

SUMMARY
Lactate is used as an energy source by producer cells or shuttled to neighboring cells and tissues. Both glucose and lactate fulfill the bioenergetic demand of neurons, the latter imported from astrocytes. The contribution of astrocytic lactate to neuronal bioenergetics and the mechanisms of astrocytic lactate production are incompletely understood. Through in vivo 1H magnetic resonance spectroscopy, 13C glucose mass spectroscopy, and electroencephalographic and molecular studies, here we show that the energy sensor AMP activated protein kinase (AMPK) regulates neuronal survival in a non-cell-autonomous manner. Ampk-null mice are deficient in brain lactate and are seizure prone. Ampk deletion in astroglia, but not neurons, causes neuronal loss in both mammalian and fly brains. Mechanistically, astrocytic AMPK phosphorylated and destabilized thioredoxin-interacting protein (TXNIP), enabling expression and surface translocation of the glucose transporter GLUT1, glucose uptake, and lactate production. Ampk loss in astrocytes causes TXNIP hyperstability, GLUT1 misregulation, inadequate glucose metabolism, and neuronal loss.
In Brief
Muraleedharan et al. demonstrate that AMPK is required for astrocytic glycolysis, lactate production, and lactate shuttle as an energy source to neurons such that AMPK loss in glia causes non-cell-autonomous neuronal loss in the mammalian and fly brain.
Graphical Abstract
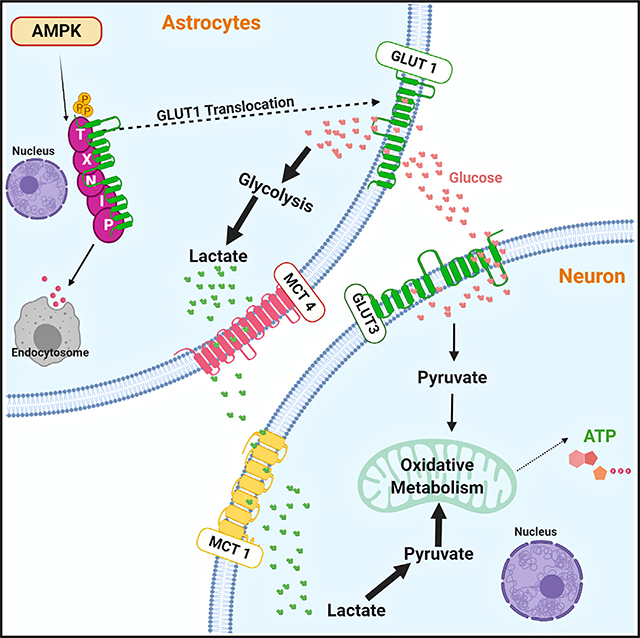
INTRODUCTION
Regulation of bioenergetics is critical for the maintenance of tissue architecture and cellular physiology. Glucose is a key bioenergetic fuel that is catabolized through glycolysis and the tricarboxylic acid (TCA) cycle. In addition, metabolites like lactate and alanine are also shuttled between cells and tissues, such as muscle fibers and neurons (Brooks, 1998, 2002, 2009). In fact, in all tissues except brain, a common blood lactate pool is shared among organs through circulation to support tissue bioenergetics that is at least as important as glucose oxidation for energy generation (Hui et al., 2017). Except when blood lactate levels are very high, very little systemic lactate crosses the blood-brain barrier (BBB) (Barros, 2013; Dienel, 2012; Hui et al., 2017; Mächler et al., 2016), and glucose remains the primary fuel for the brain. Although neurons use plenty of glucose, they seem to prefer lactate over glucose (Larrabee, 1996). The extent to which neuronal glucose oxidation contributes to its energy demand is, however, not fully understood. A large number of in vivo and in vitro studies indicate that lactate shuttled from astrocytes (known as astrocyte-neuron lactate shuttle [ANLS]) is an important source of energy and is required for various neuronal functions in the rodent and human brain (Bélanger et al., 2011; Brooks, 2018; Descalzi et al., 2019; Kasischke et al., 2004, 2011; Magistretti and Allaman, 2018; Mangia et al., 2009; Pellerin et al., 1998, 2007; Suzuki et al., 2011). However, the scope of ANLS in neuronal bioenergetics under normal physiology has been questioned (Bélanger et al., 2011; Díaz-García et al., 2017; Dienel, 2012; Yellen, 2018). In a recent ex vivo study, Díaz-García et al. (2017) demonstrated that stimulation-induced neuronal activity is independent of ANLS.
According to the ANLS hypothesis, astrocytic endfeet and perisynaptic processes sense brain metabolic microenvironment and respond to changing energy demands of neurons (Bélanger et al., 2011). Following excitation, glutamate released by neurons is taken up by glutamate transporters (GLT/GLAST1) in astrocytes, which in turn stimulates astrocytic glucose uptake and glycolysis (Bélanger et al., 2011). Lactate, which is often the glycolytic end product in astrocytes (Schurr and Payne, 2007), is then shuttled through lactate transporters in astrocytes (MCT1/4) and neurons (MCT2) (Bélanger et al., 2011), although MCT-independent lactate conduits through Pannexin and Connexin hemichannels in the brain have also been reported (Karagiannis et al., 2016). Unlike astrocytes, neurons cannot store glycogen as an energy substrate (Brown and Ransom, 2007; Brown et al., 2005) and are thought to be reliant on astrocytic lactate for ATP production, maintaining redox balance and intracellular pH, gene expression (Hashimoto et al., 2008), and lipid synthesis (Liu et al., 2017). This reliance on outsourced glycolysis seems to be conserved across species, because disruption of glycolysis in glia, but not neurons, causes neuronal death in the fly brain (Volkenhoff et al., 2015).
The molecular basis for this metabolic compartmentalization is that in neurons there is a sustained diversion of glucose to the pentose phosphate pathway for the generation of NADPH, a key reducing metabolite for the maintenance of neuronal redox. On the other hand, pyruvate dehydrogenase (PDH), the key enzyme required for the conversion of glucose-derived pyruvate to acetyl coenzyme A (CoA) to initiate glucose oxidation via the TCA cycle, is downregulated in astrocytes, both due to low expression and phosphorylation-mediated inhibition (Halim et al., 2010; Itoh et al., 2003; Laughton et al., 2007). The metabolic symbiosis between neurons and astrocytes is probably strongly selected because recent studies show that turning down aerobic glycolysis is required during neuronal differentiation, and constitutive expression of glycolytic genes, such as hexokinase or lactate dehydrogenase, causes neuronal death (Zheng et al., 2016). Fructose-2, 6-bisphosphate synthesized by the enzymes pfkb1–4 is the most powerful allosteric activator of PFK-1. Mammalian neurons do express pfkfb2, but at lower levels than astrocytes. Pfkfb3 is also expressed at much higher levels in astrocytes than neurons, and because this isoform has a considerably higher catalytic activity than pfkfb2, astrocytes are likely more glycolytic than neurons. Together, these distinguishing molecular features enable astrocytes to be more glycolytic than neurons.
Although it is appreciated that astrocytic glycolysis is important for overall brain function, little is known about the metabolic genes that control astrocytic lactate production. The AMP activated protein kinase (AMPK) is an evolutionary conserved energy sensor that regulates cellular bioenergetics. AMPK is a heterotrimeric serine/threonine kinase containing catalytic α and regulatory β and γ subunits (Dasgupta and Chhipa, 2016; Hardie, 2014). Mammals express 2α, 2β, and 3γ subunits. Specific trimeric complexes are preferentially formed in different tissues. Typically, active AMPK inhibits anabolism and activates catabolism (Dasgupta and Chhipa, 2016; Hardie, 2014). It diminishes protein synthesis by inhibiting mTORC1 (Mechanistic target of rapamycin complex 1) and lipid synthesis by inhibiting acetyl CoA carboxylase 1 (ACC1) and hydroxymethyl-glutaryl-coenzyme A reductase (HMG CoA reductase), while augmenting glucose and fatty acid oxidation through PGC1α, ACC2, and other yet unclear mechanisms. Earlier studies have shown the involvement of AMPK in glucose uptake and glucose transporter cell surface translocation in skeletal muscle (Mu et al., 2001; Sakamoto et al., 2005). AMPK-dependent phosphorylation of PFK2 has also been shown to stimulate nitric oxide-mediated glycolysis in astrocytes (Almeida et al., 2004). We have recently shown that AMPK-dependent phosphorylation of the transcription factor CREB1 regulates glycolysis in astrocytoma cells (Chhipa et al., 2018). Here, we have examined whether AMPK regulates basal glycolysis, lactate production, and the ANLS in the rodent brain. We show that AMPK regulates astroglial glycolysis. Glia-specific loss of Ampk in model organisms reduces brain lactate and affects neuronal survival through a mechanism that involves AMPK-dependent glucose transporter membrane translocation, glucose import, glycolysis, and lactate production.
RESULTS
Lactate and Other Energy Metabolites Are Reduced in the AMPK-Null Brain
The human brain represents only 2% of mass, yet it consumes 15%–20% of total energy. Although AMPK is a master regulator of energy metabolism, little is known about the metabolites that AMPK regulates at the systemic level. To this end, we first generated neural stem/progenitor cell (NPC)-specific Ampk knockout (KO) mice. Two AMPK β subunits are expressed in mammalian tissues, including the brain, and these subunits are essential for stability of the AMPK holoenzyme (Iseli et al., 2005). Earlier, we found that the Ampk β2-null mice develop metabolic phenotype under energy stress conditions (Dasgupta et al., 2012). To reduce AMPK activity in the brain, we deleted both β1 and β2 subunits in NPCs by crossing β1L/L; β2−/− mice with Nestin Cre mice. Cre recombination was efficient as revealed by substantially reduced levels of active AMPK (pAMPK) and phosphorylated ACC (pACC; a bona fide AMPK substrate and a surrogate for AMPK activity) in the Ampk β1/β2; Nestin Cre KO brain (Figure S1A). Immunohistochemistry (IHC) showed that in wild-type (WT) mice, although an abundant pACC signal was observed in oligodendrocytes of corpus callosum and in glial fibrillary acidic protein (GFAP)-positive astrocytes (red channel in Figure S1C and green channel in Figure S1I, respectively), pACC signal was below detection level in neuronal nuclei (NeuN)-positive neurons (Figures S1B–S1D). Expectedly, pACC signal was reduced in AMPKβ1/β2-null glia (compare areas between white lines in Figure S1C versus S1F, and Figure S1I versus S1L). We could not use AMPK, pAMPK, or β1/β2 antibodies for IHC because of their lack of specificity for mouse tissue and had to rely on pACC signal instead. Although lack of pACC signal in neurons precluded us from accurately evaluating Ampk deletion in these cells, we were able to assess this indirectly by examining mTORC1 signaling, which is negatively regulated by AMPK (Figures S1O and S1P).
To examine whether AMPK regulates brain metabolites, we acquired in vivo proton magnetic resonance spectroscopy (1H MRS) data from the dorsal hippocampus (a region enriched in astroglia) of post-natal day (P) 28 WT and Ampk KO mice. The spectral quality routinely achieved in this study allowed reliable quantification of 16 metabolites (Figures 1A–1D; Figure S2A; Table S1). Compared with the WT group, lactate levels were ~40% reduced in Ampk KO brains (Figures 1D and 1E). Total creatine and phosphocreatine, two other cellular energy equivalents that maintain energy homeostasis in organs of high energy turnover, were also reduced in the Ampk KO brains (Figures 1F and 1G). Myo-inositol, which is synthesized from glucose via glucose 6 phosphate, was also reduced (Figure 1H). Two amino acids, glutamate and alanine, that exchange their carbons with glucose-derived carboxylic acids of the TCA cycle through transamination reactions showed reduced abundance in the Ampk KO brain (Figures 1I and 1J). We also acquired MRS data from the midbrain, and lactate data in this region were consistent with that observed in the dorsal hippocampus (Figures S2B–S2F; Table S1). These data indicate that AMPK is likely required to maintain the levels of lactate, as well as other energy metabolites in the mammalian brain.
