G2 arrest during CDK1i treatment may enhance DNA repair in these cells, thus accounting for the reduction in immune activation following IR. To address that issue, we examined how the loss of the critical c-NHEJ repair factor Ku70 would affect systemic immune responses. Ku70-deleted B16F10 cells also failed to elicit anti-tumor immune responses to limit the growth of untreated tumors (Figures 2C and 2E) despite the importance of c-NHEJ in repairing IR-induced DNA damage. Taken together, these findings indicate that progression through mitosis is a key gatekeeper for IR-induced inflammatory signaling and anti-tumor immunity.
Figure 2. Progression of the Irradiated Tumor Cells through Mitosis Is Required for Systemic Anti-tumor Immune Responses.

(A) Scheme showing B16 melanoma model. Created with BioRender.
(B and C) Growth of WT B16 cells (untreated tumors) after injection of B16 WT cells with no treatment, 10-Gy irradiation, or 10-Gy irradiation with CDK1i treatment (B), and WT B16 cells or Ku70 KO B16 cells with no treatment or 10-Gy irradiation (C) 3 days before implantation, respectively. All mice were administrated with anti-CTLA-4 antibody (9H10) as described in (A). Animal numbers are indicated in parentheses of treatment groups.
(D and E) Statistic of tumor volumes at day 15 as measured in (B) and (C). Statistical significance is compared using a two-tailed t test. Error bars are SEM of biological replicates.
Cell Cycle Checkpoint Abrogation Accelerates Micronuclei Formation and Inflammatory Signaling
A corollary to the prior experiments is that abrogation of the G2/M checkpoint would enhance the kinetics and amplitude of micronuclei formation and inflammatory signaling. To test that possibility, we irradiated cells with 20 Gy, a dose expected to induce a strong G2/M cell cycle checkpoint, in combination with inhibitors of ATR or CHK1 kinase activity. Contrary to the limited micronuclei formation in DMSO-treated cells, approximately 50% of ATR-inhibitor (ATRi)- or CHK1-inhibitor (CHK1i)-treated cells formed micronuclei just 24 h after 20 Gy IR. A considerable portion of cells formed micronuclei in the DMSO-treated group 72 h after IR; however, their frequency was still increased after ATRi or CHK1i treatment (Figure 3A). Cell cycle analysis by flow cytometry confirmed abrogation of the G2/M checkpoint by ATRi or CHK1i (Figure 3B). In response to 20 Gy of irradiation, most cells arrested in G2. ATRi or CHK1i did not alter the cell cycle distribution in non-irradiated cells, but greatly increased G1phase populations in irradiated cells, consistent with cells traversing mitosis. To further determine whether cells progressed through mitosis after IR, we performed dye-dilution experiments (Figure S1E), which enable quantification of cell division numbers over a given interval. Reductions in fluorescence indicate cell division because of dye dilution. Irradiated MCF10A cells underwent one to two divisions over 3 days, confirming mitotic progression after IR. This change in dye content was suppressed by CDK1i administration. ATRi/CHK1i-treated cells exhibited evident STAT1 Y701 phosphorylation after 24 h, which increased strongly at three days (Figure 3C). Prevention of mitotic entry using either CDK1i or a PLK inhibitor (PLKi) abolished the enhanced inflammatory signaling (Figure 3D).
Figure 3. Disruption of the IR-Induced G2/M Checkpoint Enhances Inflammatory Signaling Activation in MCF10A Cells.

(A) MCF10A cells were irradiated or left untreated with or without the indicated treatments, followed by fixation at the indicated times. Cells with micronuclei were quantified. Mean values and SEM are plotted (n = 3).
(B) MCF10A cells irradiated or left untreated with or without indicated inhibitors were fixed and subjected to cell cycle analysis by flow cytometry. Mean values and SEM are plotted (n = 3).
(C and D) MCF10A cells irradiated with the indicated dose or left untreated in the presence of the indicated inhibitors were collected at the specified time point for western blot.
(E) MCF10A I-PpoI cells were left untreated (NIR), irradiated with 10 Gy (IR), or treated with 4-OHT and shield-1 for 5 h (I-PpoI), and then maintained in medium with or without ATR inhibitor for 3 days before collection for western blot analysis.
(F) MCF10A AsiSI cells were left untreated (NIR), irradiated with 10 Gy (IR), induced with 4-OHT and shield-1 for 5 h(AsiSI), or cultured in the presence of aphidicolin (Aph), and then maintained in medium with or without ATR inhibitor for 3 days before collection for western blot analysis. For Aph-treated cells, 2.5 μM aphidicolin was included in the medium until cell collection 3 days later.
(G) MCF10A cells left untreated or irradiated with 10 Gy were maintained for 3 days in the presence or absence of ATR inhibitor before collection for western blot analysis.
Consistently, ATR-inhibitor-treated cells, after 10 Gy of irradiation, also showed increased inflammatory signaling (Figures 3E– 3G), demonstrating that disruption of the G2 checkpoint produces similar results across a range of damage induction. Moreover, the increased inflammatory signaling was not limited to IR-induced DNA damage. ATRi treatment also increased inflammatory signaling in response to DSBs induced by two different nucleases, I-PpoI and AsiSI (Figures 3E and 3F) and in response to replication stress with aphidicolin (Figure 3F). Increased inflammatory signaling after ATRi was also largely dependent on the cGAS-STING pathway (Figure 3G), further highlighting the importance of mitotic progression for activation of this DNA-sensing axis.
Disruption of G2 and G1 Checkpoints Cooperate to Restore IR-Induced Immune Responses in c-NHEJ-Deficient Cells
The observation that G2 checkpoint disruption was able to promote micronuclei formation and inflammatory signaling encouraged us to test whether ATRi treatment can activate interferon-stimulated gene expression in irradiated cells that lack c-NHEJ repair. Unexpectedly, inhibition of ATR failed to restore micronuclei formation after 10 Gy (Figures 4A and 4B), and only slightly increased inflammatory signaling in XRCC4 KO cells (Figures 4D and 4E). In contrast, ATRi enhanced both markers in XRCC4 KO cells upon a low dose of irradiation at 2 Gy (Figures S1C and S1D). This minimal effect can be partially explained by mitotic progression in cells with excessive DNA damage leading to mitotic catastrophe (Vakifahmetoglu et al., 2008). In agreement, live cell imaging showed that 85% of XRCC4 KO cells died during mitosis upon releasing cells at the G2/M boundary after 10 Gy compared with only 30% of WT cells (Figures S2A and S2B).
