

Abstract
CpG oligodeoxynucleotides are potent toll-like receptor (TLR) 9 agonists and have shown promise as anticancer agents in preclinical studies and clinical trials. Binding of CpG to TLR9 initiates a cascade of innate and adaptive immune responses, beginning with activation of dendritic cells and resulting in a range of secondary effects that include the secretion of pro-inflammatory cytokines, activation of natural killer cells, and expansion of T cell populations. Recent literature suggests that local delivery of CpG in tumors results in superior antitumor effects as compared to systemic delivery. In this study, we utilized PRINT (particle replication in nonwetting templates) nanoparticles as a vehicle to deliver CpG into murine lungs through orotracheal instillations. In two murine orthotopic metastasis models of non-small-cell lung cancer–344SQ (lung adenocarcinoma) and KAL-LN2E1 (lung squamous carcinoma), local delivery of PRINT-CpG into the lungs effectively promoted substantial tumor regression and also limited systemic toxicities associated with soluble CpG. Furthermore, cured mice were completely resistant to tumor rechallenge. Additionally, nanodelivery showed extended retention of CpG within the lungs as well as prolonged elevation of antitumor cytokines in the lungs, but no elevated levels of proinflammatory cytokines in the serum. These results demonstrate that PRINT-CpG is a potent nanoplatform for local treatment of lung cancer that has collateral therapeutic effects on systemic disease and an encouraging toxicity profile and may have the potential to treat lung metastasis of other cancer types.
Keywords: nanoparticles, PRINT, CpG, immunotherapy, pulmonary delivery, lung cancer
Graphical Abstract
Lung cancer is the second most common cancer and in the US is the leader in cancer-related deaths. Nearly 85% of all lung cancer cases are non-small-cell lung cancer (NSCLC), of which the most common histologic subtypes are squamous cell carcinoma and adenocarcinoma. Advanced NSCLC is usually an incurable disease and is also the leading cause of cancer-related deaths globally. An emerging strategy to treat advanced stage NSCLC is immunotherapy, which aims to reactivate the adaptive immune system against cancer.1 In 2015, the FDA approved two immunotherapy drugs, nivolumab and pembrolizumab, immune check-point inhibitors targeting programmed cell death-1 (PD-1), for the treatment of lung cancer. These two approvals were landmark, paradigm-shifting events for the treatment of lung cancer. However, based on clinical evidence, only 23–28% of patients with advanced NSCLC have tumors that overexpress PD-L1, the natural ligand of PD-1, and only 15–45% of patients with PD-L1 expression respond to anti-PD-1/PD-L1 treatment.2 Since the majority of patients receiving these treatments still do not achieve long-term remission, there is a revived interest in furthering the development of cancer immunotherapies.
Dr. William Coley, known as the “father of immunotherapy”, was the first to observe tumor regression in patients treated with intratumoral injections of bacterial lysates. It was later discovered that the unmethylated cytosine-phosphate-guanine (CpG) dinucleotide motifs present in bacterial DNA were responsible for the anticancer immune activation and that the Toll-like receptor 9 (TLR9) was responsible for sensing the CpG motifs.3 TLR9 is located within the endosome of human B cells, plasmacytoid dendritic cells (pDCs), monocytes, monocyte-derived macrophages, alveolar macrophages, neutrophils, and natural killer (NK) cells.4–8 Class B CpG (CpG-B) is a potent activator of pDCs and B cells and has been widely explored for anticancer treatments. Triggering the CpG-TLR9 pathway initiates a cascade of innate and adaptive immune responses. This begins with activation of pDCs to secrete type 1 interferons (IFNs) and other inflammatory cytokines (IL-6, IL-12, etc.).9 This then initiates a range of secondary effects including the activation of NK cells and the expansion of T cell populations.9–11
There are currently three TLR9 agonists under clinical investigation for the treatment of cancer: SD-101 (Dynavax), tilsotolimod (IMO-2125, Idera), and lefitolimod (MGN1703, Mologen AG). Intratumoral injections of SD101 in combination with pembrolizumab (anti-PD-1) resulted in broad immune activation in the tumor microenvironment, resulting in an overall response rate of 78%, and an overall survival rate of 89% for patients with advanced melanoma.12 Intratumoral injections of tilsotolimod (IMO-2125) in combination with ipilimumab (an anti-CTLA4 antibody) resulted in longer-lasting responses in patients with melanoma refractory to anti-PD1 therapy.13 In both studies, all patients receiving the TLR9 agonist treatment experienced mild adverse events consisting of chills, myalgia, and injection site pain.12,13 Subcutaneous delivery of lefitolimod, as opposed to direct tumor delivery, was investigated for the treatment of small-cell lung cancer; however, it did not result in any significant improvements in progression-free or overall survival.14 Together, these studies suggest TLR9 may have therapeutic promise, and use of a local tumor delivery route may be most efficacious.
While the antitumor effects of TLR9 agonists have been optimized by direct injections into the tumor, this is a less feasible option for tumors residing in the lungs. Recent studies in mice have shown varying efficacy of a CpG monotherapy delivered into murine lungs to treat various lung tumors.15–18 However, lung delivery of soluble CpG has been documented to cause lung injury as well as a systemic inflammatory response, resulting in liver damage.19,20 Appropriate formulation of CpG may reduce its local and systemic toxicity. Sato et al. demonstrated that lung delivery of CpG adsorbed onto polyketal microparticles resulted in CpG retention at the tumor site and the promotion of tumor regression; however toxicity related to this delivery system was not investigated, nor was the microparticle–CpG formulation directly compared with soluble CpG for antitumor activity.16
Previous work from our group demonstrated that polyethylene glycol (PEG)-based PRINT (particle replication in nonwetting templates) particles delivered to the lungs via instillations remained within the lungs for 7 days without causing lung inflammation.21 In comparing PRINT particle sizes it was observed that treatment with 80 × 320 nm nanoparticles resulted in higher percentages of macrophages and dendritic cells that were positive for particle uptake as compared to larger sized PRINT particles.22 Since CpG-induced immune stimulation is dependent on TLR9 interacting with CpG intracellularly, we hypothesized that utilizing the 80 × 320 nm nanoparticle (NP) would provide an ideal platform to facilitate intracellular delivery of CpG and enhance CpG efficacy, which may result in a safer and more effective anticancer immunotherapy To test this hypothesis, CpG was conjugated to 80 × 320 nm PRINT NPs (PRINT-CpG) through a stable thioether bond and delivered via the pulmonary route for the treatment of murine models of metastatic NSCLC,.
RESULTS/DISCUSSION
Fabrication and Characterization of PRINT-CpG.
PRINT hydrogel particles (80 nm × 80 nm × 320 nm) composed of hydroxyl-terminated PEG248 acrylate, amino ethyl methacrylate, and PEG700 diacrylate were fabricated. The amine handles on the NPs were used to conjugate thiolmodified CpG to the particles through a heterobifunctional cross-linker, resulting in a “stable” thioether linkage. CpG loading was consistently maintained at 40 μg of CpG per mg of NP across all batches evaluated in animals.23 Following CpG conjugation, particles became negatively charged and remained fairly monodisperse (Figure 1A and B). CpG release from NPs was evaluated in Gamble’s solution, a surrogate for the interstitial fluid of the deep lung, and artificial lysosomal fluid (ALF) to represent the acidic intracellular environment.24 We observed less than 5% release in ALF in 168 h and time-dependent release in Gamble’s, with approximately 20% release in 168 h (Figure 1C), indicating the covalent attachment of CpG to PRINT NPs results in a stable PRINT-CpG construct that allows slow and sustained release of CpG. We measured nanoparticle dispersion stability of the PRINT-CpG construct in both Gamble’s and ALF over a 24 h time period (Figure 1D). We observed a slight increase in particle size and polydispersity index (PdI) and decrease in zeta potential (ZP) for PRINT-CpG incubated in Gamble’s (pH 7.4) as compared to particles stored in water; however this was maintained throughout all time points. Particles incubated in ALF (pH 5) showed a marked increase in size (Z-avg) and PdI and a more neutral ZP, indicative of particle aggregation. Interestingly the particle aggregation in ALF did not worsen over time.
Figure 1.
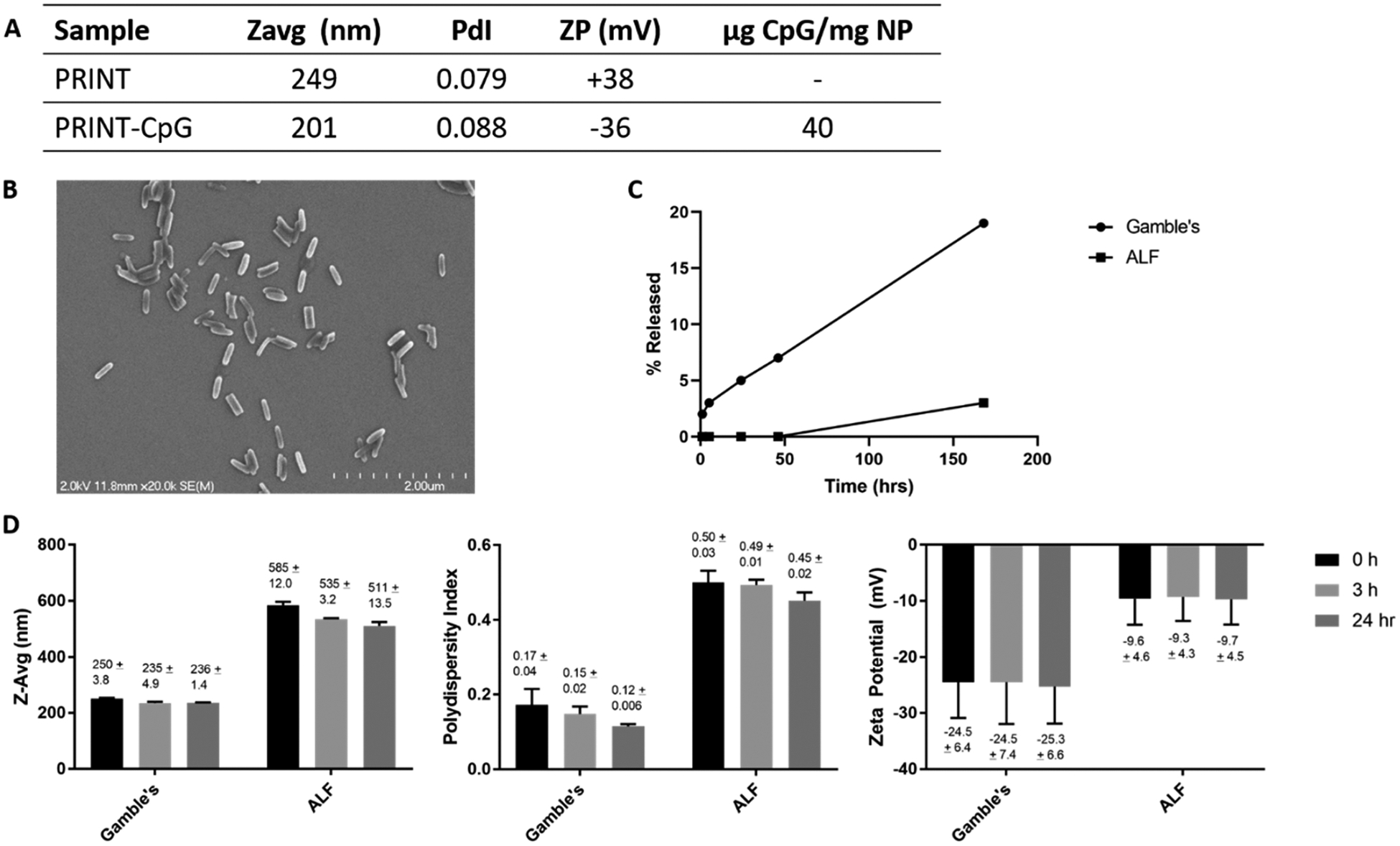
PRINT-CpG characterization. Dynamic light scattering (DLS) evaluation of blank PRINT nanoparticles (PRINT) and CpG-loaded PRINT nanoparticles (PRINT-CpG), as well as CpG loading determined via nanodrop (A). Scanning electron micrograph of PRINT-CpG nanoparticles (B). CpG release profile in Gamble’s solution (pH 7.4) and artificial lysosomal fluid (ALF) (pH 5) (C) and PRINT-CpG stability in Gamble’s and ALF over time, as measured by DLS (D).
Due to the sustained release of CpG, particle dispersion stability in artificial lung insterstial fluid mentioned above, and the previously reported extended persistence of PRINT particles in the lungs,21 we believed that utilizing PRINT NPs to deliver CpG to the lungs would result in a CpG depot in the lungs. In an effort to observe this effect, mice were inoculated with 344SQ cells in the left lung on day 0 and received a single dose (instillation) of either soluble Cy3-labeled CpG (green) or DyLight 680-labeled NPs (red) linked to Cy3-labeled CpG (green) at a dose of 20 μg of CpG per mouse on day 3. Lung pathology was performed on 344SQ tumor bearing mice either 24 or 48 h post therapeutic treatment (on day 4 and 5, respectively). Similar to previous reports of soluble CpG delivered to the lungs,16 we observed minimal soluble CpG signal in the lungs of mice sacrificed 24 h post-treatment, and no signal in mice sacrificed 48 h post-treatment, signifying that the soluble CpG was cleared quickly from the lungs (Figure 2A). Conversely, we observed strong punctate fluorescent CpG signal in the lungs of mice at both the 24 and 48 h time points after PRINT-CpG treatment (Figure 2A). Further, the CpG signal detected remained associated with the PRINT NPs up to 48 h post-treatment (Figure 2B). In an effort to evaluate where the CpG was being cleared to, a biodistribution study was conducted in which blood and major organs including spleen, kidneys, liver, and lungs were harvested at 3, 24, or 72 h following instillations. All tissues were imaged on the IVIS lumina and analyzed for CpG fluorescence; however there was no detectable fluorescence in the blood, spleen, kidneys, or liver (data not shown). The only CpG signal was detected in the lungs (Supplemental Figure 1). Similar to what we observed in the lung pathology, there was a decrease in fluorescence over time in the lungs of mice treated with soluble CpG, but extended fluorescent signal in the lungs for mice treated with PRINT-CpG (Supplemental Figure 1).
Figure 2.
