

Abstract
Kinetochores connect centromeric chromatin to spindle microtubules during mitosis. Neurons are postmitotic, so it was surprising to identify transcripts of structural kinetochore (KT) proteins and regulatory chromosome passenger complex (CPC) and spindle assembly checkpoint (SAC) proteins in Drosophila neurons after dendrite injury. To test whether these proteins function during dendrite regeneration, postmitotic RNA interference (RNAi) was performed and dendrites or axons were removed using laser microsurgery. Reduction of KT, CPC, and SAC proteins decreased dendrite regeneration without affecting axon regeneration. To understand whether neuronal functions of these proteins rely on microtubules, we analyzed microtubule behavior in uninjured neurons. The number of growing plus, but not minus, ends increased in dendrites with reduced KT, CPC, and SAC proteins, while axonal microtubules were unaffected. Increased dendritic microtubule dynamics was independent of dual leucine zipper kinase (DLK)-mediated stress but was rescued by concurrent reduction of γ-tubulin, the core microtubule nucleation protein. Reduction of γ-tubulin also rescued dendrite regeneration in backgrounds containing kinetochore RNAi transgenes. We conclude that kinetochore proteins function postmitotically in neurons to suppress dendritic microtubule dynamics by inhibiting nucleation.
INTRODUCTION
The kinetochore is a quintessential mitosis- and meiosis-specific structure that attaches chromosomes to the mitotic spindle for segregation of genetic material to daughter cells. In many species, including Drosophila, it is built on centromeric DNA that is recognized by the histone cid/Cenp-A that recruits inner kinetochore protein Cenp-C to serve as a binding site for the KMN network (Knl1, Mis12, and Ndc80 complexes; Figure 1B) (Cheeseman, 2014; Musacchio and Desai, 2017; Hinshaw and Harrison, 2018). Of these, the Mis12 complex is most central, while the remainder make up more distal parts of the structure that is involved in connecting to microtubules (Hamilton et al., 2019). Many animals have other inner kinetochore proteins, including the constitutive centromere-associated network (CCAN), but these are absent in Drosophila (Drinnenberg et al., 2016). In cycling mammalian cells, inner centromere proteins including Cenp-A, Cenp-C, and CCAN are found on the centromere in interphase as well as mitosis (Gascoigne and Cheeseman, 2011, 2013). The Mis12 complex has some nuclear staining in interphase, but it is not as clearly punctate as CENP-A (Goshima et al., 2003). In Drosophila, Cenp-A, Cenp-C, and Mis12 localize to centromeres in interphase and mitosis (Przewloka et al., 2007). In mammals and Drosophila, the other kinetochore components including the Knl1 and Ndc80 complexes are recruited to the kinetochore only in mitosis (Przewloka et al., 2007; Gascoigne and Cheeseman, 2011, 2013). Once the kinetochore is fully assembled, Nuf2 and Ndc80 in the outer kinetochore capture microtubule plus ends growing into the central spindle to allow chromosome segregation (Ciferri et al., 2007; Tanaka and Desai, 2008; Tooley and Stukenberg, 2011; Varma and Salmon, 2012).
FIGURE 1:

Kinetochore proteins are transcriptionally up-regulated after dendrite injury. (A) Schematic of experimental design for RNASeq assay. (B) Diagram of the mitotic kinetochore. Colors indicate distinct complexes: KMN network, blue; chromosome passenger complex (CPC), red; spindle assembly checkpoint (SAC), green; Rod-Zwilch-Zw10 complex (RZZ), light green; centromere-associated proteins, light blue. These colors correspond to kinetochore components in graphs throughout all the figures. (C) Average reads of kinetochore mRNA transcripts at baseline (left, gray) and 6 h after dendrite injury (right, colored). Averages and standard deviations are plotted on a log scale. N = 4 pools of 10 neurons each per condition. RNASeq data are included in Supplemental Table 1, and all data used to generate graphs are included in Supplemental Table 3.
In addition to the kinetochore proteins that bridge chromosomes to microtubules, several regulatory complexes are present at the kinetochore before final bioriented microtubule attachment. The chromosome passenger complex (CPC) targets the kinase Aurora B to the centromere, where it helps correct errors in chromosome attachment to microtubules by phosphorylation of substrates including Ndc80 (Carmena et al., 2012; Krenn and Musacchio, 2015). The targeting subunits of the CPC are borealin (or in flies borealin-related, borr), survivin (or in flies Deterin, Det), and Incenp, which are tightly associated with one another through a three-helix bundle (Carmena et al., 2012). Aurora B is also critical for recruiting spindle assembly checkpoint (SAC) proteins to unattached kinetochores (Carmena et al., 2012; Krenn and Musacchio, 2015). The major effector of the SAC is cdc20/fzy, which is a subunit of the anaphase-promoting complex or cyclosome (APC/C) (Lara-Gonzalez et al., 2012; Krenn and Musacchio, 2015; Musacchio, 2015). When bound by SAC subunits at the kinetochore, cdc20/fzy is inactive (Lara-Gonzalez et al., 2012; Musacchio, 2015). A third complex (Figure 1B), Rod1-Zw10-Zwilch (RZZ) complex, interacts with both SAC and KMN proteins (Lara-Gonzalez et al., 2012). Together these regulatory complexes ensure that anaphase does not begin until all sister chromatids are correctly attached to opposite spindle poles. Attachment causes a shift in kinase and phosphatase balance, such that Aurora B starts to lose, and “stripping” of the SAC by dynein connected to the RZZ complex is initiated (Etemad and Kops, 2016; Manic et al., 2017). As a result, cdc20/fzy is freed to activate APC/C, the ubiquitin ligase that triggers degradation of cyclin B and securin to initiate sister chromatid separation (Lara-Gonzalez et al., 2012; Musacchio, 2015). KMN network proteins remain at the kinetochore to mediate attachment of separating sister chromatids to spindle microtubules.
Most of the proteins that make up the kinetochore and its associated regulatory complexes have not been linked to functions in interphase or postmitotic cells. However, two recent studies demonstrated neuronal defects after postmitotic reduction of kinetochore proteins, suggesting that they do function in noncycling cells (Cheerambathur et al., 2019; Zhao et al., 2019). In Caenorhabditis elegans, KMN network components localize to ciliated dendrites of amphid neurons and play a role in their extension (Cheerambathur et al., 2019). Although the microtubule-binding domains of Ndc80 are required to support normal amphid development, specific defects in microtubules were not detected (Cheerambathur et al., 2019). In Drosophila, mis12 mutants were identified in a genetic screen to identify modulators of synaptic bouton structure (Zhao et al., 2019). Reduction of other KMN network proteins had similar effects on neuromuscular junction structure, and intriguingly, so did targeting the centromeric histone cid/CENP-A (Zhao et al., 2019). Knockdown of Mis12 in primary rodent hippocampal neurons increased the number of protrusions from dendrites (Zhao et al., 2019), indicating that kinetochore proteins likely function in neurons broadly across evolution. Specific alterations in the neuronal cytoskeleton were not reported in this study either. It therefore remains to be determined how kinetochore proteins impact neuronal structure.
One of the most intense structural challenges neurons face is rebuilding axons or dendrites after injury. Key regulators of axon regeneration have been identified, including sensors like the dual leucine zipper kinase (DLK) (Hammarlund et al., 2009; Xiong et al., 2010; Shin et al., 2012), epigenetic regulators like HDAC5 (Cho et al., 2015), transcription factors including fos (Xiong et al., 2010), jun (Raivich et al., 2004), and cebp-1 (Yan et al., 2009), and a myriad of downstream regeneration-associated genes (RAGS) (Fawcett and Verhaagen, 2018; Mahar and Cavalli, 2018). However, it was only recently shown that neurons survive removal of all dendrites and can regrow a new arbor (Stone et al., 2014). Only a handful of genes including Akt (Song et al., 2012) and Ror (Nye et al., 2020) have been linked to dendrite regeneration. To identify dendritic RAGs, we isolated RNA from Drosophila neurons after in vivo dendrite removal. Surprisingly, many kinetochore gene transcripts were coordinately up-regulated 6 h after dendrite injury. These included not only transcripts encoding structural kinetochore proteins in the inner kinetochore and KMN network, but also transcripts encoding regulatory proteins in the CPC, SAC, and RZZ complexes. Postmitotic RNA interference (RNAi) was used to confirm a role of some of these proteins in dendrite regeneration. In the same genetic backgrounds, no effect was seen on axon regeneration. In uninjured neurons, kinetochore protein reduction specifically increased microtubule plus-end number (also referred to as microtubule dynamics) in dendrites without altering other metrics of microtubule behavior such as polarity, speed, or minus-end density. Unexpectedly, axonal microtubule dynamics was unaffected. Therefore, kinetochore proteins act in a compartment-specific manner to suppress microtubule dynamics in dendrites. The dendritic microtubule phenotype and dendrite regeneration defect were rescued by concurrent reduction of γ-tubulin, suggesting that kinetochore proteins normally function to temper microtubule nucleation in dendrites. Together our data suggest that the KMN network and CPC, SAC, and RZZ complexes function together to promote dendrite regeneration and modulate dendritic microtubule dynamics through control of nucleation in postmitotic neurons.