Figure 4. Disruption of p53 and ATR Restores IR-Induced Inflammatory Signaling in c-NHEJ-Deficient MCF10A Cells.

(A–C) WT cells, p53 KO cells, XRCC4 KO cells, or XRCC4 P53 DKO (double knockout) cells were irradiated with 10 Gy (IR), and then maintained in medium with or without ATR inhibitor for 3 days before fixation for immunofluorescence staining. Mean values and SEM are plotted (n = 3).
(D and E) WT cells, p53 KO cells, XRCC4 KO cells or XRCC4 P53 DKO cells were irradiated with 10 Gy and then cultured for 3 days in the presence or absence of ATR inhibitor before collection for western blot analysis.
(F) Bar plot showing the normalized enrichment score (NES) and false discovery rate (FDRq) for the identified enriched biological pathways in MCF10A cells 3 days after 10 Gy irradiation.
To understand whether mitotic progression after DSB induction is sufficient to activate inflammatory signaling, we released cells from G2 arrest after irradiation and examined inflammatory signaling by immunoblot. Surprisingly, direct release of irradiated cells from G2 resulted in minimal micronuclei formation and inflammatory signaling as assessed by ISG15 (interferon-stimulated gene 15) and STAT1 Y701 phosphorylation on immunoblot at 3 days after IR (Figures S2C and S2D). The cells that exhibited micronuclei in this population were EdU+, suggesting cell cycle progression through the S phase, and a second round of mitosis had occurred (Figures S2C, S2E, and S2F). This finding prompted us to ask whether the entry into the S phase with damage in MCF10A XRCC4 KO cells is a critical barrier to inflammatory activation in addition to the G2/M checkpoint. Given the important role of ATM and p53 in the IR-induced G1/S checkpoint (Barlow et al., 1997; Canman et al., 1998; Kastan et al., 1992), we generated MCF10A p53 KO cell lines using CRISPRCas9 to test the importance of the G1/S checkpoint disruption in this inflammatory response to DNA damage. p53 loss led to increased S-phase entry, indicated by EdU incorporation (Figure S2E), and mildly higher percentages of cells with micronuclei (Figures 4A, 4B, and S2F). Despite equal cell death rates during mitosis, p53 loss increased cell viability in the subsequent interphase at 24 h after mitosis (Figure S2G), consistent with its known role in responding to DNA damage (Nikoletopoulou et al., 2013). Introduction of S-phase-entry inhibitors in cells released from G2 resulted in minimal cell populations with micronuclei at 3 days after IR in WT cells (Figures S2E and S2F). However, there were much higher percentages of cells with micronuclei in p53 KO cells, which further suggested a role of p53 in cell death during the subsequent interphase after mitotic progression with DNA damage (Figures S2E and S2F).
The preceding findings suggested that several cell cycle checkpoints affect pattern recognition in response to genotoxic stress. We, therefore, generated MCF10A cells with deletion of both XRCC4 and p53 and subjected them to 10 Gy irradiation in the presence of ATRi to understand whether the combined loss of the G1/S and G2/M checkpoints could restore inflammatory signals. p53 loss increased the cell populations accumulated at S and G2 phases as indicated by CENP-F positivity in cells at 3 days after IR (Figures 4C and 4D). Remarkably, p53 loss combined with ATRi restored micronuclei formation (Figures 4A and 4B) and inflammatory signaling (Figures 4D–4F). Similar to p53 loss, ATM inhibition cooperated with ATRi to restore inflammatory signaling in XRCC4-deficient MCF10A cells (Figures S3A and S3B). Restored inflammatory signaling was also consistently found upon disruption of ATR and p53 checkpoints in Ku70-deficient cells (Figures S3C and S3D).
ATR Inhibition Cooperates with p53 Mutation to Activate cGAS-Independent Inflammatory Signaling
Loss of cGAS or STING did not completely eliminate inflammatory signaling upon ATRi treatment (Figure 3G), leading us to speculate that the presence of additional damage-associated molecular patterns. To address that possibility, we used human telomerase reverse transcriptase (hTERT)-immortalized retinal pigmented epithelial (RPE-1) cells, a cell line that shows undetectable cGAS expression by either immunofluorescence or western blot (Figures S4A and S4B). Similar levels of STING expression were present in RPE-1 cells when compared with MCF10A cells (Figure S4B). In response to irradiation and ATRi, RPE-1 WT cells failed to discernibly activate inflammatory signaling (Figures 5A and 5B), despite approximately 50% of cells containing micronuclei (Figures 5C and 5D). Strongly increased inflammatory signaling occurred in RPE-1 WT cells upon ectopic cGAS expression but not after introduction of a cGAS-inactive mutant K411A (Figures 5A and 5B), revealing that WT cGAS promoted signaling responses to missegregated genomic DNA in RPE-1 cells, as shown in MCF10A and other independent cell lines. We also observed the RNA-sensing pattern recognition receptor RIG-I was markedly increased in RPE-1 WT cells with ectopic cGAS after IR, consistent with reports that RIG-I expression is induced by inflammatory cytokines (Liu and Gu, 2011; Yuzawa et al., 2008). Surprisingly, inflammatory signaling was strongly present in ATRi-treated RPE-1 p53 KO cells in response to 10 Gy IR, irrespective of cGAS (Figures 5B). Endogenous cGAS expression was not detected in either p53 WT or KO RPE-1 cells (Figures 5A and 5B), arguing against its involvement in damage-induced inflammatory signaling in this setting. Notably, inflammatory-stimulated gene expression remained dependent on mitotic progression because CDK1i abolished this effect (Figure 5B). We performed RNA-seq analysis to independently confirm these results and more extensively compare gene expression in each of the different settings. Activation of interferon-γ, interferon-α, and IL6-JAK-STAT3 signaling responses occurred in both RPE-1 WT cells with ectopic cGAS expression and in RPE-1 p53 KO cells that lack cGAS expression (Figure 5E).