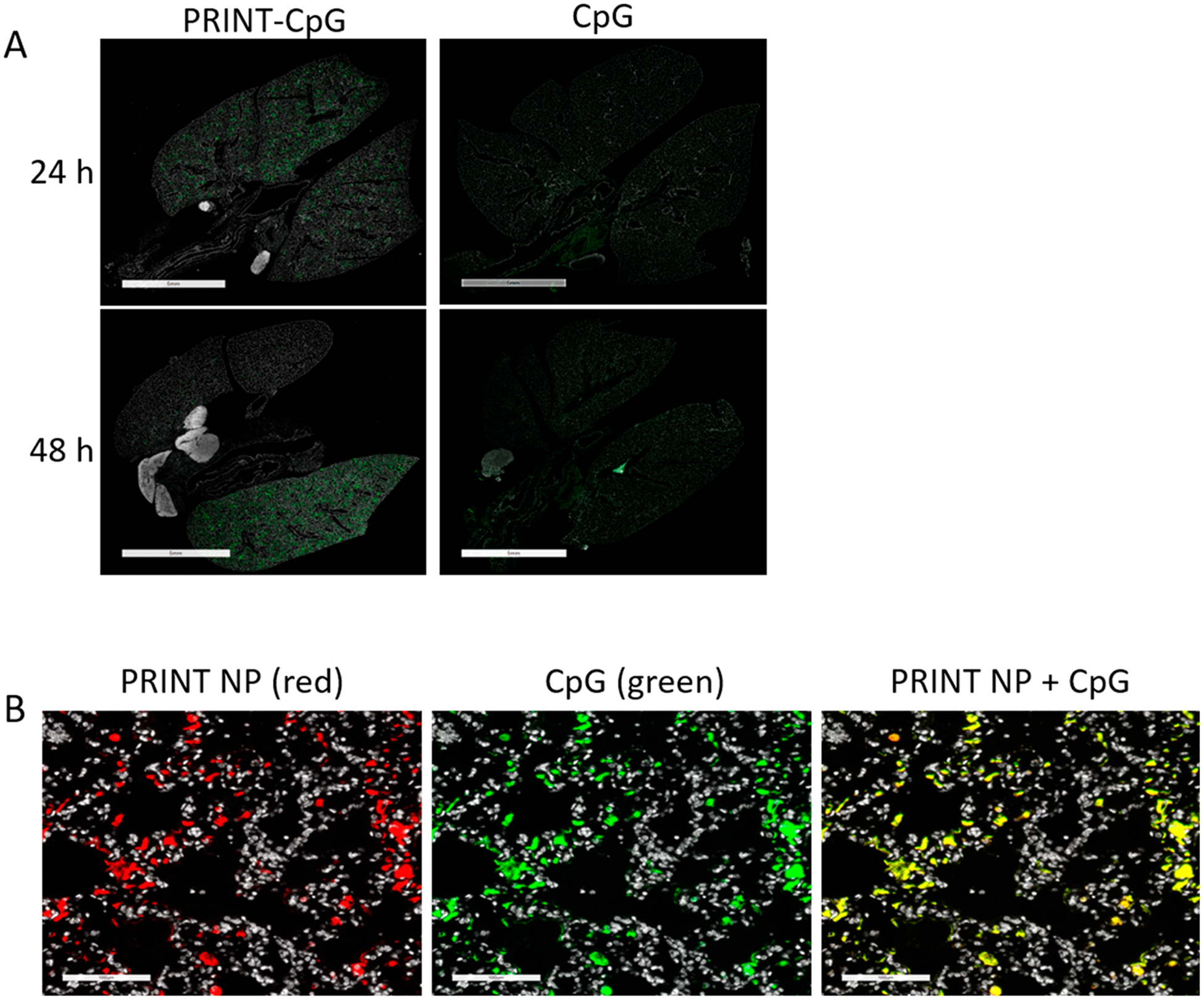
Lung distribution of instilled PRINT-CpG and CpG. Representative immunofluorescence images of lungs 24 and 48 h after instillations of PRINT-CpG or CpG; CpG was labeled with Cy3 (green) (A), scale bars represent 5 mm. Zoomed-in immunofluorescence images of PRINT-CpG in the lungs 48 h postinstillation; PRINT NPs were labeled with Dylight 680 (red), CpG was labeled with Cy3 (green), the overlay is yellow, scale bars represent 100 μm (B).
Biological Activity of PRINT-CpG.
Having confirmed that utilizing PRINT nanoparticles to deliver CpG to the lungs results in extended retention of CpG in murine lungs, we then investigated how tethering CpG to a nanoparticle affected its ability to stimulate DCs ex vivo. For this experiment CpG or PRINT-CpG was incubated in either serum or ALF for 0, 3, or 24 h and then incubated with DCs, with lipopolysacceride (LPS) serving as a positive control. Supernatants were then evaluated for the production of proinflammatory cytokines IL-6 and IL-12p40. Extending incubation of soluble CpG in serum or ALF did not result in any changes in DC proinflammatory cytokine production (Figure 3A). However, we observed limited production of proinflammatory cytokines for DCs incubated with PRINT-CpG treated with either serum or ALF for 0 h or 3 h, and elevated levels of proinflammatory cytokines after the PRINT-CpG had been incubated for 24 h in either serum or ALF (Figure 3A).
Figure 3.
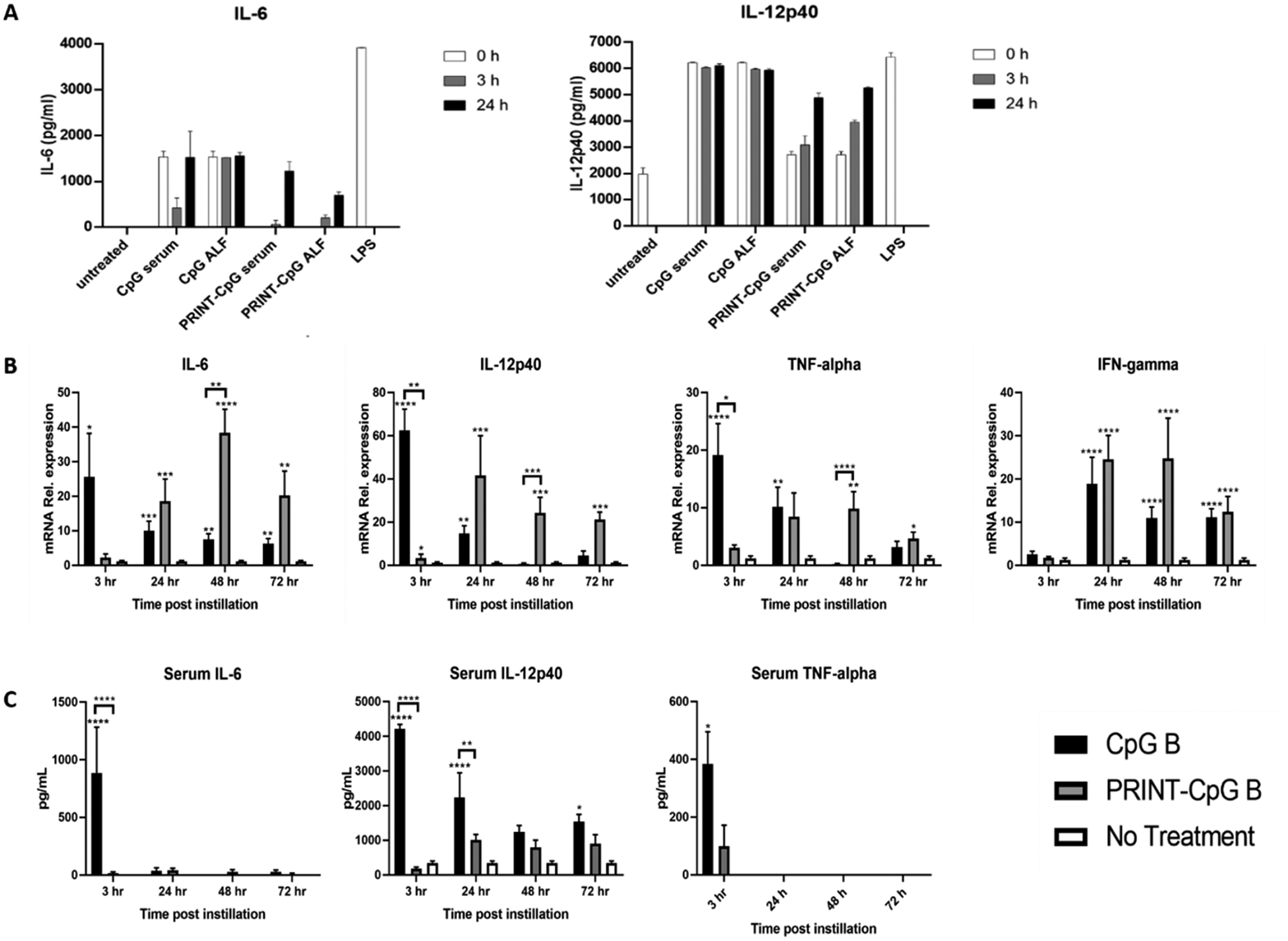
Biological function of PRINT-CpG. ELISA evaluation of proinflammatory cytokine production for PRINT-CpG or CpG incubated in ALF or serum for various time points and then incubated with BMDCs (A). qRT-pCR for mRNA relative expression of cytokines in mouse lungs homogenized at various time points after either receiving no treatment, soluble CpG, or PRINT-CpG (B); data are normalized to Gapdh mRNA and graphed as fold change over nontreated lungs. Statistics performed by 2-way ANOVA with Tukey’s multiple comparisons test of dCT values, *p < 0.05, **p < 0.001, ***p < 0.0005, and ****p < 0.0001. ELISA evaluation of serum cytokines at various time points following a single 20 μg dose of CpG or PRINT-CpG; statistics performed by 2-way ANOVA with Tukey’s multiple comparisons test, *p < 0.05, **p < 0.001, and ****p < 0.0001.
As in vitro results rarely translate in vivo, a time course study was conducted to evaluate PRINT-CpG’s ability to stimulate the production of proinflammatory cytokines in vivo. Mice were dosed with a single instillation of CpG or PRINT-CpG (20 μg dose), and lungs and serum were harvested and evaluated at 3, 24, 48, and 72 h for expression of proinflammatory cytokines IL-6, IL-12p40, and TNFα (Figure 3B and C). Three hours following instillations with soluble CpG, we observed a spike in cytokines in both the lungs and serum as compared to both untreated mice and mice treated with PRINT-CpG, which then diminished over time. Treatment with PRINT-CpG resulted in a delayed yet prolonged expression of these cytokines as compared to both CpG treated and untreated mice (Figure 3B). Interestingly, while treatment with PRINT-CpG did result in elevated levels of proinflammatory cytokines in the lungs, it did not result in elevated proinflammatory cytokines in the serum (Figure 3C).
Therapeutic Efficacy of PRINT-CpG in Orthotopic Murine Lung Cancer Models.
Studies in mice have demonstrated that local tumor delivery of CpG can coordinate the activation of both innate and adaptive immune responses and increase systemic antitumor immunity.16,17,25–27 To demonstrate anticancer efficacy of pulmonary-delivered PRINT-CpG, two orthotopic syngeneic models of NSCLC that are metastatic to the local lymph nodes and distant sites were evaluated utilizing the 344SQ lung adenocarcinoma and the KAL-LN2E1 lung squamous carcinoma cell28 lines in 129S6 and FVB mice, respectively. In each study, mice were inoculated with cancer cells in the left lung on day 0 and began receiving therapeutic CpG treatments (pulmonary instillations) on day 3.
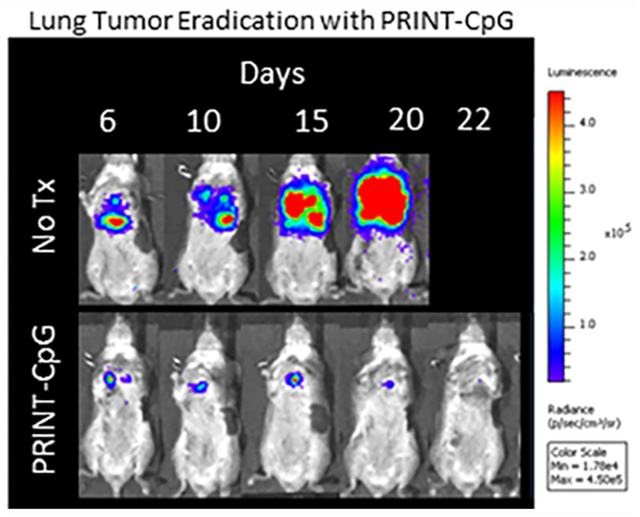
In the 344SQ model mice were treated with 20 μg of CpG every 3 days for a total of 4 doses (treatment ending on day 12). The 344SQ cell line expresses luciferase, allowing disease progression to be monitored via bioluminescence live animal imaging. Figure 4A displays luminescence images of tumor burden over time as a function of treatment. We observed a steady increase in tumor burden in untreated (No Tx) animals, whereas mice receiving CpG or PRINT-CpG continued to show a reduction in tumor burden (Figure 4A). However, only treatment with PRINT-CpG resulted in 100% survival of mice with the otherwise fatal 344SQ lung cancer (Figure 4B). Toxicity was observed following treatment with soluble CpG, as 3 out of 7 mice were euthanized due to weight loss reaching 20%, but not tumor burden (Figure 4C). We also observed a significant decrease in body weight in mice following the first treatment with PRINT-CpG; however this effect was transient, as mice did not continue to lose weight following the second dose and began to regain weight following the third dose, whereas mice receiving soluble CpG continued to lose weight following each dose (Figure 4C). We believe the difference in the toxicity profiles between soluble CpG and PRINT-CpG is the immediate spike in proinflammatory cytokines in both the lungs and serum following treatment with soluble CpG, which was not observed with PRINT-CpG (Figure 3B). It is important to note that pulmonary delivery of either CpG or PRINT-CpG is not targeted to the tumors, and while elevated levels of proinflammatory cytokines can provide antitumor effects, they can also lead to toxicity in healthy tissues. Interestingly through delivery of CpG with PRINT NPs, we are able to mitigate the toxicity of soluble CpG, while still modulating the entire lung microenvironment to be antitumor. To investigate whether PRINT-CpG-mediated tumor regression helps to induce a long-term protection against tumors, 3 months after cessation of PRINT-CpG therapy, surviving mice were rechallenged with a reinjection of 344SQ cells. Mice previously treated with PRINT-CpG remained tumor free (Figure 4D), with 100% survival (Figure 4E), indicating that a long-lasting antitumor response can be induced by lung delivery of PRINT-CpG.
Figure 4.