RESULTS
Kinetochore genes are up-regulated after dendrite injury and promote regeneration
To identify genes involved in dendrite regeneration, we used an established regeneration model in Drosophila sensory neurons. Laser microsurgery can be used to remove all dendrites of nociceptive ddaC neurons in whole, living larvae (Stone et al., 2014; Thompson-Peer et al., 2016). Robust initiation of dendrite outgrowth is observed 24 h after injury (Stone et al., 2014; Thompson-Peer et al., 2016), and by 96 h dendrites regrow to cover their former territory (Stone et al., 2014). To identify transcripts up-regulated as neurons began regrowing dendrites, we used laser capture microdissection to isolate ddaC cell bodies from larvae 6 h after dendrite removal (Figure 1A). We also isolated ddaC cell bodies from uninjured neurons. For both conditions, four pools of 10 neurons were generated and RNA was prepared from each pool. RNA was amplified, and a sequencing library was prepared. Transcript abundances in uninjured neurons and neurons 6 h after dendrite removal were compared. We originally noticed that the CPC subunit borr was among the most up-regulated transcripts after injury, and so we examined sequences of other kinetochore, CPC, SAC, and RZZ complexes. Subunits of each complex are named in Figure 1B, and each complex is color-coded. Surprisingly, transcripts from subunits of all structural and regulatory complexes were up-regulated by dendrite injury (Figure 1C and Supplemental Table 1).
As identification of transcripts encoding kinetochore proteins was surprising in postmitotic neurons, we examined localization of GFP-tagged kinetochore proteins expressed from their own promoters. GFP-cid (Henikoff et al., 2000; Brust-Mascher and Scholey, 2002) was clearly nuclear in sensory neurons (Supplemental Figure 1A), consistent with its role as a centromeric histone. Spc25-GFP (Schittenhelm et al., 2007) could also be detected in sensory neurons but was extremely dim and appeared diffusely cytoplasmic (Supplemental Figure 1B). Other tagged kinetochore proteins expressed under control of their own promoters, including Ndc80-GFP and Mis12-GFP (Schittenhelm et al., 2007), were so dim that they were difficult to separate from the cell shape marker mCD8-mCherry. We therefore switched to iBlueberry (Yu et al., 2016) as a cell shape marker as this far-red fluorophore has almost no spectral overlap with GFP. In some cells GFP puncta were seen in sensory neuron cell bodies, but these were also observed without a GFP transgene and so likely represent dim autofluorescence (Supplemental Figure 2). To determine whether informative localization might be obtained by overexpressing tagged kinetochore proteins, we examined Nuf2-GFP expressed using the Gal4-UAS system. UAS-Nuf2-GFP (Schittenhelm et al., 2007) localized in the soma and neurites with a mixture of diffuse fluorescence and small puncta, resulting in a pattern that was difficult to interpret (Supplemental Figure 1C). We conclude that kinetochore proteins are likely expressed at very low levels in sensory neurons and that it will be difficult to use localization to gain clues about their function in these cells.
To determine whether up-regulation of kinetochore transcripts by dendrite injury was biologically meaningful, we severed dendrites of ddaC neurons and assayed dendrite regrowth at 24 h. RNAi hairpins were expressed postmitotically with the ppk-Gal4 driver together with the cell shape marker mCD8-GFP and Dicer2 to enhance neuronal RNAi (Dietzl et al., 2007). A reduction in dendrite regeneration was seen in several kinetochore RNAi conditions when compared with control (Supplemental Figure 3). However, the phenotype was not strong enough to make a firm conclusion about a role in the process. In a previous study, we found that an alternate regeneration assay using proprioceptive ddaE neurons had sensitivity to specific RNAi conditions different from that of the ddaC assay (Nye et al., 2020). We therefore also tried this assay, in which the dorsal comb dendrite of the ddaE neuron is severed and branches are added to the remaining dendrites (Stone et al., 2014; Nye et al., 2020). In this cell type, postmitotic expression of RNAi hairpins, Dicer2, and mCD8-GFP was controlled with 221-Gal4. Twenty-four hours after dendrite removal, neurons were scored for regeneration by counting the new branch points that were added to the remaining dendrites (Stone et al., 2014; Nye et al., 2020). Importantly, these neurons do not add branches after the embryonic stage (Sugimura et al., 2003; Stone et al., 2014), so new branches represent a response to injury and not continued growth (Stone et al., 2014). At 24 h after injury, control dendrites add around five new branches, while knockdown of some kinetochore proteins resulted in significantly less regeneration (Figure 2B).
FIGURE 2:

Kinetochore protein knockdown reduces dendrite regeneration but does not affect dendritic morphology. (A) Example images of dendrite injury and regeneration in class 1 ddaE neurons. The severed comb dendrite (red line) degenerates and by 24 h, new dendrite branches have regenerated (green arrows) from the noncomb dendrite (green line). (B) Quantification of dendrite regeneration for control and kinetochore knockdowns. Plotted is average number of new branch points at 24 h postinjury. ***, p < 0.001; **, p < 0.01; *, p < 0.05, with a Kruskal–Wallis one-way analysis of variance (ANOVA), each experimental genotype compared with the control with Dunn’s multiple comparisons test. Numbers in columns are numbers of neurons analyzed. (C) Representative images of uninjured class 1 neurons for control and kinetochore knockdowns. The average total dendrite branch point number across genotypes is quantified in D. Numbers in columns are numbers of neurons analyzed. Error bars in graphs show SD.
Since class I ddaE neurons regenerate an arbor with the same complexity (# of branches) at 96 h postinjury (Stone et al., 2014), a low regeneration phenotype could be due to a reduced number of dendrite branches at baseline. To address whether the regeneration phenotype was an artifact of altered neuronal morphology, we quantified the baseline dendrite branch point number of ddaE neurons in larvae expressing RNAi hairpins to reduce kinetochore proteins (Figure 2C). None of the RNAi lines caused a change in morphology of uninjured neurons (Figure 2D), suggesting that low regeneration is not due to altered neuronal complexity. We conclude that both structural (color-coded blue) and regulatory (red and green) kinetochore proteins can function postmitotically in neurons and that they promote regeneration of dendrites after injury.
Kinetochore protein reduction does not affect axon regeneration
The reduction of dendrite regeneration in ddaE neurons could indicate that kinetochore proteins act generally to control growth in stressful conditions in these cells, or could indicate a specific function in dendrites. To distinguish between these two possibilities, we performed axon regeneration assays in ddaE neurons. ddaE axons were severed using laser microsurgery in intact animals in the same way as dendrites. When the axon of these cells is severed near the cell body (<∼30 μm), a new axon grows from the dendrite that has converted its microtubule polarity to plus-end out (Stone et al., 2010; Rao and Rolls, 2017). The regenerating axon extends in a wandering trajectory in the body wall (Figure 3A), and if it encounters a nerve will grow along it toward the CNS (Rao and Rolls, 2017). On average, control neurons regenerate a new axon extending more than 200 μm after 96 h, after normalization for scaled growth of the neuron during larval development (Figure 3B). Knockdown of representative kinetochore proteins from structural and regulatory complexes did not reduce regenerative growth of axons (Figure 3B). Thus, kinetochore proteins do not seem generally required for stress-induced growth in ddaE neurons, but instead are involved specifically in dendrite regeneration.
FIGURE 3:

Axon regeneration is not affected by knockdown of kinetochore proteins. (A) Class 1 ddaE axon regeneration assay is shown; labels indicate cut site (red arrow) and the regenerating axon (green line), which is a dendrite that has converted into a growing axon. Representative images of regenerating axons are shown for control and three kinetochore knockdowns, quantified in B. Normalized regeneration is calculated by the formula (Length96hr new axon – (Length0hr new axon*(Length96hr control dendrite/Length0hr control dendrite))). Numbers in columns are numbers of neurons analyzed. No genotypes were significantly different, using Kruskal–Wallis one-way ANOVA. Error bars show SD.
Kinetochore proteins are required for normal microtubule dynamics in dendrites and not axons
One major function of the structural kinetochore proteins is attachment to the microtubule plus end, and in C. elegans the microtubule-binding domains of Ndc80 are important in neurons (Cheerambathur et al., 2019). Moreover, perturbations in dendritic microtubules impair dendrite regeneration (Feng et al., 2019; Nye et al., 2020). We therefore examined microtubule organization in ddaE neurons with reduced kinetochore proteins. Neuronal microtubules are stable relative to those in other cells but have dynamic plus ends that grow and shrink in a process known as dynamic instability (Baas et al., 2016). New microtubules can be generated locally in dendrites by nucleation (Ori-McKenney et al., 2012; Nguyen et al., 2014; Weiner et al., 2020). Growing minus ends bound by Patronin are also present in dendrites (Feng et al., 2019), likely generated by severing proteins. Microtubule plus-end-binding proteins (+TIPs) bind to the plus end during the growth phase and fall off during the shrinking phase (Akhmanova and Steinmetz, 2008, 2015). End-binding (EB) proteins are the core +TIPs, and tagged EB proteins have been used to probe neuronal microtubule behavior in cultured mammalian neurons (Stepanova et al., 2003), in vivo mammalian neurons (Kleele et al., 2014; Yau et al., 2016), Drosophila neurons (Rolls et al., 2007; Stone et al., 2008), and C. elegans neurons (Goodwin et al., 2012; Maniar et al., 2012). Growing minus ends are also recognized by EB1 in neurons (Feng et al., 2019), but can be distinguished based on speed and tend to be dimmer than plus ends. Tagged +TIP proteins can be used to report on several different aspects of neuronal microtubule behavior. Direction of +TIP movement can be used to map polarity (Stepanova et al., 2003) and also provides a readout of microtubule stability. More plus ends per unit dendrite length are seen when nucleation or severing is increased (Chen et al., 2012; Tao et al., 2016), and this likely represents a transition from, on average, long microtubules to shorter microtubules. Tagged EB1-GFP can also be used to monitor growing minus ends in neurons (Tao et al., 2016). We therefore expressed EB1-GFP in control and kinetochore RNAi ddaE neurons and acquired movies of microtubule behavior.