Figure 5. p53 Loss Cooperates with ATR Inhibition to Activate cGAS-Independent Inflammatory Signaling.

(A and B) WT cells or p53 KO cells were irradiated with 10 Gy and cultured in medium with (A) or without (B) ATR inhibitor followed by collection for western blot analysis 3 days later.
(C and D) WT cells or p53 KO cells were irradiated with 10 Gy and maintained in medium with or without ATR inhibitor before fixation at indicated time. Cells with micronuclei were qualified (D), and representative images for cells with micronuclei 3 days after IR are shown in (C). Mean values and SEM (n = 2) are plotted.
(E) RNA-seq data of ectopic cGAS versus mock and p53 KO versus WT were interrogated by gene set enrichment analysis (GSEA) to identify enriched biological pathways in RPE-1 cells 3 days after 10 Gy irradiation in the presence of ATR inhibitor, respectively. Significant GSEA enrichment score curves were noted for interferon-a response, interferon-g response, and IL6-JAK-STAT3 signaling. In GSEA thumbnails, the green curve represents the enrichment score curve. Genes on the far left (red) correlated with former cells, and genes on the far right (blue) correlated with latter cells. The vertical black lines indicate the position of each gene in the studied gene set. The normalized enrichment score (NES) and false discovery rate (FDR) are shown for each pathway.
See also Figure S4.
RIG-I Mediates Inflammatory Signaling in RPE-1 p53 KO Cells with ATRi Treatment
cGAS independence after combined disruption of G1/S and G2/ M checkpoints raises the possibility that non-DNA-sensing pattern-recognition receptor(s) underlie inflammatory signaling in this setting. RIG-I has been reported to mediate inflammatory signaling to several different stimuli and has emerged as a candidate for cGAS-independent responses (Ablasser et al., 2009; Chiu et al., 2009; Nabet et al., 2017). RIG-I KO single clones were generated with the RPE-1 p53 KO cell line, and its functional deficiency was confirmed with the RIG-I-specific agonist 5’-triphosphate (50ppp)-dsRNA (Figure S5A). Importantly, STING activity was unaffected in RPE-1 p53 and RIG-I double-KO clones; 2’3’-cGAMP activated STING-mediated increases in pSTAT1, ISG56, and ISG15 (Figure S5B). RIG-I loss in more than 10 independent RPE-1 p53KO clones compromised the inflammatory signals pSTAT1, ISG56, and ISG15 to varying degrees in response to combined IR and ATRi treatment (Figures 6A and S5C). Consistently, RIG-I was also required for ISG expression in the human colon cancer cell line HCT116 p53−/− (Figure S5D), which, similar to RPE-1 cells, lacks detectable cGAS expression. Importantly, downstream effectors of the RIG-I pathway were activated, as evidenced by the presence of MAVS aggregates (Figure 6B) in RPE-1 p53 KO cells after combined treatment with IR and ATRi. This confirms RNA-sensing pathway activation in response to DNA damage, as opposed to an indirect effect of RIG-I on another pattern-recognition receptor mechanism. RIG-I has been reported to sense RNA transcripts synthesized by RNA polymerase III (Pol III) on cytoplasmic poly (dA:dT) DNA templates (Ablasser et al., 2009; Chiu et al., 2009). Therefore, we tested whether RIG-I-mediated inflammatory signaling in RPE1 p53 KO cells also involve Pol III. We observed that Pol III localized to unruptured micronuclei, but not ruptured micronuclei, as measured by EGFP-BAF signal (Denais et al., 2016) suggesting that a fraction of micronuclei are Pol III positive (Figure S5E). To explore whether Pol III has a functional role in IR+ ATRi induced inflammatory signaling, we tested whether the addition of the Pol III inhibitor (Pol IIIi) could block those signals. Consistent with previous reports, Pol IIIi significantly impaired inflammatory signaling stimulated by poly (dA:dT) DNA (Figure S5F). In contrast, Pol IIIi did not affect the inflammatory signals of pSTAT1, ISG56, and ISG15 in IR- and ATRi-treated cells. Together, these results imply that mediators other than Pol III are responsible for RNA-sensing-pathway activation in checkpoint-disrupted cells.
Figure 6. RIG-I Mediates Inflammatory Signaling in RPE-1 p53 KO Cells after IR and ATRi Treatment.

(A) RPE-1 p53 KO cells and RPE-1 p53 RIG-I DKO cells were irradiated with 10 Gy in the presence of ATRi and maintained for 3 days before collection for western blot analysis.
(B) RPE-1 p53 KO cells were irradiated with 10 Gy and subjected to culture in medium with ATRi for indicated time before crude mitochondria isolation. Equal amounts of isolated crude mitochondria were loaded into 1.5% vertical agarose gels with 0.1% SDS for SDD-PAGE analysis.
(C) Scheme showing a working model of this study. Created with BioRender.
See also Figure S5.
DISCUSSION
cGAS detects cytosolic dsDNA and triggers production of type I interferons and other inflammatory cytokines and chemokines (Sun et al., 2013). After IR-induced genotoxic stress, cells forming micronuclei stimulate cGAS-STING-mediated innate immune activation and anti-tumor immunity. Cell cycle checkpoints appear to have critical roles in this process because abolishment of mitotic progression compromises micronuclei formation and inflammatory signaling (Harding et al., 2017; Mackenzie et al., 2017). Tumors, however, develop distinct mechanisms of immune evasion, including suppressed expression of patternrecognition receptors cGAS and STING to escape immune surveillance (Beatty and Gladney, 2015; Ishizuka et al., 2019; Steven and Seliger, 2018; Xia et al., 2016).