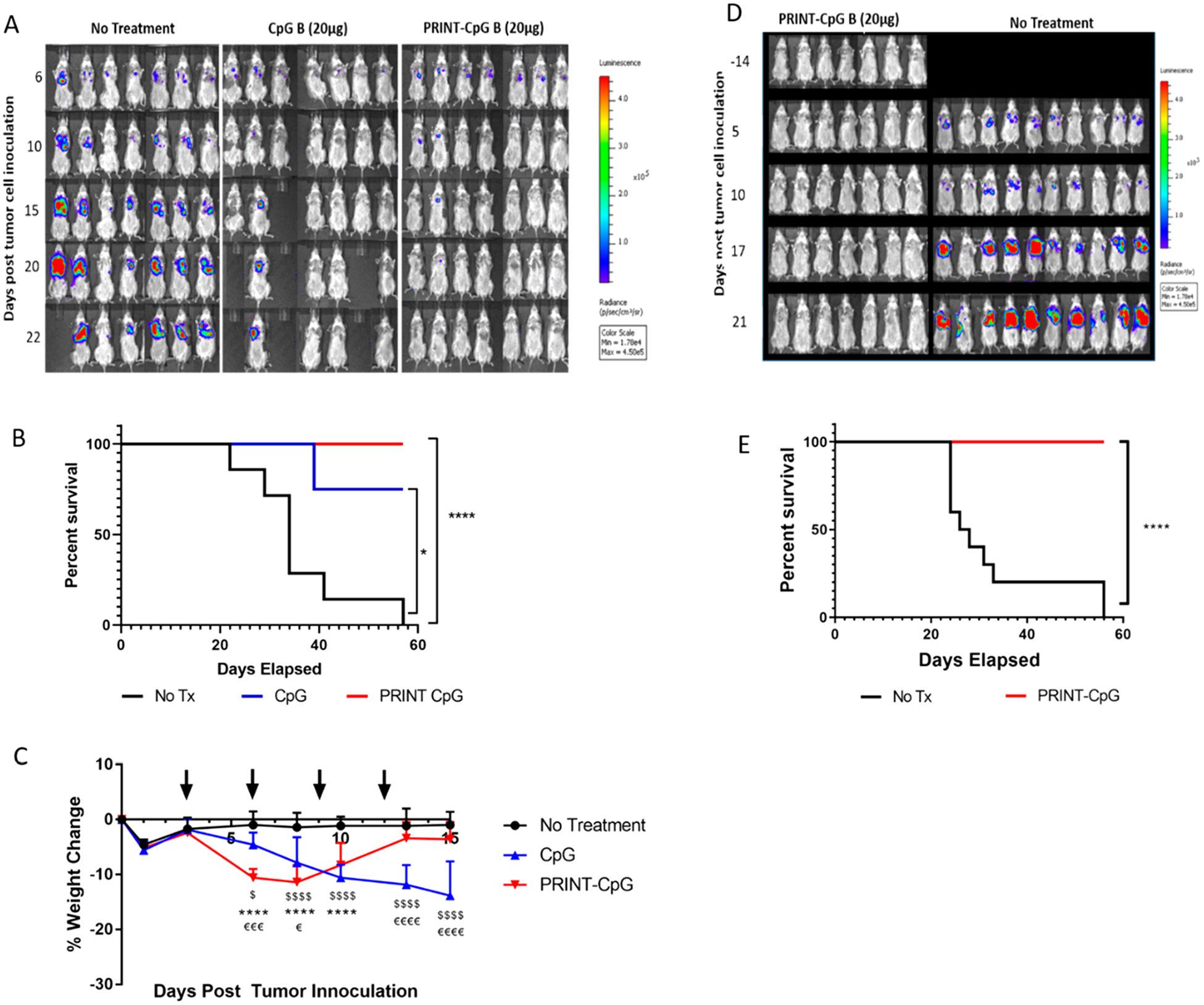
PRINT-CpG efficacy in the 344SQ model. Left hand panel: Tumor growth bioluminescence images as a function of treatment (untreated, CpG, PRINT-CpG) and days past tumor cell inoculation (A), survival data (B), and mouse body weight changes following therapeutic treatment (indicated by black arrows) (C) (n = 7 mice per treatment group). Statistics performed by 2-way ANOVA with multiple comparisons, $ = significant difference between no treatment and CpG (p < 0.05), $ = significant difference between no treatment and CpG (p < 0.0001), **** = significant difference between no treatment and PRINT-CpG (p < 0.0001), € = significant difference between CpG and PRINT-CpG (p < 0.05), €€€ = significant difference between CpG and PRINT-CpG (p < 0.0005), €€€€ = significant difference between CpG and PRINT-CpG (p < 0.0001). Right hand panel: Bioluminescence images of PRINT-CpG cured mice from (A) following tumor rechallenge, and naïve mice challenged with 344SQ tumors (D) and survival of cured mice from (A) and naïve mice following tumor cell rechallenge (E) (n = 7 for mice previously treated with PRINT-CpG, n = 10 for naïve No Tx group). Statistics performed by survival curves and log-rank (Mantel Cox) test with *p < 0.02, ****p < 0.0001.
Therapeutic efficacy was further evaluated in the 344SQ orthotopic model at a lower, 10 μg dose of CpG and with the addition of control groups such as PRINT-GpC and CpG A. GpC is a negative control for CpG, as it does not induce TLR9 activity, and GpC conjugated to PRINT NPs were evaluated to ensure that the antitumor effects were not due to the PRINT NPs. CpG A is known to form nanometer-sized multimers capable of inducing IFNα, and research suggests that conjugating CpG B to NPs produces an immune profile similar to CpG-A.29 We observed a steady increase in tumor burden (bioluminescence signal) in the control groups, untreated (No Tx), PRINT-GpC, and CpG A, whereas animals treated with either PRINT-CpG or soluble CpG resulted in extended control in tumor growth (Figure 5A) and extended survival (Figure 5B); however, treatment with PRINT-CpG resulted in significantly improved survival as compared to treatment with soluble CpG.
Figure 5.
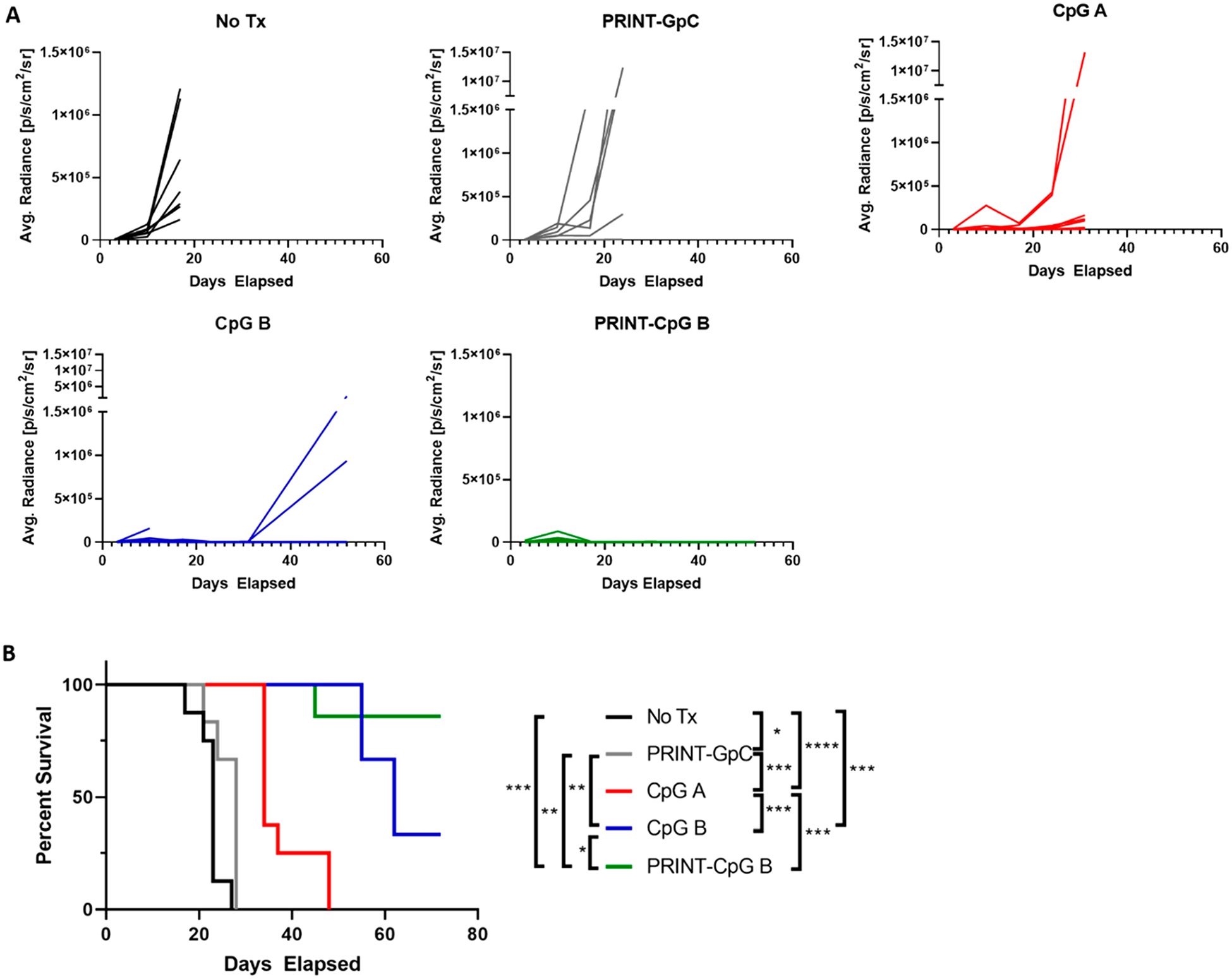
PRINT-CpG (10 μg) efficacy in the 344SQ model. Tumor growth plots as measured from bioluminescence images, for mice treated with CpG B, PRINT-CpG B, untreated (No Tx), PRINT-GpC, and CpG A (A), and survival data (B) (n = 7 mice per treatment group). Statistics performed by survival curves and log-rank (Mantel Cox) test with *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Since it has been well documented in the literature that CD8+ effector T cells and NK cells are related to direct tumor cytolysis, and memory CD8+ T cells are the major contributors to the long-term antitumor immunity,25,30,31 we depleted CD8+ and NK cells with anti-CD8 and anti-asialo GM1 antibodies, respectively, in a therapeutic study as outlined above. Surprisingly, an insignificant decrease in the survival of mice depleted of CD8+ T cells and NK cells was observed (Figure 6A), and only depletion of NK cells led to an increase in tumor burden, as visualized by bioluminescence data (Figure 6B). These results indicate that NK cells, and not CD8+ T cells, play only a partial role in the therapeutic efficacy of PRINT-CpG.
Figure 6.
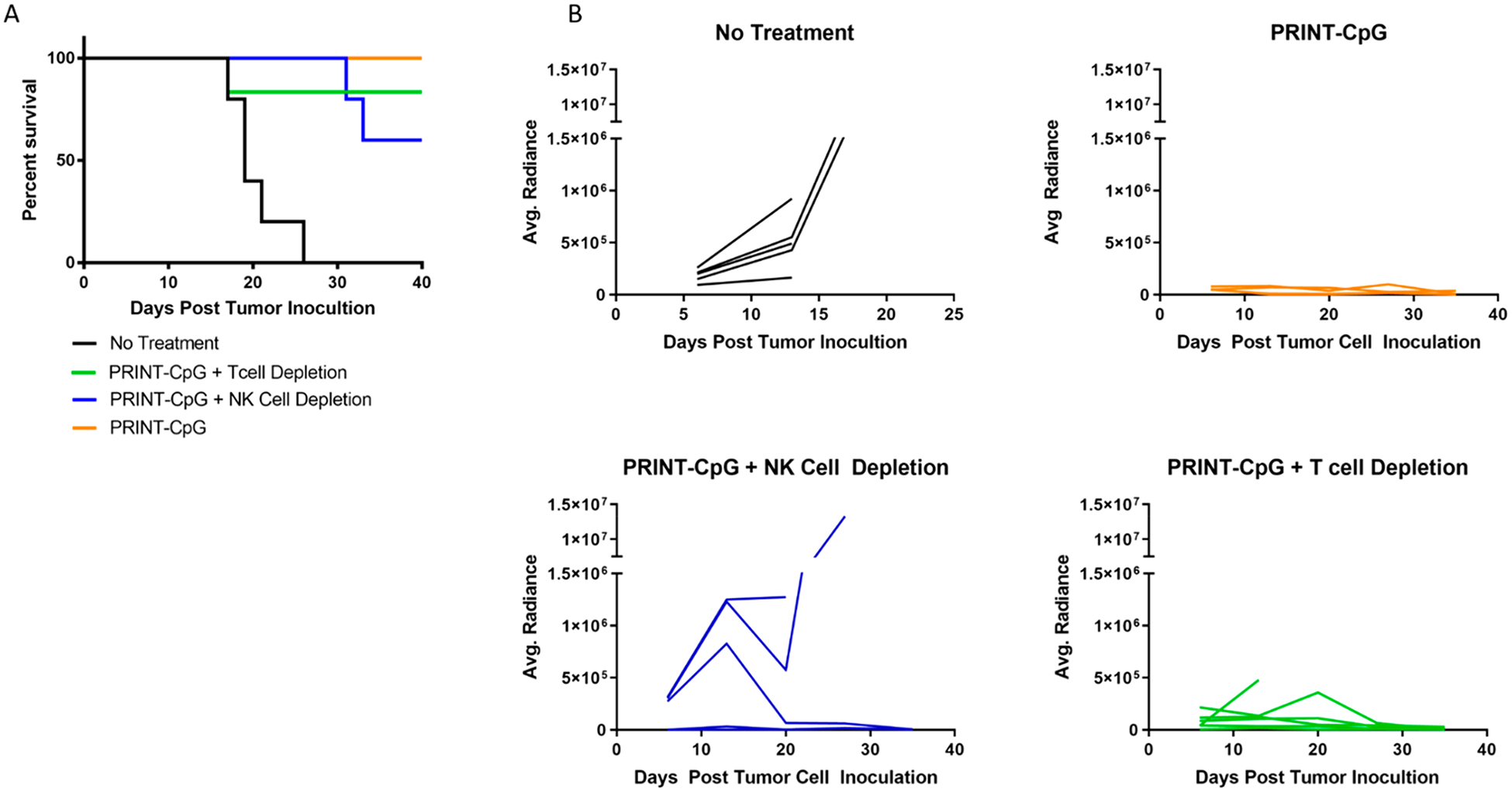
Therapeutic efficacy following depletion of T cells or NK cells. Mouse survival (A) and tumor growth plots as measured from bioluminescence images (B), for mice treated with PRINT-CpG (20 μg), untreated (No Tx), depleted of CD8 T cells and treated with PRINT-CpG (20 μg), and depleted of NK cells and treated with PRINT-CpG (20 μg) (n = 5 mice per treatment group).