In dendrites, the number of polymerizing microtubule plus ends was increased when kinetochore proteins were knocked down (Figure 4, A and B, and Supplemental Video 1), a phenotype we refer to as increased microtubule dynamics. The same phenotype was observed in dendrites of ddaC neurons (Supplemental Figure 4). We have previously identified several genetic backgrounds in which microtubule dynamics are increased in dendrites, including unc-104 and Patronin RNAi, and overexpression of neurodegeneration-causing proteins (Chen et al., 2012; Feng et al., 2019). In all of these backgrounds, the increase in dynamics was seen throughout the neuron. We therefore hypothesized that reduction of kinetochore proteins would also increase microtubule dynamics in the axon. Surprisingly, this was not the case (Figure 4C), and the compartment-specificity of the phenotype distinguished it from other conditions that increase neuronal microtubule dynamics.
FIGURE 4:

Dendritic microtubule dynamics are specifically affected by knockdown of kinetochore proteins. (A) Overview of a class 1 ddaE neuron displaying the region of analysis for dendrite (left) and axon (right) microtubule dynamics. Polymerizing microtubules are seen in this region with EB1-GFP in our videos. To convert a video into a two-dimensional image, we draw a line over the dendrite region of interest (ROI) and plot the fluorescence values of that line over time, resulting in a two-dimensional kymograph. Microtubule comets are seen as dark lines in the kymographs traveling toward the cell body over the duration of a video (300 s). (B) The total number of plus ends in the ROI is counted and divided by the length of that dendrite segment and plotted for all RNAis tested. ***, p < 0.001; **, p < 0.01; *, p < 0.05, with a Kruskal–Wallis one-way ANOVA, each experimental RNAi compared with the control with Dunn’s multiple comparisons test. The same metric was calculated for microtubule dynamics in axons and plotted in C with no significant differences found using the same statistical test as in B. (D) Diagram of possible mechanisms of increased microtubule dynamics. Left, normal conditions. Right, kinetochore protein knockdown results in increased microtubule dynamics, which could be caused by either increased severing or nucleation. (E) Kymographs of control and ndc80 knockdowns illustrating growing microtubule minus ends (green lines), which are also visible with EB1-GFP. Minus ends polymerize very slowly and in the opposite direction as plus ends, consistent with overall microtubule polarity in dendrites. These minus ends in dendrites are quantified identically to plus ends and plotted in F for kinetochore knockdowns. Numbers in/on columns represent total number of neurons analyzed per genotype in B, C, and F. Note that plus ends (B) and minus ends (F) are quantified from the same videos. Error bars are SD.
Movie S1.
Microtubule dynamics in control and Ndc80 RNAi neurons. EB1 dynamics in class I ddaE neurons expressing dicer2, EB1-GFP and control (left) or Ndc80 (right) RNAi hairpins. Note increased number of bright puncta (“comets”) moving towards the cell body with expression of Ndc80 RNAi compared to control. Videos were taken with fluorescence microscopy on a widefield microscope at a rate of one frame per second, for 300 seconds total. Videos are shown here at 30 frames per second. Data from these videos is quantified in Figure 4B, 4F, and Figure S6.
Based on the increase in microtubule dynamics in dendrites with reduced kinetochore proteins, we reasoned that microtubule turnover should also be increased in dendrites but not axons. To address this, we expressed tdEOS-αTubulin (Barlan et al., 2013; Lu et al., 2013) in class I ddaE neurons along with Dicer2 and RNAi hairpins with the 221-Gal4 driver. tdEOS normally fluoresces green but can be converted to red with exposure to 405 nm light. We photoconverted tdEOS in 5 µm regions of both axons and dendrites and measured the red fluorescence intensity. We then imaged these same regions 1 and 1.5 h later and measured how much red fluorescence intensity remained in the converted region (Supplemental Figure 5A) and normalized to the background fluorescence in a nonconverted region. Converted tubulin will be released from the area only when microtubules depolymerize back through it, and so loss of red fluorescence can be used to measure overall turnover of microtubules (Tao et al., 2016). In axons, microtubule turnover was slow and identical across genotypes. In dendrites, microtubule turnover trended toward being higher with kinetochore knockdowns but did not reach statistical significance (Supplemental Figure 5B). We conclude that kinetochore structural and regulatory proteins suppress microtubule dynamics specifically in dendrites and that overall dendritic microtubule turnover may be slightly higher when kinetochore proteins are knocked down.
To determine whether other parameters of microtubule behavior were altered in dendrites when kinetochore proteins were reduced, we assayed growth speed of microtubules, the distance each bout of growth covered, and the overall polarity of microtubules. None of these parameters differed from control neurons (Supplemental Figure 6). Most importantly, we also assayed the number of growing minus ends (Figure 4E). The extra growing plus ends observed in kinetochore RNAi neurons could be generated by either increased nucleation or increased severing by AAA ATPases like fidgetin, spastin, katanin, or kat-60L1. Severing events would be predicted to generate one new plus end and one new uncapped minus end, both of which are visible with EB1-GFP (Feng et al., 2019). In contrast, nucleation events could generate a plus end that recruited EB1-GFP without a growing minus end (Figure 4E). To determine whether nucleation or severing was more likely to increase microtubule dynamics after kinetochore protein reduction, we monitored slow-growing minus ends labeled with EB1-GFP (Figure 4E). We did not observe an increase in growing minus-end number when kinetochore proteins were knocked down (Figure 4F), suggesting that severing proteins are not mediating the increase in microtubule dynamics.
The effect of kinetochore protein knockdown on microtubule dynamics is not caused by cell stress
Increased microtubule dynamics in neurons can result from axon injury and stress in Drosophila (Chen et al., 2012) and mammals (Kleele et al., 2014). We have shown in flies that this response requires the kinases DLK and JNK and downstream transcription factor fos (Chen et al., 2012, 2016). Knockdown of the synaptic vesicle motor unc-104 or microtubule minus-end-binding protein Patronin results in similar increases in microtubule dynamics that are DLK/JNK-dependent (Chen et al., 2012; Feng et al., 2019). In the case of Patronin, the DLK inhibitor GNE3511 (Patel et al., 2015) reduced increases in microtubule dynamics without affecting other microtubule phenotypes, allowing stress-related phenotypes to be separated from other phenotypes (Feng et al., 2019). To determine whether kinetochore protein knockdown increases microtubule dynamics via a DLK-dependent cell stress pathway, we grew larvae in media supplemented with either 50 µM DLK inhibitor GNE3511 (Patel et al., 2015) or vehicle 24 h before monitoring microtubule dynamics in dendrites of class I ddaE neurons. As a control for drug efficacy, we tested whether these conditions could reduce microtubule dynamics in unc-104 RNAi neurons. Indeed, plus-end number returned to baseline in unc-104 RNAi with GNE3511 (Supplemental Figure 7). However, this treatment did not suppress elevated microtubule dynamics induced by kinetochore protein RNAi (Figure 5, A and B).
FIGURE 5:

DLK-mediated cell stress is not responsible for the microtubule phenotype caused by kinetochore knockdowns. (A) Kymographs of EB1 videos taken of ddaE neurons in larvae treated with GNE-3511, a DLK inhibitor. Compared to DMSO vehicle controls, GNE-3511 treatment did not result in a significant suppression of the microtubule dynamics phenotype (B) induced by Ndc80, Nuf2, or Bub3 RNAis. Statistics are nonparametric, Mann–Whitney t tests between DMSO and GNE-3511 treatments for each genotype separately. Numbers in columns are number of neurons analyzed. (C) puc, a negative regulator of the DLK cell stress pathway, is expressed as a function of DLK activation and accumulates in the nucleus, visible with the puc-GFP construct. Pictured are example images of control (left), severe DLK activation by unc-104 RNAi (middle), and Ndc80 RNAi (right). mCD8-RFP is used as a cell shape marker. (D) The average fluorescence intensity of puc-GFP in the nucleus is measured for each cell and normalized to the control average, which is set to 1. **, p < 0.01; *, p < 0.05, with a Kruskal–Wallis one-way ANOVA, each genotype compared with the control with Dunn’s multiple comparisons test. Error bars are SD.
Another readout of DLK-dependent cell stress is puckered (puc) expression. Puc is a MAP kinase phosphatase that negatively regulates JNK (Martin-Blanco et al., 1998). puc-LacZ and puc-GFP reporters concentrate in neuronal nuclei when DLK stress and injury signaling is active (Xiong and Collins, 2012; Stone et al., 2014). As expected, puc-GFP accumulated abundantly in nuclei of unc-104 RNAi neurons (Figure 5C). In contrast, knockdown of kinetochore proteins did not increase puc-GFP nuclear fluorescence (Figure 5, C and D). We conclude that increased microtubule dynamics is not a result of activation of DLK signaling when kinetochore proteins are reduced.