This study demonstrates that G2/M checkpoint adaptation links the activation of various inflammatory signaling pathways to anti-tumor immune responses after DNA damage. Although abrogation of the G2/M checkpoint facilitates micronuclei formation and inflammatory signaling in cells with intact DNA repair, it was not sufficient in cells that resided at the G2/M boundary for prolonged periods because of excessive, unrepaired DSBs. This scenario required combined disruption of both the G1/S and G2/ M checkpoints. Unexpectedly, loss of p53 together with ATR inhibition upregulated inflammatory signaling in several cell lines with undetectable cGAS, suggesting that cell cycle checkpoints coordinate to limit signaling through multiple pattern-recognition receptor pathways. Our findings reveal the cytosolic RNA sensor RIG-I to be an additional component of this response (Figure 6C). This is also consistent with our original report that cGAS or STING loss did not eliminate IR-induced inflammatory-stimulated gene expression (Harding et al., 2017). In further support, combined IR and ATRi significantly increased interferon production and immune infiltration in vivo compared with IR alone in TC-1 tumors with p53-inactivation from human papilloma virus (Dillon et al., 2019). Collectively, these findings imply that the combined loss of G1/S and G2/M checkpoints can enhance immune infiltration and anti-tumor responses.
How checkpoint proteins p53 and ATR cooperate to restrict inflammatory signaling warrants further consideration. A potential explanation is that their combined loss increases micronuclei formation and other structures that may attract cGAS. Our data further support that entry into the S phase and progression through a second round of mitosis enhance micronuclei formation (Figure S2). Disruption of both G1/S and G2/M checkpoints by combined p53 loss and ATR inhibition would augment this accelerated cell proliferation and cytoplasmic DNA accumulation. Interestingly, micronuclei passage through mitosis and chromosome shattering, followed by reassembly in the subsequent interphase, is thought to occur during the process of chromothripsis (Umbreit et al., 2020; Zhang et al., 2015). Such catastrophic chromosome breakage is predicted to not only promote large-scale genomic rearrangements but also to increase the number of acentric fragments that could missegregate into the cytoplasm and augment inflammatory signaling.
The role of checkpoint proteins p53 and ATR in restricting RIGI-mediated inflammatory signaling is less obvious. Notably, this response also required progression through mitosis but was not RNA Pol III dependent. An alternative possibility is that these cells experience chromatin alterations that drive changes in transcription globally. Reactivation of endogenous retroviral elements after methyltransferase inhibitors are capable of activating RNA-sensing pathways, and similar activation of endogenous RNA elements have been observed after IR (Chiappinelli et al., 2015; Ranoa et al., 2016; Roulois et al., 2015; Rudin and Thompson, 2001). Interestingly, p53 loss increases derepression of short interspersed nuclear elements (SINEs) after treatment of DSB-inducing agents (Hagan and Rudin, 2007). Whether this interplay occurs after IR treatment is an important avenue for future studies and may suggest additional routes to activate anti-tumor immune responses in p53-mutant cancers.
An additional finding in this study is the complex relationship between intrinsic DNA repair capacity and activation of anti-tumor immunity. Loss of c-NHEJ prevented anti-tumor immune responses in syngeneic melanoma models treated with radiotherapy and immune checkpoint blockade. Although c-NHEJ components DNA-PKcs and KU have been reported as DNA sensors for innate immunity (Burleigh et al., 2020; Ferguson et al., 2012; Zhang et al., 2011), we observed similar reductions in cells that have lost end ligation factors XRCC4 and Lig4, (Graham et al., 2016). XRCC4 and LIG4 are not required for DSB end recognition or signaling, making them unlikely candidates for pattern recognition. We propose the effects of c-NHEJ loss are secondary to excessive unrepaired DSBs and persistent cell cycle arrest. Consistent with this assertion, low IR doses induce slightly greater micronuclei formation and inflammatory signaling in XRCC4 KO cells as compared with high IR doses. They also predict that lower doses of DNA-damaging agents may be necessary to maximally recruit anti-tumor immune responses in the setting of DNA-repair-deficient cancers. Additional reports show that fractionated “low” doses do not induce expression of the cytoplasmic nuclease TREX1 to the extent observed for single high doses of IR (Vanpouille-Box et al., 2017). It remains unclear, however, whether that contributed to our observations.
Taken together, our work reveals the suppressive role of cell cycle checkpoints in DNAdamage-induced inflammatory signaling. These data suggest potential approaches to stratify patients based on tumor genotypes and alternative strategies to activate inflammatory signaling in tumors that harbor resistance mechanisms.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Roger Greenberg (rogergr@pennmedicine.upenn.edu)
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The RNA-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE145148 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145148).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells
MCF10A cells were obtained from ATCC and cultured in DMEM/F-12 media with 5% horse serum (Thermo Fisher Scientific), 20 ng/ml human EGF (Peprotech), 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin and 10 μg/ml recombinant human insulin (Sigma). MCF10A I-PpoI cells and MCF10A AsiSI cells were previously described (Harding et al., 2017). hTERT RPE-1 cells and hTERT RPE-1 p53 KO cells were kindly provided by Dr. Dan Durocher (Lunenfeld-Tanenbaum Research Institute) and cultured in DMEM media with 10% bovine calf serum (Hyclone). B16-F10 cells were purchased from ATCC and cultured in DMEM with 10% bovine calf serum (Hyclone). HCT116 p53−/− cells (obtained from B Vogelstein) were cultured in McCoy’s 5a media with 10% bovine calf serum (Hyclone). All cells were maintained with penicillin and streptomycin (Thermo Fisher Scientific).
RPE-1 cell lines expressing mCherry-tagged cGAS were generated using lenti-viral plasmid pLVX-mCherry as a backbone. Knockout cell lines were achieved by lenti-viral infection of LentiGuide puro-based gRNA in cells with Cas9 expression.
To determine IR-induced inflammatory signaling, chemicals was administrated in medium 1 h before IR treatment. Unless otherwise indicated, cells were collected 3 days later for subsequent analyses. Medium were changed every two days.