The therapeutic efficacy of PRINT-CpG was further evaluated in a second murine orthotopic NSCLC model, KAL-LN2E1 in FVB mice. Due to the toxicity observed in 129S6 mice treated with soluble CpG at a dose of 20 μg, a maximum tolerated dose (MTD) study was completed in the FVB mouse strain prior to efficacy studies, in which mice were treated with instilled soluble CpG at varying dosages (20, 10, and 5 μg) every 3 days for 4 doses. Mouse body weight was monitored throughout treatment (Supplemental Figure 2) as an indicator of toxicity, and the MTD was determined to be 10 μg of CpG. Therefore, in a therapeutic experiment using the KAL-LN2E1 model, mice received 10 μg doses of CpG beginning on day 3 and continuing every 3 days for a total of 3 doses. Mouse weights all declined on day 9 regardless of treatment (Figure 7A), and thus a full necropsy was completed on day 10, at which time lungs were resected, and primary tumors in the left lung, LN metastases, and distant metastases (contralateral tumors in the right lung and chest wall metastases) were carefully annotated. As shown in Figure 7B–E, there was a significant reduction in the number of primary tumors for mice treated with PRINT-CpG, as well as a significant reduction in distant metastases in mice treated with either soluble CpG or PRINT-CpG, revealing that lung-delivered PRINT-CpG can have substantial antitumor effects on local, loco-regional, and distant metastatic sites.
Figure 7.
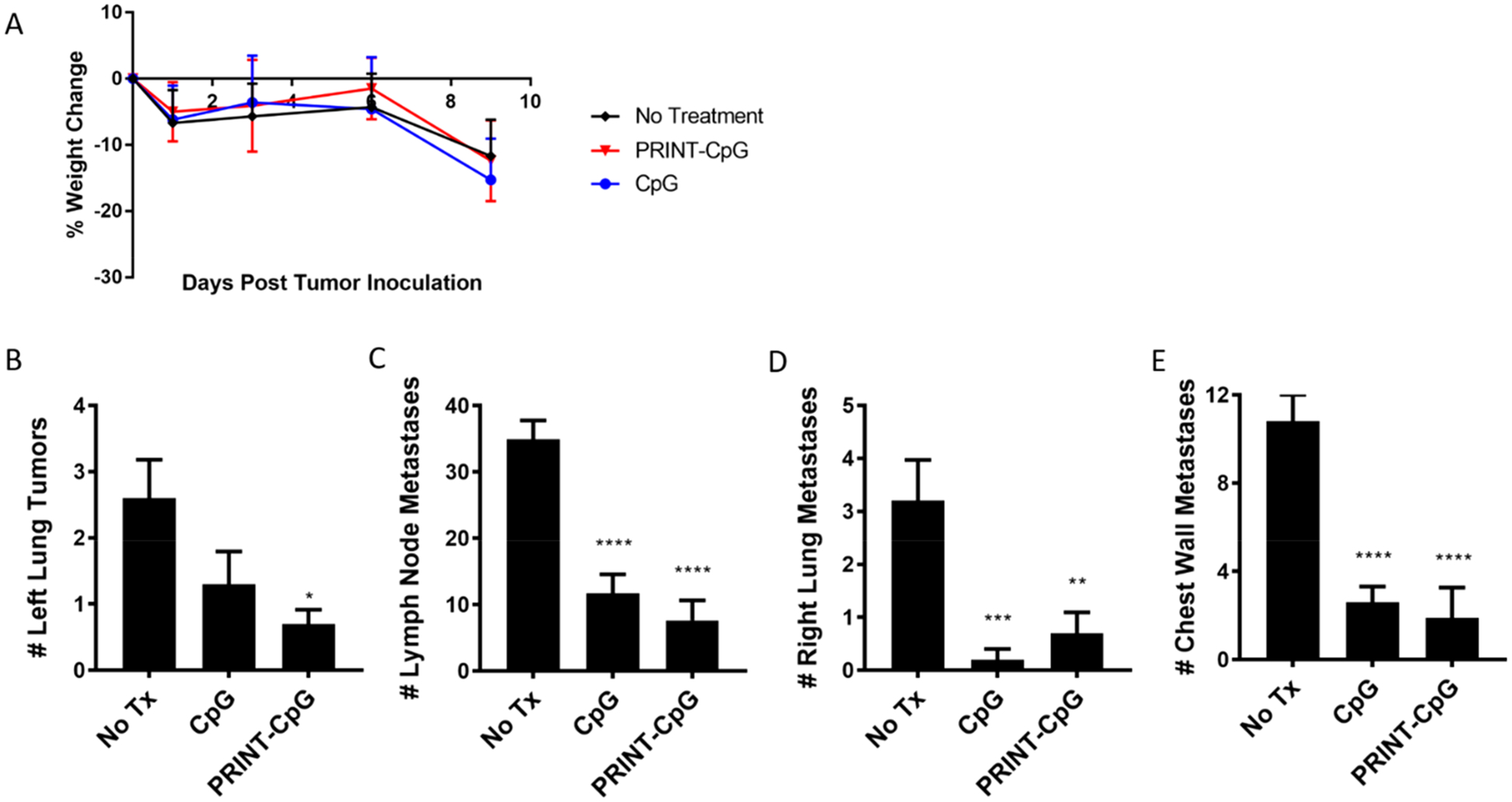
PRINT-CpG efficacy in the KAL-LN2E1 model. Tumor necropsy results from mice (n = 10 per group) inoculated with orthotopic KAL-LN2E1 tumors on day 0. Mice received no treatment, 10 μg of CpG, or PRINT-CpG on days 3, 6, and 9, and lungs/tumors were harvested on day 10. Change in mouse weights as a function of disease progression and treatment (A). Quantification of primary left lung tumors (B), lymph node tumors (C), contralateral right lung tumors (D), and chest wall tumors (E), as a function of therapeutic treatment. Statistics performed by 1-way ANOVA with Tukey’s multiple comparisons test with *p < 0.02, **p < 0.005, ***p < 0.001, ****p < 0.0001.
Instilled PRINT-CpG Leads to Enhanced Immune Cell Infiltration.
To understand the antitumor activity observed from soluble CpG and PRINT-CpG therapies, we evaluated changes in the infiltrating immune cell populations in the lungs and metastases following two treatments in the KAL-LN2E1 orthotopic lung cancer model. Of all cell types present in lung tumors, tumor-associated macrophages (TAMs) and DCs are the main targets of the PRINT-CpG immunotherapy design, as they are antigen-presenting cells (APCs) and the key mediators of the cellular immune response. The analysis was predominately focused on these immune subsets due in part to their APC function and their expression of TLR9. After activation by CpG, DCs and macrophages can trigger NK cell activation and can migrate to the mediastinal lymph nodes to interact and activate T cells.32 Representative gatings of flow cytometry histograms/dot plots are shown in Supplemental Figure 3; from this scheme DCs (CD11c+), TAMs (CD11b+ CD11c−), NK cells (TCRβ−GM1+), CD4+ T cells, and CD8+ T cells were identified. We observed elevated levels of DCs, NK cells, and CD4+ T cells in the right lungs of mice treated with either soluble CpG or PRINT-CpG, with no statistically significant changes in CD8+ T cells (Figure 8A). In the left lung (the site of primary tumor formation), we observed elevated levels of both CD4+ and CD8+ T cells for mice treated with either soluble CpG or PRINT-CpG (Figure 8B). Interestingly, treatment with soluble CpG therapy resulted in elevated levels of TAMs in the right lung and NK cells in the left lung (Figure 8A and B). There was no statistically significant differences in TAMs or DCs in the left lung nor any differences in cellularity in the metastases across all treatment groups (Figure 8B and Supplemental Figure 4). These results indicate that both soluble CpG and PRINT-CpG delivered to the lungs are capable of recruiting immune cells to the sites of disease.
Figure 8.
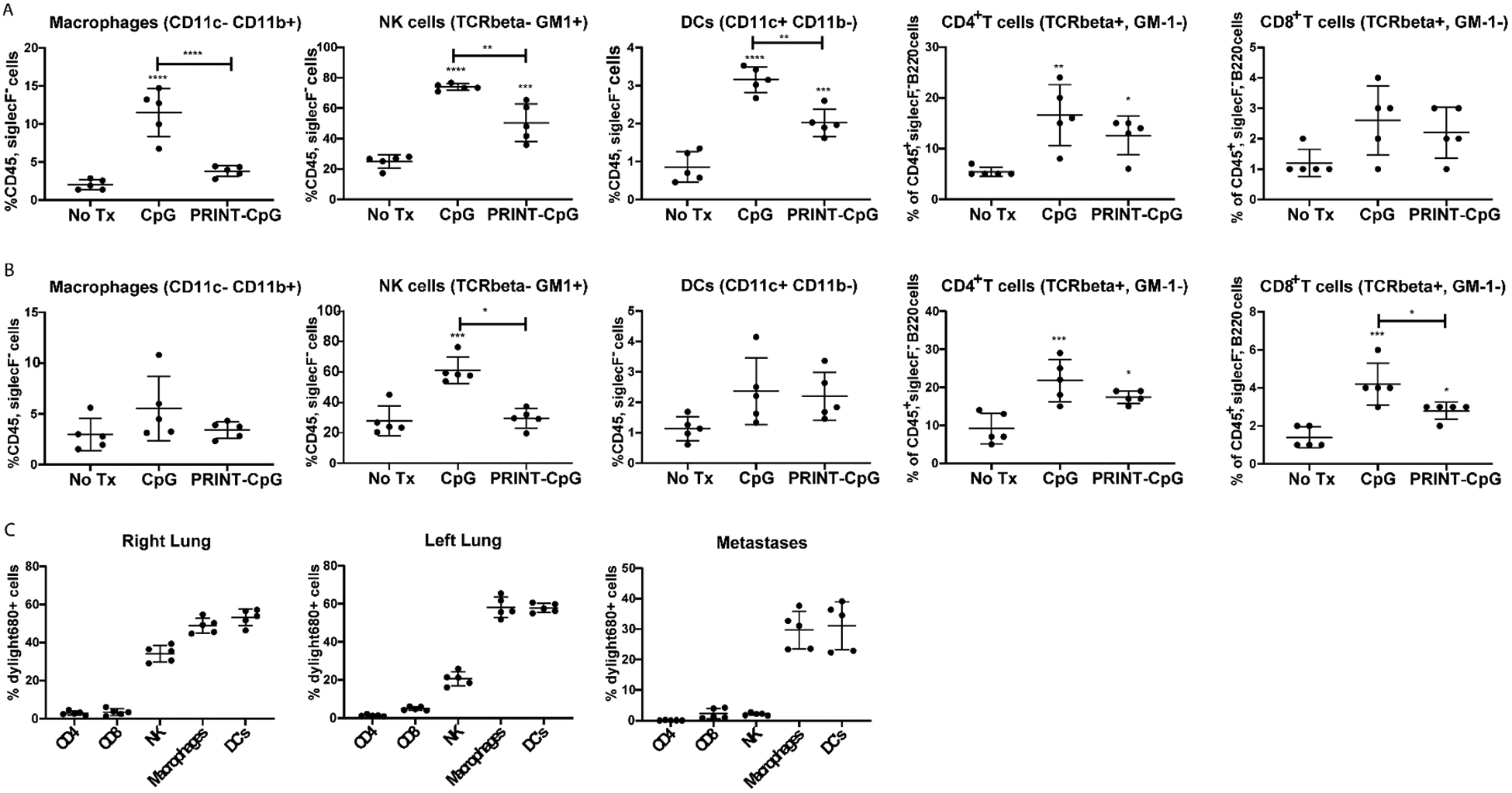
Flow cytometry quantification. Cellular makeup of the right lung (A) and left lung (containing primary tumor) (B), for mice receiving no treatment, soluble CpG, or PRINT-CpG. Statistics performed by 1-way ANOVA with Tukey’s multiple comparisons test with *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001. PRINT nanoparticle association of each cell type population was determined by flow cytometry for the right lung, left lung, and metastases (C).
We also evaluated the cells that took up DyLight 680-labeled PRINT-CpG NPs in the right lung, left lung (primary tumor included), and lymph node (LN) metastases. Results revealed that macrophages and DCs in the right and left lung readily internalized PRINT-CpG, as between 50% and 60% of these APCs were positive for NP fluorescence in both the right and left lungs, respectively (Figure 8C). Interestingly we observed that approximately 20–30% of NK cells were positive for PRINT-CpG in the left and right lungs, respectively (Figure 8C). While most literature reports indicate that activation of NK cells is a subsequent effect of the direct activation of DCs by CpG, we observed NK cells positive in NP signal in the lungs, indicating that PRINT-CpG is taken up by NK cells and can therefore directly activate NK cells that express TLR9. Furthermore, we observed that approximately 30% of macrophages and DCs in the LN metastases were positive for PRINT-CpG (Figure 8C). This observation is consistent with previous work done by our group in which NPs delivered to the lungs via instillation were trafficked to the draining lymph node within 24 h.33 Not surprisingly, we observed minimal cellular association of PRINT-CpG with CD4+ and CD8+ T cells in the lungs and metastases (Figure 8C). It is well documented that murine macrophages, DCs, and NK cells all express TLR9 and play a pivotal role in generating an anticancer response;6,34 therefore these results reveal that PRINT-CpG is being taken up by our target cells.
Instillation of PRINT-CpG Induced Increases in Anticancer Proinflammatory Cytokines and Chemokines.
The influx of immune cells into the lungs following therapeutic treatment with either CpG or PRINT-CpG suggested that this treatment may induce the production of lymphocyte-attracting chemokines within the lungs. To explore this hypothesis, the right lungs of KAL-LN2E1 tumor bearing mice were harvested on day 7, 24 h following the second dose of instilled CpG or PRINT-CpG, and the levels of cytokines and chemokines present were analyzed via qRT-PCR. As shown in Figure 9, instillations of either CpG or PRINT-CpG dramatically increased the levels of TNFα, IFNγ, and Fpr2 (formyl-peptide receptor 2) in the right lung, with soluble CpG treatment resulting in higher levels of TNFα than PRINT-CpG. However, only treatment with PRINT-CpG resulted in significantly increased levels of IL-12p40 and CXCL10 (chemokine interferon-γ inducible protein 10 kDa) versus untreated mice. Of these proinflammatory cytokines, IFNγ and TNFα have been shown in a number of other studies to directly trigger tumor cell death and perform powerful anticancer effects.35 CXCL10 is a chemoattractant for macrophages, T cells, NK cells, and DCs and is secreted by several cell types in response to IFNγ.36 The elevated levels of CXCL10 corresponds well with the observed influx of immune cells in the lungs following PRINT-CpG treatment. Fpr2 is expressed in several tissues and cell types, and its expression has been found to be lower in lung cancer tissues.37 The presence of Fpr2 has been documented to result in delayed tumor growth by limiting M2 polarization of macrophages.38 Since we observed elevated levels of Fpr2 in both the CpG and PRINT-CpG treatment groups, we then evaluated their ability to skew macrophage polarization in vitro. For these studies bone-marrow-derived macrophages were treated with IL-4 to induce an M2 phenotype or treated with IFNγ and LPS to an M1 phenotype. The M2 macrophages were then incubated with CpG or PRINT-CpG. Twenty-four hours later, cells or cell supernatants were tested for RNA or cytokines that are indicative of M2 (Fizz1, Ym1, and Mrc1) and M1 (IL-6, TNFα) polarization. Interestingly we found that PRINT-CpG, but not soluble CpG, was able to efficiently skew macrophage polarization from M2 to M1, as treatment with PRINT-CpG resulted in a reduction of M2 biomarkers and an increase in M1 cytokines (Figure 10). Taken together, these findings revealed that lung treatment with both soluble CpG and PRINT-CpG are capable of eliciting a potent anticancer, proinflammatory cytokine response in the lungs.