Kinetochore proteins influence microtubule dynamics and regeneration by modulating nucleation
Increases in the number of growing microtubule plus ends in dendrites can be mediated by up-regulation of microtubule nucleation or activation of severing proteins (Chen et al., 2012; Tao et al., 2016). The absence of a concurrent increase in minus ends to match the increase in plus ends when kinetochore proteins are knocked down (Figure 4C) suggests that nucleation is responsible in this case. We first reasoned that reduction of kinetochore proteins could lead to a higher than normal concentration of γ-tubulin, the key subunit of the γ-tubulin ring complex (γTuRC), in dendrites, which could lead to extra nucleation of microtubules. In ddaE neurons, γ-tubulin localizes robustly to dendrite branch points (Figure 6A) when tagged and overexpressed or analyzed at endogenous levels (Nguyen et al., 2014; Weiner et al., 2020). When kinetochore proteins were knocked down, we did not observe an increase in γ-tubulin localization in dendrites (Figure 6, A and B), suggesting that increased levels of γ-tubulin in dendrites do not account for the increase in microtubule dynamics. The assay for localization of nucleation sites to dendrite branch points relies on overexpressed γ-tubulin-GFP (green fluorescent protein), and while it reflects localization of endogenous γ-tubulin (Nguyen et al., 2014; Weiner et al., 2020), it is not a good readout of overall levels of γ-tubulin. To determine whether kinetochore proteins might impact overall levels of γ-tubulin, we used γ-tubulin tagged at the endogenous locus with sfGFP (superfolder) (Tovey et al., 2018) and compared levels in control neurons and neurons expressing Ndc80 and borr RNA hairpins. We saw no significant difference between conditions (Supplemental Figure 8). We conclude that it is unlikely that kinetochore proteins regulate levels or localization of the core nucleation protein γ-tubulin.
FIGURE 6:

γTub-GFP localization is not affected by knockdown of kinetochore proteins, and a single γTub null allele specifically blocks increases in dynamics due to nucleation. (A) Example images displaying the enriched localization of γTub-GFP to dendrite branch points in control and cid RNAi class 1 ddaE neurons. Inset images are close-ups of the branch point labeled by the white arrow in each image. (B) The average fluorescence intensity of branch point regions was measured and normalized to non–branch point regions for each genotype tested; which expressed different RNAi hairpins. No significant difference in localization was detected using a Kruskal–Wallis one-way ANOVA. (C) Example kymographs display microtubule dynamics at baseline (left), 30 min after dendrite injury (middle), and 8 h after axon injury (right) for control (top) and γTub23CA15-2 heterozygous (bottom) genotypes. (D) Microtubule dynamics in dendrites 8 h after axon injury are plotted for control and γTub23CA15-2 heterozygotes; HPA = hours post-axotomy. ***, p < 0.001, in an unpaired t test with Welch’s correction between control uninjured and 8 h HPA data. γTub23CA15-2 uninjured and 8 h HPA data are not different with the same statistical test. (E) Microtubule dynamics in dendrites 30 min after the dendrite has been severed proximally to the cell body are plotted. ***, p < 0.001, in an unpaired t test with Welch’s correction between control uninjured and 30 min post-dendrotomy (HPD) and between γTub23CA15-2 uninjured and 30 min postdendrotomy. Note that control uninjured and γTub23CA15-2 uninjured are the same in D and E and are the same as control data in Figure 7B. In all graphs numbers of cells analyze is shown in the bars and error bars are SD.
To test the hypothesis that reduction of kinetochore proteins increases microtubule dynamics by activating nucleation, we wanted to identify a genetic background that specifically eliminates nucleation-dependent plus-end increases but does not affect increases in plus ends that result from severing. We previously showed that animals heterozygous for a null allele of γTubulin cannot increase dynamics in response to axon injury, although microtubule dynamics at baseline is normal (Chen et al., 2012). To combine loss of one copy of γTubulin with kinetochore RNAi, we made a line that had the γTub23CA15-2 allele with 221-Gal4, UAS-Dcr2, and UAS-EB1-TagRFP-T (Feng et al., 2019) and crossed this to RNAi lines. We first made sure that this genetic background could specifically suppress increases in microtubule dynamics that were due to up-regulated nucleation. In uninjured neurons, one mutant copy of γTubulin had no effect on the number of growing microtubules in dendrites (Figure 6, C and D). In contrast, up-regulation of dynamics in response to axon injury, which depends on increasing nucleation (Chen et al., 2012), was completely blocked (Figure 6, C and D). To test whether this background would dampen all increases in microtubule dynamics, we assayed microtubule plus ends in dendrites separated from the cell body by laser microsurgery. In severed dendrites, an increase in microtubule plus-end number occurs at early time points before overt signs of degeneration (Tao et al., 2016). This increase in dynamics depends on the microtubule severing protein fidgetin (Tao et al., 2016), but we did not previously determine whether a mutant copy of γTubulin might also affect it. Unlike the increase in plus-end number after axon injury, the increase in severed dendrites was not suppressed in γTubulin heterozygous animals (Figure 6, C and E).We conclude that one mutant allele of γTubulin can be used to distinguish nucleation- and severing-dependent increases in microtubule dynamics and should therefore be useful for determining whether kinetochore reduction increases plus-end number through nucleation or some other mechanism like severing.
To assess whether increased nucleation is responsible for the kinetochore RNAi phenotype, we introduced RNAi hairpins into animals with wild-type γTubulin or animals heterozygous for the γTubulin null allele. With control hairpins, plus-end number was similar in animals with one or two copies of functional γTubulin, as expected (Figure 7, A and B). RNAi hairpins targeting transcripts of kinetochore proteins resulted in increased plus ends in dendrites (Figure 7, A and B). However, the increased microtubule dynamics phenotype was completely suppressed in heterozygous γTubulin animals (Figure 7, A and B, and Supplemental Video 2). This result strongly suggests that kinetochore proteins regulate microtubule dynamics in dendrites by regulating nucleation of microtubules (Figure 7C).
FIGURE 7:

Normal levels of the core microtubule nucleation protein γTub23C are required for the up-regulation of microtubule dynamics induced by kinetochore protein knockdown. (A) A null mutant allele of γTub23C (γTub23CA15-2) was combined with EB1-TagRFP-T, which was used as a cell shape marker and labels polymerizing microtubules similarly to EB1-GFP, to generate a tester line. An example region of analysis for a ddaE neuron is shown (green line). Control and kinetochore RNAi flies were crossed to a control tester line or a tester line containing the γTub23C mutant. Kymographs were generated from 300 s (120 frames) videos. Left, kymograph shows microtubule dynamics with a control RNAi and heterozygous null allele for γTub23C. Kymographs of microtubule dynamics in Ndc80 RNAi with normal levels of γTub and one mutant allele are also shown. (B) Quantification of microtubule dynamics in ddaE dendrites for each genotype with and without the γTub23C null allele. ***, p < 0.001; **, p < 0.01, with a nonparametric, Mann–Whitney t test performed between RNAi alone and RNAi+ γTub23CA15-2 for each RNAi knockdown. Note that the microtubule dynamics of kinetochore RNAi with EB1-TagRFP-T but without γTub23CA15-2 in B is very similar to that with EB1-GFP in Figure 4B. Numbers on bars are neurons analyzed, and error bars show SD. (C) Diagrammatic summary of the results is shown.
Movie S2.
Microtubule dynamics in Ndc80 RNAi neurons with normal and one mutant copy of γTub23C. EB1 dynamics in class I ddaE neurons expressing dicer2, EB1-Tag-RFP-T and Ndc80 RNAi, with (right) and without (left) a heterozygous null allele for γTub23C. Note decreased number of comets when Ndc80 RNAi is combined with the γTub23C mutant. Videos were taken with fluorescence microscopy on a confocal microscope at a rate of one frame per 2.5 seconds, for 300 seconds total (120 frames). Videos are shown here at 12 frames per second. Data from these videos is quantified in Figure 7B.
The suppression of the microtubule dynamics phenotype by reduction of γ-tubulin suggests that one function of kinetochore proteins is to reduce nucleation activity in dendrites. To test whether this function is related to the defect in dendrite regeneration in kinetochore RNAi neurons, we tested whether the γTubulin null mutant could also rescue reduced dendrite regeneration. Introduction of the γTubulin null allele into control animals did not significantly affect dendrite regeneration (Figure 8), although stronger reduction of γ-tubulin does hinder regeneration (Weiner et al., 2020). When paired with borr or Bub3 RNAi, one copy of the γTubulin null allele was able to rescue regeneration RNAi (Figure 8). This suppression suggests that the deficit in dendrite regeneration in kinetochore RNAi neurons is due to dysregulated microtubule nucleation.
