

Abstract
The Oryza officinalis complex is the largest species group in Oryza, with more than nine species from four continents, and is a tertiary gene pool that can be exploited in breeding programs for the improvement of cultivated rice. Most diploid and tetraploid members of this group have a C genome. Using a new reference C genome for the diploid species O. officinalis, and draft genomes for two other C genome diploid species Oryza eichingeri and Oryza rhizomatis, we examine the influence of transposable elements on genome structure and provide a detailed phylogeny and evolutionary history of the Oryza C genomes. The O. officinalis genome is 1.6 times larger than the A genome of cultivated Oryza sativa, mostly due to proliferation of Gypsy type long-terminal repeat transposable elements, but overall syntenic relationships are maintained with other Oryza genomes (A, B, and F). Draft genome assemblies of the two other C genome diploid species, Oryza eichingeri and Oryza rhizomatis, and short-read resequencing of a series of other C genome species and accessions reveal that after the divergence of the C genome progenitor, there was still a substantial degree of variation within the C genome species through proliferation and loss of both DNA and long-terminal repeat transposable elements. We provide a detailed phylogeny and evolutionary history of the Oryza C genomes and a genomic resource for the exploitation of the Oryza tertiary gene pool.
Keywords: rice, C genome, wild rice, transposon, polyploid, reference genome
Introduction
Rice is the second most produced crop (Ray et al. 2013), and its production is growing fastest, especially in sub-Saharan Africa (Toriyama 2005). Wild Oryza species are adapted to diverse habitats (Atwell et al. 2014) and encompass 15 Myr of evolutionary history (Jacquemin et al. 2014), culminating in independent domestications on two continents. These genomes are a rich resource for breeding improved cultivated rice to meet the demands caused by the pressures of increasing population and climate change. Oryza genomes are classified on the ability of their chromosomes to pair correctly in interspecies hybridizations (Kurata and Omura 1984). The five wild and two domesticated A genome species, Oryza sativa and Oryza glaberrima, can be crossed relatively easily. The A genome diversified roughly 3 Ma (Brozynska et al. 2017), however, some of the 14 non-A genome Oryza species dating from the divergence of Oryza brachyantha (roughly 15 Ma) have received less attention. Seven wild Oryza genomes were recently characterized by the International Oryza Map Alignment Project (Stein et al. 2018), along with five cultivated rice genomes, comprising five wild AA genomes, the BB genome of Oryza punctata, and the closest non-Oryza species from a sister clade, Leersia perrieri, used as an outgroup. Characterization of further non-A genome species of Oryza promises to make available further reserves of genetic and phenotypic diversity from this tertiary gene pool that can contribute to higher yielding, resilient and environmentally sustainable rice production.
Oryza comprises ∼21 wild species composed of 10 different genome types (A–H, J, and K; Ge et al. 1999), and the species are classified into several complexes based on their characteristics; the largest of these is the Oryza officinalis complex (with B–E genomes in diploids and allotetraploids), and we aimed to further characterize these species based on a newly constructed C genome reference. The Oryza C genome is at the heart of the O. officinalis complex, as shown in figure 1A—nine species possess a C genome, comprising three diploid species and six allotetraploid species (O. malampuzhaensis not shown). The three diploid C genome Oryza species, O. eichingeri, O. officinalis, and O. rhizomatis, are distributed in Africa and Asia (fig. 1B). Oryza officinalis has the widest distribution across Asia, whereas O. rhizomatis is distributed in a very limited area in Sri Lanka (Vaughan 1994). Uniquely, O. eichingeri is found both in Africa and in Asia, although the Asian distribution is limited to a small region in Sri Lanka. These three species, along with O. punctata (B genome diploid, Africa) and Oryza australiensis (E genome diploid, Oceania), comprise the diploid members of the O. officinalis complex (Vaughan et al. 2003). They are joined by several allotetraploid C genome containing species: Oryza minuta, O. punctata (also called Oryza schweinfurthiana), and Oryza malampuzhaensis (O. minuta—Philippines; O. punctata—Africa; O. malampuzhaensis—India; all with B and C genomes); and Oryza alta, Oryza grandiglumis, and Oryza latifolia (South and Central America, with C and D genomes). Zou et al. (2015) showed that at least three hybridizations have resulted in three allotetraploid BBCC genome species. In hybridizations resulting in O. malampuzhaensis and O. minuta, a B genome diploid was the maternal parent, whereas for O. punctata (schweinfurthiana), a C genome diploid was the maternal parent. The D genome progenitor of CCDD allotetraploids is presumed extinct, but the D genome is closest to the diploid E genome of Oryza australiensis (Wang et al. 2009). These species together are distributed in most tropical and subtropical areas of the world (fig. 1B), which is reflected in their wide phenotypic diversity. Traits linked to favorable ecological adaptations and disease resistance have been isolated from the O. officinalis genome (Ishimaru et al. 2010; Zhang et al. 2014). Among C genome species, O. officinalis is the most widely distributed, across South and South-east Asia. Therefore, we targeted the O. officinalis genome to define the evolutionary status and explore genomic structural divergence in the O. officinalis complex. We provide a chromosome-level assembly of the C genome diploid O. officinalis, along with scaffold-level assemblies of the two other diploid C genome species, O. eichingeri and O. rhizomatis. Based on these genomes and their annotations, and on resequencing data from 77 further National Bioresource Project accessions, we estimate the timing of origin of the O. officinalis complex, consider the impact of transposable elements (TEs) on genome structure, and examine the possibility of additional hybridization events in the origin of O. punctata (schweinfurthiana).
Fig. 1.

—Species relationships in Oryza and distribution of the O. officinalis complex. (A) Phylogenetic tree based on Oryza chloroplast sequences illustrates species relationships in the Oryza genus. Leersia perrieri is used as an outgroup. Allotetraploid species are illustrated with circles at the tips. The O. officinalis complex comprises at least ten species; most contain a C genome. The B genome diploid O. punctata is placed in the O. officinalis complex but represents a sister clade to the A genome diploid species in the Oryza sativa complex. Three species in the O. sativa complex are the progenitors of the three species/subspecies of cultivated rice, illustrated with blue arrows. Neighbor-Joining tree based on mapping Illumina short reads to the O. sativa japonica Nipponbare chloroplast genome. Bootstrap support is shown as node labels. Representative accessions were used as follows: Oryza rufipogon W1943, Oryza nivara W0106, Oryza barthii W0042, Oryza glumaepatula W1169, Oryza meridionalis W1297, Oryza longistaminata W1504, O. punctata (BB) W1514, O. minuta W1319, O. punctata schweinfurthiana (BBCC) W1564, O. officinalis W0002, O. eichingeri W1525, O. rhizomatis W1808, O. latifolia W1539, O. grandiglumis W0613, O. alta W0017, O. australiensis W0008, Oryza meyeriana W1348, Oryza granulata W0003, Oryza ridleyii W0001, Oryza longiglumis W1215, and O. brachyantha W1711. (B) Distribution of O. officinalis complex species in tropical and subtropical regions around the world. The C genome occurs in America, Africa, and Asia; the B genome occurs in Africa and Asia. The D genome is limited to American CCDD allotetraploids. The E genome only occurs in Australasia.
Materials and Methods
Oryza officinalis Complex Plant Material
Wild Oryza accessions used in this study were collected and have been maintained in the National Institute of Genetics (NIG) for several decades (Nonomura et al. 2010). Eighty accessions selected from the O. officinalis complex were grown, and leaf DNA was extracted for sequencing as shown in supplementary table S16, Supplementary Material online.
Sequencing Data
Sequencing instruments and libraries used to construct the O. officinalis reference genome and the draft O. eichingeri and O. rhizomatis genomes are detailed in supplementary table S1, Supplementary Material online. For O. officinalis and O. rhizomatis, PacBio SMRT Bell libraries were constructed following PacBio standard protocols and sequenced on a PacBio RSII instrument.
For resequencing of wild rice accessions, short-read Illumina sequencing was performed using an Illumina Hiseq 2500 instrument, using Truseq DNA PCR-Free library preparation, with paired reads of 2× 150 bp and an insert size of 350 bp.