Figure 1. Neurochemical Profiling of the Ampk-Null Brain Using In Vivo 1H Magnetic Resonance Spectroscopy Shows Reduced Lactate.

(A–D) Representative magnetic resonance (MR) images and in vivo 1H MR spectra acquired from the brain of the wild-type (WT; A and B) (n = 6) and Ampk β1lox/lox; β2−/−; Nestin Cre (Ampk knockout [KO]) 28-day-old mice (C and D) (n = 6) (STEAM, echo time [TE] = 2 ms, repetition time [TR] = 5 s, nontarget [NT] = 240). Axial fast spin-echo images show the typical selection of the 3.6 μL volume of interest [VOI] in dorsal hippocampus. Arrows show the direction of changes in myo-inositol, taurine, total creatine (Cr+PCr), glutamate, alanine, and lactate in the Ampk KO group, relative to the WT group.
(E–J) The bar diagrams show the differences in metabolite concentrations between the WT and Ampk KO groups (n = 6/genotype). Error bars are mean ± SD. Significance of two-tailed, unpaired t tests is shown.
Ala, alanine; Asc, ascorbate; Cr, creatine; GABA, γ-aminobutyric acid; Gln, glutamine; Glu, glutamate; GPC, glycerophosphocholine; GSH, glutathione; Ins, myo-inositol; Lac, lactate; NAA, N-acetylaspartate, PC, phosphocholine; PCr, phosphocreatine; Tau, taurine. See also Figures S1 and S2 and Table S1.
Post-natal Neuronal Loss and Reduced Cortical Thickness in AMPK-Null Mice
Development of the multi-layered murine cerebral cortex is completed by P28. Because we observed misregulated metabolite levels in P28 Ampk KO; Nestin Cre mice, we examined cortical histology at this stage. Nissl staining revealed that Ampk KO brain exhibited thinner cerebral cortex and cortical plate (Figures 2A–2C), a feature that persisted throughout the life of the animals (Figures 2D–2I). At 2 years of age, cortical thickness and brain volume of Ampk KO mice were substantially reduced (Figures 2G–2L). At 24 months, the number of GFAP-positive astrocytes was greatly increased in the KO brain (Figures S3A–S3C), suggestive of reactive gliosis that occurs in response to chronic CNS pathology. The reactive nature of these astrocytes in AMPK-deficient brains was confirmed by combined IHC-in situ hybridization that showed increased expression of LCN2 (lipocalin 2) and C3 (complement C3), the two most upregulated genes in reactive astrocytes (Figures S3D–S3G). Consistent with diminished cortical thickness, the number of NeuN-positive neurons was significantly reduced at P28, 15-month-old, and 24-month-old Ampk KO brains (Figures 2M–2S). IHC of Cux1/2 (layers II/III and IV) and FoxP2 (layer VI) showed that neuronal loss was not layer specific (Figures S3H–S3M). Interestingly, at P28, although the number of GFAP-positive astrocytes was slightly reduced (Figures S3N–S3P), Olig2-positive oligodendrocytes were increased in the AMPK KO brain (Figures S3Q–S3S). In addition, although very few cleaved caspase-positive apoptotic cells were observed in the WT cortex, a 2-fold increase in apoptotic cells was observed in the Ampk KO cortex (Figures 2T–2V). However, reduction in cortical thickness or NeuN-positive neurons was not observed in the embryonic day (E) 18.5 Ampk KO cortex (Figures 2W–2Y; Figures S3T–S3V). Collectively, our data indicate that AMPK deletion in NPCs leads to specific loss of neurons in the post-natal murine cortex.
Figure 2. Ampk Deletion in NPCs Reduces Neuronal Survival and Cortical Thickness and Volume in Adult Mice.

(A–I) IHC of 1- (A and B), 15- (D and E), and 24-month-old (G and H) control and Ampk β1lox/lox; β2−/−; Nestin Cre (Ampk KO) cortices stained with Nissl stain. Scale bars: 50 μm. Quantification of the cortical thickness at 1, 15, and 24 months is shown in (C), (F), and (I), respectively (n = 3–8 mice/genotype).
(J and K) Representative whole-mount images of 24-month-old brains from WT and Ampk KO animals.
(L) Quantification of the cortical surface area of 24-month-old animals (n = 2/genotype).
(M–R) IHC of 1- (M and P), 15- (N and Q), and 24-month-old (O and R) WT and Ampk KO cortices stained with antibody to NeuN (green), a neuronal marker, and DAPI (blue). Scale bars: 50 μm.
(S) Quantification of the number of NeuN+ cells in 1- (n = 5 mice/genotype), 15- (n = 5 mice/genotype), and 24-month-old (n = 3 mice/genotype) cortices. Error bars are mean ± SEM. Significance of two-way ANOVA is shown.
(T–V) IHC of WT and Ampk KO cortices stained for cleaved caspase-3 (CC3; green) and DAPI (blue) at 1 month (T and U), and quantification of the number of CC3+ cells in the cortex (V) (n = 3 mice/genotype). Scale bars: 50 μm.
(W and X) Nissl stain images of WT and Ampk KO cortices at E18.5, and quantification of cortical thickness (y) (n = 3 mice/genotype). Scale bars: 50 μm.
Error bars are mean ± SEM. Significance of two-tailed, unpaired t tests is shown. See also Figure S3.
AMPK-Null Astrocytes Are Deficient in Glycolysis and Lactate Production
Astrocytic lactate supports neuronal bioenergetics and function in multiple ways, including energy generation through lactate oxidation, lipid synthesis, and synaptic remodeling. We hypothesized that misregulation of lactate production in Ampk KO astrocytes could be causally related to reduced metabolic support and neuronal loss in the Ampk KO cortex. To test this, we measured lactate production in cultured cortical astrocytes and neurons. Lactate released by neurons was undetectable at earlier time points, and three times the number of neurons than astrocytes had to be used to detect lactate at later time points. Although there was no difference in lactate release between WT and Ampk KO neurons, Ampk KO astrocytes were significantly deficient in lactate production (Figures 3A and 3B). Growing astrocytes in serum or in G5 serum supplement did not alter our findings. When mitochondrial oxidative phosphorylation is inhibited, glycolytic cells maximize glycolysis (glycolytic capacity) to compensate for mitochondrial ATP loss. Glycolysis stress test showed that Ampk KO astrocytes were deficient in glucose-stimulated glycolysis and glycolytic capacity (Figures 3C and 3D). To assess the rate of glucose flux, we incubated astrocytes with the glucose isotopomer [U-13C] glucose and examined 13C-labeled metabolites by mass spectrometry. Consistent with our in vivo results, Ampk KO astrocytes showed significant reduction in labeled lactate, fructose 6 phosphate, fructose 1,6 bisphosphate, 2 phosphoglycerate/3 phosphoglycerate, ribose 5 phosphate (a metabolite of the pentose phosphate pathway that originates from glycolysis), and citrate (Figures 3E–3J). Citrate reduction raises the possibility that pyruvate-derived acetyl CoA entry into the TCA cycle is also reduced in Ampk KO astrocytes. Together, these results confirm that AMPK is required for optimal glycolysis and lactate production in astrocytes.
Figure 3. Ampk Deficiency in Astrocytes Reduces Glycolysis, Cortical Thickness, and Neuronal Number.

(A and B) Lactate released into the media by WT or Ampk-null neurons (A) and astrocytes (B). Error bars are mean ± SEM. n = 3 biological replicates.
(C and D) WT and Ampk KO astrocytes were tested for their glycolytic capacity and ECAR (extracellular acidification rate) on a Seahorse XFe96 Analyzer. A representative plot of ECAR over time of cells with addition of glucose (10 mM), oligomycin (1 μM), and 2-DG (50 mM), as indicated (C). Quantitative analysis of glycolysis, glycolytic capacity, and glycolytic reserve of WT and Ampk KO astrocytes (D). Error bars are mean ± SEM. Significance of two-tailed, unpaired t tests is shown. n = 3 biological replicates.
(E–J) Kinetic flux analysis of glycolysis using U13C glucose isotopomer followed by high-performance liquid chromatography (HPLC)/mass spectrometry in WT and Ampk-null astrocytes. Abundance of 13C-labeled metabolites measured after 12-h incubation with U13C glucose. Error bars are mean ± SEM. Significance of two-tailed, unpaired t tests is shown. n = 4 biological replicates.
(K–M) Nissl-stained 1-month-old cortices of control and Ampk lox/lox; Gfap Cre ER mouse (K and L) and quantification of the cortical thickness (M) (n = 5–7 mice/genotype). Scale bars: 50 μm. Error bars are mean ± SEM. Significance of two-tailed, unpaired t tests is shown.
(N–P) IHC of 1-month-old control and Ampk lox/lox; Gfap Cre ER mouse cortices stained with NeuN antibody (green) and DAPI (blue) (N and O), and quantification of NeuN+ cells (P) (n = 4 mice/genotype). Scale bars: 50 μm. Error bars are mean ± SEM. Significance of two-tailed, unpaired t tests is shown.
See also Figures S4 and S5.
Astrocytic AMPK Regulates Neuronal Survival in a Non-Cell-Autonomous Manner
Nestin Cre recombinase is active in NPCs; therefore, AMPK is expected to be deleted in both glia and neurons. Although our in vitro experiments suggested that the cortical phenotype could potentially be caused by diminished astrocytic lactate support, to be definitive, we made astrocyte-specific or neuron-specific Ampk KO mice by crossing β1L/L; β2−/− mice with Gfap Cre ER or Thy1 Cre mice, respectively. The Thy1 Cre recombinase is constitutively active and is expressed in neurons of the post-natal cortex and hippocampus. To delete AMPK in post-natal brain astrocytes, we induced Gfap Cre by injecting tamoxifen during P2–P5. pACC IHC showed that staining was reduced from about 80% of GFAP-positive astrocytes, indicating efficient Cre recombination (Figures S4A–S4G). Similar to Ampk KO; Nestin Cre mice, cortical thickness and NeuN-positive neurons were reduced in Ampk KO; Gfap Cre mice (Figures 3K–3P), indicating a non-cell-autonomous role of astrocytic AMPK in the maintenance of post-natal cortical neurons.
Next, to determine the cell-autonomous consequences of neuronal Ampk loss, we examined cortical histology of Ampk KO; Thy1 Cre mice. Although we used pACC IHC as a surrogate for AMPK activity in astrocytes, basal pACC level in neurons was too low to determine AMPK deletion in these cells. AMPK inhibits mTORC1 (Figure 4A); therefore, AMPK loss increases phosphorylation of mTORC1 downstream substrate ribosomal protein S6 (Figure 4A). Therefore, we used pS6 IHC as a surrogate for AMPK activity in these cells. Compared with control mice, neurons in the Ampk KO; Thy1 Cre cortex demonstrated considerably elevated pS6 levels (Figures S5A–S5D), confirming AMPK deletion in neurons. However, unlike Ampk KO; Gfap Cre ER mice, no reduction of cortical thickness or neuronal loss was observed in 1-month-old Ampk KO; Thy1 Cre mice. In fact, the cortical thickness and NeuN-positive neurons were increased in these mice relative to controls (Figures S5E–S5J). Gleaning together the cortical neuronal phenotype in Nestin Cre, Gfap Cre ER, and Thy1 Cre Ampk KO mice, our results indicate astrocytic AMPK and perhaps AMPK-regulated metabolic support by astrocytes are essential for post-natal neuronal survival. Second, only in the presence of this metabolic support, mTORC1 overdrive in neurons (as in Thy1 Cre mice) increases neuronal number and cortical thickness in the post-natal rodent brain.
Figure 4. Activation of mTORC2 Feedback Loop Partially Rescues Neuronal Survival in Ampk KO Mice.

(A) Schematic showing the regulation of mTORC1 by AMPK and feedback activation of mTORC2 and AKT upon mTORC1 inhibition.
(B and C) IHC of WT mouse brain showing active AMPK (pACC staining) and active mTORC1 (pS6 staining) in astrocytes and neurons. Scale bars: 50 μm (B) and 20 μm (C).