Figure 9.
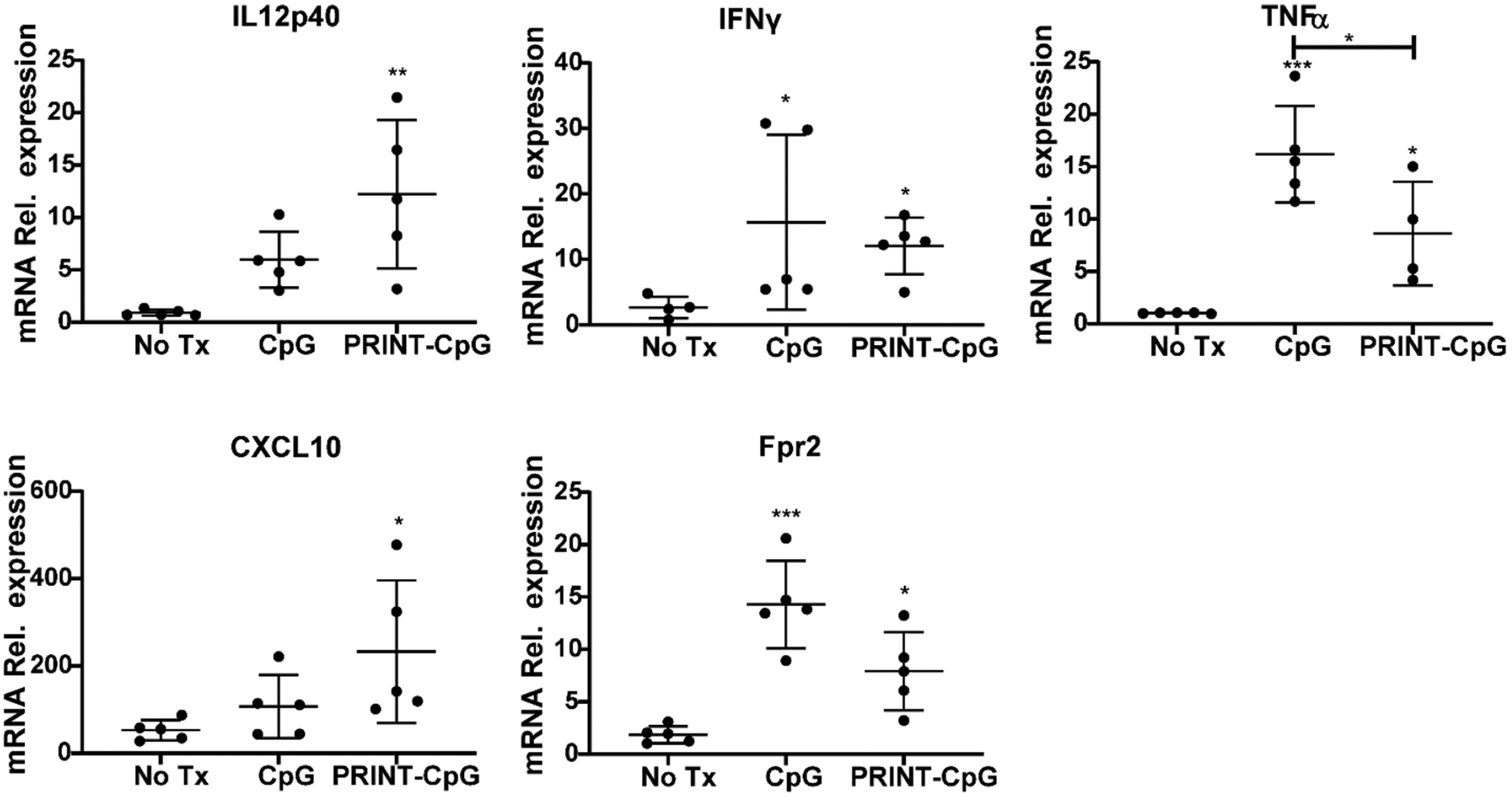
Evaluation of lung cytokines. qRT-pCR for mRNA relative expression of IL12p40, IFNγ, TNFα, Fpr2, CXCL10, and Fpr2 by mouse right lungs homogenized on day 7, 24 h after receiving either no treatment or 2 treatments of soluble CpG or PRINT-CpG. Data are normalized to Gapdh mRNA and graphed as fold change over non-tumor-bearing lungs. Statistics performed by 1-way ANOVA with Tukey’s multiple comparisons test, *p < 0.05, **p < 0.001, ***p < 0.0005.
Figure 10.
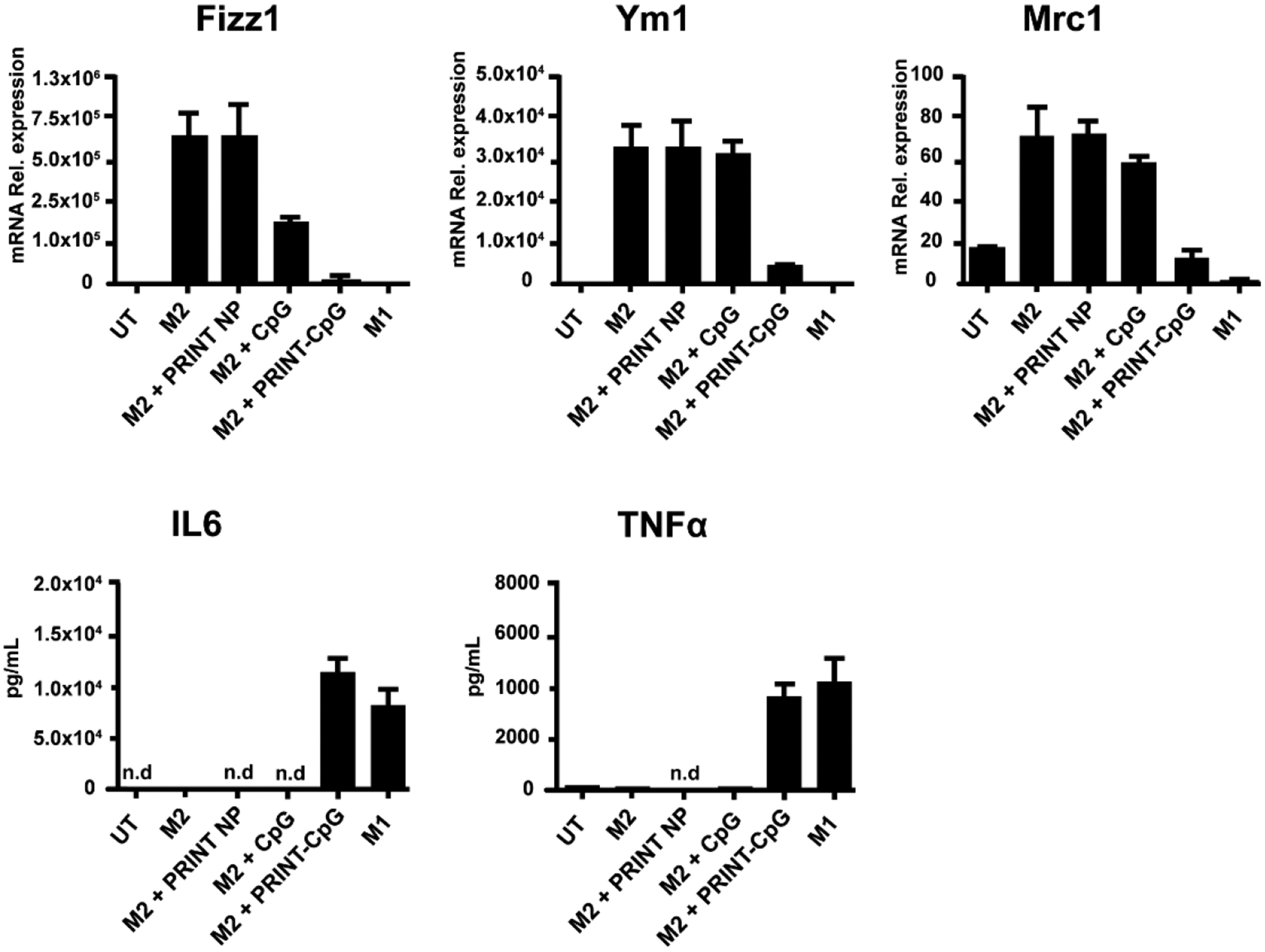
Macrophage repolarization with PRINT-CpG in vitro. PRINT NP conjugated CpG caused an M2 to M1 switch. Mouse bone marrow macrophages were cultured in IL-4 (25 ng/mL) to induce an M2 phenotype (M2(+)) or LPS (500 ng/mL)+IFNγ (25 ng/mL) to induce an M1 phenotype (M1(+)). M2 condition resulted in increased M2 biomarkers, Fizz1, Ym1, and Mrc-1. The addition of NP alone had little effect. Soluble CpG added to M2-inducing condition (M2+sol. CpG) caused a modest reduction in Fizz1 and Mrc-1, while NP-conjugated CpG (M2+NP-CpG) greatly reduced these biomarkers. Additionally, even under an M2-inducing condition, NP-CpG resulted in the induction of IL-6 and TNF, which are M1 cytokines.
Serum Cytokine and Clinical Chemistry Evaluations.
Previous reports have characterized the lung administration of CpG as causing a systemic inflammatory response, resulting in hypercytokinemia and liver damage.19 Therefore, peripheral blood analysis was included to provide a systemic view of the immune response induced on day 7, 24 h following 2 doses of CpG or PRINT-CpG (10 μg per dose). At the 10 μg dose, neither the soluble CpG nor the PRINT-CpG therapeutic lung instillations resulted in elevated levels of IL-6, IL-12, or TNFα in the blood (Figure 11A). Due to the previous claims of liver damage by systemic soluble CpG, clinical chemistry tests were conducted to evaluate liver and kidney function through measuring the levels of alanine transaminase (ALT), albumin (ALB), and creatinine (CREAT) in the serum. While we did not observe any significant changes in serum levels of ALB or CREAT in either of the therapeutic treatment groups, the serum levels of ALT were significantly higher for mice treated with soluble CpG (Figure 11B), indicative of liver damage.
Figure 11.
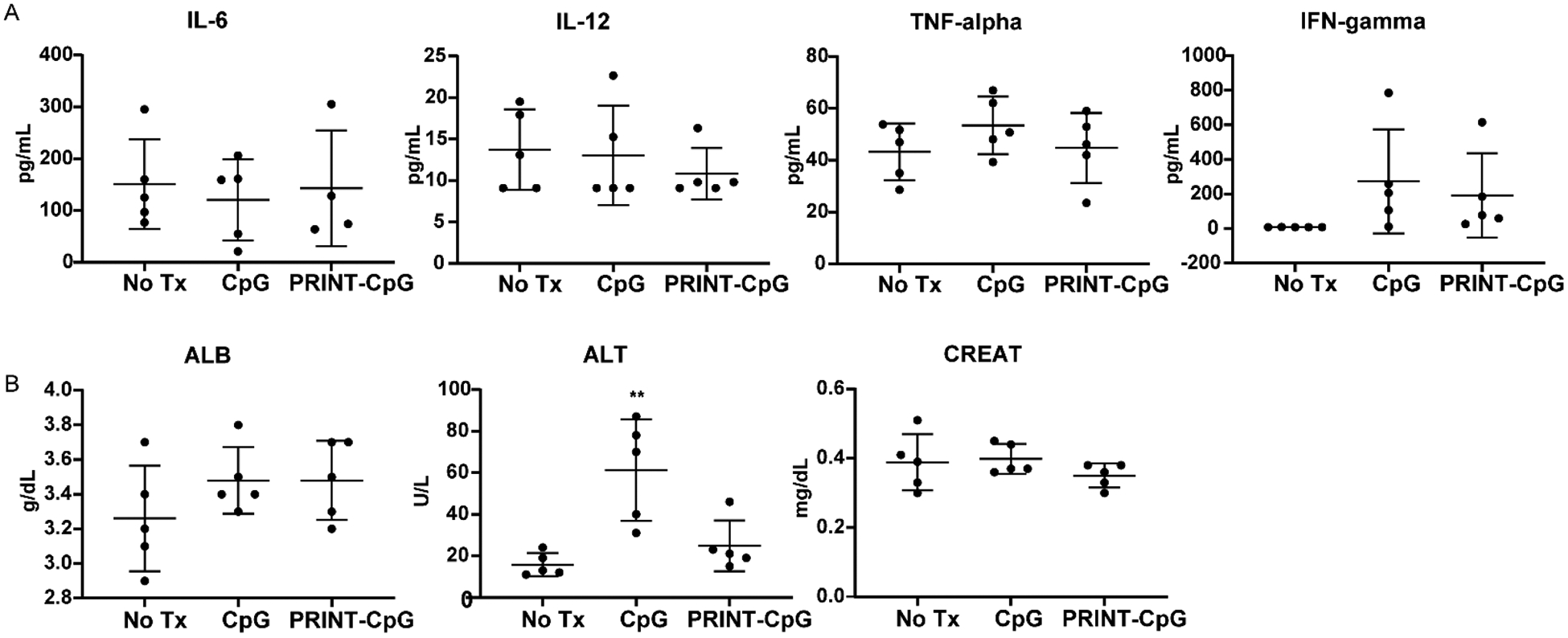
Serum cytokine and clinical chemistry analysis. Evaluation of serum cytokines (A) and serum clinical chemistry evaluation of albumin (ALB), alanine aminotransferase (ALT), and creatinine (CREAT) (B) 24 h post treatment. Statistics performed by 1-way ANOVA with Tukey’s multiple comparisons test, **p < 0.001.