FIGURE 8:
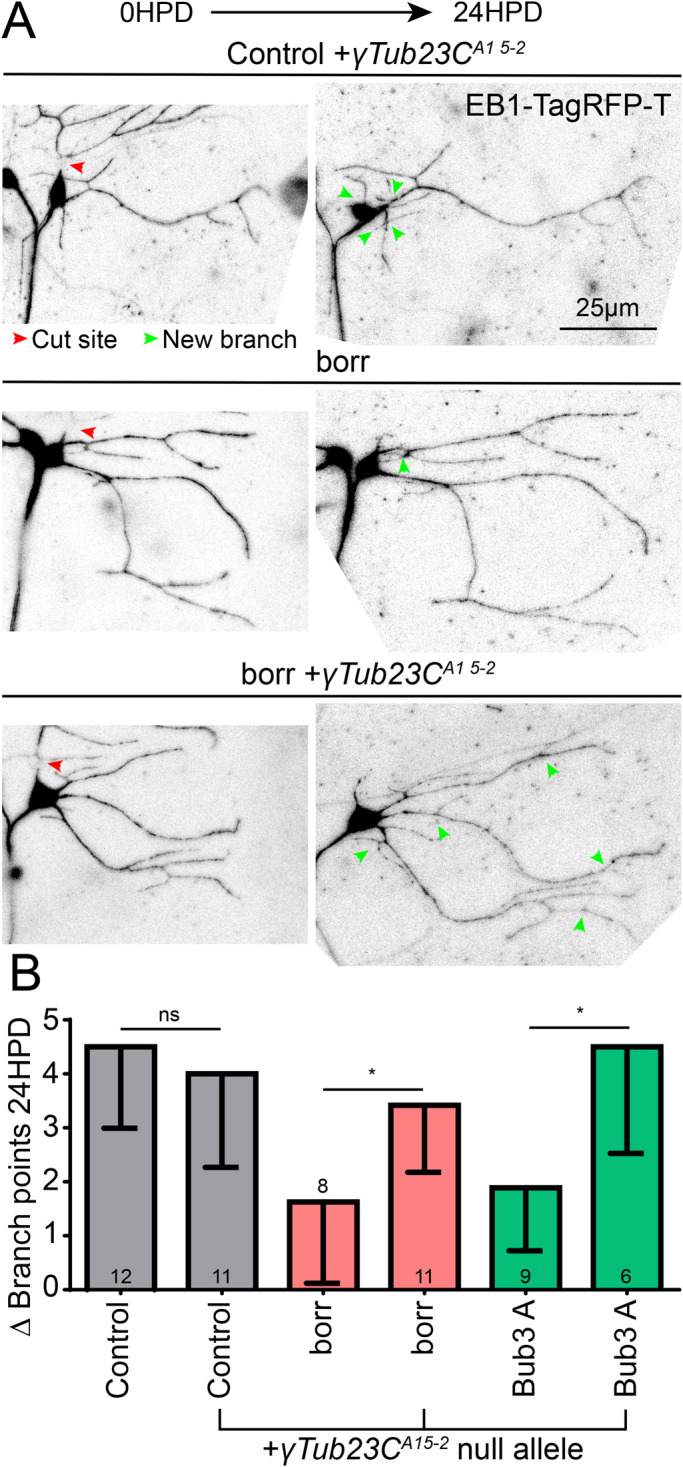
Normal levels of γTub23C are also required for the reduced dendrite regeneration phenotype induced by kinetochore protein knockdown. (A) The γTub23CA15-2 null mutant tester line generated for the preceding figure was used for a dendrite regeneration assay. Borr and Bub3 RNAis were crossed to this tester line, and a dendrite regeneration assay was performed. Top, example images of control RNAi at 0 and 24 h with the γTub23CA15-2 null allele; middle, example images of borr RNAi without the γTub23CA15-2 null allele; bottom, example images of borr RNAi with the γTub23CA15-2 null allele. Image brightness of tif files was increased linearly to make the signal visible. (B) Quantification of dendrite regeneration at 24 h postdendrotomy. Control vs. control + γTub23CA15-2 was not significant with an unpaired Mann–Whitney t test. The same test was performed between borr and borr + γTub23CA15-2, and between Bub3 and Bub3 + γTub23CA15-2, with borr almost reaching significance and Bub3 having a p value of 0.014 (*). Numbers in columns are numbers of neurons analyzed. Error bars show SD.
DISCUSSION
On the basis of the surprising appearance of transcripts of structural and regulatory kinetochore proteins in RNASeq data from injured neurons, we investigated their role in dendrite regeneration. We found that regeneration after dendrite, but not axon, injury was impaired when they were reduced using postmitotic RNAi. Because kinetochore proteins function to attach, and monitor attachment, to microtubules in mitotic spindles, we surveyed microtubule behavior in neurons in which they were reduced. RNAi knockdowns of many kinetochore proteins caused an up-regulation of dendritic microtubule dynamics, an effect not seen in axons. Other parameters of microtubule behavior, including number of growing minus ends, were not affected. The increase in plus-end, but not minus-end, number suggested that nucleation rather than severing was responsible. The involvement of nucleation was confirmed by rescue of the plus end and regeneration phenotypes in neurons with reduced γ-tubulin. The increase in microtubule dynamics when kinetochore proteins were reduced suggests that they normally function to dampen nucleation of dendritic microtubules in postmitotic neurons.
Two recent studies indicate that kinetochore proteins are likely to function broadly in different neuron types. In one study, the authors identified Mis12 mutants in a Drosophila forward genetic screen. Other components of the KMN network complexes (Ndc80, Knl1, and Nnf1a), as well as centromeric protein Cenp-A (cid), had similar neuromuscular junction phenotypes and reduced neuropil in the CNS (Zhao et al., 2019). The authors went on to show that mammalian neurons with reduced Mis12 also had structural defects, this time in dendrites (Zhao et al., 2019). The group reported some localization of KMN network proteins to spots in peripheral nerves and neuropil in the CNS (Zhao et al., 2019), but the pattern did not help to suggest how the proteins might function in neurons.
The other study demonstrated that Cenp-C, Ndc80, and Nuf2 localize postmitotically to the ciliated dendrites of amphid neuron bundles in C. elegans. When these proteins were knocked down with an elegant GFP-degrader system, amphid dendrite extension was impaired (Cheerambathur et al., 2019). Effects on egg laying and fertility were also seen. Notably, a deletion of only the microtubule-binding domains of Ndc80 was able to phenocopy the knockdowns, indicating that an interaction with microtubules likely mediates the phenotypes. Degradation of GIP2, an essential γTuRC subunit, showed the same neuron extension deficits, supporting the hypothesis that microtubules in these neurons are affected by KMN network protein knockdown, although the authors were unable to pinpoint any specific changes in the microtubule cytoskeleton (Cheerambathur et al., 2019).
The two previous studies on neuronal roles for kinetochore proteins focused on the structural components of the kinetochore, including inner centromere components Cenp-A and C and KMN network proteins (Cheerambathur et al., 2019; Zhao et al., 2019). We add the three regulatory complexes—CPC, RZZ, and SAC—to the list of kinetochore proteins with neuronal function. Moreover, we find that players in all structural and regulatory complexes have similar roles in controlling microtubule dynamics. This suggests that, as in mitosis, these complexes function together in a single pathway in neurons. It is therefore likely that they are not only involved in attaching microtubules to something, but are also acting as sensors of microtubule behavior. If they have a similar role in neurons as in mitosis, they may recognize plus-end arrival at a specific cellular location. Again, in analogy with mitosis, plus-end arrival could trigger release of regulatory complexes and free them to send signals to other cellular locations about the status of the microtubule cytoskeleton. These signals could involve ubiquitination mediated by association of Cdc20/fzy with an E3 ligase complex, or phosphorylation by Aurora B. One target of either output signal could be suppression of γTuRC activity.
One intriguing aspect of our findings is that we only observed changes in the microtubule cytoskeleton in dendrites, and dynamics in the axon was unaffected. While phenotypes in C. elegans and mammalian neurons occurred in dendrites (Cheerambathur et al., 2019; Zhao et al., 2019), changes in motor axon terminals were seen in Drosophila (Zhao et al., 2019). One possible way to reconcile the axonal and dendritic phenotypes would be if kinetochore proteins are important throughout dendrites but function specifically at synaptic regions of axons. There are several hints that microtubule nucleation might be particularly important near presynaptic sites. In Drosophila motor neurons, concentrations of γ-tubulin were seen in large synaptic boutons at the neuromuscular junction (Nguyen et al., 2014). In cultured mammalian neurons, γ-tubulin also concentrates at presynaptic sites (Qu et al., 2019), and increases in neuronal activity lead to increases in EB comet formation at these sites (Qu et al., 2019), suggesting that regulation of nucleation is important in axons. It is therefore possible that kinetochore proteins regulate presynaptic nucleation in addition to dendritic nucleation.
In mitotic cells, kinetochore proteins localize to tight spots around centromeric DNA, and their localization has provided invaluable clues to their function. So far, localization patterns in neurons have not helped pin down specific sites of action. In C. elegans, expression levels in amphid neurons were high enough to detect tagged proteins expressed at endogenous levels, and they were seen throughout the linear ciliated dendrite region (Cheerambathur et al., 2019). In contrast, expression seems low in Drosophila neurons, and beyond being present in neuropil, a pattern has not been discernible (Zhao et al., 2019). Our attempts to visualize meaningful localization in sensory neurons also did not provide any insights (Supplemental Figures 1 and 2). Use of amplification systems like SunTag (Tanenbaum et al., 2014) may be required to acquire meaningful information about where structural and regulatory kinetochore proteins function in neurons.