(D–G) IHC of control and Ampk β1lox/lox; β2−/−; Nestin Cre (Ampk KO) cortices stained with NeuN and pS6 antibodies. DAPI (blue) was used to stain nuclei (n = 5 mice/genotype). Scale bars: 50 μm. Insets show co-localization of pS6 with NeuN.
(H–K) IHC of control and Ampk β1lox/lox; β2−/−; Nestin Cre (Ampk KO) cortices stained with GFAP and pS6 antibodies (n = 5 mice/genotype). Scale bars: 50 μm. Insets: co-localization of pS6 with GFAP.
(L and M) Nissl-stained cortical sections from the rapamycin-treated control and Ampk β1lox/lox; β2−/−; Nestin Cre (Ampk KO) mice. Scale bars: 50 μm.
(N) Quantification of the cortical thickness in vehicle- or rapamycin-treated groups (n = 3 mice/genotype). Error bars are mean ± SEM. Significance of one-way ANOVA is shown.
(O–Q) IHC of cortices stained with NeuN antibody (O and P) and quantification of NeuN+ cells (Q) (n = 3 mice/genotype). Error bars are mean ± SEM. Significance of one-way ANOVA is shown.
(R–U) IHC of cortices showing pAKTSer473 staining in rapamycin- or vehicle-treated control and Ampk β1lox/lox; β2−/−; Nestin Cre (Ampk KO) animals. Scale bars: 50 μm.
CC, corpus callosum; DG, dentate gyrus; HC, hippocampus.
Rapamycin Partially Rescues Cortical Thickness in AMPK KO; Nestin Cre Mice through Feedback Activation of mTORC2
Our data so far underscore that as long as astrocytes are proficient in AMPK, neuronal AMPK loss (as in Ampk KO: Thy1 Cre animals) is insufficient to reduce cortical thickness in mice. In the Ampk KO; Nestin Cre mice, where both astrocytes and neurons are AMPK deficient, we also observed hyperactive mTORC1 in neurons. We were curious whether any alternative mechanism could rescue the cortical phenotype in these mice even in the absence of optimal metabolic (lactate) support from astrocytes. In some contexts, activation of mTORC1 (that contains RAPTOR) inhibits mTORC2 (that contains RICTOR) through a S6 kinase-IRS1/2-phosphatidylinositol 3-kinase (PI3K) feedback loop (Figure 4A). Therefore, inhibiting hyperactive mTORC1 can activate the mTORC2. Earlier studies showed that active mTORC2 regulates neuronal growth, and overactivation of mTORC2 can counter neuronal death (Creus-Muncunill et al., 2018; Thomanetz et al., 2013). Therefore, we tested whether blocking mTORC1 by rapamycin activates mTORC2 and rescues cortical phenotype of Ampk KO; Nestin Cre mice.
First, it is important to reiterate that in the WT mouse brain, we observed a clear and unambiguous difference in the magnitude of active AMPK and mTORC1 signal between neurons and glia. Although high levels of pACC (a surrogate for AMPK activity) were present in astrocytes of the cerebral cortex (Figure 4B), glial progenitors and transit-amplifying cells in the dentate gyrus (Figure 4C), and oligodendrocytes in corpus callosum (Figure 4B), pS6 (a surrogate for mTORC1 activity) was more evident in cortical and hippocampal neurons (Figures 4B and 4C). Accordingly, loss of Ampk in Nestin Cre mice increased mTORC1 activity (high pS6) in neurons, similar to that in Thy1 Cre mice (Figures 4D–4G). Interestingly, Ampk KO GFAP-positive astrocytes in the Nestin Cre mice did not show a similar increase in pS6 signal (Figures 4H–4K). These data suggest that perhaps neuronal AMPK phosphorylates ACC to a much lower degree than other AMPK substrates. Alternatively, basal AMPK activity is low in neurons but more sensitive to Ampk loss compared with astrocytes. Therefore, AMPK loss robustly increases mTORC1 activity in neurons, but not as much in astrocytes.
Chronic rapamycin exposure in post-natal mice (P2–P28) led to stunted overall growth, but the brain was spared. In fact, rapamycin slightly increased cortical thickness of WT mice (Figure 4N). Remarkably, rapamycin partially rescued both cortical thickness (Figures 4L–4N, compare pink and green bars) and neuronal numbers (Figures 4O–4Q) in the Ampk KO; Nestin Cre mice. To test our hypothesis if this rescue correlates with mTORC2 activation, we examined pAKTSer473 signal because mTORC2 directly phosphorylates AKT at this site. Rapamycin-treated brains showed robust activation of mTORC2 both in WT and in Ampk KO; Nestin Cre mice (Figures 4R–4U). Although we could not examine whether rapamycin also activated mTORC2 in astrocytes and whether that modulated lactate levels in vivo, our results suggest that at least in this context, neuronal mTORC2 overdrive can partly counter neuronal loss even in the face of inadequate astrocytic lactate support.
Loss of AMPK Causes Increased Baseline Brain Activity and Increased Susceptibility to Seizures in Mice
AMPK phosphorylates and activates the Tuberous Sclerosis Complex 2 (TSC2) (Inoki et al., 2003), which in turn inhibits mTORC1 though RHEB (Figure 4A). Loss-of-function mutation of the TSC1/2 complex in people or its deletion in the mouse brain causes epileptogenic seizure through unregulated mTORC1 activity (Nabbout et al., 2018; Tsai et al., 2012), which could be mitigated by rapamycin treatment. To examine whether increased mTORC1 activity in Ampk KO neurons heightens neuronal excitability, we quantified cortical electroencephalographic (EEG) signal using a wireless 24/7 video-EEG recording system in WT and Ampk KO; Nestin Cre mice. Representative traces of EEG recordings from both genotypes are shown in Figure 5A. At baseline, total EEG power across all frequencies (Figure 5B), as well as gamma power (Figure 5C), were significantly increased during the night in Ampk-null mice compared with WT mice, suggesting overall increased neuronal excitability. Similar trends were observed during the day. Neuronal hyperexcitability in Ampk-null mice was confirmed by assessing seizure susceptibility with a kainic acid challenge. Ampk-null mice showed a significantly reduced latency to seizure onset after intraperitoneal (i.p.) injection of kainic acid (Figure 5D). These results are consistent with earlier studies underscoring the role of the AMPK-TSC1/2-mTORC1 pathway in regulating neuronal excitability.
Figure 5. Ampk KO Mice Demonstrated Increased EEG Power and Seizure Susceptibility.

(A) Representative baseline EEGs from control and Ampk β1lox/lox; β2−/−; Nestin Cre (Ampk KO) mice.
(B) EEG power analysis showed a significant increase in total EEG power in Ampk KO mice over 3 consecutive nights compared with controls (Wilcoxon rank-sum test, *p(N1) = 0.03, *p(N2) = 0.05, and *p(N3) = 0.03; WT, n = 4; KO, n = 5).
(C) Gamma waveform analysis showed significantly increased gamma power during the same night periods in Ampk KO mice compared with controls (Wilcoxon rank-sum test, *p(N1) = 0.015, *p(N2) = 0.031, and *p(N3) = 0.015; WT, n = 4; KO, n = 5).
(D) Seizure-onset analysis showed significantly reduced latency to kainic acid (15 mg/kg)-induced seizure in Ampk KO mice (*p = 0.05; WT, n = 3; KO, n = 3, one-sample t test) compared with controls. Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and p values were determined by Wilcoxon rank-sum test.
Glia-Specific AMPK Silencing in the Fly Brain Recapitulates Neuronal Loss and Reduces Lifespan
We took a step further and asked whether the non-cell-autonomous role of AMPK in neuronal survival is evolutionarily conserved across species. In Drosophila, signaling from glia to neurons, including lactate shuttle, is important to coordinate neuronal development, metabolism, and survival (Fernandes et al., 2017; Volkenhoff et al., 2015). Volkenhoff et al. (2015) found that genetic suppression of glycolysis specifically in fly glia, but not in neurons, caused neuronal death. To test whether glia-specific Ampk deletion causes non-cell-autonomous death of fly neurons, we used a temperature-sensitive Gal80 variant that allowed tissue-specific RNAi only in the adult fly brain (McGuire et al., 2003). Unlike mammals, only one AMPK α, β, or γ subunit is expressed in the fly. We therefore used a glia-specific promoter (repo) or a neuron-specific promoter (elav) to silence either Ampk α, β, or γ subunit in the adult fly glia or neurons. Compared with control RNAi (repo>>-Cherry-dsRNA [double-stranded RNA]), we observed holes in the cortex region of the fly brain when AMPK subunits were silenced in the glia (Figures 6A–6D). These holes are reminiscent of the phenotype observed in glia-specific inhibition of glycolysis in the Trehalase RNAi fly (Figures 6E and 6F) and are indicative of neuronal loss. In contrast, Ampk RNAi using the neuron-specific elav promoter did not cause neuronal loss (Figures 6G–6J). Remarkably, quite similar to Trehalase or Pyruvate Kinase RNAi in fly glia (Volkenhoff et al., 2015), Ampk RNAi in the glia significantly reduced survival in flies (Figure 6K). In contrast, Ampk RNAi in neurons did not reduce survival (Figure 6L). Together, these intriguing results underscore an important and evolutionarily conserved non-cell-autonomous role of the energy sensor AMPK in neuronal energy supply and survival.
Figure 6. Glia-Specific Ampk Deletion Causes Neuronal Death and Reduces Lifespan in Drosophila.

(A–D) Semi-thin sections of brains (one hemisphere of the central brain and one optic lobe) of 50-day-old adult flies. Compared with controls (mCherry-dsRNA), AMPK knockdown in adult glial cells (using repo-Gal4, tub-Gal80ts) induces holes in the cortex region indicating neurodegeneration (arrows). Scale bars: 50 μm.
(E and F) Semithin sections of brains of 18-day-old adult flies. Adult glia-specific, but not adult neuron-specific, knockdown of the glycolytic gene Trehalase (Treh) induces neurodegeneration. AMPK knockdown albeit has a later onset and induces a similar phenotype. Scale bars: 50 μm.
(G-J) Semithin sections of brains of 50-day-old adult flies. Adult neuron-specific knockdown (using elav-Gal4, tub-Gal80ts) of AMPK does not alter brain structure. Scale bars: 50 μm.
(K and L) Kaplan-Meier survival curves of flies with AMPK knockdown in adult glial cells (K) or neurons (L), showing reduced lifespan of flies specifically with an adult glia-specific AMPK knockdown. n = 5 independent experiments; n > 300 flies/genotype. Statistics were performed using a Kaplan-Meier estimator in combination with a log rank test. Multiple comparison was performed using the Holm-Sidak method.
AMPK Loss Causes TXNIP (Thioredoxin-Interacting Protein) Stability, Glut1 Degradation, and Mislocalization and Diminished Glucose Uptake in Astrocytes
To understand why glycolysis is diminished in Ampk-null astrocytes, we first measured glucose import using the fluorescent glucose analog 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-D-glucose (2-NBDG). We found that glucose import was significantly reduced in Ampk-null astrocytes (Figure 7A). This could result from mislocalization of glucose transporters from the perinuclear space to the cell membrane. Indeed, following glucose feeding, plasma membrane translocation of the glucose transporter GLUT1 was markedly reduced in Ampk-null astrocytes (Figures 7B–7E). Although a significant amount of GLUT1 was membrane bound in WT astrocytes, nearly all GLUT1 remained mislocalized to the perinuclear space in Ampk-null astrocytes (Figures 7F and 7G). In mouse fibroblasts, GLUT1 membrane translocation is enabled by autophagy (Roy et al., 2017), a process enhanced through ULK1 phosphorylation by AMPK. Expectedly, autophagy was defective in Ampk-null astrocytes (Figure S6A). However, membrane localization of GLUT1 was similar in WT and Ulk1/2-null astrocytes (Figures S6B and S6C), indicating a lesser role of autophagy in astrocytic GLUT1 regulation.