TLR agonists are immunomodulators that are in the early stages of clinical development as anticancer agents. They have gained much attention for their ability to evoke innate immune responses and regulate adaptive immunity. CpG-B is a TLR9 agonist and has been shown by many investigators to activate the immune response and stimulate both local and systemic antitumor responses after intratumoral injections25,27,39–41 or via the pulmonary route for the treatment of murine models of lung cancers and lung metastases of other tumor types.15–17 In this current study, we assessed the efficacy of CpG-B immunotherapy against immune-competent, orthotopic metastasis models of lung cancer. Consistent with previous reports, we observed strong anticancer responses in mice treated with lung-delivered CpG. However, as has been reported elsewhere, we also observed weight loss, lung inflammation, and a systemic inflammatory response resulting in liver damage for mice treated with soluble CpG.19,20 Importantly, we found that covalently attaching the CpG to PRINT nanoparticles resulted in a antitumor potency and long-term memory (as demonstrated by the tumor rechallenge study) and perhaps more importantly led to markedly diminished toxicity associated with the lung delivery of soluble CpG. Our studies showed that PRINT-CpG was able to extend the residence time of CpG in the lungs for at least 2 days, while soluble CpG was cleared from the lungs within 24 h. Furthermore, we found that the PRINT-CpG immunotherapy targeted the innate immune cells in the lungs effectively and resulted in the production of sustained levels of anticancer cytokines in the lungs.
CpG stimulation of macrophages and DCs results in the release of soluble mediators (cytokines/chemokines) that can directly mediate an antitumor response and orchestrate the migration of immune cells to the affected tissues.42,43 Specifically, CpG treatment is documented to increase the expression of pro-inflammatory cytokines IL-6 and IL-12.44,45 IL-12 stimulation of lymphoid cells elicits the production of IFNγ and TNFα.46,47 IFN-γ stimulates myeloid cells, which induce the secretion of CXCL10 to attract immune cells as well as induce tumor cell apoptosis and necroptosis, while TNFα can induce hemorrhagic necrosis in tumors. Delivery of both CpG and PRINT-CpG to the lungs of mice resulted in the production of proinflammatory cytokines (IL-6, IL-12, IFNγ, and TNFα); however the production of IL-6, IL-12, and TNFα was delayed from 3 h post-treatment to 24 h post-treatment when CpG was delivered utilizing PRINT nano-particles.
As mentioned above, IL-12 plays a critical role in orchestrating the CpG-induced immune response against cancer.44 IL-12 is produced mainly by APCs, such as monocytes, macrophages, and dendritic cells. IL-12 has a stimulatory effect on NK cells; it also activates cytotoxic CD8+ T cells, differentiates CD4+ lymphocytes, and primes macrophages for nitric oxide production. Unfortunately, therapies that result in elevated serum levels of IL-12 can be problematic due to the potential to develop lethal inflammatory syndrome.48 In our studies, 3 h post-treatment we observed elevated levels of IL-12p40 in both the lungs and serum of mice treated with soluble CpG (20 μg), as compared to both untreated and PRINT-CpG-treated mice. Further, serum levels remained elevated out to 24 h post-treatment with soluble CpG, whereas serum levels for mice treated with PRINT-CpG were not statistically different from untreated mice across all time points investigated. Since we also observed toxicity-related deaths of mice treated with 20 μg of soluble CpG, we believe that the elevated serum levels of IL-12p40 were the main contributor, as when the dose of CpG was reduced to 10 μg, elevated levels of IL-12p40 were no longer detected in the serum and mice survived the full treatment course. On the other hand, reduction of soluble CpG to 10 μg also resulted in no appreciable increase of IL-12p40 in the lungs as compared to untreated mice, partially explaining the reduced efficacy in the 344SQ model. In contrast, treatment with PRINT-CpG at 10 μg resulted in elevated levels of IL-12p40 in the lungs of mice 24 h post-treatment as compared to untreated mice, and this dosing level still maintained efficacy in the 344SQ model.
Studies by Müller et al. revealed that CpG stimulation in combination with IFNγ can skew macrophages to a proinflammatory (antitumor) M1 phenotype.49 In vitro studies with bone-marrow-derived macrophages resulted in macrophage repolarization (from M2 to M1) following treatment with PRINT-CpG and not soluble CpG. Since our studies were conducted in the absence of additional IFNγ, it is plausible that the IFNγ in the lungs induced by CpG and PRINT-CpG was able to skew macrophages to an M1 phenotype. This was further confirmed through evaluating levels of Fpr2 (a classical chemoattractant expressed in several cell types) in the lungs, as Fpr2 was recently identified as a distinct M1 macrophage marker50 and has been shown to play a role in antitumor immunity by sustaining macrophages in an M1 phenotype.38 Consistent with these findings, we observed low levels of Fpr2 in untreated tumor-bearing mice and elevated levels of Fpr2 in mice treated with CpG and PRINT-CpG, further indicating that CpG and PRINT-CpG treatment result in M1 macrophage (antitumor) polarization.
As mentioned previously, IFNγ secretion is known to trigger the production of CXCL10, which results in recruitment of immune cells to the site of inflammation.36,43 At 24 h following therapeutic treatment with PRINT-CpG, we observed elevated levels of CXCL10 in the right lung, corresponding well with the elevated levels of DCs, NK cells, and CD4+ T cells in the right lung. Interestingly, at this time point, treatment with soluble CpG did not result in elevated levels of CXCL10; however we did observe an increase in macrophages, DCs, NK cells, and CD4+ T cells in the right lung. Overall, treatment with soluble CpG and not PRINT-CpG resulted in elevated levels of immune cells infiltrating the right lung, potentially the source of the observed lung inflammation.
In addition to mediating antitumor cellular responses, IFNγ has direct, potent, anticancer properties. It can promote tumor cell apoptosis, inhibit angiogenesis, and induce ischemia in the tumor stroma, as well as blood vessel destruction and necrosis. Furthermore, investigators showed that IFNγ sensitizes cancer cells to the effects of TNFα.51 Although TNFα shows potent antitumor activity in various animal cancer models, it binds nonselectively to tumor cells and normal cells, resulting in nonspecific damage to various cell types.35 In fact, experiments conducted by Campell et al. determined that TNFα was the central mediator of CpG-induced toxicity in mice, resulting in weight loss and lung pathology.20 While both soluble CpG and PRINT-CpG resulted in elevated levels of TNFα, treatment with PRINT-CpG resulted in significantly lower levels of TNFα than soluble CpG. We therefore believe that the elevated levels of TNFα in the lungs and serum following treatment with soluble CpG is another major source of toxicity associated with this therapy.
CONCLUSION
In conclusion, we showed that local pulmonary administration of PRINT-CpG nanoformulation was less toxic and more effective than soluble CpG at reducing tumor burden in syngeneic murine models of metastatic lung cancer. This study shows the benefits of using NPs as a local lung delivery platform for immunomodulators to generate an antitumor immune environment, while simultaneously reducing both local and systemic toxicities. Our results suggest the potential for further development of our NP platform to treat other cancers that metastasize to the lung.
METHODS/EXPERIMENTAL
Materials.
Dimethylformamide (DMF), sterile water, and phosphate-buffered saline (PBS, pH 7.4), and succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate) (SMCC) were obtained from Fisher Scientific. Poly(ethylene glycol) diacrylate (Mn 700) (PEG700DA), 2-aminoethyl methacrylate hydrochloride (AEM), diphenyl (2,4,6-trimethylbenzoyl)phosphine oxide (TPO), thiolmodified CpG 1826 (C6-S-S-C6-tccatgacgttcctgacgtt), thiol-modified GpC (C6-S-S-C6-tccatgagcttcctgagctt), and DNase- and RNase-free sterile water were purchased from Sigma-Aldrich. Tetraethylene glycol monoacrylate (HP4A) was synthesized in house. PRINT molds (80 nm × 320 nm) were obtained from Liquidia Technologies. DNA grade NAP-10 columns were purchased from GE Healthcare. RPMI1640 medium, penicillin, streptomycin, L-glutamine, and fetal bovine serum (FBS) were all from Life Technologies. Dylight 680 was from Pierce. CpG A 1585 was from InvivoGen.
Methods.
PRINT Nanoparticle Fabrication and Characterization.
The PRINT particle fabrication process has been described in detail previously.23 Briefly, a preparticle solution (PPS) was prepared by dissolving 3.5 wt % of the various reactive monomers in isopropyl alcohol (IPA). The PPS was composed of 69 wt % HP4A, 10 wt % PEG700DA, 20 wt % AEM, and 1 wt % TPO. A thin film of preparticle solution was drawn onto corona-treated PET with a # 3 Mayer rod, using a custom-made roll-to-roll lab line (Liquidia Technologies) running at 12 feet per minute. The solvent was evaporated by heat guns. Then the mold was laminated to a delivery sheet and passed through a nip (80 psi, 12 feet per minute). After delamination, particles were cured in the mold by passing it through UV LED lamp (λmax = 395 nm, 3 psi N2, 12 feet per minute; Phoseon). The filled mold was laminated against a PVA harvesting sheet and passed through a heated nip (320 °C, 80 psi). Particles were harvested by dissolving a sacrificial harvesting layer of PVA into water (2 mL of water per 10 feet of harvesting sheet). To remove excess PVA, particles were spun down at 14 000 rpm (Eppendorf Thermomixer R) for 25 min and resuspended into sterile water. This purification procedure was repeated 3 times.
Concentrations of particles were determined by thermogravimetric analysis (TGA) using a TA Instrument’s Discovery TGA. Aluminum sample pans were tared before loading the sample. A 10 μL amount of the stock nanoparticle solution was loaded onto the pan. Samples suspended in water were heated at 30 °C/min to 130 °C, followed by a 10 min isotherm at 130 °C. All samples were then cooled at 30 °C/min to 30 °C, followed by a 2 min isotherm at 30 °C.
Particle size and ZP were measured in sterile water by dynamic light scattering (DLS) using the cumulant analysis method on a Zetasizer Nano ZS (Malvern Instruments, Ltd.).
We utilized amine groups from AEM to conjugate NPs to the SMCC linker. The SMCC linker (mg) was dissolved in DMF. NPs were incubated with the SMCC linker for 2 h in a shaker plate. The particles fabricated contain free primary amine groups, which were used as functional handles to react with SMCC. The particles (1 mg NPs in 840 μL of 1× PBS and 0.1% PVA) were reacted with SMCC (0.26 mg in 160 μL of DMF) for 2 h at room temperature on a shaker plate (Eppendorf Thermomixer R, 1400 rpm). After 2 h, unconjugated SMCC was removed from the NPs via two centrifugation washes (Eppendorf Centrifuge 5417g) with sterile water. For both CpG and GpC conjugation, the C6-S-S-C6 CpG 1826 or C6 S-S-C6 GpC (Sigma) was reduced with a 100 mM DTT solution in sodium phosphate buffer (pH 8.0) and purified by gel filtration chromatography using a Sephadex NAP-10 column. Concentration of CpG/GpC was measured by evaluating absorption at 260 nm by using a NanoDrop 2000 spectrophotometer. NPs (1 mg of NP in 1× PBS) were reacted with 100 μg of CpG/GpC overnight at room temperature on a shaker plate (Eppendorf Thermomixer R, 1400 rpm). The next day unreacted CpG/GpC was removed from the NPs via a centrifugation wash. The supernatant was evaluated via the NanoDrop to determine the amount of unreacted CpG/GpC. NPs were resuspended in 500 μL of 10× PBS to remove electrostatically bound CpG/GpC. NPs were again centrifuged to remove unconjugated CpG/GpC. Again the supernatant was evaluated via the NanoDrop to evaluate unreacted CpG/GpC. The concentration of CpG/GpC was determined by subtracting the amount of CpG/GpC removed in the supernatants from the 100 μg of CpG/GpC originally loaded on the NPs.
CpG release was evaluated in two buffers: Gamble’s solution that simulates lung fluid and artificial lysosomal fluid that simulates lysosomal environment. PRINT-CpG particles were incubated in either Gamble’s or ALF at a concentration of 1 mg of PRINT-CpG/mL, under continuous shaking at 37 °C, 300 rpm. Samples were removed at specific time points (2, 5, 24, 48, and 168 h) and centrifuged to pellet out PRINT-CpG. Supernatants were removed and evaluated on the NanoDrop to determine the amount of CpG released. Percent of release was calculated as the amount of CpG loaded on the NPs minus CpG released, divided by the amount of CpG loaded.
Endotoxin level of PRINT-CpG was evaluated using the Pierce LAL chromogenic endotoxin quantitation kit, following the manufacturer’s instructions. All PRINT-CpG preparations had an endotoxin level of <0.1 EU/mg NP, ensuring <0.1 EU endotoxin injected per mouse per dose in the in vivo studies.
Dendritic cell (DC) activation by PRINT-CpG was evaluated ex vivo utilizing bone-marrow-derived DCs (BMDCs). BMDCs were prepared and cultured as published previously.52 Briefly, bone marrow cells were collected from femurs and tibias of C57BL/6 mice. After lysing erythrocytes using ACK lysis buffer, cells were grown in RPMI 1640 supplemented with 10% FBS, 50 μM 2-mercaptoethanol, 10 ng/mL GM-CSF, and 10 ng/mL of IL-4 (R&D Systems). On day 6, BMDCs were harvested and further purified for CD11c+ DCs with mouse CD11c microbeads (Miltenyi Biotec) following the manufacturer’s instructions. To examine the stability of CpG associated with PRINT particles, PRINT-CpG or free CpG were incubated in ALF or mouse serum for 0, 3, or 24 h at 37 °C. Purified BMDCs were dosed with these samples at 1 μg/mL CpG equivalent, for 24 h at 37 °C. Conditioned media were then tested for IL-6 and IL-12p40 secreted using cytokine ELISAs (BD) following the manufacturer’s instructions.
Animals.
All studies were conducted in accordance with National Institutes of Health guidelines for the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee at the University of North Carolina (UNC). Female 129S6 mice were purchased from Taconic Farms. Female FVB mice were purchased from Jackson Laboratories. All animals were maintained in pathogen-free facilities at UNC and were 6–10 week of age at the beginning of studies.
Tumor Inoculation, Treatments, and Tumor Rechallenge.