Preprocessing of Illumina Short Reads
Quality control of the reads detailed in supplementary table S1, Supplementary Material online, was performed by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/; last accessed March 12, 2020). Removal of duplicated read pairs was performed with an in-house C++ program available at https://github.com/meiji-bioinf/NGS_pre-process (last accessed March 12, 2020). Adapter sequences were removed from the reads with Cutadapt (version 1.5) (Martin 2011). The low-quality reads were filtered out by an empirically optimized custom C++ program available at https://github.com/meiji-bioinf/NGS_pre-process. Its filters are as follows: 1) both ends of each read should have QV ≥ 10 (if it is not, the end base will be trimmed away until QV ≥ 10 is exposed), 2) each read should have average QV ≥ 17, 3) final length of each read should be ≥20 bp, 4) each read should have low-quality positions (QV < 10) no more than 10% of final length, and 5) each read should not contain any N bases. Short reads containing base call error were purged by using k-mer frequencies. At first, we counted the frequencies of all 17 base k-mers among the short reads with Jellyfish (version 2.1.3) (Marçais and Kingsford 2011) and plotted a histogram. Low frequency k-mers were probably derived from base call error and the reads containing them were filtered out with a custom C++ program (available at https://github.com/meiji-bioinf/NGS_pre-process). Error correction of single-pass PacBio reads was performed using the Sprai pipeline (version 0.9.9) (http://zombie.cb.k.u-tokyo.ac.jp/sprai/; last accessed March 12, 2020).
De Novo Assembly
At first, we performed de novo assembly, scaffolding, and gap closing with preprocessed short reads using Platanus (version 1.2.1) (Kajitani et al. 2014). Next, gaps contained in the scaffolds were closed with preprocessed PacBio reads by using PBJelly (version 14.1.14) (English et al. 2012). We then performed an additional five rounds of scaffolding with the mate-pair libraries using SSPACE (version 3.0) (Boetzer et al. 2011). Finally, gaps contained in the scaffolds were closed with the short reads by using GapCloser (version 1.12) (http://sourceforge.net/projects/soapdenovo2/files/GapCloser/; last accessed March 12, 2020).
Pseudomolecule Assembly
Scaffolds were arranged into pseudomolecules with reference to the O. officinalis BAC library physical contigs and end-sequence from the Oryza Map Alignment Project (OMAP). The genome-wide 101,091 BAC end DNA sequences of O. officinalis (IRGC accession 100896 = NIG Wild Rice Collection accession W0065), generated by Sanger sequencing technology, were downloaded from the NCBI GenBank GSS records, referring to the information on the OMAP website (http://www.omap.org/cgi-bin/status/status.cgi; last accessed March 12, 2020). A BLAST search was performed using the combined O. officinalis scaffolds as a reference and the BAC end sequences (BESs) as queries in order to avoid nonspecific alignments due to repetitive DNA sequences. BESs, which had multiple hits among the O. officinalis scaffolds, were excluded. Scaffolds were associated with OMAP Finger Print Contig (FPC) contigs by comparing the number and order of BES blast hits on each scaffold. Scaffolds were assigned to the physical contig where they had the highest number of correctly ordered BES hits. If correctly ordered BES hits from different FPCs were found on the same scaffold, the scaffold was disassembled into smaller contigs, and the parts were assigned to the correct FPC. Unambiguously located scaffolds, along with remaining BESs not used to assign scaffolds to contigs, were then aligned on the physical coordinates of pseudomolecules according to their FPC locations available on the OMAP website. The consequent 12 DNA sequences (i.e., the number of O. officinalis chromosomes) with gaps represented as N-arrays, along with several unplaced FPCs, were considered as the initial pseudomolecules. Next, the remaining scaffolds created by GapCloser were homology searched against the BES in the initial pseudomolecules employing BLAT with appropriate options (-t=dna -q=dna -fastMap -out=blast9), and each BLAT best hit was adopted as the physical location of the scaffold. The orientations of scaffolds on the pseudomolecule coordinate were determined as far as possible, based on their double-anchoring information onto multiple BESs if it was available. Each scaffold DNA sequence was then incorporated into the pseudomolecule at the appropriate location. As a consequence, the final pseudomolecule assembly was generated, and the scaffolds that did not align on the pseudomolecule coordinate were gathered in the “unanchored” scaffold category.
De Novo Assembly of O. eichingeri and O. rhizomatis
Draft de novo assemblies were also created for the two other diploid C genome Oryza species, O. eichingeri and O. rhizomatis. For O. eichingeri, 3-, 5-, and 8-kb mate-pair libraries and paired-end Illumina libraries with 350- and 500-bp insert sizes were prepared, whereas for O. rhizomatis, 3-, 5-, and 8-kb mate-pair libraries and paired-end Illumina libraries with 350- and 500-bp insert sizes and PacBio SMRT Bell libraries were prepared. Assemblies were produced using the same procedure as for O. officinalis up to the pseudomolecule assembly step.
Annotation
The baseline repeat annotation of the assembly was obtained merging the output of RepeatMasker (http://www.repeatmasker.org/; last accessed March 12, 2020, v. 3.3.0) and Blaster (a component of the REPET package; Flutre et al. 2011). The two softwares were run using nucleotidic libraries (PReDa and RepeatExplorer) from RiTE-db (Copetti et al. 2015) and an in-house curated collection of TE proteins, respectively. Reconciliation of the masked repeats was carried out using custom Perl scripts and formatted in gff3 files.
To determine the abundance of TEs and nongenic repeats in the native genome and in the assembly, raw O. officinalis reads and simulated reads were aligned to a curated TE/repeat library, and hits to each category were tallied (Copetti and Wing 2016).
Unrooted phylogenetic trees were constructed aligning coding regions of Copia, Gypsy, CACTA, and Mutator elements with ClustalX (Larkin et al. 2007) or Muscle (Edgar 2004). If required, alignments were manually edited and trees were constructed with FastTree 2 (Price et al. 2010). For Copia and Gypsy, the reverse transcriptase of accession CAD40165.2 from residues 781 to 872 and CAH66235.1 from 471 to 632, respectively, was used. For CACTA and Mutator elements, the DDE domains defined by Yuan and Wessler (2011) were selected. To assign O. officinalis elements to families or subclades, known element sequences were added (Lisch 2002; Domingues et al. 2012; Buchmann et al. 2014). To estimate the relative timing of proliferation between species, heuristic trees with elements from all fully sequenced Oryza species (Stein et al. 2018) were generated, aligning the same region for a subset of sequences proportional to the abundance in each species.
Infernal (Nawrocki and Eddy 2013) was adopted to identify noncoding RNAs using the Rfam library Rfam.cm.1_1. Hits above the e-value threshold of 1e−5 were filtered, as well as results with scores lower than the family-specific gathering threshold. When loci on both strands were predicted, only the hit with the highest score was kept. Transfer RNAs were also predicted using tRNAscan-SE (Schattner et al. 2005) at default parameters.
Transcribed sequences were assembled with Trinity (r2013-02-25) (Grabherr et al. 2011) using spikelet RNA-Seq data (bioproject PRJDB2848) and leaf, panicle, and root from PRJNA239525. The assembled transcripts were merged in a unique EST file. Reference-guided transcriptome assembly was obtained with Tuxedo (Trapnell et al. 2012; Tophat v2.0.6 and Cufflinks v2.0.2) using the spikelet sample.
Gene models were predicted with MAKER2 (v2.31.8; Holt and Yandell 2011) using the assembled transcript and repeat libraries evidence from above and using Exonerate (v. 2.2.0; Slater and Birney 2005), SNAP (v. 2006-07-28; Korf 2004), and Augustus (v. 3.1; Stanke and Waack 2003) for the ab initio gene prediction. EST and protein evidence from other species were aligned with Blast (2.31.8; Camacho et al. 2009). Gene models containing TE domains were removed, and models with nonconventional start and stop codons were flagged.