Figure 7. AMPK Preserves GLUT1 and Its Surface Translocation by Destabilizing TXNIP.

(A) Glucose uptake in Ampk β1lox/lox; β2+/+;Adeno empty (control) and Ampk β1lox/lox; β2−/−;Adeno Cre-infected (Ampk KO) cortical astrocytes. Error bars are mean ± SEM. Significance of two-tailed, unpaired t tests is shown. n = 3 biological replicates.
(B-E) Distribution of GLUT1 (red) in control and Ampk KO astrocytes. GFAP immunostaining (green) indicative of astrocytes. Scale bars: 100 μM.
(F and G) Quantification of GLUT1 intensity in plasma membrane versus perinuclear space in control and Ampk-null astrocytes.
(H) Immunoblot of TXNIP, GULT1, and other indicated proteins in WT and Ampk-null astrocytes. Vinculin was used as loading control.
(I and J) Immunoblots showing TXNIP levels following AMPK activation by 2-DG (I) and hypoxia (J) in WT and null astrocytes.
(K) Immunoblot showing GLUT1 expression in AMPK KO astrocytes relative to WT astrocytes exposed to hypoxia.
(L) Immunoblot showing TXNIP stability in cycloheximide (CHX)-treated WT and AMPK KO astrocytes.
(M) Quantification of TXNIP levels in (I) at indicated times.
(N) Immunoblot of human astrocytes expressing AMPKβ1 shRNA (short hairpin RNA) or nontarget shRNA showing TXNIP levels after AMPK activation by 2-DG. (O-Q) Immunostaining of astrocytes expressing nontarget or TXNIP shRNA using TXNIP and GLUT1 antibodies. Scale bars: 100 μm.
(R) Glucose uptake in WT and Ampk-null astrocytes treated with nontarget or TXNIP shRNA. Error bars are mean ± SEM. n = three biological replicates.
(S) Viability analysis of Ampk β1lox/+; β2+/−; Nestin Cre (considered WT control) and Ampk β1lox/lox; β2−/−; Nestin Cre (Ampk KO) neurons grown in stated conditions. n = three biological replicates. Error bars are mean ± SEM. Significance of two-tailed, unpaired t tests is shown. Western blots represent data from two to three independent repeats. Unprocessed blots are available in Data S1.
See also Figures S6 and S7.
The arrestin superfamily protein TXNIP is an evolutionarily conserved glucose-inducible protein that is involved in human pathology, such as diabetes and diabetic retinopathy (Bodnar et al., 2002; Chutkow et al., 2008; Parikh et al., 2007; Singh, 2013). TXNIP directly sequesters Glut1, degrading it in endocytic vesicles, and indirectly reduces GLUT1 mRNA. Thus, TXNIP overexpression represses glucose uptake and its downregulation increases GLUT1 expression and glucose uptake (Wu and Wei, 2012; Wu et al., 2013). AMPK was shown to phosphorylate TXNIP, a modification that signals TXNIP degradation and improved GLUT1 expression (Wu et al., 2013). Relative to neurons, TXNIP is highly expressed in mouse and human astrocytes (Figure S6D). Protein analysis showed higher (stabilized) TXNIP and reduced GLUT1 levels in Ampk-null astrocytes (Figure 7H). Expression of GLUT3 remained unchanged (Figure S6E). Activation of AMPK by the glucose analog 2-deoxy-D-glucose (2-DG) or hypoxia caused TXNIP degradation in WT astrocytes, but not in Ampk-null astrocytes (Figures 7I and 7J). Although hypoxia activated AMPK and increased GLUT1 level, this effect was considerably diminished in Ampk-null astrocytes (Figure 7K). Further, a cycloheximide chase experiment showed that TXNIP is hyperstable in Ampk-null astrocytes (Figures 7L and 7M). Importantly, TXNIP and GLUT1 regulation by AMPK was conserved in normal human astrocytes (NHAs). In these cells, 2-DG degraded TXNIP in control NHA-expressing nontarget shRNA, but not in Ampkβ1-silenced NHA (Figure 7N).
We asked whether reducing TXNIP levels in Ampk-null astrocytes could reverse GLUT1 surface expression. Txnip shRNA (#1) was effective in reducing TXNIP levels (Figure S6F). We titrated lentivirus and reduced TXNIP level in Ampk-null astrocytes to match the levels in WT astrocytes. Whereas GLUT1 was restricted to the perinuclear space in cells expressing non-target shRNA, GLUT1 was redistributed to the plasma membrane in cells expressing Txnip shRNA (Figures 7O–7Q). This redistribution of GLUT1 by Txnip shRNA rescued glucose uptake in Ampk-null astrocytes (Figure 7R; Figures S6G–S6I). Collectively, our data imply that loss of AMPK causes TXNIP stabilization, GLUT1 sequestration, and mislocalization and reduced glucose uptake in astrocytes.
Lactate Supplementation or Ampk-Proficient Astrocytes Rescue Neuronal Death
Tissue glucose, including brain glucose concentration, is significantly lower than glucose used in cell culture (Fellows et al., 1992; Li et al., 2009). Therefore, we tested whether death of glucose-limited neurons is rescued by lactate in vitro. WT and Ampk-null cortical neurons were switched to either 5 (physiological) or 0.75 mM (limited) glucose for 24 h, and cell viability was monitored. Significant cell death was observed in both WT and Ampk-null neurons in 0.75 mM glucose medium, which was completely rescued by the highly metabolizable lactate stereoisomer L-lactate, but to a lesser extent by D-lactate that is metabolized at a much slower rate (Figure S6J). Given that L-lactate completely rescued neuronal death, we next tested whether this could be rescued by astrocyte conditioned medium (CM). We collected CM either from untreated astrocytes or astrocytes treated either with the lactate dehydrogenase A (LDHA) inhibitor oxamate or LDHA shRNA. Both oxamate and LDHA shRNA substantially reduced lactate production (Figures S6K–S6M). Culture media of WT or Ampk KO neurons were replaced with conditioned media from either WT or Ampk KO astrocytes (grown in 5 mM glucose with above treatments), and neuronal viability was determined after 24 h. CM from untreated WT astrocytes, but not AMPK KO astrocytes, rescued neuronal death (Figure 7S; Figure S6N, compare 7th bar with 10th bar). This rescue was abolished in CM collected from LDHA shRNA or oxamate-treated WT astrocytes (Figure 7S; Figure S6N, compare 10th bar with 11th bar). LDHA shRNA or oxamate-treated WT astrocyte CM caused significant death of WT neurons, an effect that was blunted by exogenous lactate (Figure 7S; Figure S6N, compare 1st, 2nd, and 3rd bars). These results confirm that lactate derived from WT astrocytes played a specific role in neuronal viability in vitro.
Next, we examined whether lactate-proficient WT astrocytes could provide similar protection in a brain-mimetic co-culture system. Astrocytes were grown in 5 mM glucose and switched to 0.75 mM glucose. Neurons (grown in coverslips with studs) were then layered over astrocytes such that they remained separated by a thin film of media. This culture condition allowed metabolic contact but prevented any physical contact of neurons with astrocytes. After 24 h, we observed a significant increase in the number of cleaved caspase-positive WT and Ampk KO neurons when co-cultured with Ampk KO astrocytes relative to WT neuron-WT astrocyte control co-cultures (Figures S7A, S7B, S7D, and S7E). Co-culturing Ampk KO neurons with WT astrocytes reversed neuronal death (Figures S7C and S7E). Collectively, these findings underscore the importance of AMPK in lactate production in astrocytes and the role of astrocyte-derived lactate in providing metabolic support for neurons.
DISCUSSION
Lactate, which is 10–50 times more abundant than pyruvate in healthy cells (Barros, 2013; Barros and Weber, 2018), feeds the TCA cycle and is freely released into circulation as a ready source of energy shared between all organs except the brain (Hui et al., 2017). Under normal physiology, the brain almost exclusively uses glucose; however, glucose catabolism is distinct in neurons and glia. As brains became more complex and glia arose, neuronal energy supply was partly outsourced to glia. An efficient glial glycolysis machinery catabolizes glucose to lactate and shuttles it to neurons for ATP production through the neuronal TCA cycle. This energy compartmentalization known as ANLS hypothesis is, however, not irrefutable (Bélanger et al., 2011; Díaz-García et al., 2017; Dienel, 2012; Yellen, 2018). In ex vivo studies, Díaz-García et al. (2017) showed that neuronal metabolic responses to stimulation in brain slices do not depend on astrocytic stimulation by glutamate release, nor do they require neuronal uptake of lactate; instead they reflect increased direct glucose consumption by neurons. On the other hand, ex vivo and in vivo evidence showed that a lactate gradient from astrocytes to neurons plays an important role in neuronal energy metabolism, fate, and function (Barros, 2013; Descalzi et al., 2019; Mächler et al., 2016; Pellerin et al., 1998, 2007; Schurr et al., 1988; Suzuki et al., 2011; Wyss et al., 2011). Lactate measurement using a genetically encoded FRET sensor called Laconic in combination with two-photon microscopy showed lactate gradient from astrocytes to neurons in vivo (Mächler et al., 2016). More recently, Descalzi et al., (2019) reported that lactate from astrocytes fuels learning-induced mRNA translation in excitatory and inhibitory neurons. Our results demonstrate that insufficient lactate from AMPK KO astrocytes could at least be partly causal to neuronal death.
We provide genetic evidence that the cellular bioenergetic sensor AMPK regulates neuronal energy metabolism in the adult mammalian and fly brain by augmenting glial glycolysis and lactate production. We show that the absence of AMPK in mammalian astrocytes or neural stem cells or in fly glia reduces neuronal viability. Through MRS, we provide in vivo genetic-metabolic evidence that AMPK deletion considerably reduces lactate levels (by about 40%) in the post-natal rodent brain. Cerebral blood volume in rodents is only 2%–5% and is about 2% in the hippocampus (Hua et al., 2019). Therefore, the lactate we measured is primarily local because the contribution of blood lactate to the hippocampus is not significant under normal physiological conditions. AMPK-null brains also showed reduced levels of creatine and phosphocreatine, indicating lower energy reserves. Myo-inositol, which has antiosmotic properties, was also reduced; this may reflect a potential osmotic imbalance in the Ampk-null brain.
Collectively, decreased concentrations of lactate and glutamate in Ampk KO mice indicate reduced glycolytic and TCA cycle flux, respectively, and reduction in energy metabolism is in agreement with the current understanding of AMPK function. Because MRS does not provide data at cellular resolution, we performed in vitro studies and confirmed that AMPK-null astrocytes, but not neurons, are deficient in glycolysis and lactate production. A previous study reported that AMPK negatively regulates glycolysis in oncogenic mouse lymphocytes (Faubert et al., 2013). However, other studies showed that AMPK supports glycolysis in various other contexts (Almeida et al., 2004; Chhipa et al., 2018; Doménech et al., 2015; Wu and Wei, 2012). Nitric oxide was found to rapidly induce glycolysis specifically in mouse astrocytes through AMPK phosphorylation-dependent activation of a key glycolytic enzyme phosphofructokinase 1 (Almeida et al., 2004), and we recently showed that AMPK regulates the glycolysis transcriptional program in human astrocytoma cells (Chhipa et al., 2018).