Luciferase-expressing 344SQ cells and KAL-LN2E1 cells were maintained in RPMI1640 medium supplemented with 10% FBS, 1 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. To establish orthotopic lung tumors, lung cancer cells were implanted via intrathoracic injection into either female 129S6 mice (344SQ cells) or female FVB mice (KAL-LN2E1 cells). For time course studies, 129S6 mice were inoculated with 2 × 103 344SQ cells on day 0 and received a single dose of either CpG or PRINT-CpG (20 μg). Mice were sacrificed at 3, 24, 48, and 72 h following treatment. Blood was collected via cardiac puncture in BD Microtainer SST tubes for cytokine ELISA evaluation, and lungs were harvested for PCR analysis (see details below in Serum Analysis). For efficacy studies, mice were inoculated with either 2 × 103 344SQ cells or 5 × 105 KAL-LN2E1 cells (1:1 ratio of Dulbecco’s PBS and BD Matrigel), and disease progression was monitored by weight gain/loss and overall health of the mouse, as well as bioluminescence for the 344SQ model. NP and control formulations were delivered to the lungs of anesthetized mice through an orotracheal instillation in a 50 μL volume. In the 344SQ model, CpG was dosed at 20 μg, starting 3 days after tumor cell inoculations and continuing every 3 days for a total of 4 doses. Disease progression was monitored primarily by bioluminescence imaging (IVIS Lumina), as well as weight loss and overall health of the mouse. Three months after initial CpG treatment, all surviving 129S6 animals were imaged (IVIS Lumina) to confirm tumor eradication and then rechallenged with 2 × 103 344SQ cells in the lung. A secondary survival study was conducted in the 344SQ model in the same manner except mice were doesed with 10 μg of CpG/PRINT-CpG, CpG-A, and PRINT-GpC every 3 days for a total of 4 doses. Mice were again monitored by IVIS imaging, weight loss, and overall health of the mouse. For the KAL-LN2E1 model, CpG was dosed at 10 μg on day 3 and continued every 3 days for a total of 3 doses. Twenty-four hours following the final CpG dose, mice were sacrificed and tumor burden was assessed by weighing the lungs and metastases, as well as counting the primary, contralateral, chest wall, and lymph node metastases.
Depletion Assays.
Female 129S6 mice were inoculated with 2 × 103 luciferase-expressing 344SQ cells on day 0. On day 1 mice received 200 μg of IP injections of either anti-CD8 (BioXcell) or antiasialo GM1 (VWR) to deplete mice of CD8 T cells or NK cells, respectively. Depletion injections were repeated every 4 days, until the termination of the study. On day 3 mice received PRINT-CpG treatment, which continued every 3 days for a total of 4 doses. Mice were monitored for disease progression by weight gain/loss and overall health of the mouse, as well as tumor burden via bioluminescence imaging (IVIS Lumina).
Immunofluorescence.
Immunofluorescence was performed on formalin-fixed, paraffin-embedded lung sections (5 μm thickness) by UNC’s Tissue Pathology Laboratory. Lung sections were stained with DAPI (gray), Cy3 (green), and Cy5 (red) to visualize nuclei, CpG, and PRINT NPs, respectively. High-resolution images were acquired with the Aperio ScanScope FL (Leica).
Biodistribution.
Healthy 129S6 mice were dosed 1× with 20 μg of Cy3-CpG or PRINT-Cy3-CpG via instillation. Mice were sacrificed at various time points postinstillation, at which time blood was collected via cardiac puncture and stored in heparized Eppendorf tubes (Milian, USA), and lungs, liver, spleen, and kidneys were removed and placed in 12-well plates for fluorescence analysis with an IVIS Lumina imager (Caliper Life Science); excitation and emission filters were set to 550 and 570 nm, respectively. Heparinized blood was pipetted in 100 μL aliquots into black 96-well plates and imaged on the IVIS Lumina.
Flow Cytometry and Quantitative Real-Time RT-PCR.
Six- to eight-week-old mice were inoculated with KAL-LN2E1 cells as described above on day 0 and dosed with 10 μg of CpG per mouse on day 6 and day 9. On day 10 blood was collected (via submandibular bleeds), and mice were sacrificed and necropsied. A small (~10 μg) right lung sample was saved for PCR analysis, and the remaining right lung, left lung (containing primary tumor), and metastases were prepared for flow cytometry. Tissues were resected, minced with scissors, and transferred to 15 mL tubes containing 5 mL of RPMI with 10% heat-inactivated FBS and 10 mM HEPES buffer on ice. The tissue suspensions were then supplemented with 2 mg/ML collagenase D and 20 μg/mL DNase I and incubated under rotation at 37 °C for 30 min. Dissociated cells were washed with 5 mL of PBS and incubated with 2 mL of ACK lysis buffer for 1 min at room temperature to remove red blood cells. Cells were resuspended in FACS buffer (0.5% BSA and 2 mM EDTA in PBS) at a concentration of ~106 cells/100 μL. Samples were incubated with the following antibodies: CD8 (FITC-conjugated, #100706 Biolegend), TCRb (PE-conjugated, #109208 Biolegend), CD25 (PE/Cy7-conjugated, #102016 Biolegend), GM1 (Primary, #986–10001 Wako followed by goat anti-rabbit AF594, #A11012 Invitrogen), CD4 (APC/Cy7-conjugated #100414, Biolegend), B220 (Pacific blue-conjugated, #103227 Biolegend), F4/80 (PE-efluor 610 conjugated, #61-4801-82 eBioscience), MHC II (PE-conjugated, #107608 Biolegend), CD11b (PE/Cy5-conjugated, #101210 Biolegend), CD11c (PE/Cy7-conjugated, #128018 Biolegend), CD45 (APC-conjugated, #103112 Biolegend), CD206 (BV785-conjugated, Biolegend, clone C068C2), Siglec-F (BV421-conjugated, #562681 BD Bioscience), and LIVE/DEAD Fixable Violet Dead Cell Stain (# L34963 ThermoFisher Scientific).
Approximately 0.2 μg of antibody was used for every 106 cells. Cells were incubated with antibody for 30 min on ice, in the dark. Cells were then washed two times with FACS buffer and taken for flow cytometry analysis on an LSR II. The collected data were analyzed using FCS Express 6. When performing FACS on lung tissues, Siglec-F positive cells were included with dead cells to remove alveolar macrophages and/or eosinophils. In the T-cell/NK cell panel, B220 positive cells were also included with dead and Siglec-F positive cells to remove B cells. Dendritic cells were identified as CD45+/MHCII +/CD11b+/CD11c+ cells, macrophages as CD45+/MHCII +/CD11b+/CD11c− cells, natural killer cells as CD45+/GM1+/TCRb− cells, CD4 T cells as CD45+/TCRb+/GM1−/CD4+/CD8− cells, and CD8 T cells as CD45+/TCRb+/GM1−/CD4−/CD8+ cells. Print CpG expression was determined using unstained spleen samples to set the negative, and its fluorescence was collected as AF700.
For mRNA quantification, total RNA was extracted from lung tissues using TRIzol (Invitrogen), followed by DNA removal with the DNA-free kit (Ambion) according to manufacturer’s instructions. cDNA was synthesized from 1000 ng of RNA utilizing iScript Reverse Transcription Supermix (BIO-RAD) as per the manufacturer’s instructions. Analysis of mRNA levels was performed on a real-time PCR system (Applied Biosystems). Specific murine primers for IL-12p40 (forward) CAGAAGCTAACCATCTCCTGGTTTG, (reverse) TCCGGAGTAATTTGGTGCTTCACAC; TNFα (forward) CCCTCACACTCAGATCATCTTCT, (reverse) GCTACGAC-GTGGGCTACAG; IFNγ (forward) TCAAGTGGCATAGATGTG-GAAGAA, (reverse) TGGCTCTGCAGGATTTTCATG; CXCL10 (forward) TGCTGGGTCTGAGTGGGACT, (reverse) GGATAGGCTCGCAGGGATGA; Fpr2 (forward) TTACATC-TACCACAATGTGAACTA, and (reverse) TCTACCATCTCCAGAGTTCTGTGG were used for SYBR Green-based real-time PCR, with murine glyceraldehyde 3-phosphate dehydrogenase (GAPDH) used as a housekeeping gene. PCR was done with reverse-transcribed RNA, 1 μL each of 20 μM forward and reverse primers, and PowerUp SYBR Green Master Mix in a total volume of 13 μL for mRNA studies. Reactions were run in duplicate. The delta-delta Ct method was used to calculate fold change.
Macrophage Polarization.
Murine bone-marrow-derived macrophages (BMDMs) were isolated from 8-week-old C57BL/6 wild-type mice as previously described.53 Briefly, bone marrows from femurs and tibia were flushed out by injecting DMEM medium (Gibco). The cell suspension was filtered by 70 μm cell strainers (BD Falcon), centrifuged, and then lysed by ACK buffer (Gibco). BMDMs were cultured in macrophage-growth medium (MGM: DMEM + 10% FBS + 30% L929 medium). On day 6, cells were stimulated with LPS (500 ng/mL) + IFN-γ (25 ng/mL) for M1 polarization (M1(+)) or IL-4 (25 ng/mL) for M2 polariation (M2(+)), or left as untreated (UT) control. On day 7, M2 cells were treated with 1 μg/mL of CpG delivered as soluble or PRINT-CpG or an equivalent amount of blank PRINT-NP. On day 8, all cells were harvested for gene expression analysis (Fizz1, Ym1, and Mrc1) by using TaqMan primer/probe sets and master mix (Applied Biosystems); supernatants were collected for IL-6 and TNF cytokine detection by ELISA (BD).
Serum Analysis.
Blood was collected (via submandibular bleeds) into BD microtainer SST tubes prior to harvesting tissues for the flow cytometry and PCR analysis. Tubes were spun at 1200g for 10 min to separate the serum. Cytokines in the serum samples were assayed with cytokine ELISA kits (BD) following manufacturer’s instructions. Clinical chemistry testing for ALB, ALT, and CREAT was conducted by UNC’s Animal Histopathology and Lab Medicine Core.
Statistical Analysis.
Statistical comparison in all different experimental conditions was done with the Prism software (version 8.0, GraphPad Software Inc.) using two-way ANOVA. Survival was plotted using a Kaplan–Meier survival curve, and statistical significance was determined by the log-rank (Mantel-Cox) test. A P value of less than 0.05 was considered significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Muhmad Alzpair for his help with harvesting and characterizing PRINT particles, as well as the University of North Carolina Animal Studies Core, specifically Charlene Santos and Mark Ross, for their assistance with all animal studies. Additionally we thank Liquida Technologies for providing PRINT molds used in the fabrication of the PRINT nanoparticles, the Translational Pathology Lab, for tissue sectioning and immunofluorescence evaluations of lung samples, and the Animal Histopathology and Laboratory Medicine Core for clinical chemistry tests and multiplexed biomarker immunoassays. The authors would like to especially thank the UNC Biomedical Research Imaging Center and the UNC Flow Cytometry Core Facility. This research was supported by the Carolina Center for Cancer Nanotechnology Excellence (U54-CA151652) and by the University Cancer Research Fund. C.V.P. was supported in part by the National Institutes of Health R01-CA215075, R01-CA042978, and U54-CA198999, a Mentored Research Scholar Grants in Applied and Clinical Research (MRSG-14-222-01-RMC) from the American Cancer Society, the Jimmy V Foundation Scholar award, the UCRF Innovator Award, the Stuart Scott V Foundation/Lung Cancer Initiative Award for Clinical Research, the University Cancer Research Fund, the Lung Cancer Research Foundation, the Free to Breathe Metastasis Research Award, and the Susan G. Komen Career Catalyst Award. E.B.H. was supported in part by a grant from the National Cancer Institute of the National Institutes of Health under award number T32CA196589. The UNC Flow Cytometry Core Facility and Lineberger Comprehensive Cancer Center Animal Histopathology and Animal Studies Cores are all supported in part by a National Cancer Institute Center Core Support Grant (CA016086) to the UNC Lineberger Comprehensive Cancer Center. The UNC Flow Cytometry Core Facility is also supported in part by the North Carolina Biotech Center Institutional Support Grant 2012-IDG-1006.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.0c02207.
Figures showing lung distribution of soluble Cy3-CpG and PRINT-Cy3-CpG, maximum tolerated dose of CpG in FVB mouse strain, gating strategy and immunologic effects of PRINT-CpG, and flow cytometry analysis of KAL-LN2E1 metastases (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsnano.0c02207
The authors declare the following competing financial interest(s): PRINT-CpG has been licensed by Liquidia, resulting in royalty funds
Contributor Information
Jillian L. Perry, UNC Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, North Carolina 27599, United States.
Shaomin Tian, Department of Microbiology & Immunology, University of North Carolina, Chapel Hill, North Carolina 27599, United States.
Nisitha Sengottuvel, Curriculum in Genetics and Molecular Biology, University of North Carolina, Chapel Hill, North Carolina 27599, United States.
Emily B. Harrison, UNC Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, North Carolina 27599, United States
Balachandra K. Gorentla, UNC Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, North Carolina 27599, United States
Chintan H. Kapadia, UNC Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, North Carolina 27599, United States
Ning Cheng, UNC Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, North Carolina 27599, United States.
J. Christopher Luft, UNC Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, North Carolina 27599, United States.
Jenny P.-Y. Ting, Department of Microbiology & Immunology, Department of Genetics, Institute for Inflammatory Diseases, and Center for Translational Immunology, University of North Carolina, Chapel Hill, North Carolina 27599, United States; Department of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, North Carolina 27695, United States
Joseph M. DeSimone, UNC Lineberger Comprehensive Cancer Center, UNC Eshelman School of Pharmacy, Department of Chemistry, and Institute for Nanomedicine, University of North Carolina, Chapel Hill, North Carolina 27599, United States; Department of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, North Carolina 27695, United States;.