Phylogenetic Analyses
Phylogenetic analyses using whole-genome single-nucleotide polymorphisms (SNPs) were performed as follows. Illumina short reads were mapped to the appropriate reference (O. punctata [Stein et al. 2018], O. officinalis [this study], or the concatenation of the two for BBCC tetraploids) using bwa mem (Li and Durbin 2009). SNPs were called using bcftools (Li 2011) with the filter -e QUAL < 20‖DP < 3. Where the combined reference was used, the vcf files were subsetted using bcftools so that the appropriate chromosomes could be compared across samples (i.e., all B genomes in both diploids and tetraploids and all C genomes in both diploids and tetraploids). The all B genomes data set was 6,133,964 SNPs and the C genome set comprised 13,961,216 SNPs. The SNPhylo pipeline (Lee et al. 2014) was used to extract representative SNPs based on linkage disequilibrium and to construct maximum-likelihood trees. Population structure analysis was conducted using the fastStructure software (Raj et al. 2014), implemented using the wrapper script Structure threader (Pina‐Martins et al. 2017) and admixture (Alexander et al. 2009). Whole-genome SNPs called as above were used for the analysis.
For analysis of chloroplast sequences, resequencing Illumina data (containing substantial numbers of chloroplast-derived reads) were mapped to the Oryza sativa japonica Nipponbare chloroplast sequence (GenBank: AC092750.2). SNPs were called as above, and a Neighbor-Joining tree was constructed using the R package ape (Paradis and Schliep 2019).
k-Mer Analysis
Reads for each genome were selected as follows. For diploid B, C, E, and F genome species, reads that did not align to mitochondrial or chloroplast sequences were selected. Mapping and filtering were performed as for the whole-genome phylogenetic analysis. For tetraploid BBCC species, the reads were further selected that did not align to the B genome O. punctata reference (Stein et al. 2018) for the C genome sample, and for those that did not align to the O. officinalis C genome for the B genome sample. For CCDD tetraploids, reads mapping to the C genome reference were taken as the C genome sample. Unmapped reads were used as the D genome sample. The software kwip (Murray et al. 2017) was used to calculate the k-mer weighted inner product and estimate pairwise similarity between the short-read data sets.
Divergence Time
The aligned single-copy orthologs from Oryza species described in Stein et al. (2018) were identified from the O. officinalis MAKER-P (Campbell et al. 2014) gene annotation using blat (Kent 2002). Following Stein et al. (2018), the coding regions of each locus were aligned at the nucleotide level using PRANK v.140110 (Löytynoja and Goldman 2005) with the -F setting. For each chromosome, alignments containing sequences from each genome were concatenated to create 12 supermatrix alignments, and maximum-likelihood phylogenies were inferred using GARLI version 2.01 (Zwickl 2006) with bootstrapping replicates at the CIPRES Science Gateway (Miller et al. 2010). Estimation of divergence time was performed using PATHd8 (Britton et al. 2007) for each chromosome as in Stein et al. (2018) using a fixed age of 15 Myr for the origin of the Oryza genus.
Enrichment of GO Terms in O. officinalis Orthologs
IPRScan (Jones et al. 2014) was used to classify annotated genes with Gene Ontology (GO) terms. Enrichment analyses using hypergeometric tests were performed using the R package GOstats (Falcon and Gentleman 2007).
CAFÉ Analysis
Gene families in six Oryza species (O. brachyantha, O. officinalis, O. eichingeri, O. rhizomatis, and O. sativa japonica) were identified using OrthoMCL. A rooted ultrametric tree was derived using the aligned single-copy orthologs from Stein et al. (2018) as for divergence time above, and rapidly evolving gene families were identified. Rapidly evolving families defined by PFAM annotations or GO terms were identified in the same way.
Reticulate Phylogeny of Allotetraploid BBCC Genome Species
The gene sequences of single-copy orthologs from Stein et al. (2018) were inferred in allotetraploid BBCC genome species representative of each group of accessions. bcftools consensus was used to output the exon sequences based on mapping of their short reads to the concatenated C genome reference (this study) and B genome O. punctata reference (Stein et al. 2018). Regions with zero coverage in mapping were excluded from the analysis. Orthologs were aligned using Garli 2.01 (Zwickl 2006) with 200 bootstrap replicates for each gene. The resulting 1,652 phylogenetic trees were then used to infer network phylogenies using Phylonet software (Wen et al. 2018), specifying the hybrid species and their potential progenitor diploid species, and the maximum number of additional hybridizations/reticulations in each case (InferNetwork_ML method).
Results
Genome Sequencing, Assembly, and Pseudomolecule Construction
We developed a chromosome-level assembly for the key Oryza C genome species O. officinalis using short- and long-read technologies, with support from a large-insert BAC library and physical map (Wing et al. 2005; Ammiraju et al. 2006; supplementary fig. S1, Supplementary Material online). The 584-Mb genome comprised 12 chromosome pseudomolecules totaling 577 Mb, with 7 Mb of unplaced contigs, made up of 5,421 scaffolds with an N50 of ∼0.5 Mb. The pseudomolecules were verified by mapping reads back to the assembly, wherein more than 96% of the short reads were correctly mapped and paired and 96% of the long reads were mapped. To confirm the order and orientation of the assembled scaffolds, and thus the quality of the overall assembly, nonrepetitive paired BESs were mapped to the pseudomolecules. The vast majority (98%) of the BES mapped as paired in the correct orientation and at a distance between 25 and 300 kb (the expected size of a BAC clone). The assembly was further verified by comparison with other sequenced Oryza genomes and by comparison with the short arm of O. officinalis chromosome 3 (Fan et al. 2008), previously assembled using Sanger sequencing data, using LASTZ (Harris 2007; supplementary fig. S2, Supplementary Material online). Our assembly maintains the overall structure of the short arm of chromosome 3, though some gaps are present in repetitive regions. We employed PacBio sequencing on a set of O. officinalis BAC clones, resulting in fully assembled BAC sequences. These were highly repetitive sequences that supported the structure of our pseudomolecules (supplementary fig. S3, Supplementary Material online).
Genome Characteristics
Oryza officinalis accession W0002 (IRGC100878, Bangkok, Thailand; Nonomura et al. 2010) has a haploid nuclear genome of ∼597 Mb as estimated by flow cytometry. Our assembly at ∼584 Mb is larger than those of neighboring diploid species in the Oryza phylogeny, and about 1.6 times larger than O. sativa (International Rice Genome Sequencing Project 2005). According to our annotation, 9.16% of the genome is composed of exons, whereas repeats and TEs make up 51.09%, with 29.69% of the genome left uncharacterized (table 1 and supplementary table S3, Supplementary Material online). Most of the genome size increase compared with other Oryza genomes consists of repetitive sequences, and more than half of the genome consists of TEs and other high copy number sequences. Gypsy retroelements constitute the vast majority of TEs (65.75%) occupying 31.8% of the entire assembly (supplementary table S5, Supplementary Material online). Although the C genome of O. officinalis is greatly expanded, overall synteny within Oryza is maintained (fig. 2). Identification of conserved eukaryotic and plant genes with BUSCO (Simão et al. 2015) showed that the O. officinalis assembly is highly complete (98% with BUSCO, supplementary table S3, Supplementary Material online).
Table 1.
Assembly and Annotation Statistics of Diploid Oryza C Genome Species
| Species [genome type] | Assembly Size (Mb) | Repeats (%) | Annotated Loci | ScaffoldN50 (kb) |
|---|---|---|---|---|
| O. officinalis [CC] | 584 | 51.09 | 29,930 | 508 |
| O. rhizomatis [CC] | 559 | 57.96 | 32,083 | 82 |
| O. eichingeri [CC] | 471 | 50.10 | 31,030 | 64 |
Fig. 2.

—Syntenic relationships of O. officinalis with other Oryza species. The O. officinalis reference genome (y-axis) was aligned to three other diploid reference genomes from the Oryza genus—O. brachyantha, O. punctata, and O. rufipogon using SynMap2 (Haug-Baltzell et al. 2017). Although the O. officinalis genome is increased in size by ∼1.6 times, chromosome-level synteny is maintained.
The gene annotation identified 29,930 protein coding loci, occupying about one quarter of the genome. Only 30 kb (0.01% of the genome sequence) contained noncoding RNA genes (supplementary table S5, Supplementary Material online). With a total of 44,838 predicted isoforms, there are on average 51 genes per Mb of sequence, with chromosomes 10 and 11 having around 43 genes/Mb and chromosomes 1–3 having around 60 genes/Mb (supplementary table S4, Supplementary Material online). Corresponding with the larger size of the C genome, the density of genes along the chromosome is reduced, while the content of long-terminal repeat retrotransposons (LTR-RTs) is higher. Enrichment of LTR-RTs toward the center of the chromosomes is less obvious than in other diploid Oryza species (supplementary fig. S4, Supplementary Material online).