We observed that genetic deletion of AMPK β1 and β2 subunits (that knock out AMPK activity) in either neural stem cells or astrocytes reduces neuronal numbers as early as 1 month of age. This neuronal loss did not recover because the cerebral cortex of older (2 years) Ampk-null mice was still considerably smaller and contained fewer neurons. Our results clarify a previous report where AMPK deletion was shown not to adversely affect short-term (up to 3 days post-natal) neuronal survival (Williams et al., 2011). We could not provide direct in vivo evidence that deficient lactate production by Ampk-null astrocytes causes neuronal loss. We transplanted GFP-labeled WT astrocytes in the early post-natal brain of Ampk-null animals to test rescue, but the majority of these astrocytes did not survive. Efforts are underway to optimize a technically challenging method to continuously infuse and maintain a higher steady-state lactate level in the brain throughout the first few post-natal weeks. Astrocytic AMPK can certainly have additional roles relevant to neuronal survival in vivo, and we do not intend to draw parallels between in vivo and in vitro conditions. However, our data indicate that in our experimental setup, astrocytic lactate was key to survival of Ampk KO neurons. First, data from Nestin-Cre, GFAP-Cre ER, and Thy1 Cre mice were reproduced in the fly brain, where knocking out Ampk α, β, or γ subunits in adult glia, but not in neurons, caused neuronal death. These data are relevant to our studies because glycolysis deficiency in fly glia, but not neurons, resulted in neuronal loss in the fly brain (Volkenhoff et al., 2015). Second, providing exogenous lactate or conditioned media from WT, but not LDHA-inhibited, astrocytes protected Ampk KO neurons in vitro.
Ampk deletion in adult cortical neurons did not cause neuronal loss. Previous studies demonstrated that AMPK phosphorylation of the GABAb R2 receptor in vivo plays an important role in neuronal survival after ischemia, and AMPK activation is necessary during synaptic activation in vitro (Kuramoto et al., 2007; Williams et al., 2011). The role of neuronal AMPK may be revealed when Ampk KO; Nestin Cre and Thy1 Cre mice are subjected to energy stress such as hypoxia-ischemia or during enhanced neuronal firing when energy demand peaks. Indeed, EEG studies revealed that Ampk deletion in neurons raised basal neuronal excitability and reduced latency to seizure upon kainic acid injection. These results could be attributed to unregulated mTORC1 activity in Ampk-deficient neurons, which is consistent with previous studies in Tsc KO mice (Tsai et al., 2012). mTORC1 overdrive causes hypertrophy, and this may be a possible reason for the increase in cortical thickness of Ampk KO; Thy1 Cre mice. Treatment of post-natal mice with the mTORC1 allosteric inhibitor rapamycin dramatically augmented mTORC2 and AKT activity in cortical neurons following a feedback loop involving IRS phosphorylation by the mTORC1 substrate S6K1 (Marinangeli et al., 2018). mTORC2 overdrive can prevent neuronal death (Wan et al., 2007), and we predict that hyperactive neuronal AKT countered neuronal death in rapamycin-treated Ampk KO; Nestin Cre mice.
The mechanisms by which AMPK regulates glucose metabolism is contentious. Being part of a tumor suppressor pathway, AMPK was shown to inhibit glycolysis in lymphoma cells (Faubert et al., 2013). In contrast, AMPK activation was required for glucose uptake during muscle contraction and to augment glycolysis through various mechanisms (Almeida et al., 2004; Chhipa et al., 2018). We found that AMPK promotes glycolysis in astrocytes by protecting and promoting surface translocation of the glucose transporter GLUT1. In the absence of AMPK, GLUT1 levels were reduced and sequestered inside cells away from the plasma membrane. This was due to the loss of AMPK-mediated phosphorylation and increased stability of TXNIP that sequesters and internalizes GLUT1 in endosomes for lysosomal degradation (Wu et al., 2013). Consequently, glucose uptake, glycolysis, and lactate production were compromised in AMPK-null astrocytes. We show that during metabolic stress that activates AMPK, TXNIP is degraded and GLUT1 is upregulated in AMPK-proficient astrocytes. This biological response is largely abolished in AMPK-deficient astrocytes. Partial silencing and reducing TXNIP in AMPK-deficient astrocytes rescued GLUT1 cell surface localization and glucose uptake. Importantly, we found that GLUT1 regulation by AMPK via TXNIP degradation is conserved in human astrocytes.
In summary, we present genetic, metabolic, and biochemical data to show that under basal conditions, AMPK-mediated glycolysis and lactate production through TXNIP regulation are required for neuronal energy metabolism in the adult brain, and that this principle is conserved across species.
Limitations of Study
We cultured astrocytes either in the presence of serum or in serum-free G-5-supplemented medium. We determined purity of astrocytes by GFAP immunocytochemistry. This culture method usually yields 90%–95% astrocytes. Most of the remaining cells are fibroblasts, with a minority of cells being oligodendroglial cells and microglia. We did not select astrocytes by any method, and any contribution by non-astrocytic cells to the phenotype we describe in this manuscript remains unaccounted for and is a limitation of our study.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, Biplab DasGupta (Biplab.dasGupta@cchmc.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate new deposited data.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Husbandry and Generation of Brain specific AMPK-knockout mice
The experiments were performed on C57BL/6J (wild-type) and Transgenic AMPK Knockout mice males and females of E18.5 to 25 Months. Mice were housed at groups of four per cage with water and food ad libitum on a 12:12 h light cycle (lights on at 7 a.m.). All experiments were conducted in accordance with animal protocols approved by the Institutional Animal Care and Use Committee of Cincinnati Children’s Hospital. AMPKβ1lox/lox mice obtained from the Sanger Center and published (Chhipa et al., 2018) and AMPKβ2−/− mice that were generated in our lab (Dasgupta et al., 2012). Nestin-Cre (JAX stock #003771) and Thy1-cre (JAX stock #006143) mice were obtained from the Jackson Laboratory. GFAP-CreER were a kind gift from Lionel M. L. Chow, Dayton Children’s Hospital, Ohio. GFAP-CreER mice were injected with tamoxifen (75 mg / kg body weight, intraperitoneal) at 2 days of age every day for three consecutive days. Recombination efficiency was determined by western blot and immunocytochemistry using AMPKβ 1/2 antibodies. Genotyping was carried out using standard PCR protocols. Genotyping primers are provided in Key Resources Table. Rapamycin injections starting at 3 days of age, mice were injected intraperitoneally (i.p.) with 4 mg/kg body weight rapamycin (LC Laboratories) or vehicle every other day, according to a previously established protocol (Anderl et al., 2011), and were euthanized at P28.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE Antibodies | SOURCE | IDENTIFIER |
|---|---|---|
| phospho AMPK Thr172 | Cell Signaling Technology | RRID: 2535 |
| AMPK | Cell Signaling Technology | RRID: 2532 |
| AMPK β1./β2 | Cell Signaling Technology | RRID: 4150 |
| phospho ACC Ser79 | Cell Signaling Technology | RRID: 3661 |
| ACC | Cell Signaling Technology | RRID: 3676 |
| Cleaved Caspase-3 | Cell Signaling Technology | RRID: 9661 |
| pULK1 | Cell Signaling Technology | RRID: 5869 |
| ULK1 | Cell Signaling Technology | RRID: 4773 |
| LC3 | Cell Signaling Technology | RRID: 2775 |
| GLUT1 | EMD Millipore | RRID: 07–1401 |
| Ki-67 | Cell Signaling Technology | RRID: 9449 |
| NeuN | EMD Millipore | RRID: MAB_377 |
| GFAP | Cell Signaling Technology | RRID: 3670 |
| GFAP (for combined In situ/IHC) | DAKO | RRID: Z0334 |
| Phospho-S6 Ribosomal Protein (Ser235/236) | Cell Signaling Technology | RRID: 2211 |
| Phospho-Akt (Ser473) | Cell Signaling Technology | RRID: 9271 |
| TUJ1 | Covance | RRID: PRB_435P |
| CUX 1/2 | Santa Cruz | RRID: Sc_13024 |
| FOXP2 | Abcam | RRID: Ab_16046 |
| TXNIP | Novusbio | RRID: NBP1_54578SS |
| Critical Commercial Assays | ||
| Lactate Colorimetric/Fluorometric Assay Kit | Bio vision | RRID: K607 |
| 2-NBDG Glucose Uptake Assay Kit | Abcam | RRID: AB_235976 |
| Experimental Models: Cell Lines | ||
| Normal Human Astrocytes | Lonza | CC-2565 |
| Mouse Astrocytes | DasGupta Lab | N/A |
| Primary Mouse Neuron | DasGupta Lab | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | The Jackson Laboratory | OOO664 |
| B6.Cg-Tg(Nes-cre)1Kln/J | The Jackson Laboratory | OO3771 |
| FVB/N-Tg(Thy1-cre)1Vln/J | The Jackson Laboratory | OO6143 |
| GFAP-CreER | Chow et.al | N/A |
| AMPKβ1lox/lox | Sanger Center | N/A |
| AMPKβ2−/− | DasGupta et.al | N/A |
| AMPKα-dsRNA attP2 | Vienna Drosophila Resource Centre (VDRC) | N/A |
| alc-dsRNA | Vienna Drosophila Resource Centre (VDRC) | VDRC 104489 |
| SNF4Aγ-dsRNA | TRIP collection (Bloomington) | 26291 |
| Cherry-dsRNA | TRIP collection (Bloomington) | 35785 |
| Treh-dsRNA | Vienna Drosophila Resource Centre (VDRC) | VDRC 30730 |
| repo4.3-Gal4 | Sepp and Auld, 1999 | N/A |
| repo-Gal4 | Sepp and Auld, 1999 | N/A |
| elav-Gal4 | TRIP collection (Bloomington) | 8765 and 8760 |
| tub-Gal80ts | TRIP collection (Bloomington) | 7018 and 7019 |
| Oligonucleotides | ||
| AMPKb1 wildtype Forward | ATGACTGCGTCTGTTCCCC/ | N/A |
| AMPKb1 wildtype Reverse | AGCTCTCAAGCAAACCCTGC | N/A |
| AMPKb1 lox Forward | AGCTCTCAAGCAAACCCTGC/ | N/A |
| AMPKb1 lox Reverse | TCGTGGTATCGTTATGCGCC | N/A |
| AMPKb2 wildtype Forward | CGACTCGAGCTGCAGCCATGGGA | N/A |
| AMPKb2 wildtype Reverse | GGAATTAGCACCCAGCATC | N/A |
| AMPKb2 null Forward | AGGCTGAGGCCTGGTGAGGCCAAGTT | N/A |
| AMPKb2 null Reverse | CGACTCGAGCTGCAGCCATGGGA | N/A |
| Cre Forward | AGCGATCGCTGCCAGGAT | N/A |
| Cre Reverse | ACCAGCGTTTTCGTTCTGCC | N/A |
| LCN2–5′ | GCTGTCGCTACTGGATCAGA | Amplicon: 505 bp, Anneal:60 |
| LCN2–3′ | ATTAACCCTCACTAAAGGTGGTGGTGTTAAGACAGGTGG | N/A |
| CompC3–5′ | TGGGCAAGACAGTCGTCATC | Amplicon: 592 bp, Anneal:60 |
| CompC3–3′ | ATTAACCCTCACTAAAGGTGGATCTGGTACGGGGAAGT | N/A |
| Txnip shRNA | AGCATCTGTATTAGCGCATTT | TRCN0000347096 |
| Txnip shRNA | CTCAAGACAGCCCTATCTTTA | TRCN0000262800 |
| PRKAB1 shRNA | CCGGGCCTGGCTATGGAACTAAATAC TCGAGTATTTAGTTCCATAGCCAGGCTTTTT |
TRCN0000004770 |
| Ldha shRNA | CCAGCAAAGACTACTGTGTAA | TRCN0000308635 |
| Ldha shRNA | GTTCCCAGTTAAGTCGTATAA | TRCN0000308704 |
| Ldha shRNA | CGTGAACATCTTCAAGTTCAT | TRCN0000308638 |
| Ldha shRNA | CGTGAACATCTTCAAGTTCAT | TRCN0000041744 |
| Ldha shRNA | GTTCCCAGTTAAGTCGTATAA | TRCN0000041743 |
| Software and Algorithms | ||
| GraphPad Prism 8 | Software | RRID: SCR_002798 https://www.graphpad.com/ |
| ImageJ | Software | ImageJ; RRID: SCR_003070 https://imagej.nih.gov/ij/download.html |
| FlowJo | Software | Version 10 https://www.flowjo.com/solutions/flowio/downloads |
Fly Husbandry and Generation of Transgenic Lines
Flies used in experiments were kept at 25°C and 70% humidity raised on cornmeal-based feed. The following fly strains were used: UAS-dsRNA strains were obtained from Vienna Drosophila Resource Centre (VDRC) or the TRIP collection (Bloomington). dsRNA strains used: AMPKα-dsRNA attP2, alc-dsRNA (P{KK109325}VIE-260B, VDRC 104489), SNF4Aγ-dsRNA (P{TRiP.JF02060}attP2, Bloomington 26291), Treh-dsRNA (P{GD5118}v30730, VDRC 30730) and Cherry-dsRNA (P{VALIUM20-mCherry}attP2, Bloomington 35785). Driver lines used: repo4.3-Gal4, repo-Gal4 (Sepp and Auld, 1999), elav-Gal4 (Bloomington 8765 and 8760). tub-Gal80ts (Bloomington 7018 and 7019). elav-Gal4 (8765) and tub-Gal80ts (7019) were recombined to generate elav-Gal4, tub-Gal80ts (II) and repo-Gal4 and tub-Gal80ts (7018) were recombined to generate repo-Gal4, tub-Gal80ts (III).