One possible model for neuronal kinetochore protein function is that, in analogy with mitosis, the structural and regulatory complexes colocalize at a specific cellular site, perhaps in the cell body, when microtubule plus ends are absent or in low abundance. Upon plus-end arrival, the RZZ complex in conjunction with dynein could transport regulatory proteins toward microtubule minus ends. Dendrites contain minus-end-out microtubules, so this would mean that regulatory proteins could be transported outward into dendrites to suppress microtubule dynamics distally after plus-end arrival. This type of mechanism could allow for global homeostatic control of microtubule dynamics in dendrites.
MATERIALS AND METHODS
Drosophila stocks
Many of the RNAi lines used in this study were acquired from the Bloomington Drosophila Stock Center (National Institutes of Health [NIH] P40OD018537) and the Vienna Drosophila Resource Center. Tester lines that we have generated previously and used in this study include (UAS-Dcr2; 221-Gal4, UAS-EB1-GFP), (UAS-Dcr2; 221-Gal4, UAS-mCD8-GFP), (UAS-Dcr2, UAS-mCD8-RFP; 221-GAL4, puc-GFP), (γTub15-2/CyO-GFP; 221-Gal4, UAS-EB1-TagRFP-T, UAS-Dcr2), (UAS-Dicer2; 221-Gal4, UAS-γTub -GFP, UAS-mCD8-RFP), (477-Gal4, UAS-EB1-GFP/CyO; UAS-Dcr2/TM6), (UAS-Dcr2; ppk-Gal4, UAS-mCD8-GFP), (221-Gal4, UAS-EB1-TagRFP-T, UAS-Dcr2 on chromosome 3), and (UAS-Dcr2; 221-Gal4, UAS-tdEOS-αTub/TM6).
To obtain larvae of desired genotypes, virgin female flies from tester lines were mated to males from RNAi lines in bottles containing a cap of media at the bottom. Caps with larval offspring were collected every 24 h. These caps were aged at 25°C for 72 h before use. Fly food media (ingredients for 10 l) 45 g agar, 259 g sucrose, 517 g dextrose, 155 g yeast, 858 g cornmeal, 40 ml 10% tegosept in ethanol, 60 ml proprionic acid. For a complete list of Drosophila lines used, see Supplemental Table 2.
Larval neuron selection
Fly larvae have eight abdominal (A) body segments, with each segment containing a dorsal cluster of neurons that includes the class I (ddaE) and class IV (ddaC). For all assays, we used one neuron per animal within the A2–A4 segments.
Localization of tagged kinetochore proteins in class I neurons
Males from transgenic lines expressing GFP-tagged kinetochore proteins (see Supplemental Table 2 for genotypes) were crossed to females from a tester line with either mCD8-mCherry or iBlueberry as cell shape markers together with 221-Gal4. Larvae ∼3 d old were selected for imaging. No anesthetic was used. Images were taken with a two-track, sequential scan on an LSM800 confocal microscope. Microscope settings (laser power, gain, dwell time, and averaging) for the green channel were adjusted for each genotype as brightness and visibility of the constructs varied. The “no-GFP” representative image was acquired using the same microscope settings as Mis12-GFP and eGFP-Ndc80 images.
Class I dendrite regeneration assay
Three-day-old larvae reared at 25°C were removed from their food cap and gently washed in water. They were then mounted on a glass microscope slide with a pad of dried 3% agar (for stability) and immobilized with a coverslip (40 × 22 mm; VWR) held down by tape. Larvae had the comb/main dendrite of one ddaE neuron severed with a MicroPoint pulsed UV laser (Oxford Instruments/Andor, USA) and were imaged on either a Zeiss confocal LSM700, LSM800, or widefield microscope. Postinjury and -imaging, the larvae were returned to a small piece of fly media in a cap and kept at 25°C for 24 h. Larvae were remounted and reimaged using a Zeiss LSM800 confocal or widefield microscope. Regeneration was quantified as (total branch points at 24 h postinjury) – (total branch points on remaining dendrite at time of injury). For experiments in the heterozygous γTub23C mutant background, a red cell shape marker was used, while other experiments included a green cell shape marker. When using the red cell shape marker, some images of injured neurons were too dim to accurately quantify and were excluded from analysis.
Class IV dendrite regeneration assay
Three-day-old larvae reared at 25°C were used for ddaC dendrite regeneration assays. A pulsed UV laser was used to cut all dendrites from one ddaC neuron proximal to the first branch points. Larvae were then placed in a cap with fly food and kept at 25°C for 24 h. The regenerated dendritic arbor was imaged using a Zeiss LSM800 confocal or widefield microscope. Regeneration was quantified as the largest-diameter line that can be drawn across the dendrite arbor.
Class I axon regeneration assay
Two-day-old larvae reared at 25°C were used for the axon regeneration assay. A pulsed UV laser was used to sever the axon of the ddaE neuron within 20 μm of the soma. If possible, we spared the axon of the neighboring class I ddaD neuron. Neurons were imaged with a Zeiss LSM700 confocal or widefield microscope. After injury, larvae were returned to a cap with fly food and kept at 20°C for 96 h. After 96 h, larvae were imaged on a Zeiss LSM800 confocal or widefield microscope. When the axon is cut proximally, a dendrite converts itself to be a new axon rather than grow a new axon from the stump or soma. Axon regeneration is quantified as follows to get a measurement of growth from the dendrite tip that accounts for expansion of the entire dendrite arbor during animal growth:
Class I neuronal morphology analysis
Images of class I ddaE dendrite arbors were taken on either a Zeiss LSM800 confocal or widefield microscope, and a Z-stack was generated with FIJI software (NIH). The total number of dendritic branch points was quantified.
Class I EB1 dynamics assay (dendrite)
Three-day-old larvae reared at 25°C were mounted on glass under a coverslip held down tightly with tape. Five-minute videos of single neurons were taken on a Zeiss widefield microscope at one frame per second. EB1-GFP is easily visible on the polymerizing plus ends and minus ends of microtubules and provides the basis for quantification.
Plus ends: total number of EB1-labeled plus ends (referred to as comets) polymerizing for more than 1 μm were counted in the region of the comb/main dendrite that was in focus, usually from branch points 1 to 4 or 5. The region between the cell body and first branch point was not included in the analysis. Since EB1 activity dwindles farther up the dendritic tree, distal regions with no EB1 activity were not included in the analysis even if in focus. The total number of comets was then divided by the length of the dendritic segment to get a comets per micron measurement for each neuron.
Minus ends: slowly polymerizing minus ends are also visible with EB1-GFP and easily distinguishable from plus ends based on speed. The total number of polymerizing minus ends was counted for in-focus dendrite, similar to plus-end quantification, in each video to result in a minus ends per micron measurement.
EB1 comet speed: kymographs were generated from three to six videos per genotype, and the distance per time (x distance: microns, y distance: seconds) of each visible comet was quantified for at least 19 comets per genotype.
EB1 comet run length: kymographs used for comet speed were also used to quantify run length, for 30–70 comets per genotype. Only comets with a defined start and end point visible in the kymograph were measured.
Plus ends (with γTub23C mutant and EB1-TagRFP-T): videos were taken on a Zeiss LSM800 confocal microscope at approximately one frame per 2.5 s for 5 min (120 frames). The quantification method was identical to EB1-GFP plus-end methods.
Class IV EB1 dynamics assay (dendrite)
Three-day-old larvae reared at 25°C were mounted similarly to those for the class I EB1 dynamics assay, and again 5-min videos were taken at one frame per second on a Zeiss widefield microscope. As the class IV neuron covers the whole dorsal region of a larval hemisegment, some areas are in focus and some are not. For quantification, we chose three segments of proximal dendrite per neuron that were in focus and counted the comets in each segment, dividing by the length of the respective segment. For each neuron, the three comets/micron regional measurements were averaged into a single number; these were then averaged across neurons to obtain a comets/micron measurement per genotype.
Class I EB1 dynamics assay (axon)
Three-day-old larvae were mounted in the same manner as in other EB1 dynamics assays. Neurons in the A2–A4 segments that had the longest section of axon in focus were chosen for analysis. Five-minute videos were taken at one frame per second on a Zeiss widefield microscope. The total number of comets were counted in the region of axon that was in focus and could be distinguished from the neighboring ddaD axon, which bundles with the ddaE axon. A comets/micron measurement was generated with the length of axon from the cell body to the end of the focal range.
Class I puc assay
A tester line with puc-GFP (Morin et al., 2001; Stone et al., 2014), UAS-Dcr2, UAS-mCD8-RFP, and 221-GAL4 was crossed individually to RNAi lines for analysis. Larvae were reared at 25°C for 3 d and then imaged on a Zeiss LSM800 inverted confocal microscope. Z-stacks were generated with FIJI software. Puc-GFP is packaged neatly in the nucleus, so a circle was drawn around the nucleus and the fluorescence intensity of the green channel was measured in FIJI. Only images with at least three images in the z-stack containing the nucleus were used.
Class I EB1 dynamics assay with DLK inhibitor
GNE-3511 was obtained from EMD Millipore and diluted in dimethyl sulfoxide (DMSO). The drug was added at a 50 μM concentration to normal media in small batches. Larvae were aged 48 h at 25°C in normal media. Experimental animals were then transplanted into media containing a 50 μM DLK inhibitor GNE-3511 and aged at 25°C for 24 h. Vehicle control animals were transplanted into media containing the same amount of DMSO (0.5% by volume) but no drug, and aged at 25°C for 24 h. At this point, the larvae were imaged and quantified in an identical manner to the normal class I EB1 dynamics assay.