Contribution of TEs to O. officinalis Complex Genome Evolution
As more than half of the O. officinalis assembly comprises (TEs) and repeats (table 1 and supplementary tables S3 and S5, Supplementary Material online), we characterized them in better detail. LTR-RTs occupy about two-thirds of the repeat/TE space, with the rest mostly being DNA transposons (DNA-Ts). Compared with other Oryza species (Stein et al. 2018 and fig. 3A), there is an increased proportion of retroelements in the O. officinalis complex (represented by O. officinalis and O. punctata). Class I TEs (especially LTR-RTs) were responsible for the observed genome size variation in Oryza because their proportional representation in the genome correlates with genome size across the diploid Oryza species. The DNA-T amount, although variable, did not correlate with genome size (supplementary fig. S5, Supplementary Material online). Proliferation patterns of TE-coding sequences in Oryza vary both among superfamilies and families, and across time, and participate in shaping the genome. We observed three different patterns of TE behavior: 1) An element was active before speciation and maintained a steady and/or low activity level (fig. 3B, I), resulting in a TE tree with long branches and species intermingled with each other (e.g., Copia Retro family; fig. 3C, I and supplementary fig. S6, Supplementary Material online). 2) An element was active since before speciation and maintained different paces of duplication in different species and TE subfamilies, and in some cases the element was lost or silenced (fig. 3B, II and III exemplified by the Gypsy Athila family, fig. 3C, II; and Gypsy Atlantys family, fig. 3C, III; respectively; supplementary fig. S7, Supplementary Material online). 3) During species divergence, one or very few copies diverged substantially and continued evolving, also duplicating at different paces. In an extreme case, a new subfamily was originated that escaped host control for a time (fig. 3B, IV, exemplified by a new CACTA family, fig. 3C, IV; supplementary fig. S8, Supplementary Material online). The MuDR2 family (fig. 3B, V and fig. 3C, V and supplementary fig. S9, Supplementary Material online) showed branch-specific acceleration and deceleration of transposition, probably a consequence of escape or reactivation of host TE suppression.
Fig. 3.

—Proportion of different repeat classes in sequenced diploid Oryza genomes and L. perrieri. (A) The repeat annotation was performed consistently across all assemblies and thus highlights actual differences among assemblies. The greater number of repeats reflects the larger genome size of O. officinalis. Copia, Gypsy retroelements, and CACTA DNA-Ts appear to have proliferated much more in the BB and CC genome types compared with other species. (B) Simplified representation of different TE behaviors. TE family gene trees are drawn (black lines) within a representative species tree (yellow shadow), depicting the changes occurring to the type and abundance of TEs for each family. Line thickness is a proxy of TE copy number, finely dashed lines are dead/silenced lineages, and dashed lines represent a distantly related family. Stars indicate TE burst, Xs TE inactivation, or loss. Each bifurcation represents the origination of a different subfamily. Each panel I–V represents the pattern observed in (C, I–V). (C) Examples of TE behavior across multiple closely related species. (I) Subtree of the Copia retro family, (II) subtree of the Gypsy Athila family, (III) subtree of the Gypsy Atlantys family, (IV) subtree of a newly found Cacta family, related to the known Eric and Grover elements, and (V) subtree of the Mutator MuDR2 family.
Fig. 5.
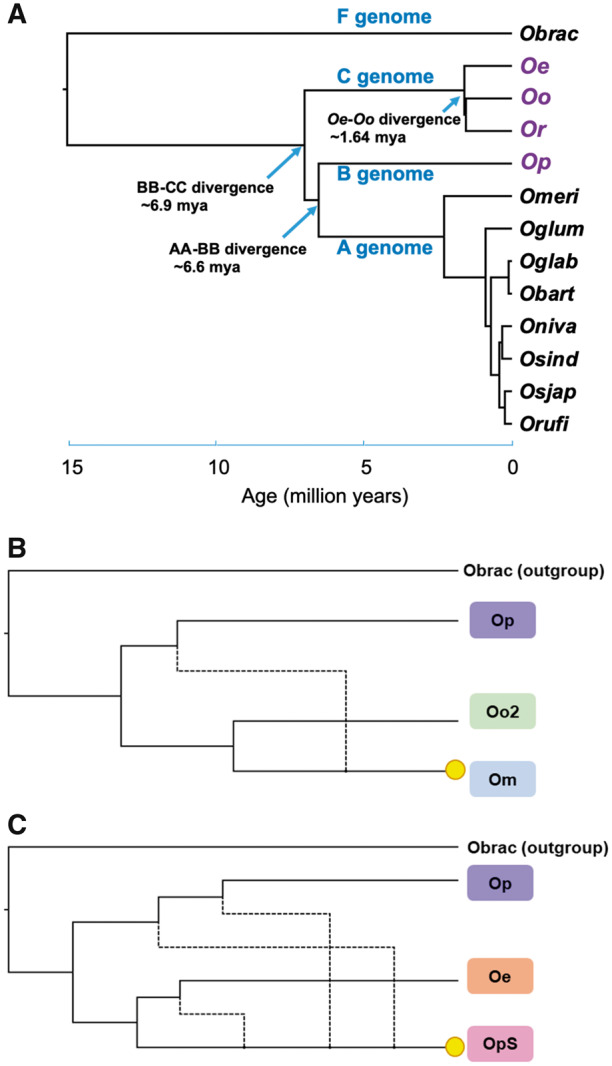
—Evolutionary history of the Oryza officinalis complex. (A) Orthologous gene sequences from 13 Oryza species and the outgroup species L. perrieri (Stein et al. 2018) were aligned and maximum-likelihood trees calculated. Divergence times were estimated according to the crown age of 15 Myr for Oryza. All nodes had >99% bootstrap support. Phylogenies and divergence times calculated for each chromosome supermatrix are shown in supplementary figure S16, Supplementary Material online. (B) Orthologous gene sequences were inferred for representative C and B genome species, and individual maximum-likelihood gene trees were constructed. Phylogenetic networks were inferred using the InferNetwork_ML method in Phylonet (Wen et al. 2018). Yellow circles highlight tetraploid species. A single hybridization between B and C genome diploids resulted in O. minuta (Om), whereas at least two hybridizations resulted in O. punctata (schweinfurthiana) (OpS). The networks with the highest likelihoods are displayed. Further details are in supplementary figure S17, Supplementary Material online. Obrac, O. brachyantha; Oe, O. eichingeri; Oo, O. officinalis; Or, O. rhizomatis; Op, O. punctata (diploid); Omeri, Oryza meridionalis; Oglum, Oryza glumaepatula; Oglab, Oryza glaberrima; Obart, Oryza barthii; Oniva, Oryza nivara; Osind, Oryza sativa indica; Orufi, O. rufipogon; Osjap, O. sativa japonica; OpS, tetraploid O. punctata (schweinfurthiana).
Using short reads to compare the completeness of the native genome and the assembly (Copetti and Wing 2016), we demonstrated that for all repeat/TE categories, the content in the O. officinalis assembly is lower than that estimated in the native genome (supplementary table S6, Supplementary Material online).
Quantitatively, Copia, Gypsy, CACTA, Mutator TEs, and tandemly repeated DNA sequences are more underrepresented in the assembly. Despite many efforts in sequencing and assembly with different platforms and software, more than 25%—or 90 Mb—of the native O. officinalis TEs and repeated sequences are still missing from the final assembly (supplementary table S6, Supplementary Material online), although in most cases, the amount represented in the assembly is roughly proportional to their total occurrence. The only repeat class significantly underrepresented in the assembly is tandemly repeated sequences with homology to rice centromeric sequences, after a nonparametric test (Rodriguez-Brito et al. 2006). Most of them are highly similar to the clusters CL1, CL7, and CL227 produced by a RepeatExplorer (Novak et al. 2013) analysis on the raw reads (Copetti et al. 2015). The motifs are not similar to each other, as shown when assembled regions of these three motifs are aligned and displayed as a dot plot (supplementary fig. S10, Supplementary Material online). CL1 is by far the most abundant centromeric sequence and is the most underrepresented in the genome assembly (the native genome has twice as many hits as the assembly), whereas about one quarter of the CL7 and CL227 sequences are not assembled (supplementary table S8, Supplementary Material online). Because they could not be assembled in long stretches, their distribution on the chromosome pseudomolecules was very fragmented and could not be associated with putative centromeric or pericentromeric regions.