METHOD DETAILS
Rapamycin Injection
Beginning at day 3 of age, mice were injected intraperitoneally with 4 mg/kg rapamycin (LC Laboratories) or vehicle every other day and maintained for one month. Briefly, a stock solution of rapamycin (50 mg/ml) was prepared in 100% ethanol and stored at −20°C. Rapamycin was then diluted in vehicle (DMSO) before injection. The vehicle control consisted of the same volume of ethanol.
In vivo 1H MR Spectroscopy
Wild-type and AMPK β−/− knockout mice (N = 6 per group) were studied on postnatal day 28. MRI and 1H MRS experiments were performed using a 9.4T/31cm horizontal bore magnet (Varian/Magnex Scientific; Yarnton, UK) equipped with a 15-cm gradient/shim coil (Resonance Research, Inc., Billerica, MA, USA) and interfaced to a DirectDrive console (Agilent/Varian; Palo Alto, CA) (Rao et al., 2003; Tkác et al., 2003). Uniform temperature was maintained inside the magnet using circulating warm water in tubes. MR spectra were collected from spontaneously breathing pups under inhalational anesthesia (isoflurane, 3% for induction and 1%–2% for maintenance in a 50:50 mixture of N2O and O2). The depth of anesthesia was monitored using continuous respiratory rate monitoring. Multi-slice fast spin-echo (FSE) MR imaging in axial and sagittal orientation (slice thickness = 0.8 mm) was used for precise positioning of the 3.6 μl (2.0 × 1.2 × 1.5 mm3) volume of interest (VOI) centered in the dorsal hippocampus. The B0 magnetic field homogeneity was adjusted by FASTMAP shimming. In vivo 1H MRS data were acquired using ultra-short echo-time STEAM (TE = 2 ms, TR = 5 s) localization sequence combined with VAPOR water suppression. Metabolites were quantified using LCModel with the spectrum of fast relaxing macromolecules (MM) acquired in vivo included in the basis set and unsuppressed water signal as the internal reference, assuming 80% brain water content. Only metabolites that were consistently quantified with the Cramèr-Rao lower bounds below 50% were included for the further analysis.
Cell lines and Culturing Conditions
Primary mouse Astrocytes were cultured as described previously (Chhipa et al., 2018; Dasgupta and Gutmann, 2005), with minor modifications. In brief, for the preparation of astrocytes, postnatal mice of either sex (P2-P3, control Ampk β1 null mice, or ULK1/2 null mice) were decapitated and cortices were removed and separated from the meninges and surrounding tissue. After enzymatic digestion of the cortices with 0.25% trypsin (Invitrogen) for 10 minutes, then tissue was mechanically triturated and centrifuged. The resulting cell pellet was resuspended in 1 mL of astrocyte medium (DMEM; Invitrogen) with 10% v/v FBS (Sigma), or G-5 Supplement (# 17503012, Thermofisher), and 1% v/v penicillin/streptomycin (Invitrogen). Astrocytes in G-5 supplemented cultures were never exposed to serum. The single-cell suspension obtained was added to 10 mL of astrocyte medium in T-75 flasks. The cells were grown for at least 7 days in vitro (DIV) at 37°C with 5% v/v CO2, and a complete medium change was performed every second day. 24 h before the preparation of neurons was undertaken (see below), astrocytes were replaced with Neurobasal media. AMPK deletion was achieved by transduction of cultures with Adeno Cre or control Adenovirus (Adeno-GFP). Efficiency of recombination was confirmed by western blotting with AMPK β1/β2 and α antibodies.
A mixed primary culture of cortical neurons was prepared from E16.5 embryos derived from timed pregnant control and Ampk β1lox/lox; β2−/−; Nestin Cre (AMPK KO) mice and maintained in culture at 37°C and 5% CO2 as previously described (Loktev and Jackson, 2013) with slight modifications. Briefly, the cortical region of the brain was dissected in ice cold HBSS (HyClone Cat# SH30268.01) containing penicillin-streptomycin solution (MP Biomedicals Cat# 0916700) and sodium pyruvate (GIBCO Cat# 11360070) and was subjected to enzymatic desegregation for 10 min at 37°C in 0.25% w/v trypsin (Invitrogen). The tissue was then mechanically disaggregated into isolated cells in a Neurobasal medium (GIBCO Cat# A1371201) with 5% (v/v) FBS, Glutamax-I (GIBCO Cat# 35050–061), and B27 supplement (GIBCO Cat# A3582801). The cells (1.0 × 106 cells/cm2) were plated in wells of 6-well plates that were coated with 0.1 mg/mL poly-D-lysine (Sigma-Aldrich Cat# P7280). After 5 h, medium was replaced with Neurobasal medium supplemented with B27, Glutamax-I, and penicillin-streptomycin solution. Half the medium was replaced every 3 days, and the cells were maintained in culture for 7–10 days. A competitive LDHA inhibitor, sodium oxamate (O2751), was purchased from Sigma-Aldrich.
Cell viability endpoint
For Neuronal viability, Presto Blue cell viability assay (Life Technologies) was performed following the manufacturer’s instructions. Briefly, Presto Blue cell viability reagent was diluted 1:10 in culture media and incubated with aggregates for 40 min. at 37°C and 5% CO2. Fluorescence readings were made using a Cytation3 multimode plate reader (BioTek, Winooski, VT, USA) set to ex/em of 530/590 nm.
Lentivirus production and transduction
shRNA vectors were obtained from the Sigma Mission RNAi shRNA library, the sequences are listed in Key Resources Table. For knockout of Human AMPKβ1 and mouse Txnip and Ldha, four shRNA targeting corresponding genes were designed. Lentiviral particles were prepared by co-transfection of shRNA, (4.5 μg), VSVG (500 ng) and CDNL (4.5 mg) vectors in 2 million 293T cells. JETPRIME (10 μl/plate) was used according to manufacturer’s instruction. Six hours post transfection, supernatant was removed, and complete medium was added. 24h later, media was removed, and the cells were washed with PBS. DMEM/F12 medium with G-5 Supplement were added. Supernatant was harvested after 24 – 72 hours and filtered at 0.22 mm and Concentrated using Lenti-X Concentrator (#631232, TakaraBio) following manufactures protocol. After 12 hours lentivirus was removed and replaced with fresh medium. Efficacy of knockdown was assayed by WB.
Western Blot Analysis
Cells were grown as described above. Following removal of the growth media, cells were washed in ice-cold PBS and lysed on ice using RIPA buffer (#R0278, Sigma) with Protease Inhibitor Cocktail (#P8340, Sigma) and Phosphatase Inhibitor Cocktail 2 (#P5726, Sigma). Lysates were centrifuged at 14,000 rpm for 10 min at 4°C and protein concentrations were quantified using BCA Protein Assay Kit (71285-M, Sigma). Denatured lysates were separated by PAGE on 4%–12% Bis-Tris gradient gels (Invitrogen) along with Precision Plus Protein Standards (Bio-Rad) using SDS NuPAGE Running Buffer (Bio-Rad) on ice at 200 V for 45 min. Gels were transferred to immobilin-FL PVDF membranes (Millipore) using Transfer Buffer (Bio-Rad) on ice at 30 V for 1.5 hr before being washed 3× 5 min. with 5% Tween-TBS (TTBS, Amresco & Invitrogen) at room temperature (RT) with agitation and blocked with 10% Milk (Sigma) in TTBS for 1 hr at RT with agitation. Blots were again washed 3× 5 min in TTBS at RT before being incubated in primary antibody in 5% BSA in TTBS overnight at 4°C. Blots were washed 3x for 5 min at RT with agitation and then transferred to HRP-conjugated secondary antibody in 5% BSA in TTBS and incubated at RT for 1 hr. See Key Resources Table for primary antibodies and HRP-conjugated secondary antibodies used in western blots. Blots were washed again as previously described and visualized using SuperSignal West Pico Chemiluminescent HRP Substrate Kit (Thermo Scientific) and CL-X Posure Film (Thermo Scientific).
Immunohistochemistry
Brains were removed and post-fixed in 10% paraformaldehyde overnight at 4°C followed by cryopreservation in 30% (w/v) sucrose in PBS. Serial coronal sections of 10–12 um thickness were cut using a freezing cryostat (Thermo Scientific, Microm HM550). Coronal sections beginning at bregma 1.50 to 3.50 mm through the dorsal hippocampus encompassing the dentate gyrus region and cortex were chosen from both Wild-type and experimental animals. Antigen retrieval was performed with citrate buffer (pH 6.2) and blocked with 10% BSA, 0.1% Triton X-100, in PBS for 2 hours. Sections were then incubated in primary antibody for overnight at 4°C. Secondary antibodies used included anti-mouse and Anti-Rabbit anti-chicken Alexa Fluor 488 (1:400); and anti-mouse and anti-Rabbit Alexa Fluor 594 (1:400). Sections were mounted with DAPI containing Hard Set anti-fade mounting medium (Vectashield, Vector Laboratories, CA, USA) and stored in the dark at 4°C. Images from the stained slides were acquired for fluorescence co-labeling under inverted Leica fluorescence microscope (DMI 3000B) and Nikon C2-Confocal Microscope using Nikon Elements software (Nikon Instruments Inc.). The images were analyzed, and background corrected by ImageJ software and Nikon Elements software as described in user manual. For quantification, cell counting in cortical and hippocampal region for NeuN positive cells were performed in coronal sections. For the measurement of cortical area, a single cerebral hemisphere was traced from a dorsally oriented whole-mount image. For relative thickness of cortical layers, the thickness of the major layers was measured relative to the overall thickness of the cortex (from the pial surface to the white matter). For cell counts in cortical layers, all cells in a 250-μm-wide columnar area from the white matter to the pial surface were counted. For cell counts of embryonic progenitors, all cells in a 250-μm-wide columnar area from the ventricular surface to the pial surface were counted. Any cell that the morphology was not clearly visible was excluded from analysis. At least 5 sections in each group, were analyzed.
Combined In situ hybridization and IHC
In situ hybridization was essentially carried out as described (Kohli et al., 2018; Toresson et al., 1999) except levamisole was added to blocking solution on day 2 and followed with PBT washes before the addition of anti-digoxigenin antibody (1:2,500, Roche). Primers used to generate probes are given in Key Resources Table. For combinatory in situ hybridization/IHC with LCN2 and C3 RNA probes and the rabbit-GFAP antibody (DAKO, 1:2,000), slides were washed in phosphate buffered saline following the in situ hybridization development in BM purple (Roche) and IHC was completed as above.