For live cell imaging, cells were arrested at G2 phase using 9 uM CDK1i (RO-3306) for 24 h. G2 arrested cells were then irradiated on ice followed by washing twice with PBS and replaced with medium containing 0.5 μM siR-DNA.
MICE
We used 5–7 weeks old female C57BL/6 mice. Animals were sequentially subcutaneously injected with untreated and treated tumors followed by anti-CTLA4 antibody administration. Untreated tumor volumes were then measured every other day. The animal experiments were performed according to protocols approved by the Institute of Animal Care and Use Committee of the University of Pennsylvania (IACUC).
METHOD DETAILS
Plasmid construction
pLVX-mCherry-cGAS was generated as previously described (Harding et al., 2017). pLVX-mCherry-cGAS K411A was generated by site-directed mutagenesis. LentiGuide-puro targeting p53 and RIG-I was generated by inserting targeting duplex oligos into LentiGuide-puro at BsmBI site.
Irradiation and drug treatments
Cells were seeded at densities of 40%–50% at the time of treatment. Cells were irradiated using a Cs-137 Gammacell irradiator (Nordion) with ~0.76 Gy/min. Inhibitors were administrated 1 h prior to irradiation and maintained till collection unless otherwise stated. Media with or without inhibitors were changed every 2 days after treatment. Medium containing Pol IIIi were changed every day. Inhibitors were used at following concentrations: CDK1i (9 μM, RO-3306, Selleck Chemical), ATRi (2.5 μM, VE-821, Selleck Chemical), ATMi (10 μM, Ku55933, Selleck Chemicals), and Pol IIIi (40 μM, ML-60218, Millipore). Agonists were transfected using Avalanche-Omni (EZT-OMNI-1, EZ Biosystems) and incubated for 18 hours before collection. Agonist were used as following concentrations: 5’ppp-dsRNA (2 μg/ml, tlrl-3prna, Invivogen) and 2’3’-cGAMP (10 μg/ml, tlrl-nacga23, Invivogen). For AsiSI or I-PpoI nuclease induced DSBs, cells were treated with Shield-1 (1 μM, AOB1848, Aobious) and 4-OHT (2 μM, H7904, Sigma) for 5 hours before washing with PBS and adding back media.
CRISPR–Cas9 knockout
Lentiviruses of LentiCas9-blast and LentiGuide-Puro (gifts from F. Zhang, Addgene #52962, Addgene#52963) were produced and concentrated as previously described (Kutner et al., 2009). Cells were infected with lentiviruses overnight in the presence of 8 ug/ml polybrene. Cells infected with LentiCas9 were subjected to 10 μg/ml blasticidin (Invivogen) until control cells without infection were all killed. Knockout clones were generated by infecting cells with lentivirus using lentiGuide-Puro followed by selection in 2 μg/ml puromycin (Sigma) for 2 days. Single colonies were achieved by seeding one cell per 96-well. p53 knockout clones were validated by western blotting using anti-p53 antibody (DO-1, santa cruz). RIG-I knockout clones were validated by western blotting using anti-RIG-I antibody (D33H10, CST).
B16-F10 Ku70 knockout clones were created as previously described (Benci et al., 2016). Ku70 knockout clones were validated by western blotting using anti-Ku70 antibody (A302–624A, bethyl antibody).
Western blotting
Western blotting was performed using standard methods. In brief, cells were collected by trypsinization, washed in PBS and lysed in NETN buffer (150 mM NaCl, 1% NP-40 alternative, 50 mM Tris pH 7.4) with turbo nuclease (Accelagen) in the presence of 1 mM MgCl2, protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (5 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM sodium pyrophosphate decahydrate, 1 mM β-glycerophophate) on ice for 30 min. Protein was quantified using Bradford method. Equal amount of protein was separated on 4%–12% Bis-Tris gels (Thermo Fisher Scientific) using MOPS/MES buffer and transferred to 0.2 μM Amersham Protran Premium Nitrocellulose Western Blotting Membranes (10600004, GE) at 350 mA for 2 h in ice cold transfer buffer (20% methanol, 191 mM glycine, 25 mM tris-base, 0.1% SDS, pH 8.3). Membrane were blocked in 3% non-fat milk in PBST (0.1% Tween 20) and incubated overnight at 4°C in primary antibody diluted with PBS containing 1% BSA. Membrane were washed in PBST for 20 min and incubated at room temperature for 2 h in secondary antibody (Amersham ECL HRP, GE) diluted with PBST containing 3% non-fat milk. Blots were developed using immobilon forte western HRP substrate (WBLUF0100, Millipore).
In vivo MAVS aggregation assay
In vivo MAVS aggregation assay was performed according to a published protocol (Hou et al., 2011). Briefly, crude mitochondria isolated using mitochondria isolation kit (89874, Thermo) were resuspended in 1x sample buffer (0.5 x TBE, 10% glycerol, 2% SDS) supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (5 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM sodium pyrophosphate decahydrate, 1 mM β-glycerophophate) followed by protein concentration quantification using BCA method. Equal amounts of crude mitochondria were mixed with 1/10 volume gel loading dye (7025, NEB) and subjected to Semi-Denaturing Detergent Agarose Gel Electrophoresis (SDD-AGE). Samples were loaded onto a homemade vertical 1.5% agarose gel prepared using Novex empty gel cassettes (NC2015, Thermo), empty gel cassette combs (NC3515, Thermo) and Bio-Rad agarose (1613101, BioRad). After electrophoresis in the running buffer (1 x TBE and 0.1% SDS) for 40 min with a constant voltage of 100 V at 4°C, the proteins were transferred to 0.2 μM Amersham Protran Premium Nitrocellulose Western Blotting Membranes (10600004, GE) at 400 mA for 3 h in ice cold transfer buffer (10% methanol, 191 mM glycine, 25 mM tris-base, 0.1% SDS, pH 8.3) followed by immunoblotting.