Genome Evolution among the C Genome Diploid Oryza Species
We also produced draft genomes for the two other diploid Oryza C genome species, O. eichingeri and O. rhizomatis, using paired-end short-read, mate-pair, and PacBio sequencing libraries (table 1 and supplementary table S1, Supplementary Material online). Compared with the 584-Mb assembly of O. officinalis, the O. rhizomatis assembly comprised 559 Mb with a scaffold N50 of 82 kb, whereas the O. eichingeri assembly was 471 Mb, with a scaffold N50 of 64 kb. The smaller size of the O. eichingeri assembly is consistent with previous estimates of the genome size (Nonomura et al. 2010). We also applied the MAKER-P pipeline (Campbell et al. 2014) to these assemblies, resulting in 32,082 and 31,030 annotated genes for O. rhizomatis and O. eichingeri, respectively, compared with 29,930 for O. officinalis. The larger numbers of genes annotated for the two draft assemblies may be due to their increased proportion of fragmented gene loci, as suggested by the higher proportion of fragmented models detected from the BUSCO (Simão et al. 2015) analysis (supplementary table S9, Supplementary Material online).
We compared the O. officinalis C genome reference and the O. rhizomatis and O. eichingeri draft genome assemblies using the nucmer tool from the MUMmer software package (Kurtz et al. 2004). We then compiled statistics on polymorphic sequences and other rearrangements using Assemblytics (Nattestad and Schatz 2016; supplementary fig. S11, Supplementary Material online). In pairwise comparisons of O. rhizomatis or O. eichingeri with the O. officinalis reference, there were large numbers of insertions and deletions of lengths around 250 bp and around 350 bp. These sequences were similar with each other and many appear to be related to DNA-Ts, especially Harbinger-MITE (miniature inverted-repeat transposable element) sequences.
Among 2,049 sequences present in O. officinalis and deleted in O. rhizomatis, 757 of 1,208 with a BLAST hit in the RiTE (Copetti et al. 2015) repeat database (63%) were MITE sequences. Similarly, of 1,204 sequences with a significant match, deleted in O. eichingeri relative to O. officinalis, 750 (63%) were MITE sequences. For sequences present in O. officinalis and deleted in both O. eichingeri and O. rhizomatis, 177/250 (71%) were similar to MITE sequences.
Considering sequences absent from O. officinalis, 1,031 of 1,759 O. rhizomatis polymorphisms (59%), and 733 of 1,191 polymorphisms (62%) in O. eichingeri were MITEs. There were 154 polymorphic MITE-related sequences common to O. eichingeri and O. rhizomatis but absent in O. officinalis. Of these, 26 had a significant hit in the repeat database and 13 (50%) were MITEs (supplementary table S10, Supplementary Material online). The number of MITE sequences may be underestimated because the RiTE database is lacking in MITEs from non-A genome species.
Thus, although DNA-Ts make up a smaller proportion of the Oryza C genome compared with LTR transposons (fig. 3), they may still have significant effects on genome evolution. Furthermore, MITE sequences are generally associated with genic regions (Jiang et al. 2004), and indeed, most of the detected insertions and deletions occurred in locations within 1 kb of a gene annotated by the MAKER-P pipeline (supplementary table S10, Supplementary Material online).
We attempted to examine the impact on gene sequences of Oryza C genome evolution. Using gene annotations of O. officinalis (present study) and O. punctata (Stein et al. 2018), we examined the enrichment of GO terms in the different species and using hypergeometric tests, we tested for overrepresentation of particular GOslim terms in the O. officinalis complex (supplementary table S11, Supplementary Material online) and in O. officinalis (supplementary table S12, Supplementary Material online), compared with the whole genus. Several GO terms related to organo-nitrogen processes were overrepresented in O. officinalis. We did not find any overrepresented GO terms in O. eichingeri or O. rhizomatis relative to O. officinalis. On the other hand, when we checked for enrichment of GO terms in genes affected by MITE insertions or deletions, there were some terms (e.g., GO0006807; nitrogen compounds metabolic process) that were both enriched in O. officinalis and enriched in MITE affected genes (supplementary tables S13 and S14, Supplementary Material online). To further explore gene family representation in the O. officinalis complex, we employed the CAFÉ algorithm (De Bie et al. 2006) to investigate birth and death evolution of gene families. There were a number of gene families, PFAM domains and GO terms annotated as rapidly evolving in O. officinalis, notably GO:0006952; defense response (supplementary table S15 and fig. S12, Supplementary Material online).
Phylogenetic and Evolutionary Relationships in the O. officinalis Complex
Using reference genomes and annotations for the C genome diploid species (O. officinalis, O. eichingeri, and O. rhizomatis, this study) and the O. punctata B genome (Stein et al. 2018), we explored the evolutionary history and diversity in the O. officinalis complex, resequencing 77 diverse accessions from different species (supplementary table S16, Supplementary Material online). In an unrooted phylogenetic tree of diploid C genome species (supplementary fig. S13, Supplementary Material online), the five O. eichingeri accessions form a single bifurcated group, most closely related to the two O. rhizomatis accessions. The 15 O. officinalis accessions show more variation, perhaps reflecting their wider geographical distribution. Naredo et al. (2017) showed two major population groups in O. officinalis (supplementary fig. S14, Supplementary Material online). Only two of our O.officinalis accessions, W0065 and W1830, appear to belong to the south Asian group, the remainder originating from Malesia. W1830 is from China, but W0065 originates from Thailand, although it clearly belongs to a different population than do the other Thai O. officinalis accessions, W0002 and W1930.
To characterize BBCC genome species, we concatenated the O. officinalis genome with the diploid BB genome O. punctata reference (Stein et al. 2018). No reference is available for the D genome, whose diploid progenitor is presumed to be extinct, but resequencing reads were mapped to the C genome reference to allow C genome analyses in these CCDD genome tetraploid species. Of the BBCC genome tetraploids, O. minuta and O. malampuzhaensis resulted from hybridization between diploid B genome female parent and a C genome male parent (Zou et al. 2015), as evidenced by the similarity of their chloroplast genome sequences with extant B genome species (supplementary fig. S15, Supplementary Material online). On the other hand, tetraploid O. punctata chloroplast genomes resemble those of extant C genome diploid species.
Figure 4A shows phylogenetic relationships among the C genomes of the diploid and tetraploid species. The O. minuta accessions clearly arose from a separate polyploidization event than the tetraploid O. punctata (schweinfurthiana) accessions, as they have different cytoplasm, their chloroplast sequences resembling extant diploid B genome O. punctata (supplementary fig. S15, Supplementary Material online). The phylogenetic analysis of all diploid and tetraploid C genome species shows three major groups (fig. 4A). Group CI possesses diploid CC genome species of O. eichingeri composed of two separate groups (O. eichingeri 1 and O. eichingeri 2), and two groups of the tetraploid BBCC genome species, O. punctata (O. punctata S1 and O. punctata S2), both distributed in Africa. Group CII contains all three allotetraploid CCDD genome species of O. alta, O. latifolia, and O. grandiglumis, all found in South and Central America. Group CIII contains two diploid CC genome species, O. officinalis (Malesian and SE Asian, O. officinalis 1 and O. officinalis 2), O. rhizomatis and one tetraploid species with BBCC genome, O. minuta together, all of Asian habitat. The allotetraploid BBCC species O. minuta was formed by hybridization of a B genome O. punctata female with a C genome male (Zou et al. 2015), possibly a C genome parent from SE Asia, from O. officinalis 2. The substantial separation of a group of four BBCC O. punctata accessions (O. punctata S1 in CI in fig. 4A) suggests that they are diverged from the rest of the BBCC tetraploids with C genome type cytoplasm, although both groups C genomes originate from O. eichingeri. Their B genomes are also separated from most of the tetraploid O. punctata accessions, forming a separate clade in figure 4B.
Fig. 4.