Bioenergetics Experiments
Seahorse Assay
Metabolic profiles over time were analyzed using Seahorse technology. Astrocytes were seeded at 25,000 cells/well into Seahorse XFp 96cell culture plates in a final volume of 80 μL, briefly spun at 500 RPM, and allowed to settle overnight at 37°C and 5% CO2. 200 μL of XF calibrant (pH 7.4) was added to an XFp extracellular flux cartridge and left at 37°C in the absence of CO2. The next day, cells were washed twice with 200 μL of glucose-free Seahorse base medium supplemented with 1.0mM sodium pyruvate, 4mM glutamine, pH 7.4. After washing, each well was incubated in 180 μL of this Seahorse base medium. At this point, the wells were imaged using the IncuCyte to obtain mKate2+ live cell counts that were used for normalization. The cell culture plate was allowed to incubate at 37°C in the absence of CO2 for one hour prior to the beginning of the experiment. Test compounds were prepared at 10x in Seahorse medium in a final volume of 20 mL in the XFp extracellular flux cartridge to be injected later in the experiment. The plate was transferred to the XF flux analyzer for measurement of the extracellular acidification rate change (ΔpH/min). The order of additions for the assay was 1) Glucose (10mM), 2) Oligomycin (inhibitor of ATP synthase/complex V) (1mM), and 3) 2-deoxyglucose (2-DG) (10mM).
Glucose Import: 2NBDG Uptake Assay
Glucose uptake was assayed by incubating cells with 100 μM 2-[N-(7-nitrobenz-2-oxa1,3-diazol-4-yl) amino]-2-deoxy-D-glucose (2-NBDG) at 37°C in glucose-free RPMI 1640 (GIBCO) containing G-5-supplement, 2 mM L-glutamine. Cells were plated at 1× 106 cells/well l in 6-well plates and incubated for 24hrs. 2-NBDG was added at final concentration of 100 μM to the medium. Control cells were not incubated with 2-NBDG. After 1 hr of incubation cells were washed once with ice-cold PBS, then trypsinized and centrifuged for 5 min at 250 rcf. Cells were washed twice more with ice-cold PBS and resuspended in 1 mL of PBS for flow cytometry (C6 Accuri Flow Cytometer, BD Bioscience) using 488 nm excitation a 533/30nm emission filter to measure 2-NBDG uptake. For each measurement, data from 10,000 single-cell events were collected. Data were normalized to constant cell number and analyzed using FlowJo software (Version 10.2).
Extracellular Lactate Assay
Lactate was assayed using a colorimetric test in cell supernatants deproteinized by filtration with 10 kDa MWCO spin. The soluble fraction was assayed using a Lactate Assay Kit (Bio-vision).
Extracellular Citrate Assay
Citrate was assayed using a colorimetric test in cell supernatants deproteinized by filtration with 10 kDa MWCO spin. The soluble fraction was assayed using a Lactate Assay Kit (Bio-vision).
Stable Isotope Tracing Experiments
For glucose isotope tracing experiment, Astrocytes were cultured in 10 cm tissue culture dishes for 24 h in a custom made DMEM labeled medium containing the following stable isotopes (Cambridge Isotope Laboratories, Inc): 13C6-glucose (CLM-1396–0). At the time of extraction, spent medium was collected and cells were rapidly washed twice with cold phosphate buffered saline and flash freeze using liquid Nitrogen. For metabolomics studies, astrocytes were analyzed by liquid chromatography-high resolution mass spectrometry (LC-HRMS).
Real-Time qPCR
Total cellular RNAs were extracted using QIAGEN RNeasy Mini Kit (# 74104) according to the manufacturer’s instructions, and RNA purity was confirmed by analysis of the ratio of absorbance at 260 nm to that at 280 nm absorption. cDNAs were prepared from 1 g total RNAs using SuperScript II Reverse Transcriptase for qPCR (Invitrogen, #18064022) according to the manufacturer’s instructions. qRT-PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems, # 4344463) and QuantiTect Primer (QIAGEN) in an ABI PRISM 7900 Sequence Detection System (Applied Biosystems). Relative mRNA expression was calculated using the comparative Ct method after normalization to a loading control. Samples were run in triplicates with a primer-limited probe for the reference gene. Primers are provided in Key Resources Table.
Electrode Implantation and Electroencephalography
6–8 weeks old male AMPK KO and littermate control mice were implanted with wireless transmitters for EEG monitoring (Tiwari et al., 2019) following a method described (Tse et al., 2014). Briefly, mice were anesthetized with 4% isoflurane in medical grade oxygen in a closed chamber, and then maintained at 0.7%–1.5% isoflurane throughout the surgery and monitored for pattern of respiration. Single channel wireless EEG transmitters, (TA11ETA-F10, Data Science International (DSI), St, Paul, MN, USA) were used. The head was first shaved and disinfected using Dermachlor (2% Chlorhexidine) and carprofen (0.1 ml) was administered subcutaneously. The skull was exposed by making an incision along the midline. Dorsoventral coordinates were measured from the bregma and two holes were drilled at AP = −2.5 mm, L= ± 2.0 mm. 1 mm length of each of the leads of the transmitter was carefully inserted into the cortex and sealed using GLUture (Abbott Laboratories, IL, USA). The wireless transmitter was then placed subcutaneously by creating a pocket behind the neck. A screw was attached to the back of the skull and a slurry of dental cement (Patterson Dental, OH, USA) was applied to secure the assembly and the cement was allowed to dry. The open skin at the incision was closed using surgical sutures (Covidien, MN, USA) and later sealed with GLUture. Post-surgery, the mice were injected with 1 mL saline, placed on a heating pad and monitored until recovery.
Video-EEG Recording
Post recovery, the mice were housed in individual static cages placed on wireless receiver plates (RPC1; DSI). DATAQUEST A.R.T software was used for recording of EEG data received from the telemetry system. EEG frequency was recorded between 1 and 1500 Hz and video was continuously recorded (Axis 221, Axis communication) in parallel. Baseline EEG was monitored for one week to measure the effect of genotype on EEG waveforms. Post one week of baseline EEG recording the mice were treated with kainic acid to measure seizure susceptibility (see below).
Spectral Power Analysis
The EEG data was analyzed using NeuroScore software (DSI, MN, USA). For power analysis the raw EEG signal was exported in 10 s epochs and subjected to Fast Fourier Transform (FFT) to generate power bands. One 5-minute period of recording between 12pm and 2pm (day) and 12am and 2 am (night) for each day of recording per mice was selected for analysis. Times of excessive moving or grooming were excluded from the analysis. Total EEG power across all frequencies as well as the gamma power band (24–80 Hz) were analyzed, and cumulative EEG power within the 5-minute periods were reported.
Kainic Acid Injection
Kainic acid injection was performed after one week of EEG baseline recording. Mice were injected intraperitoneally with 15mg/kg kainic acid (2mg/ml solution in sterile Ringers) and returned immediately to the recording platform to measure seizure onset. After 90 minutes of EEG-video recording, seizures were terminated by a subcutaneous injection of diazepam (15mg/kg). Seizure onset was manually scored and defined as a behavioral seizure in the recorded video accompanied by a twofold change in the amplitude and frequency of baseline EEG signal.
Experiments in Drosophila
Lifespan
To address AMPK requirement in adult neurons or glial cells the TARGET system was used (McGuire et al., 2004). Elav-Gal4, tub-Gal80ts; elav-Gal4 or repo4.3-Gal4; repo-Gal4, tub-Gal80ts animals were crossed to the respective dsRNA strains at 18°C to prevent dsRNA-expression. After hatching 0–6 days old females were collected and kept at 29°C to allow dsRNA-expression (in batches of 20 flies). The animals were transferred onto new vials every 2–3 days. Survival of the animals was monitored. Data is presented as Kaplan-Meier survival curves. Statistics were performed using a Kaplan-Meier estimator in combination with a log-rank test. Multiple comparison was performed using the Holm-Sidak method. The survival of flies was considered significantly different if p < 0.05. > 300 animals per genotype were analyzed (5 experiments).
Semi-thin sections
Semi-thin Sections
Crosses were set up and animals were treated as for the lifespan assay. After 50 days (for AMPK knockdowns and respective controls) or 18 days (for Trehalase knockdown and the respective control) at 29°C the flies’ heads were embedded in Epon as described in Stork et al. (2008). 1 mm semi-thin sections were made using a Leica EM UC7 Ultra Microtome, stained with toluidine blue, and imaged using a Zeiss Axiophot.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details of the statistical analysis are provided in Figure Legends and in STAR Methods. Plots were generated using GraphPad Prism version 8.4.2 for PC (GraphPad Software, La Jolla California USA) or R (R Core Team, R: A Language and Environment for Statistical Computing (Version 3.5.0, R Foundation for Statistical Computing, Vienna, 2018; https://www.R-project.org/). Animals were assigned to groups based on genotype. Age-matched littermates were used as controls in all experiments. No randomization was used, and no animals were excluded from analyses. No statistical methods were used to predetermine sample sizes. Each experiment was done by analyzing three to seven brains of each genotype. For all experiments, data distribution was assumed to be normal, but this was not formally tested. Two groups of data were assessed for significant differences using a two-tailed unpaired Student’s t test with a cutoff for significance of p < 0.05 (Prism software, GraphPad). The data are presented as the mean ± s.d. For smaller sample size, it’s difficult to assess the underlying assumptions of parametric statistical tests such as a t test and ANOVA. Therefore, for EEG studies, non-parametric test, the Kruskal-Wallis test was applied (using R software, 64-bit version 3.4.3), and data were presented in graphs showing median and interquartile ranges. A value of p < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
AMPK deletion reduces energy metabolites, including lactate, in the rodent brain
AMPK regulates astrocytic glycolysis and astrocyte-neuron lactate shuttle (ANLS)
Glia or neural stem cell AMPK deletion causes neuronal loss in rodent and fly brains
AMPK destabilizes TXNIP to enable GLUT1 localization, glucose import, and glycolysis
ACKNOWLEDGMENTS
We would like to thank Shabnam Pooya and Janvi Gandhi for assisting with western blots, Oluwadamilola Omojola for performing genotyping PCR, and Ashwini Hinge for assistance with fluorescence-activated cell sorting (FACS) analysis. We thank Matthew Kofron of CCHMC Microscopy Core for assistance with imaging. This work was supported by National Institutes of Health (NIH) grants R01 NS075291 and R01 NS099162 (to B.D.), R01 MH115058 (to M.K.), and R01 NS092705 (to C.G.). The Center for Magnetic Resonance Research at the University of Minnesota is supported by NIH grants P41 EB015894 and P30 NS076408.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108092.