Class I EB1 dynamics after axon injury assay
Three-day-old larvae of desired genotypes grown on normal media were subjected to an axon injury paradigm: a MicroPoint laser on a Zeiss LSM800 confocal microscope was used to sever axons of class I neurons. Larvae were returned to normal media for 8 h at 20°C. Then, neurons with successful axon cuts had 5-min EB1 videos of the comb dendrite taken on a Zeiss LSM800 confocal microscope. EB1 dynamics were quantified in a manner identical to that of the normal EB1 assay, also excluding the region proximal to the first branch point.
Class I γTub localization assay
Flies from the tester line dicer2; 221-GAL4, UAS- γTub-GFP, UAS-mCD8-RFP were crossed to males from RNAi lines, and larvae were aged at 25°C for 72 h. Single neurons were imaged with a Zeiss LSM800 confocal microscope. γTub-GFP localizes mainly to dendrite branch points. For quantification, the average fluorescence intensity of γTub-GFP was measured in each branch point as well as in between regions in the comb dendrite of class I neurons. Average branch point fluorescence intensity was divided by average non–branch point fluorescence intensity for a normalized reading of γTub-GFP localization to branch points.
Class I γTub expression assay
Males from RNAi lines were crossed to a line containing γTub tagged at its endogenous locus with sfGFP (Tovey et al., 2018) and 221-Gal4, UAS-iBlueberry. Because Dcr2 is not present in this tester line, we used experimental RNAi lines that do not require dicer (Ndc80: val20 [Bloomington 38260]; borr: val20 [Bloomington 56942]). Three-day-old larvae were imaged on an LSM800 confocal microscope.
Microtubule turnover assay
Flies from the tester line UAS-dicer2; 221-GAL4, UAS-tdEOS-αTubulin were crossed to control and kinetochore RNAi lines. Three-day-old larvae were mounted on a glass slide for imaging on an LSM800 inverted confocal microscope. Four sequential prebleach images are taken to keep the neuron in focus, after which the microscope performs photoconversion for a 5-μm circle on either the proximal dendrite or the axon. For photoconversion, a 405-nm-wavelength light was used at 5% power. The fifth and later frames of the image contain the converted region and adjacent regions of dendrite or axon for quantification. For each cell, both the dendrite and the axon undergo photoconversion sequentially, in two separate acquisitions. For quantification, a shape is drawn around the converted region of dendrite or axon, and a similar area shape is drawn around an adjacent, nonconverted region in both 0 and 1/1.5 h time points. Average fluorescence intensity in the red channel is measured for these four regions. The formula for fluorescence intensity remaining is
(FL intensity of converted region(1hr) – FL intensity of nonconverted region(1hr))/(FL intensity of converted region(0hr) – FL intensity of nonconverted region(0hr))
for both axons and dendrites separately. The same formula is used for the 1.5 h experiment.
Image acquisition and analysis
All images were acquired with Zen software on Zeiss microscopes. The following systems were used in this study:
LSM800 inverted confocal on an Axio Observer Z1 stand and equipped with GaAsP detectors and a Zeiss Plan-APOCHROMAT 63x DIC (oil, 1.4 NA) objective.
LSM800 upright confocal on an Axio Imager.Z2 stand and equipped with GaAsP detectors. Zeiss Plan-APOCHROMAT 63x DIC (oil, 1.4 NA) and Zeiss Plan-APOCHROMAT DIC (UV) VIS-IR 40x (oil, 1.3 NA) objectives.
LSM700 inverted confocal on an Axio Observer.Z1 stand equipped with standard detectors.
Zeiss Plan-APOCHROMAT 63x DIC (oil, 1.4 NA) objective; Zeiss Axio Imager.M2 widefield equipped with an AxioCam 506 mono camera.
Zeiss Plan-APOCHROMAT 63x DIC (oil, 1.4 NA); Zeiss EC Plan NEO FLUAR 40x (oil, 1.3 NA) objectives.
Pre-injury images and EB1 videos were taken with a 63 × 1.4 NA objective; postinjury images were taken with a 40 × 1.4 NA objective. All image analysis was performed with FIJI software (NIH). For injury, morphology, γTub-GFP localization, tdEOS and puc experiments, Z-stacks/time series were made into maximum-intensity projections with the z-project plug-in. If larval movement during imaging rendered maximum intensity projections impossible, the stitching plug-in (max intensity option) was used to make a complete image. For EB1 videos, the bleach correction plug-in was used to make videos easier to see, and the template matching plug-in was used to align all images per video to make generation of kymographs possible (multi kymograph plug-in, line width = 3).
RNA isolation, sequencing, and bioinformatics
Detailed methods have been described (Nye et al., 2020). In summary, at time 0 dendrite injury (or no injury) was performed on eight GFP-labeled ddaC neurons in each animal and 6 h later the animals were fixed and flash frozen in optimal cutting temperature compound. The dorsal region of each animal was cryosectioned, and cell bodies were isolated based on GFP fluorescence using a Zeiss PALM laser capture microdissection system. Pools of 10 cells were generated, and RNA was extracted from four pools for each condition. RNA was then used for preparation of libraries for sequencing with Nextera reagents, and sequencing was performed on an Illumina HiSeq instrument with 150-base-pair single-end reads.
A bioinformatics pipeline similar to that described (Nye et al., 2020) was used. Base quality and Nextera adapters were assessed using FASTQC. Trimmomatic (Bolger et al., 2014) was used for adaptor and quality trimming. Base quality trimming was performed using a sliding window of 4 and Phred quality of 20 for cutoff values, with a minimum read length of 20 for retention. Successful adaptor trimming was confirmed with a second FASTQC assessment on trimmed .fastq files. Trimmed reads were aligned to the Flybase dmel 6.11 release of the Drosophila genome using the HISAT2 aligner (Kim et al., 2015) with standard parameters. Samtools was used to convert .sam files to .bam files and perform sorting and indexing of .bam files. A raw count table was then generated using featureCounts in conjunction with the dmel 6.11 .gtf file. Differential expression analysis was performed using the DESeq2 package (Love et al., 2014).
Statistical analysis
GraphPad Prism software was used for all statistical analysis and graph generation. Methods used are specified in the figure legends. Significance is shown as *, p < 0.05, **, p < 0.01, ***, p = 0001, with error bars representing SD.
Supplementary Material
Acknowledgments
We thank the Bloomington Drosophila Stock Center (National Institutes of Health [NIH] P40OD018537) and the Vienna Drosophila Resource Center for providing fly strains used in this study. Kevin Janes, University of Virginia, provided very helpful advice about isolating and sequencing RNA from small numbers of cells. We are very grateful to Christian Lehner, University of Zurich, for providing fly lines. Funding for this work was provided by the NIH, R01 GM085115.
Abbreviations used:
- APC
anaphase promoting complex
- CPC
chromosome passenger complex
- ddaC
dorsal dendritic arborization C
- ddaE
dorsal dendritic arborization E
- GFP
green fluorescent protein
- HPA
hours post-axotomy
- HPD
hours post-dendrotomy
- KMN
Knl1, Mis12, Ndc80 network
- KT
kinetochore
- OCT
optimal cutting temperature
- RFP
red fluorescent protein
- RZZ
Rod-Zwilch-Zw10 complex
- SAC
spindle assembly checkpoint
- γTuRC
γ-tubulin ring complex.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-04-0237-T) on July 16, 2020.