—Phylogenetic and diversity analyses based on whole-genome SNPs identified by resequencing and mapping to the C genome and B genome reference sequences. Species group abbreviations are as follows: Oo1, O. officinalis group 1 (Malesian); Oo2, O. officinalis group 2 (S.E. Asian); Oe1, O. eichingeri group 1; Oe2, O. eichingeri group 2; Or, O. rhizomatis; Op, O. punctata (diploid); OpS1, tetraploid O. punctata (schweinfurthiana) group 1; OpS2, tetraploid O. punctata (schweinfurthiana) group 2; Om, O. minuta; Oal, O. alta; Ola, O. latifolia; Ogr, O. grandiglumis. Taxa labeled with yellow circles are allotetraploids. (A) Maximum-likelihood tree based on mapping of all Oryza C genome containing accessions to the O. officinalis reference. The procedure in Lee et al. (2014) was followed to obtain a representative SNP set and compute a maximum-likelihood tree. Colored bars indicate the group assignments obtained in fastStructure and admixture analysis with cluster number set at K = 4. (B) Maximum-likelihood tree based on mapping of all Oryza B genome containing accessions to the O. punctata reference. A representative SNP set and maximum-likelihood tree was obtained using the same procedure as for (A).
We employed the fastStructure (Raj et al. 2014) and admixture (Alexander et al. 2009) methods to infer the number of population groups. The optimum number of clusters was K = 4 for both the B genome and C genome accessions. For the C genomes, fastStructure supported the clades described above, but placed O. rhizomatis separately from O. officinalis (fig. 4A), admixture additionally highlighting the differentiation of O. eichingeri 2. For the B genomes, fastStructure and admixture discriminated O. minuta (BIII), diploid O. punctata (BI), and the two groups of tetraploid O. punctata (O. punctata S1 and O. punctata S2) from each other. This analysis suggests that the BBCC tetraploid species O. punctata (schweinfurthiana) may have evolved by two separate hybridizations of C genome O. eichingeri female parents with different B genome diploid O. punctata male parents in Africa. An alternative explanation could involve a more complex story of admixture and hybridization; this may be supported by the fastStructure and admixture analysis, and the unclear group assignment of accessions W0614 and W1564 in group BII of figure 4B.
We used gene sequences of single-copy orthologs described in Stein et al. (2018), and their identified orthologs in O. officinalis, O. eichingeri, and O. rhizomatis, to estimate the timing of the differentiation of the O. officinalis complex (fig. 5A and supplementary table S19, Supplementary Material online) based on branch lengths in maximum-likelihood phylogenetic trees of concatenated orthologous gene sequences for each chromosome. We calibrated the timing using an age of 15 Myr for the root of the Oryza genus, as in Stein et al. (2018). According to this analysis, the O. officinalis complex B and C genomes diverged around 6.9 Ma, and the common ancestor of O. officinalis and O. eichingeri diverged at ∼1.64 Myr (fig. 5A, supplementary fig. 16 and table S19, Supplementary Material online). Our estimate for the divergence of the B and A genomes is slightly more recent than that of Stein et al. (2018), 6.58 versus 6.76 Ma.
To try to confirm the origins of BBCC allotetraploid species, we inferred orthologous gene sequences of representative members of each group (O. minuta, O. punctata S1, O. punctata S2 as well as the O. punctata S1 accessions W0614 and W1564) using the same single copy orthologous loci from Stein et al. (2018) as above. Using 1,652 gene trees of these orthologs, we inferred a species network in the context of incomplete lineage sorting and introgression using the maximum-likelihood InferNetwork_ML method in Phylonet, specifying the maximum number of hybridizations, to identify the most likely network. Based on this analysis, O. minuta resulted from a single hybridization between B and C genome hybrids (fig. 5B and supplementary fig. S17A, Supplementary Material online), as only one reticulation was predicted independent of the number of specified hybridizations. Meanwhile O. punctata S1 and O. punctata S2 likely resulted from successive hybridizations or introgressions (fig. 5C and supplementary fig. S17B, Supplementary Material online). The O. punctata (schweinfurthiana) accession W1564 is suggested to result from additional hybridization or introgression compared with O. punctata S1 (supplementary fig. S17C, Supplementary Material online), whereas the accession W0614 is suggested to result from an additional hybridization or introgression involving O. officinalis as well as O. eichingeri (supplementary fig. S17D, Supplementary Material online)—suggesting a possibility of up to four B genome—C genome hybridizations in O. punctata (schweinfurthiana) (fig. 5B and supplementary fig. S17, Supplementary Material online). Further sampling of accessions similar with W0614 and W1564 might provide greater certainty.
We used k-mer analysis to compare all resequenced accessions including CCDD allotetraploids. First, we estimated the genome size of the resequenced CC diploid accessions by observing the k-mer representation in the sequencing reads (supplementary table S17, Supplementary Material online). According to this analysis, after O. rhizomatis diverged from its common ancestor with O. eichingeri (extant genome size of O. eichingeri: 484–521 Mb), the net change in genome size resulted in an increase to 592–603 Mb. The two accessions W0065 (Thailand) and W1830 (China) have somewhat smaller genomes (563 and 542 Mb, respectively), whereas the other O. officinalis accessions are similar with O. rhizomatis at 574–599 Mb (average 589 Mb).
There is considerable structural variation in the C genome of tetraploid wild Oryza species (Li et al. 2001). From previous estimates and from our draft genome assembly, we know that the O. eichingeri genome is roughly 100 Mb smaller than the O. officinalis genome, so it might not be surprising if the C genome of O. minuta (C genome donor O. officinalis) was larger than that of tetraploid O. punctata (C genome donor O. eichingeri), and we found that to be the case. However, based on short-read mapping rates to each genome, there seems to be substantial variation in the relative sizes of the B and C genomes within tetraploid Oryza punctata (supplementary table S18, Supplementary Material online).
Because reference genomes are not available for the D and E genomes, we investigated the diversity of CCDD tetraploids and the whole O. officinalis complex based on their shared representation of k-mer sequences in raw sequencing reads (Murray et al. 2017). Using the software pipeline provided in Murray et al. (2017), we calculated the k-mer weighted inner product for all of the resequenced accessions in the O. officinalis complex and performed clustering analysis to visualize the phylogenetic relationships (supplementary fig. S18, Supplementary Material online). The derived unrooted tree supported not only the previous results concerning BBCC tetraploids but also a single hybridization between a C genome diploid and a D genome diploid being the origin of the three CCDD tetraploid species, as the topology of the D and C genome trees for these accessions was very similar. The D genome has been suggested to be most similar to the E genome among Oryza genomes; while not contradicted by this analysis, we could not provide further confirmation for it based on raw sequence data analysis.
Discussion
The pressures of climate change and demand for increased crop production require genome innovation by breeding of crop species. Wild species, including those of the O. officinalis complex can provide the necessary variation, and the Oryza C genome may be particularly dynamic among Oryza species.
We can explain the increased size of the Oryza C genome by its higher proportion of TEs and tandem repeats (fig. 3 and supplementary fig. S5, Supplementary Material online)—apart from changes in ploidy, these sequences are the main player in genome size variation (Flavell et al. 1974; Lee and Kim 2014). We also observed a clearly higher activity of retroelements O. officinalis (and in O. punctata) markedly deviating from the general trend of a continuous increase in LTR-RT activity from the more basal to the more recent Oryza species. Another snapshot of the C genome dynamism was represented by the abundant presence of terminal branches with very short length (or even length 0) in some TE trees (supplementary figs. S6–S9, Supplementary Material online), entailing a very rapid and recent or still ongoing proliferation of elements.
Draft C genomes from O. rhizomatis and O. eichingeri further underline the dynamic nature of the Oryza C genome. First, the large difference in size (∼110 Mb or 19%) between O. officinalis and O. eichingeri implies rapid loss and/or gain of genomic material—the obvious candidate being LTR retrotransposons—in the common ancestor of O. officinalis/O. rhizomatis and/or O. eichingeri since the divergence of their common C genome ancestor (Zang et al. 2011). Furthermore, although their proportion in the genome and as a percentage of repeat sequences is not large, MITEs appear to have a significant effect on genome evolution, as evidenced by their high polymorphism (especially in proximity of genes) when comparing the three diploid C genome species.