Immunofluorescence
Cells were seeded onto coverslips in 24-well plate one day before treatment and fixed with 3% paraformaldehyde (PFA). Cells were then washed with PBS, permeabilized in 0.5% Triton X-100 solution for 1 min at room temperature and processed for immunostaining using the indicated antibodies. Coverslips were mounted in VECTASHIELD Antifade Mounting Medium with DAPI (H-1200–10, Vector Labs). Images were taken using a Nikon Eclips 80i microscope with a Coolsnap Myo camera (Photometrics) and Nikon NIS-Elements software. Images were prepared using FIJI (NIH). Cells with aberrant nuclear structure in cytoplasm were visualized by DAPI staining and counted manually.
Live-cell imaging
MCF10A cells were seeded onto falcon 24-well plates (08–772-1H, Fisher) and cultured in medium as described above in which DMEM/F-12 base medium was replaced by 1:1 mixed FluoroBrite DMEM (A1896701, Thermo) and phenol red free F12 medium (HFL05–500ML, Caisson Labs). CDK1i arrested cells were irradiated on ice, washed, replaced with medium containing 0.5 μM siR-DNA (CY-SC007, Cytoskeleton), a cell permeable far-red probe for DNA, and then immediately subjected to live-cell imaging using EVOS FL Auto Imaging System (Thermo). Imaging was carried out in a 37°C humidified chamber equilibrated with 5% CO2 using a 20x air objective. Images were acquired every 20 min for 20–24 h.
Flow cytometry
Cells were dissociated into single cells with trypsin, washed once in PBS, resuspended in 300 μL PBS, and fixed by dropwise addition of 700 μL pre-chilled (‒20°C) 100% ethanol. After fixation at 4°C or storage at 20°C, cells were washed once with 6 mL 1% BSA/PBS and pelleted by centrifuge. Cell pellets were loosed and resuspended by flick in 100 μL 1% BSA/PBS with 1 μL Alexa Fluor 488 conjugated phospho-Histone H3 (Ser10) antibody (DC28, Cell Signaling). After incubation at room temperature for 1 hour with occasional flick, cells were washed once with 6 mL 1% BSA/PBS and resuspended in PBS containing 10 μg/ml propidium iodide (Santa Cruz) and 100 μg/ml RNase A (QIAGEN). Flow cytometry was performed on a BD FACSCalibur and analyzed with FlowJo software. Single cell population and G1/S/G2 population were manually gated.
Modified RadVax procedure
Experiment was performed as previously described (Harding et al., 2017). In brief, five- to seven-week-old female C57BL/6 mice were obtained from Charles River and maintained under pathogen-free conditions. Mice were divided randomly into cages upon arrival and were randomly injected and measured without blinding. All animal experiments were performed according to protocols approved by the Institute of Animal Care and Use Committee of the University of Pennsylvania (IACUC). The minimal number of animals was used based on prior experience to yield consistent measurements. On the day before the experiment, B16-F10 cells were treated in vitro with 10 Gy of ionizing radiation and cultured with or without CDK1i. CDK1i was added 1h before irradiation and maintained until cell isolation and injection on day 2. On day 0 untreated B16-F10 cells (5 × 104) in 50 μL of PBS were mixed with an equal volume of Matrigel (356237, Corning) and injected into the right flank. On day 2, 5 × 105 IR treated cells including B16-F10 parental cells, parental cells with CDK1i treatment and Ku70 knockout cells were mixed with Matrigel and injected on the opposite flank. On days 5, 8 and 11 anti-CTLA4 antibody (9H10; BioXCell) was administered interperitoneally at 200 μg per mouse. Volumes were measured using calipers starting at day 11 and calculated using the formula I × w2 × 0.52, where I is the longest dimension and w is perpendicular to l. Animals were euthanatized when either tumor reached 1.5 cm in the largest dimension according to IACUC guidelines.
RNA purification, RNA-seq library preparation and analysis
Total RNA for RNA-seq library was purified using miRNeasy Mini Kit (217004, QIAGEN) and validated on an agilent RNA 6000 nano chip (Agilent Technologies) with a RNA integrity number (RIN) > 8. RNA-seq libraries were prepared using the TruSeq Stranded mRNA Library Prep (20020594, Illumina), TruSeq RNA Single Indexes Set A (20020492, Illumina) and TruSeq RNA Single Indexes Set B (20020493, Illumina) according to standard Illumina library preparation procedure. In brief, 0.5 μg of purified RNA was poly-A selected and fragmented followed by first and second strand cDNA synthesis. Double-stranded cDNA was processed from end-repair to PCR amplification according to library construction steps. Libraries were purified using AMPure XP beads (A63880, Beckman Coulter) and validated for appropriate size on a 2100 Bioanalyzer High Sensitivity DNA chip (Agilent Technologies, Inc.). The DNA library was quantitated using Qubit (Thermo Fisher) and normalized to a concentration of 4 nM prior to pooling. Libraries were pooled in an equimolar fashion and diluted to a final concentration of 2 pM. Library pools were clustered and run on a Nextseq500 platform with single-end reads of 75 bases (20024906, Illumina), according to the manufacturer’s recommended protocol (Illumina Inc.). Differential gene expression analysis between the target and reference sets of treatments were determined using DESeq2 (Love et al., 2014) (https://bioconductor.org/packages/release/bioc/html/DESeq.html). Gene Set Enrichment Analysis (GSEA) was performed with the Molecular Signatures Database (MSigDB) hallmark gene set (Liberzon et al., 2015; Subramanian et al., 2005). Both DESeq2 and MSigDB are provided by the Broad Institute.