REFERENCES
- Akhmanova A, Steinmetz MO. (2008). Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol , 309–322. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz MO. (2015). Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol , 711–726. [DOI] [PubMed] [Google Scholar]
- Baas PW, Rao AN, Matamoros AJ, Leo L. (2016). Stability properties of neuronal microtubules. Cytoskeleton , 442–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlan K, Lu W, Gelfand VI. (2013). The microtubule-binding protein ensconsin is an essential cofactor of kinesin-1. Curr Biol , 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics , 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust-Mascher I, Scholey JM. (2002). Microtubule flux and sliding in mitotic spindles of Drosophila embryos. Mol Biol Cell , 3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmena M, Wheelock M, Funabiki H, Earnshaw WC. (2012). The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol , 789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheerambathur DK, Prevo B, Chow TL, Hattersley N, Wang S, Zhao Z, Kim T, Gerson-Gurwitz A, Oegema K, Green R, Desai A. (2019). The kinetochore-microtubule coupling machinery is repurposed in sensory nervous system morphogenesis. Dev Cell , 864–872.e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM. (2014). The kinetochore. Cold Spring Harb Perspect Biol , a015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Nye DM, Stone MC, Weiner AT, Gheres KW, Xiong X, Collins CA, Rolls MM. (2016). Mitochondria and caspases tune Nmnat-mediated stabilization to promote axon regeneration. PLoS Genet , e1006503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Stone MC, Tao J, Rolls MM. (2012). Axon injury and stress trigger a microtubule-based neuroprotective pathway. Proc Natl Acad Sci USA , 11842–11847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Sloutsky R, Naegle KM, Cavalli V. (2015). Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell , 691. [DOI] [PubMed] [Google Scholar]
- Ciferri C, Musacchio A, Petrovic A. (2007). The Ndc80 complex: hub of kinetochore activity. FEBS Lett , 2862–2869. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature , 151–156. [DOI] [PubMed] [Google Scholar]
- Drinnenberg IA, Henikoff S, Malik HS. (2016). Evolutionary turnover of kinetochore proteins: a ship of Theseus? Trends Cell Biol , 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etemad B, Kops GJ. (2016). Attachment issues: kinetochore transformations and spindle checkpoint silencing. Curr Opin Cell Biol , 101–108. [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Verhaagen J. (2018). Intrinsic determinants of axon regeneration. Dev Neurobiol , 890–897. [DOI] [PubMed] [Google Scholar]
- Feng C, Thyagarajan P, Shorey M, Seebold DY, Weiner AT, Albertson RM, Rao KS, Sagasti A, Goetschius DJ, Rolls MM. (2019). Patronin-mediated minus end growth is required for dendritic microtubule polarity. J Cell Biol , 2309–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Cheeseman IM. (2011). Kinetochore assembly: if you build it, they will come. Curr Opin Cell Biol , 102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Cheeseman IM. (2013). CDK-dependent phosphorylation and nuclear exclusion coordinately control kinetochore assembly state. J Cell Biol , 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin PR, Sasaki JM, Juo P. (2012). Cyclin-dependent kinase 5 regulates the polarized trafficking of neuropeptide-containing dense-core vesicles in Caenorhabditis elegans motor neurons. J Neurosci , 8158–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima G, Kiyomitsu T, Yoda K, Yanagida M. (2003). Human centromere chromatin protein hMis12, essential for equal segregation, is independent of CENP-A loading pathway. J Cell Biol , 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton G, Dimitrova Y, Davis TN. (2019). Seeing is believing: our evolving view of kinetochore structure, composition, and assembly. Curr Opin Cell Biol , 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. (2009). Axon regeneration requires a conserved MAP kinase pathway. Science , 802–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Platero JS, van Steensel B. (2000). Heterochromatic deposition of centromeric histone H3-like proteins. Proc Natl Acad Sci USA , 716–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw SM, Harrison SC. (2018). Kinetochore function from the bottom up. Trends Cell Biol , 22–33. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods , 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleele T, Marinkovic P, Williams PR, Stern S, Weigand EE, Engerer P, Naumann R, Hartmann J, Karl RM, Bradke F, et al (2014). An assay to image neuronal microtubule dynamics in mice. Nat Commun , 4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn V, Musacchio A. (2015). The Aurora B kinase in chromosome bi-orientation and spindle checkpoint signaling. Front Oncol , 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. (2012). The spindle assembly checkpoint. Curr Biol , R966–R980. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol , 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Fox P, Lakonishok M, Davidson MW, Gelfand VI. (2013). Initial neurite outgrowth in Drosophila neurons is driven by kinesin-powered microtubule sliding. Curr Biol , 1018–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahar M, Cavalli V. (2018). Intrinsic mechanisms of neuronal axon regeneration. Nat Rev Neurosci , 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniar TA, Kaplan M, Wang GJ, Shen K, Wei L, Shaw JE, Koushika SP, Bargmann CI. (2012). UNC-33 (CRMP) and ankyrin organize microtubules and localize kinesin to polarize axon-dendrite sorting. Nat Neurosci , 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manic G, Corradi F, Sistigu A, Siteni S, Vitale I. (2017). Molecular regulation of the spindle assembly checkpoint by kinases and phosphatases. Int Rev Cell Mol Biol , 105–161. [DOI] [PubMed] [Google Scholar]
- Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A. (1998). puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev , 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin X, Daneman R, Zavortink M, Chia W. (2001). A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci USA , 15050–15055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A. (2015). The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol , R1002–R1018. [DOI] [PubMed] [Google Scholar]
- Musacchio A, Desai A. (2017). A molecular view of kinetochore assembly and function. Biology (Basel) , 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MM, McCracken CJ, Milner ES, Goetschius DJ, Weiner AT, Long MK, Michael NL, Munro S, Rolls MM. (2014). Gamma-tubulin controls neuronal microtubule polarity independently of Golgi outposts. Mol Biol Cell , 2039–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nye DMR, Albertson RM, Weiner AT, Hertzler JI, Shorey M, Goberdhan DCI, Wilson C, Janes KA, Rolls MM. (2020). The receptor tyrosine kinase Ror is required for dendrite regeneration in Drosophila neurons. PLoS Biol , e3000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori-McKenney KM, Jan LY, Jan YN. (2012). Golgi outposts shape dendrite morphology by functioning as sites of acentrosomal microtubule nucleation in neurons. Neuron , 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Cohen F, Dean BJ, De La Torre K, Deshmukh G, Estrada AA, Ghosh AS, Gibbons P, Gustafson A, Huestis MP, et al (2015). Discovery of dual leucine zipper kinase (DLK, MAP3K12) inhibitors with activity in neurodegeneration models. J Med Chem , 401–418. [DOI] [PubMed] [Google Scholar]
- Przewloka MR, Zhang W, Costa P, Archambault V, D’Avino PP, Lilley KS, Laue ED, McAinsh AD, Glover DM. (2007). Molecular analysis of core kinetochore composition and assembly in Drosophila melanogaster. PLoS One , e478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Kumar A, Blockus H, Waites C, Bartolini F. (2019). Activity-dependent nucleation of dynamic microtubules at presynaptic boutons controls neurotransmission. Curr Biol , 4231–4240.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, Nateri AS, Makwana M, Riera-Sans L, Wolfer DP, et al (2004). The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron , 57–67. [DOI] [PubMed] [Google Scholar]
- Rao KS, Rolls MM. (2017). Two Drosophila model neurons can regenerate axons from the stump or from a converted dendrite, with feedback between the two sites. Neural Dev , 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls MM, Satoh D, Clyne PJ, Henner AL, Uemura T, Doe CQ. (2007). Polarity and compartmentalization of Drosophila neurons. Neural Dev , 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schittenhelm RB, Heeger S, Althoff F, Walter A, Heidmann S, Mechtler K, Lehner CF. (2007). Spatial organization of a ubiquitous eukaryotic kinetochore protein network in Drosophila chromosomes. Chromosoma , 385–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, Diantonio A. (2012). Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron , 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Ori-McKenney KM, Zheng Y, Han C, Jan LY, Jan YN. (2012). Regeneration of Drosophila sensory neuron axons and dendrites is regulated by the Akt pathway involving Pten and microRNA bantam. Genes Dev , 1612–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N. (2003). Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein). J Neurosci , 2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Albertson RM, Chen L, Rolls MM. (2014). Dendrite injury triggers DLK-independent regeneration. Cell Rep , 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Nguyen MM, Tao J, Allender DL, Rolls MM. (2010). Global up-regulation of microtubule dynamics and polarity reversal during regeneration of an axon from a dendrite. Mol Biol Cell , 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Roegiers F, Rolls MM. (2008). Microtubules have opposite orientation in axons and dendrites of Drosophila neurons. Mol Biol Cell , 4122–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura K, Yamamoto M, Niwa R, Satoh D, Goto S, Taniguchi M, Hayashi S, Uemura T. (2003). Distinct developmental modes and lesion-induced reactions of dendrites of two classes of Drosophila sensory neurons. J Neurosci , 3752–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka TU, Desai A. (2008). Kinetochore-microtubule interactions: the means to the end. Curr Opin Cell Biol , 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. (2014). A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell , 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Feng C, Rolls MM. (2016). The microtubule-severing protein fidgetin acts after dendrite injury to promote their degeneration. J Cell Sci , 3274–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson-Peer KL, DeVault L, Li T, Jan LY, Jan YN. (2016). In vivo dendrite regeneration after injury is different from dendrite development. Genes Dev , 1776–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooley J, Stukenberg PT. (2011). The Ndc80 complex: integrating the kinetochore’s many movements. Chromosome Res , 377–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovey CA, Tubman CE, Hamrud E, Zhu Z, Dyas AE, Butterfield AN, Fyfe A, Johnson E, Conduit PT. (2018). gamma-TuRC heterogeneity revealed by analysis of Mozart1. Curr Biol , 2314–2323.e2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma D, Salmon ED. (2012). The KMN protein network —chief conductors of the kinetochore orchestra. J Cell Sci , 5927–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner AT, Seebold DY, Torres-Gutierrez P, Folker C, Swope RD, Kothe GO, Stoltz JG, Zalenski KK, Barbera DJ, Patel MA, et al (2020). Endosomal Wnt signaling proteins control microtubule nucleation in dendrites. PLoS Biol , e3000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Collins CA. (2012). A conditioning lesion protects axons from degeneration via the Wallenda/DLK MAP kinase signaling cascade. J Neurosci , 610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA. (2010). Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol , 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y. (2009). The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell , 1005–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau KW, Schatzle P, Tortosa E, Pages S, Holtmaat A, Kapitein LC, Hoogenraad CC. (2016). Dendrites in vitro and in vivo contain microtubules of opposite polarity and axon formation correlates with uniform plus-end-out microtubule orientation. J Neurosci , 1071–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Dong Z, Gustafson WC, Ruiz-Gonzalez R, Signor L, Marzocca F, Borel F, Klassen MP, Makhijani K, Royant A, et al (2016). Rational design of a monomeric and photostable far-red fluorescent protein for fluorescence imaging in vivo. Protein Sci , 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Oztan A, Ye Y, Schwarz TL. (2019). Kinetochore proteins have a post-mitotic function in neurodevelopment. Dev Cell , 873–882.e874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.