Besides proliferation and subsequent elimination of TEs, polyploidization is the major driver of plant genome diversity, and Oryza C genomes have participated in a minimum of three tetraploidization events. As discussed by Zou et al. (2015), polyploidization may have contributed to survival in rapidly changing climatic conditions within the last 900,000 years. The survival of ongoing transposon activity in the C genome may also be driven in part by these pressures. One complication in studying the BBCC tetraploid Oryza species is difficulty in their identification, as well as confusion in their taxonomy. In particular, O. punctata comprises both diploid BB and tetraploid BBCC forms (the tetraploid form also being called O. schweinfurthiana). O. eichingeri, currently usually regarded as a C genome diploid, has at other times been treated as a BBCC genome tetraploid. Finally, there also exists confusion between O. officinalis and O. malampuzhaensis, sometimes recorded as a tetraploid O. officinalis. For these reasons, accessions are sometimes incorrectly labeled in germplasm collections, as previously noted by Zou et al. (2015). Some accessions were incorrectly assigned in our database (http://shigen.nig.ac.jp/shigen/about/database.jsp; last accessed March 12, 2020) and will be reclassified as a result of this work (see supplementary table S16, Supplementary Material online). Also, O. malampuzhaensis was lacking in our collection, so was not examined. Despite these difficulties, using a network phylogeny approach, we demonstrate the possibility of up to four different hybridizations leading to the formation of BBCC tetraploid O. punctata (schweinfurthiana), although as the W0614 and W1564 case rely on single individuals, and it is also possible that large-scale introgression rather than polyploidization would result in similar patterns, more data would be desirable for confirmation. Sampling of O. malampuzhaensis would also be informative. A representative population sample from these species is difficult to achieve because many of the original habitats have been destroyed, and only genebank samples remain.
The tetraploid CCDD species accessions have also sometimes been characterized as different species. Our phylogenetic analysis resolves three groups, and three species are recorded. All O. grandiglumis accessions were grouped together (fig. 4B), but two other groups both contained accessions labeled as O. alta and O. latifolia. Likely, new species assignment of several accessions based on their C genome sequence data and on phenotype data from Oryzabase (http://shigen.nig.ac.jp/shigen/about/database.jsp) is shown in supplementary table S16, Supplementary Material online.
Our new C genome reference and subsequent analyses allows us to confirm and expand on previous studies in the O. officinalis complex (Wang et al. 2009; Zang et al. 2011; Zou et al. 2015; Naredo et al. 2017) by resequencing and mapping to the reference genome. Based on these and previous results, we can summarize the evolutionary history of the C genome as follows: The common ancestor of the B and C genomes arose around 6.9 Ma and the divergence of the B and C genomes occurred relatively soon afterward (6.58–6.76 Ma, Stein et al. 2018; this study; fig. 5A). Subsequently, the common ancestor of O. rhizomatis and O. officinalis diverged from O. eichingeri ∼1.64 Ma (fig. 5A). Oryza officinalis occurs only in Asia, whereas O. eichingeri is found mainly in Africa and also occurs in Sri Lanka. Much more recently, the BBCC tetraploid species in the O. officinalis complex proliferated (within the last 800,000 years; Zou et al. 2015), perhaps surviving because genome innovation conferred an advantage in the context of changes in climate or environmental conditions. Oryza minuta, formed in Asia, inherited its cytoplasm from an Asian B genome diploid (see chloroplast phylogeny in supplementary fig. S15, Supplementary Material online), and its male parent was most likely O. officinalis (see the C genome tree in fig. 4A). Meanwhile, tetraploid Oryza punctata (schweinfurthiana) inherited a C genome diploid species-like cytoplasm from O. eichingeri (supplementary fig. S15, Supplementary Material online, and fig. 4A) and its male parent was likely O. punctata, which is extant in Africa (fig. 4B). This hybridization has occurred more than once, and there may be a complex history of introgressions between BB, CC, and BBCC genome species in Africa that might be revealed with further sampling. The CCDD genome species of South and Central America remain mainly unexplored, although the C genome may derive from a common ancestor of O. officinalis and O. eichingeri. The D genome is not closely related to any extant species. It seems that the history of the three species likely shares a single hybridization event that resulted in tetraploidization.
Oryza officinalis complex species are noted for their stress tolerance, and it is possible that both the prevalence of tetraploid species in the genus and the proliferation of TEs in their genomes have been a result of selection for resilience to environmental change during the earlier history of the genus, which did not act so strongly on the A genome species of the O. sativa complex. Supporting this idea, we found certain GO terms were enriched in O. officinalis, and similar terms were also enriched in genes that showed insertions or deletions related to MITE transposons (supplementary tables S13 and S14, Supplementary Material online). However, it is difficult to make strong conclusions using this kind of analysis based on gene numbers. As shown by Kitazumi et al. (2018), the real potential of wild crop relative genomes may be in network complementation, wherein introduced alleles can connect or augment existing signaling pathways resulting in improved crop performance.
For the purpose of exploiting the stress tolerance characteristics of the O. officinalis complex, the C genome reference is now available as a tool to aid in molecular breeding of cultivated rice. Introgressions from wild rice species in the O. officinalis complex have already made significant contributions to rice improvement (Multani et al. 1994; Jairin et al. 2009), and the genome sequence will aid in the more efficient utilization of diverse wild rice resources in rice breeding for further improvements. The Oryza C genome appears dynamic, based on recent variation caused by TEs and genome innovations associated with polyploidization. In the context of plant breeding, this relative instability provides us with diverse genetic resources that will be invaluable in future rice improvement.
Supplementary Material
Acknowledgments
This work was supported by the National Bioresource Project (NBRP) Genome Information Upgrading Program (NBRP MEXT, Japan) for N.K., the National Bioresource Project (NBRP AMED, Japan) for Y.S., and the Systems Functional Genetics Project of the Transdisciplinary Research Integration Center, Research Organization of Information and Systems (ROIS), Japan, for N.K. MS would like to thank Chikako Miura for constant and invaluable support during this project.
Literature Cited
- Alexander DH, Novembre J, Lange K.. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19(9):1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammiraju JSS, et al. 2006. The Oryza bacterial artificial chromosome library resource: construction and analysis of 12 deep-coverage large-insert BAC libraries that represent the 10 genome types of the genus Oryza. Genome Res. 16(1):140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwell BJ, Wang H, Scafaro AP.. 2014. Could abiotic stress tolerance in wild relatives of rice be used to improve Oryza sativa? Plant Sci. 215–216:48–58. [DOI] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W.. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics (Oxford, England) 27(4):578–579. [DOI] [PubMed] [Google Scholar]
- Britton T, Anderson CL, Jacquet D, Lundqvist S, Bremer K.. 2007. Estimating divergence times in large phylogenetic trees. Syst Biol. 56(5):741–752. [DOI] [PubMed] [Google Scholar]
- Brozynska M, et al. 2017. Sequencing of Australian wild rice genomes reveals ancestral relationships with domesticated rice. Plant Biotechnol J. 15(6):765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchmann JP, Löytynoja A, Wicker T, Schulman AH.. 2014. Analysis of CACTA transposases reveals intron loss as major factor influencing their exon/intron structure in monocotyledonous and eudicotyledonous hosts. Mobile DNA 5(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10(1):421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MS, Holt C, Moore B, Yandell M.. 2014. Genome annotation and curation using MAKER and MAKER-P. Curr Protoc Bioinf. 48(1):4.11.1–4.11.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copetti D, et al. 2015. RiTE database: a resource database for genus-wide rice genomics and evolutionary biology. BMC Genomics. 16(1):538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copetti D, Wing RA.. 2016. The dark side of the genome: revealing the native transposable element/repeat content of eukaryotic genomes. Mol Plant 9(12):1664–1666. [DOI] [PubMed] [Google Scholar]
- De Bie T, Cristianini N, Demuth JP, Hahn MW.. 2006. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 22(10):1269–1271. [DOI] [PubMed] [Google Scholar]
- Domingues S, et al. 2012. Natural transformation facilitates transfer of transposons, integrons and gene cassettes between bacterial species. PLoS Pathog. 8(8):e1002837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5(1):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AC, et al. 2012. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One 7(11):e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R.. 2007. Using GOstats to test gene lists for GO term association. Bioinformatics 23(2):257–258. [DOI] [PubMed] [Google Scholar]
- Fan C, et al. 2008. The subtelomere of Oryza sativa chromosome 3 short arm as a hot bed of new gene origination in rice. Mol Plant 1(5):839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell RB, Bennett MD, Smith JB, Smith DB.. 1974. Genome size and the proportion of repeated nucleotide sequence DNA in plants. Biochem Genet. 12(4):257–269. [DOI] [PubMed] [Google Scholar]
- Flutre T, Duprat E, Feuillet C, Quesneville H.. 2011. Considering transposable element diversification in de novo annotation approaches. PLoS One 6(1):e16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Sang T, Lu B-R, Hong D-Y.. 1999. Phylogeny of rice genomes with emphasis on origins of allotetraploid species. Proc Natl Acad Sci U S A. 96(25):14400–14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7):644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS. 2007. Improved pairwise alignment of genomic DNA [Ph.D thesis]. 2007 September 24; Penn State University. Available from: https://etda.libraries.psu.edu/catalog/7971 (accessed November 22, 2018).