REFERENCES
- Almeida A, Moncada S, and Bolaños JP (2004). Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat. Cell Biol 6, 45–51. [DOI] [PubMed] [Google Scholar]
- Anderl S, Freeland M, Kwiatkowski DJ, and Goto J (2011). Therapeutic value of prenatal rapamycin treatment in a mouse brain model of tuberous sclerosis complex. Hum. Mol. Genet 20, 4597–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros LF (2013). Metabolic signaling by lactate in the brain. Trends Neurosci 36, 396–404. [DOI] [PubMed] [Google Scholar]
- Barros LF, and Weber B (2018). CrossTalk proposal: an important astrocyte-to-neuron lactate shuttle couples neuronal activity to glucose utilisation in the brain. J. Physiol 596, 347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, and Magistretti PJ (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14, 724–738. [DOI] [PubMed] [Google Scholar]
- Bodnar JS, Chatterjee A, Castellani LW, Ross DA, Ohmen J, Cavalcoli J, Wu C, Dains KM, Catanese J, Chu M, et al. (2002). Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat. Genet 30, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks GA (1998). Mammalian fuel utilization during sustained exercise. Comp. Biochem. Physiol. B Biochem. Mol. Biol 120, 89–107. [DOI] [PubMed] [Google Scholar]
- Brooks GA (2002). Lactate shuttles in nature. Biochem. Soc. Trans 30, 258–264. [DOI] [PubMed] [Google Scholar]
- Brooks GA (2009). Cell-cell and intracellular lactate shuttles. J. Physiol 587, 5591–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks GA (2018). The science and translation of lactate shuttle theory. Cell Metab 27, 757–785. [DOI] [PubMed] [Google Scholar]
- Brown AM, and Ransom BR (2007). Astrocyte glycogen and brain energy metabolism. Glia 55, 1263–1271. [DOI] [PubMed] [Google Scholar]
- Brown AM, Sickmann HM, Fosgerau K, Lund TM, Schousboe A, Waagepetersen HS, and Ransom BR (2005). Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J. Neurosci. Res 79, 74–80. [DOI] [PubMed] [Google Scholar]
- Chhipa RR, Fan Q, Anderson J, Muraleedharan R, Huang Y, Ciraolo G, Chen X, Waclaw R, Chow LM, Khuchua Z, et al. (2018). AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol 20, 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chutkow WA, Patwari P, Yoshioka J, and Lee RT (2008). Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J. Biol. Chem 283, 2397–2406. [DOI] [PubMed] [Google Scholar]
- Creus-Muncunill J, Rué L, Alcalá-Vida R, Badillos-Rodríguez R, Romaní-Aumedes J, Marco S, Alberch J, Perez-Otaño I, Malagelada C, and Pérez-Navarro E (2018). Increased Levels of Rictor Prevent Mutant Huntingtin-Induced Neuronal Degeneration. Mol. Neurobiol 55, 7728–7742. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, and Chhipa RR (2016). Evolving lessons on the complex role of AMPK in normal physiology and cancer. Trends Pharmacol. Sci 37,192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, and Gutmann DH (2005). Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J. Neurosci 25, 5584–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Ju JS, Sasaki Y, Liu X, Jung S-R, Higashida K, Lindquist D, and Milbrandt J (2012). The AMPK β2 subunit is required for energy homeostasis during metabolic stress. Mol. Cell. Biol 32, 2837–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descalzi G, Gao V, Steinman MQ, Suzuki A, and Alberini CM (2019). Lactate from astrocytes fuels learning-induced mRNA translation in excitatory and inhibitory neurons. Commun. Biol 2, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-García CM, Mongeon R, Lahmann C, Koveal D, Zucker H, and Yellen G (2017). Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab 26, 361–374.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA (2012). Brain lactate metabolism: the discoveries and the controversies. J. Cereb. Blood Flow Metab 32, 1107–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doménech E, Maestre C, Esteban-Martínez L, Partida D, Pascual R, Fernández-Miranda G, Seco E, Campos-Olivas R, Pérez M, Megias D, et al. (2015). AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell Biol 17, 1304–1316. [DOI] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. (2013). AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17, 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellows LK, Boutelle MG, and Fillenz M (1992). Extracellular brain glucose levels reflect local neuronal activity: a microdialysis study in awake, freely moving rats. J. Neurochem 59, 2141–2147. [DOI] [PubMed] [Google Scholar]
- Fernandes VM, Chen Z, Rossi AM, Zipfel J, and Desplan C (2017). Glia relay differentiation cues to coordinate neuronal development in Drosophila. Science 357, 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim ND, Mcfate T, Mohyeldin A, Okagaki P, Korotchkina LG, Patel MS, Jeoung NH, Harris RA, Schell MJ, and Verma A (2010). Phosphorylation status of pyruvate dehydrogenase distinguishes metabolic phenotypes of cultured rat brain astrocytes and neurons. Glia 58, 1168–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2014). AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr 34, 31–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Hussien R, Cho H-S, Kaufer D, and Brooks GA (2008). Evidence for the mitochondrial lactate oxidation complex in rat neurons: demonstration of an essential component of brain lactate shuttles. PLoS ONE 3, e2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua J, Liu P, Kim T, Donahue M, Rane S, Chen JJ, Qin Q, and Kim S-G (2019). MRI techniques to measure arterial and venous cerebral blood volume. Neuroimage 187, 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Zhan L, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, and Guan K-L (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. [DOI] [PubMed] [Google Scholar]
- Iseli TJ, Walter M, van Denderen BJW, Katsis F, Witters LA, Kemp BE, Michell BJ, and Stapleton D (2005). AMP-activated protein kinase beta subunit tethers alpha and gamma subunits via its C-terminal sequence (186–270). J. Biol. Chem 280, 13395–13400. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Esaki T, Shimoji K, Cook M, Law MJ, Kaufman E, and Sokoloff L (2003). Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc. Natl. Acad. Sci. USA 100, 4879–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis A, Sylantyev S, Hadjihambi A, Hosford PS, Kasparov S, and Gourine AV (2016). Hemichannel-mediated release of lactate. J. Cereb. Blood Flow Metab 36, 1202–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, and Webb WW (2004). Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305, 99–103. [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Lambert EM, Panepento B, Sun A, Gelbard HA, Burgess RW, Foster TH, and Nedergaard M (2011). Two-photon NADH imaging exposes boundaries of oxygen diffusion in cortical vascular supply regions. J. Cereb. Blood Flow Metab 31, 68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli V, Nardini D, Ehrman LA, and Waclaw RR (2018). Characterization of Glcci1 expression in a subpopulation of lateral ganglionic eminence progenitors in the mouse telencephalon. Dev. Dyn 247, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, et al. (2007). Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron 53, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrabee MG (1996). Partitioning of CO2 production between glucose and lactate in excised sympathetic ganglia, with implications for brain. J. Neurochem 67, 1726–1734. [DOI] [PubMed] [Google Scholar]
- Laughton JD, Bittar P, Charnay Y, Pellerin L, Kovari E, Magistretti PJ, and Bouras C (2007). Metabolic compartmentalization in the human cortex and hippocampus: evidence for a cell- and region-specific localization of lactate dehydrogenase 5 and pyruvate dehydrogenase. BMC Neurosci 8, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Ahn CH, Shutter LA, and Narayan RK (2009). Toward real-time continuous brain glucose and oxygen monitoring with a smart catheter. Biosens. Bioelectron 25, 173–178. [DOI] [PubMed] [Google Scholar]
- Liu L, MacKenzie KR, Putluri N, Maletić-Savatić M, and Bellen HJ (2017). The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab 26, 719–737.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loktev AV, and Jackson PK (2013). Neuropeptide Y family receptors traffic via the Bardet-Biedl syndrome pathway to signal in neuronal primary cilia. Cell Rep 5, 1316–1329. [DOI] [PubMed] [Google Scholar]
- Mächler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martín A, Romero-Gómez I, et al. (2016). In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab 23, 94–102. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, and Allaman I (2018). Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci 19, 235–249. [DOI] [PubMed] [Google Scholar]
- Mangia S, Simpson IA, Vannucci SJ, and Carruthers A (2009). The in vivo neuron-to-astrocyte lactate shuttle in human brain: evidence from modeling of measured lactate levels during visual stimulation. J. Neurochem 109 (Suppl 1), 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinangeli C, Didier S, Ahmed T, Caillerez R, Domise M, Laloux C, Bégard S, Carrier S, Colin M, Marchetti P, et al. (2018). AMP-Activated Protein Kinase Is Essential for the Maintenance of Energy Levels during Synaptic Activation. iScience 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire SE, Le PT, Osborn AJ, Matsumoto K, and Davis RL (2003). Spatiotemporal rescue of memory dysfunction in Drosophila. Science 302, 1765–1768. [DOI] [PubMed] [Google Scholar]
- McGuire SE, Mao Z, and Davis RL (2004). Spatiotemporal gene expression targeting with the TARGET and gene-switch systems in Drosophila. Sci. STKE 2014, pl6. [DOI] [PubMed] [Google Scholar]
- Mu J, Brozinick JT Jr., Valladares O, Bucan M, and Birnbaum MJ (2001). A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol. Cell 7, 1085–1094. [DOI] [PubMed] [Google Scholar]
- Nabbout R, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, Dahlin M, D Amato L, d’Augères GB, de Vries PJ, et al. ; TOSCA Consortium and TOSCA Investigators (2018). Epilepsy in tuberous sclerosis complex: Findings from the TOSCA Study. Epilepsia Open 4, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJ, et al. (2007). TXNIP regulates peripheral glucose metabolism in humans. PLoS Med 4, e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL, Stella N, and Magistretti PJ (1998). Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci 20, 291–299. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore A-K, Aubert A, Serres S, Merle M, Costalat R, and Magistretti PJ (2007). Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55, 1251–1262. [DOI] [PubMed] [Google Scholar]
- Rao R, Tkac I, Townsend EL, Gruetter R, and Georgieff MK (2003). Perinatal iron deficiency alters the neurochemical profile of the developing rat hippocampus. J. Nutr 133, 3215–3221. [DOI] [PubMed] [Google Scholar]
- Roy S, Leidal AM, Ye J, Ronen SM, and Debnath J (2017). Autophagy-Dependent Shuttling of TBC1D5 Controls Plasma Membrane Translocation of GLUT1 and Glucose Uptake. Mol. Cell 67, 84–95.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, and Alessi DR (2005). Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 24, 1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, and Payne RS (2007). Lactate, not pyruvate, is neuronal aerobic glycolysis end product: an in vitro electrophysiological study. Neuroscience 147, 613–619. [DOI] [PubMed] [Google Scholar]
- Schurr A, West CA, and Rigor BM (1988). Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 240, 1326–1328. [DOI] [PubMed] [Google Scholar]
- Sepp KJ, and Auld VJ (1999). Conversion of lacZ enhancer trap lines to GAL4 lines using targeted transposition in Drosophila melanogaster. Genetics 151, 1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh LP (2013). Thioredoxin interacting protein (TXNIP) and pathogenesis of diabetic retinopathy. J. Clin. Exp. Ophthalmol 4, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork T, Engelen D, Krudewig A, Silies M, Bainton RJ, and Klämbt C (2008). Organization and function of the blood-brain barrier in Drosophila. J. Neurosci 28, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, and Alberini CM (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomanetz V, Angliker N, Cloëtta D, Lustenberger RM, Schweighauser M, Oliveri F, Suzuki N, and Rüegg MA (2013). Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol 201, 293–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari D, Brager DH, Rymer JK, Bunk AT, White AR, Elsayed NA, Krzeski JC, Snider A, Schroeder Carter LM, Danzer SC, and Gross C (2019). MicroRNA inhibition upregulates hippocampal A-type potassium current and reduces seizure frequency in a mouse model of epilepsy. Neurobiol. Dis 130, 104508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkác I, Rao R, Georgieff MK, and Gruetter R (2003). Developmental and regional changes in the neurochemical profile of the rat brain determined by in vivo 1H NMR spectroscopy. Magn. Reson. Med 50, 24–32. [DOI] [PubMed] [Google Scholar]
- Toresson H, Mata de Urquiza A, Fagerström C, Perlmann T, and Campbell K (1999). Retinoids are produced by glia in the lateral ganglionic eminence and regulate striatal neuron differentiation. Development 126, 1317–1326. [DOI] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, and Sahin M (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse K, Puttachary S, Beamer E, Sills GJ, and Thippeswamy T (2014). Advantages of repeated low dose against single high dose of kainate in C57BL/6J mouse model of status epilepticus: behavioral and electroencephalographic studies. PLoS ONE 9, e96622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkenhoff A, Weiler A, Letzel M, Stehling M, Klämbt C, and Schirmeier S (2015). Glial glycolysis is essential for neuronal survival in Drosophila. Cell Metab. 22, 437–447. [DOI] [PubMed] [Google Scholar]
- Wan X, Harkavy B, Shen N, Grohar P, and Helman LJ (2007). Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 26, 1932–1940. [DOI] [PubMed] [Google Scholar]
- Williams T, Courchet J, Viollet B, Brenman JE, and Polleux F (2011). AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc. Natl. Acad. Sci. USA 108, 5849–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S-B, and Wei Y-H (2012). AMPK-mediated increase of glycolysis as an adaptive response to oxidative stress in human cells: implication of the cell survival in mitochondrial diseases. Biochim. Biophys. Acta 1822, 233–247. [DOI] [PubMed] [Google Scholar]
- Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen C-H, Wen J, Asara J, McGraw TE, et al. (2013). AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 49, 1167–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss MT, Jolivet R, Buck A, Magistretti PJ, and Weber B (2011). In vivo evidence for lactate as a neuronal energy source. J. Neurosci 31, 7477–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G (2018). Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol 217, 2235–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L, Ma L, Hamm M, Gage FH, and Hunter T (2016). Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 5, e13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new deposited data.