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were carried out using GraphPad Prism (version 7.00). For quantitative data (Figures 2B–2E, 3A and 3B, 4B and 4C, 5D, S1D–S2F, and S3A), data was presented as mean ± SEM and statistical significance was analyzed using two-tailed unpaired Student’s t test. A p value less than 0.05 is statistically significant. Other statistical parameters such as number of experiment repeats and n values can be found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| RIG-I (D33H1G) | CST | Cat# 42GGS; RRID:AB_21757G6 |
| GAPDH (14C1G) | CST | Cat# 2118S; RRID:AB_1G693448 |
| pSTAT1 Y7G1 (58D6) | CST | Cat# 9167S; RRID:AB_1G86GG71 |
| cGAS (D1D3G) | CST | Cat# 151G2S; RRID:AB_2799712 |
| STING (D2P2F) | CST | Cat# 13647; RRID:AB_2799947 |
| IFITM1 | Proteintech | Cat# 11727–3-AP; RRID:AB_2122G83 |
| ISG54 | Proteintech | Cat# 126G4–1-AP |
| ISG56 | Thermo | Cat# PA3–848; RRID:AB_1958733 |
| XRCC4 | BD | Cat# 6115G6; RRID:AB_398966 |
| CENP-F | BD | Cat# 61G768; RRID:AB_398G91 |
| gH2AX (JBW3G1) | Millipore | Cat# G5–636; RRID:AB_3G9864 |
| RIG-I (1C3) | Millipore | Cat# MABF297; RRID:AB_265G546 |
| p53 (DO-1) | Santa Cruz | Cat# sc-126; RRID:AB_628G82 |
| p21 (F-5) | Santa Cruz | Cat# sc-6246; RRID:AB_628G73 |
| Pol III RPC32 (H-9) | Santa Cruz | Cat# sc-48365; RRID:AB_2165718 |
| MAVS (E-3) | Santa Cruz | Cat# sc-166583; RRID:AB_2G123GG |
| Ku7G | Bethyl | Cat# A3G2–624A; RRID:AB_1G554672 |
| CTLA4 (CD152) | BioXCell | Cat# BEG131; RRID:AB_1G95G184 |
| pH3Ser1G (D2C8) | CST | Cat# 3465S; RRID:AB_1G69586G |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2'3'-cGAMP | InvivoGen | Cat# tlrl-nacga23 |
| 5'ppp-dsRNA | InvivoGen | Cat# tlrl-3prna |
| RO-33G6 | Selleck Chemical | Cat# S7747 |
| SiR-DNA | Cyotskeleton | Cat# CY-SCGG |
| Ku55933 | Selleck Chemical | Cat# S1G92 |
| VE-821 | Selleck Chemical | Cat# S8GG7 |
| CHIR-124 | Selleck Chemical | Cat# S2683 |
| LY26G3618 | Selleck Chemical | Cat# S2626 |
| BI2536 | Selleck Chemical | Cat# S11G9 |
| ML-6G218 | Millipore | Cat# 5574G3 |
| Shield-1 | Aobious | Cat# AOB1848 |
| 4-OHT | Sigma | Cat# H79G4 |
| Critical Commercial Assays | ||
| TruSeq® Stranded mRNA Library Prep | Illumina | Cat# 2GG2G594 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | Cat# 2GG249G6 |
| Mitochondria isolation kit | Thermo | Cat# 89874 |
| miRNeasy Mini Kit | QIAGEN | Cat# 217GG4 |
| Deposited Data | ||
| RNA-seq data | This paper | GEO: GSE145148 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice, female | Charles River | Strain code: G27 |
| Oligonucleotides | ||
| p53#1 F | IDT DNA | CACCGATCCACTCACAGTTTCCAT |
| p53#1 R | IDT DNA | aaacATGGAAACTGTGAGTGGATC |
| p53#2F | IDT DNA | CACCGAGCACATGACGGAGGTTGTG |
| p53#2 R | IDT DNA | aaacCACAACCTCCGTCATGTGCTC |
| RIG-I#A F | IDT DNA | CACCGCTAGGGCATCCAAAAAGCCA |
| RIG-I#A R | IDT DNA | aaaclGGCIIIIIGGAIGCCCI AGC |
| RIG-I#B F | IDT DNA | CACCGAGATCAGAAATGATATCGGT |
| RIG-I#B R | IDT DNA | aaacACCGATATCATTTCTGATCTC |
| Ku70 F | IDT DNA | CACCGGAGTCCTACTACAAAACTG |
| Ku70 R | IDT DNA | aaacCAGTTTT GTAGTAGGACTCC |
| Recombinant DNA | ||
| LentiGuide Puro | Addgene | Cat# 52963 |
| LentiCas9 Blast | Addgene | Cat# 52962 |
| PX459 Puro | Addgene | Cat# 62988 |
| pLVX-mCherry-cGAS | This paper | N/A |
| Software and Algorithms | ||
| Graphpad Prism | GraphPad Software | https://www.graphpad.com/ |
| FlowJo | FlowJo Software | https://www.flowjo.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/index.html |
Highlights.
Prolonged arrest before mitosis prevents inflammation and anti-tumor immunity
Disruption of cell-cycle checkpoints restores inflammation in NHEJ-deficient cells
Combined loss of p53 and ATR activates cGAS- and RIG-I-dependent pattern recognition
ACKNOWLEDGMENTS
We thank all members of the Greenberg laboratory for critical discussion. We are grateful to D. Durocher (Lunenfeld-Tanenbaum Research Institute) for providing hTERT RPE-1 cells and hTERT RPE-1 p53 KO cells, and to B. Vogelstein (Johns Hopkins University Medical School) for providing HCT116 p53 KO cells. This work was supported by NIH grants GM101149 and CA17494 and a V Foundation for Cancer Research, United States team convergence award (to R.A.G.), who is also supported by funds from the Penn Center for Genome Integrity and the Basser Center for BRCA. J.C. was supported by the Michael Brown Penn-GSK postdoctoral fellowship.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108080.