- Haug-Baltzell A, Stephens SA, Davey S, Scheidegger CE, Lyons E.. 2017. SynMap2 and SynMap3D: web-based whole-genome synteny browsers. Bioinformatics 33(14):2197–2198. [DOI] [PubMed] [Google Scholar]
- Holt C, Yandell M.. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12(1):491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Rice Genome Sequencing Project 2005. The map-based sequence of the rice genome. Nature 436(7052):793–800. [DOI] [PubMed] [Google Scholar]
- Ishimaru T, et al. 2010. A genetic resource for early-morning flowering trait of wild rice Oryza officinalis to mitigate high temperature-induced spikelet sterility at anthesis. Ann Bot. 106(3):515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemin J, et al. 2014. Fifteen million years of evolution in the Oryza genus shows extensive gene family expansion. Mol Plant 7(4):642–656. [DOI] [PubMed] [Google Scholar]
- Jairin J, et al. 2009. Development of rice introgression lines with brown planthopper resistance and KDML105 grain quality characteristics through marker-assisted selection. Field Crops Res. 110(3):263–271. [Google Scholar]
- Jiang N, et al. 2004. Using rice to understand the origin and amplification of miniature inverted repeat transposable elements (MITEs). Curr Opin Plant Biol. 7(2):115–119. [DOI] [PubMed] [Google Scholar]
- Jones P, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30(9):1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajitani R, et al. 2014. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24(8):1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res. 12(4):656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazumi A, et al. 2018. Potential of Oryza officinalis to augment the cold tolerance genetic mechanisms of Oryza sativa by network complementation. Sci Rep. 8(1):16346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata N, Omura T.. 1984. Chromosome analysis In: Tsunoda S, Takahashi N, editors. Developments in crop science: Biology of rice. Vol. 7 Amsterdam: Elsevier. p. 305–320. [Google Scholar]
- Kurtz S, et al. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5(2):R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948. [DOI] [PubMed] [Google Scholar]
- Lee S-I, Kim N-S.. 2014. Transposable elements and genome size variations in plants. Genomics Inform. 12(3):87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T-H, Guo H, Wang X, Kim C, Paterson AH.. 2014. SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics. 15(1):162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C-B, Zhang D-M, Ge S, Lu B-R, Hong D-Y.. 2001. Identification of genome constitution of Oryza malampuzhaensis, O. minuta, and O. punctata by multicolor genomic in situ hybridization. Theor Appl Genet. 103(2-3):204–211. [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27(21):2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D. 2002. Mutator transposons. Trends Plant Sci. 7(11):498–504. [DOI] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N.. 2005. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 102:10557–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G, Kingsford C.. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics (Oxford, England) 27(6):764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17(1):10. [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T.. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: 2010 Gateway Computing Environments Workshop (GCE). New Orleans (LA): IEEE. p. 1–8. doi:10.1109/GCE.2010.5676129.
- Multani DS, et al. 1994. Development of monosomic alien addition lines and introgression of genes from Oryza australiensis Domin. to cultivated rice O. sativa L. Theor Appl Genet. 88(1):102–109. [DOI] [PubMed] [Google Scholar]
- Murray KD, Webers C, Ong CS, Borevitz J, Warthmann N.. 2017. kWIP: the k-mer weighted inner product, a de novo estimator of genetic similarity. PLoS Comput Biol. 13(9):e1005727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naredo MEB, et al. 2017. Genetic diversity patterns in ex situ collections of Oryza officinalis Wall. ex G. Watt revealed by morphological and microsatellite markers. Genet Resour Crop Evol. 64(4):733–744. [Google Scholar]
- Nattestad M, Schatz MC.. 2016. Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32(19):3021–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki EP, Eddy SR.. 2013. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29(22):2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonomura K-I, et al. 2010. The wild Oryza collection in National BioResource Project (NBRP) of Japan: history, biodiversity and utility. Breed Sci. 60(5):502–508. [Google Scholar]
- Novak P, Neumann P, Pech J, Steinhaisl J, Macas J.. 2013. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29(6):792–793. [DOI] [PubMed] [Google Scholar]
- Paradis E, Schliep K.. 2019. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35(3):526–528. [DOI] [PubMed] [Google Scholar]
- Pina‐Martins F, Silva DN, Fino J, Paulo OS.. 2017. Structure_threader: an improved method for automation and parallelization of programs structure, fastStructure and MavericK on multicore CPU systems. Mol Ecol Resour. 17:e268–e274. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP.. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5(3):e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Stephens M, Pritchard JK.. 2014. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197(2):573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray DK, Mueller ND, West PC, Foley JA.. 2013. Yield trends are insufficient to double global crop production by 2050. PLoS One 8(6):e66428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Brito B, Rohwer F, Edwards RA.. 2006. An application of statistics to comparative metagenomics. BMC Bioinformatics 7(1):162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattner P, Brooks AN, Lowe TM.. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33(Web Server):W686–W689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212. [DOI] [PubMed] [Google Scholar]
- Slater GSC, Birney E.. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Waack S.. 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl 2):ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Stein JC, et al. 2018. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat Genet. 50(2):285–296. [DOI] [PubMed] [Google Scholar]
- Toriyama K. 2005. Rice is life scientific perspectives for the 21st century. Los Baños, Laguna, Philippines: International Rice Research Institute. [Google Scholar]
- Trapnell C, et al. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7(3):562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan DA. 1994. The wild relatives of rice: a genetic resources handbook. Los Baños, Laguna, Philippines: International Rice Research Institute. [Google Scholar]
- Vaughan DA, Morishima H, Kadowaki K.. 2003. Diversity in the Oryza genus. Curr Opin Plant Biol. 6(2):139–146. [DOI] [PubMed] [Google Scholar]
- Wang B, et al. 2009. Polyploid evolution in Oryza officinalis complex of the genus Oryza. BMC Evol Biol. 9(1):250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D, et al. 2018. Inferring phylogenetic networks using phylonet. Syst Biol. 67(4):735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing RA, et al. 2005. The Oryza Map Alignment Project: the golden path to unlocking the genetic potential of wild rice species. Plant Mol Biol. 59(1):53–62. [DOI] [PubMed] [Google Scholar]
- Yuan Y-W, Wessler SR.. 2011. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc Natl Acad Sci U S A. 108(19):7884–7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang L-L, Zou X-H, Zhang F-M, Yang Z, Ge S.. 2011. Phylogeny and species delimitation of the C-genome diploid species in Oryza. J Syst Evol. 49(5):386–395. [Google Scholar]
- Zhang W, et al. 2014. Small brown planthopper resistance loci in wild rice (Oryza officinalis). Mol Genet Genomics. 289(3):373–382. [DOI] [PubMed] [Google Scholar]
- Zou X-H, et al. 2015. Multiple origins of BBCC allopolyploid species in the rice genus (Oryza). Sci Rep. 5(1):14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwickl DJ. 2006. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence data sets under the maximum-likelihood criterion [Ph.D thesis]. 2006; The University of Texas at Austin. Available from: https://repositories.lib.utexas.edu/handle/2152/2666 (accessed April 11, 2019).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.