Chad V. Pecot, UNC Lineberger Comprehensive Cancer Center, Curriculum in Genetics and Molecular Biology, and Department of Medicine, University of North Carolina, Chapel Hill, North Carolina 27599, United States
REFERENCES
- (1).Carbone DP; Gandara DR; Antonia SJ; Zielinski C; Paz-Ares L Non-Small Cell Lung Cancer: Role of the Immune System and Potential for Immunotherapy. J. Thorac. Oncol 2015, 10, 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tsiara A; Liontos M; Kaparelou M; Zakopoulou R; Bamias A; Dimopoulos M-A Implementation of Immunotherapy in the Treatment of Advanced Non-Small Cell Lung Cancer (NSCLC). Ann. Transl. Med 2018, 6, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Adamus T; Kortylewski M The Revival of CpG Oligonucleotide-Based Cancer Immunotherapies. Contemp. Oncol. (Pozn) 2018, 22, 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Juarez E; Nuñez C; Sada E; Ellner JJ; Schwander SK; Torres M Differential Expression of Toll-Like Receptors on Human Alveolar Macrophages and Autologous Peripheral Monocytes. Respir. Res 2010, 11, 2–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bourke E; Bosisio D; Golay J; Polentarutti N; Mantovani A The Toll-Like Receptor Repertoire of Human B Lymphocytes: Inducible and Selective Expression of TLR9 and TLR10 in Normal and Transformed Cells. Blood 2003, 102, 956. [DOI] [PubMed] [Google Scholar]
- (6).Roda JM; Parihar R; Carson WE CpG-Containing Oligodeoxynucleotides Act through TLR9 to Enhance the NK Cell Cytokine Response to Antibody-Coated Tumor Cells. J. Immunol 2005, 175, 1619–1627. [DOI] [PubMed] [Google Scholar]
- (7).Schreibelt G; Tel J; Sliepen KHEWJ; Benitez-Ribas D; Figdor CG; Adema GJ; de Vries IJM Toll-Like Receptor Expression and Function in Human Dendritic Cell Subsets: Implications for Dendritic Cell-Based Anti-Cancer Immunotherapy. Cancer Immunol. Immunother 2010, 59, 1573–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hayashi F; Means TK; Luster AD Toll-Like receptors stimulate human neutrophil function. Blood 2003, 102, 2660. [DOI] [PubMed] [Google Scholar]
- (9).Krieg AM Toll-Like Receptor 9 (TLR9) Agonists in the Treatment of Cancer. Oncogene 2008, 27, 161. [DOI] [PubMed] [Google Scholar]
- (10).Engel AL; Holt GE; Lu H The Pharmacokinetics of Toll-Like Receptor Agonists and the Impact on the Immune System. Expert Rev. Clin. Pharmacol 2011, 4, 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jin B; Sun T; Yu X-H; Yang Y-X; Yeo AET The Effects of TLR Activation on T-Cell Development and Differentiation. Clin. Dev. Immunol 2012, 2012, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ribas A; Medina T; Kummar S; Amin A; Kalbasi A; Drabick JJ; Barve M; Daniels GA; Wong DJ; Schmidt EV; Candia AF; Coffman RL; Leung ACF; Janssen RS SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase 1b, Multicenter Study. Cancer Discovery 2018, 8, 1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Diab A; Haymaker C; Bernatchez C; Johnson DH; Andtbacka R; James M; Johnson D; Markowitz J; Murthy R; Puzanov I; Shaheen M; Rahimian S; Geib J; Chunduru S; Hwu P The Safety and Efficacy of Intratumoral Injection of the TLR9 Agonist Tilsotolimod (IMO-2125) in Combination with Ipilimumab in Patients with PD-1 Inhibitor Refractory Metastatic Melanoma: An Analysis of Efficacy in Injected and Uninjected Lesions Poster Presented at ESMO Congress; Munich, Germany, October 2018; Abstract 5845. [Google Scholar]
- (14).Thomas M; Ponce-Aix S; Navarro A; Riera-Knorrenschild J; Schmidt M; Wiegert E; Kapp K; Wittig B; Mauri C; Dómine Gómez M; Kollmeier J; Sadjadian P; Fröhling KP; Huber RM; Wolf M IMPULSE study Team. Immunotherapeutic Maintenance Treatment with Toll-Like Receptor 9 Agonist Lefitolimod in Patients with Extensive-Stage Small-Cell Lung Cancer: Results From the Exploratory, Controlled, Randomized, International Phase II IMPULSESstudy. Ann. Oncol 2018, 29, 2076–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gallotta M; Assi H; Degagné É; Kannan SK; Coffman RL; Guiducci C Inhaled TLR9 Agonist Renders Lung Tumors Permissive to PD-1 Blockade by Promoting Optimal CD4+ and CD8+ T-Cell Interplay. Cancer Res. 2018, 78, 4943–4956. [DOI] [PubMed] [Google Scholar]
- (16).Sato T; Shimosato T; Ueda A; Ishigatsubo Y; Klinman DM Intrapulmonary Delivery of CpG Microparticles Eliminates Lung Tumors. Mol. Cancer Ther 2015, 14, 2198–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sfondrini L; Sommariva M; Tortoreto M; Meini A; Piconese S; Calvaruso M; Van Rooijen N; Bonecchi R; Zaffaroni N; Colombo MP; Tagliabue E; Balsari A Anti-Tumor Activity of CpG-ODN Aerosol in Mouse Lung Metastases. Int. J. Cancer 2013, 133, 383–393. [DOI] [PubMed] [Google Scholar]
- (18).Le Noci V; Tortoreto M; Gulino A; Storti C; Bianchi F; Zaffaroni N; Tripodo C; Tagliabue E; Balsari A; Sfondrini L Poly(I:C) and CpG-ODN Combined Aerosolization to Treat Lung Metastases and Counter the Immunosuppressive Microenvironment. Oncoimmunology 2015, 4, No. e1040214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tasaka S; Kamata H; Miyamoto K; Nakano Y; Shinoda H; Kimizuka Y; Fujiwara H; Hasegawa N; Fujishima S; Miyasho T; Ishizaka A Intratracheal Synthetic CpG Oligodeoxynucleotide Causes Acute Lung Injury with Systemic Inflammatory Response. Respir. Res 2009, 10, 84–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Campbell JD; Cho Y; Foster ML; Kanzler H; Kachura MA; Lum JA; Ratcliffe MJ; Sathe A; Leishman AJ; Bahl A; McHale M; Coffman RL; Hessel EM CpG-Containing Immunostimulatory DNA Sequences Elicit TNF-Alpha-Dependent Toxicity in Rodents but Not in Humans. J. Clin. Invest 2009, 119, 2564–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Roberts RA; Shen T; Allen IC; Hasan W; DeSimone JM; Ting JPY Analysis of the Murine Immune Response to Pulmonary Delivery of Precisely Fabricated Nano- and Microscale Particles. PLoS One 2013, 8, No. e62115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shen TW; Fromen CA; Kai MP; Luft JC; Rahhal TB; Robbins GR; DeSimone JM Distribution and Cellular Uptake of PEGylated Polymeric Particles in the Lung towards Cell-Specific Targeted Delivery. Pharm. Res 2015, 32, 3248–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kapadia CH; Tian S; Perry JL; Sailer D; Christopher Luft J; DeSimone JM Extending Antigen Release from Particulate Vaccines Results in Enhanced Antitumor Immune Response. J. Controlled Release 2018, 269, 393–404. [DOI] [PubMed] [Google Scholar]
- (24).Marques M; Löbenberg R; Almukainzi M Simulated Biological Fluids with Possible Application in Dissolution Testing. Dissolution Technol. 2011, 18, 15–28. [Google Scholar]
- (25).Heckelsmiller K; Rall K; Beck S; Schlamp A; Seiderer J; Jahrsdörfer B; Krug A; Rothenfusser S; Endres S; Hartmann G Peritumoral CpG DNA Elicits a Coordinated Response of CD8 T Cells and Innate Effectors to Cure Established Tumors in a Murine Colon Carcinoma Model. J. Immunol 2002, 169, 3892. [DOI] [PubMed] [Google Scholar]
- (26).Sagiv-Barfi I; Czerwinski DK; Levy S; Alam IS; Mayer AT; Gambhir SS; Levy R Eradication of Spontaneous Malignancy by Local Immunotherapy. Sci. Transl. Med 2018, 10, eaan4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shirota Y; Shirota H; Klinman DM, Intratumoral Injection of CpG Oligonucleotides Induces the Differentiation and Reduces the Immunosuppressive Activity of Myeloid-Derived Suppressor Cells. J. Immunol 2012, 188, 1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Porrello A; Leslie PL; Harrison EB; Gorentla BK; Kattula S; Ghosh SK; Azam SH; Holtzhausen A; Chao YL; Hayward MC; Waugh TA; Bae S; Godfrey V; Randell SH; Oderup C; Makowski L; Weiss J; Wilkerson MD; Hayes DN; Earp HS; Baldwin AS; Wolberg AS; Pecot CV Factor XIIIA—Expressing Inflammatory Monocytes Promote Lung Squamous Cancer through Fibrin Cross-Linking. Nat. Commun 2018, 9, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hanagata N Structure-Dependent Immunostimulatory Effect of CpG Oligodeoxynucleotides and Their Delivery System. Int. J. Nanomed 2012, 7, 2181–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Alizadeh D; Zhang L; Brown CE; Farrukh O; Jensen MC; Badie B Induction of Anti-Glioma Natural Killer Cell Response following Multiple Low-Dose Intracerebral CpG Therapy. Clin. Cancer Res 2010, 16, 3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Baines J; Celis E Immune-Mediated Tumor Regression Induced by CpG-Containing Oligodeoxynucleotides. Clin. Cancer Res 2003, 9, 2693. [PubMed] [Google Scholar]
- (32).Cook PC; MacDonald AS Dendritic Cells in Lung Immunopathology. Semin. Immunopathol 2016, 38, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Fromen CA; Rahhal TB; Robbins GR; Kai MP; Shen TW; Luft JC; DeSimone JM Nanoparticle Surface Charge Impacts Distribution, Uptake and Lymph Node Trafficking by Pulmonary Antigen-Presenting Cells. Nanomedicine 2016, 12, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Schneberger D; Caldwell S; Kanthan R; Singh B Expression of Toll-Like Receptor 9 in Mouse and Human Lungs. J. Anat 2013, 222, 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Shen J; Xiao Z; Zhao Q; Li M; Wu X; Zhang L; Hu W; Cho CH Anti-Cancer Therapy with TNFα and IFNγ: A Comprehensive Review. Cell Proliferation 2018, 51, No. e12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Liu M; Guo S; Stiles JK The Emerging Role of CXCL10 in Cancer. Oncol. Lett 2011, 2, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Cattaneo F; Guerra G; Parisi M; Lucariello A; De Luca A; De Rosa N; Mazzarella G; Bianco A; Ammendola R Expression of Formyl-Peptide Receptors in Human Lung Carcinoma. Anticancer Res. 2015, 35, 2769–2774. [PubMed] [Google Scholar]
- (38).Liu Y; Chen K; Wang C; Gong W; Yoshimura T; Liu M; Wang JM Cell Surface Receptor FPR2 Promotes Antitumor Host Defense by Limiting M2 Polarization of Macrophages. Cancer Res. 2013, 73, 550–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kunikata N; Sano K; Honda M; Ishii K; Matsunaga J; Okuyama R; Takahashi K; Watanabe H; Tamura G; Tagami H; Terui T Peritumoral CpG Oligodeoxynucleotide Treatment Inhibits Tumor Growth and Metastasis of B16F10 Melanoma Cells. J. Invest. Dermatol 2004, 123, 395–402. [DOI] [PubMed] [Google Scholar]
- (40).Wang S; Campos J; Gallotta M; Gong M; Crain C; Naik E; Coffman RL; Guiducci C Intratumoral Injection of a CpG Oligonucleotide Reverts Resistance to PD-1 Blockade by Expanding Multifunctional CD8+ T Cells. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E7240–E7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sato-Kaneko F; Yao S; Ahmadi A; Zhang SS; Hosoya T; Kaneda MM; Varner JA; Pu M; Messer KS; Guiducci C; Coffman RL; Kitaura K; Matsutani T; Suzuki R; Carson DA; Hayashi T; Cohen EE Combination Immunotherapy with TLR Agonists and Checkpoint Inhibitors Suppresses Head and Neck Cancer. JCI Insight 2017, 2, No. e93397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Vilgelm AE; Richmond A Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Front. Immunol 2019, 10, DOI: 10.3389/fimmu.2019.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Decalf J; Fernandes S; Longman R; Ahloulay M; Audat F; Lefrerre F; Rice CM; Pol S; Albert ML Plasmacytoid Dendritic Cells Initiate a Complex Chemokine and Cytokine Network and Are a Viable Drug Target in Cronic HCV Patients. J. Exp. Med 2007, 204, 2423–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Yin P; Liu X; Mansfield AS; Harrington SM; Li Y; Yan Y; Dong H CpG-Induced Antitumor Immunity Requires IL-12 in Expansion of Effector Cells and Down-Regulation of PD-1. Oncotarget 2016, 7, 70223–70231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Pesce I; Monaci E; Muzzi A; Tritto E; Tavarini S; Nuti S; De Gregorio E; Wack A Intranasal Administration of CpG Induces a Rapid and Transient Cytokine Response followed by Dendritic and Natural Killer Cell Activation and Recruitment in the Mouse Lung. J. Innate Immun 2010, 2, 144–159. [DOI] [PubMed] [Google Scholar]
- (46).Tugues S; Burkhard SH; Ohs I; Vrohlings M; Nussbaum K; Vom Berg J; Kulig P; Becher B New Insights into IL-12-Mediated Tumor Suppression. Cell Death Differ. 2015, 22, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ma X TNF-α and IL-12:a Balancing Act in Macrophage Functioning. Microbes Infect. 2001, 3, 121–129. [DOI] [PubMed] [Google Scholar]
- (48).Wang P; Li X; Wang J; Gao D; Li Y; Li H; Chu Y; Zhang Z; Liu H; Jiang G; Cheng Z; Wang S; Dong J; Feng B; Chard LS; Lemoine NR; Wang Y Re-Designing Interleukin-12 to Enhance Its Safety and Potential as an Anti-Tumor Immunotherapeutic Agent. Nat. Commun 2017, 8, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Müller E; Christopoulos PF; Halder S; Lunde A; Beraki K; Speth M; Øynebråten I; Corthay A Toll-Like Receptor Ligands and Interferon-γ Synergize for Induction of Antitumor M1Macrophages. Front. Immunol 2017, 8, 1383–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Jablonski KA; Amici SA; Webb LM; Ruiz-Rosado J d. D; Popovich PG; Partida-Sanchez S; Guerau-De-Arellano M Novel Markers to Delineate Murine M1 and M2Macrophages. PLoS One 2015, 10, No. e0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Roberts NJ; Zhou S; Diaz LA Jr.; Holdhoff M Systemic Use of Tumor Necrosis Factor Alpha as an Anticancer Agent. Oncotarget 2011, 2, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kapadia CH; Tian S; Perry JL; Luft JC; DeSimone JM Reduction Sensitive PEG Hydrogels for Codelivery of Antigen and Adjuvant to Induce Potent CTLs. Mol. Pharmaceutics 2016, 13, 3381–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Cheng N; Watkins-Schulz R; Junkins RD; David CN; Johnson BM; Montgomery SA; Peine KJ; Darr DB; Yuan H; McKinnon KP; Liu Q; Miao L; Huang L; Bachelder EM; Ainslie KM; Ting JPY A Nanoparticle-Incorporated STING Activator Enhances Antitumor Immunity in PD-L1-Insensitive Models of Triple-Negative Breast Cancer. JCI Insight. 2018, 3, No. e120638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.