REFERENCES
- Ablasser A, and Hur S. (2020). Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol. 21, 17–29. [DOI] [PubMed] [Google Scholar]
- Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, and Hornung V. (2009). RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham RT (2001). Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15, 2177–2196. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Brown KD, Deng CX, Tagle DA, and Wynshaw-Boris A. (1997). Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat. Genet. 17, 453–456. [DOI] [PubMed] [Google Scholar]
- Beatty GL, and Gladney WL (2015). Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 21, 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. (2016). Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167, 1540–1554.e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, Xu B, Ye D, Nathanson KL, June CH, et al. (2019). Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell 178, 933–948.e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh K, Maltbaek JH, Cambier S, Green R, Gale M Jr., James RC, and Stetson DB (2020). Human DNA-PK activates a STING-independent DNA sensing pathway. Sci. Immunol. 5, eaba4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX, and Auh SL (2011). The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 71, 2488–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, and Siliciano JD (1998). Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- Chen YA, Shen YL, Hsia HY, Tiang YP, Sung TL, and Chen LY (2017). Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat. Struct. Mol. Biol. 24, 1124–1131. [DOI] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. (2015). Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162, 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YH, Macmillan JB, and Chen ZJ (2009). RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquel F, Silva MJ, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz AL, et al. (2018). SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61. [DOI] [PubMed] [Google Scholar]
- Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, and Lammerding J. (2016). Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. (2014). STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon MT, Bergerhoff KF, Pedersen M, Whittock H, Crespo-Rodriguez E, Patin EC, Pearson A, Smith HG, Paget JTE, Patel RR, et al. (2019). ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin. Cancer Res. 25, 3392–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson BJ, Mansur DS, Peters NE, Ren H, and Smith GL (2012). DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 1, e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, et al. (2018). Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 24, 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, and Ablasser A. (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 19, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TG, Walter JC, and Loparo JJ (2016). Two-stage synapsis of DNA ends during non-homologous end joining. Mol. Cell 61, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan CR, and Rudin CM (2007). DNA cleavage and Trp53 differentially affect SINE transcription. Genes Chromosomes Cancer 46, 248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM (2018). Nuclear envelope rupture: little holes, big openings. Curr. Opin. Cell Biol. 52, 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, and Hetzer MW (2013). Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, and Chen ZJ (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. (2019). Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, and Fornace AJ Jr. (1992). A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71, 587–597. [DOI] [PubMed] [Google Scholar]
- Kutner RH, Zhang XY, and Reiser J. (2009). Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 4, 495–505. [DOI] [PubMed] [Google Scholar]
- Kwon J, and Bakhoum SF (2020). The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 10, 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau L, Gray EE, Brunette RL, and Stetson DB (2015). DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571. [DOI] [PubMed] [Google Scholar]
- Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, Beckett M, Sharma R, Chin R, Tu T, et al. (2009). Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood 114, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Deng L, Chmura S, Burnette B, Liadis N, Darga T, Beckett MA, Lingen MW, Witt M, Weichselbaum RR, and Fu YX (2013). Radiation-induced equilibrium is a balance between tumor cell proliferation and T cell-mediated killing. J. Immunol. 190, 5874–5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, and Gu J. (2011). Retinoic acid inducible gene-I, more than a virus sensor. Protein Cell 2, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Liu S, Kwon M, Mannino M, Yang N, Renda F, Khodjakov A, and Pellman D. (2018). Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 561, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet BY, Qiu Y, Shabason JE, Wu TJ, Yoon T, Kim BC, Benci JL, DeMichele AM, Tchou J, Marcotrigiano J, and Minn AJ (2017). Exosome RNA unshielding couples stromal activation to pattern recognition receptor signaling in cancer. Cell 170, 352–366.e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoletopoulou V, Markaki M, Palikaras K, and Tavernarakis N. (2013). Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 1833, 3448–3459. [DOI] [PubMed] [Google Scholar]
- Patel SA, and Minn AJ (2018). Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity 48, 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, et al. (2012). Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 366, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranoa DR, Parekh AD, Pitroda SP, Huang X, Darga T, Wong AC, Huang L, Andrade J, Staley JP, Satoh T, et al. (2016). Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 7, 26496–26515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney S, Chaudhuri J, and Alt FW (2004). The role of the non-homologous end-joining pathway in lymphocyte development. Immunol. Rev. 200, 115–131. [DOI] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. (2015). DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, and Thompson CB (2001). Transcriptional activation of short interspersed elements by DNA-damaging agents. Genes Chromosomes Cancer 30, 64–71. [PubMed] [Google Scholar]
- Santaguida S, Richardson A, Iyer DR, M’Saad O, Zasadil L, Knouse KA, Wong YL, Rhind N, Desai A, and Amon A. (2017). Chromosome mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev. Cell 41, 638–651.e635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven A, and Seliger B. (2018). The role of immune escape and immune cell infiltration in breast cancer. Breast Care (Basel) 13, 16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. (2015). Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbreit NT, Zhang CZ, Lynch LD, Blaine LJ, Cheng AM, Tourdot R, Sun L, Almubarak HF, Judge K, Mitchell TJ, et al. (2020). Mechanisms generating cancer genome complexity from a single cell division error. Science 368, eaba0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakifahmetoglu H, Olsson M, and Zhivotovsky B. (2008). Death through a tragedy: mitotic catastrophe. Cell Death Differ. 15, 1153–1162. [DOI] [PubMed] [Google Scholar]
- Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, and Demaria S. (2017). DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 8, 15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, and Barber GN (2016). Recurrent Loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 76, 6747–6759. [DOI] [PubMed] [Google Scholar]
- Xu B, Kim ST, Lim DS, and Kastan MB (2002). Two molecularly distinct G2/M checkpoints are induced by ionizing irradiation. Mol. Cell. Biol. 22, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, and Chen ZJ (2017). cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 114, E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzawa E, Imaizumi T, Matsumiya T, Yoshida H, Fukuhara R, Kimura H, Fukui A, Tanji K, Mori F, Wakabayashi K, et al. (2008). Retinoic acidinducible gene-I is induced by interferon-g and regulates CXCL11 expression in HeLa cells. Life Sci. 82, 670–675. [DOI] [PubMed] [Google Scholar]
- Zhang X, Brann TW, Zhou M, Yang J, Oguariri RM, Lidie KB, Imamichi H, Huang DW, Lempicki RA, Baseler MW, et al. (2011). Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 186, 4541–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, and Pellman D. (2015). Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE145148 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145148).