

Supplemental Digital Content is available in the text.
Keywords: antidepressant, fast onset of action, intravenous administration, major depressive disorder
Abstract
This 7-day randomized, double-blind, placebo-controlled fixed-dose study (NCT03766867) explored the potential for accelerating the onset of antidepressant efficacy of single-dose intravenous (IV) vortioxetine at oral vortioxetine treatment initiation. Patients (ages 18–65 years) hospitalized per standard-of-care with major depressive disorder, who were currently treated with a selective serotonin reuptake inhibitor or serotonin-norepinephrine reuptake inhibitor for a major depressive episode [Montgomery–Åsberg Depression Rating Scale (MADRS) total score ≥ 30], received one dose of single-blind IV placebo (1-day placebo lead-in period) before being randomly switched to either single-dose IV vortioxetine 25 mg plus daily oral vortioxetine 10 mg (n = 39), or IV placebo plus daily oral placebo (n = 41). In the placebo lead-in period, patients improved slightly by 0.6 MADRS-6 point; however, at day 1 after randomization, both treatment groups had improved by approximately 3 MADRS-6 points (mean difference = −0.8; P = 0.263), the study thus not meeting its primary endpoint. Similar results were seen for other outcomes except a numerically larger improvement in anxiety symptoms with vortioxetine vs placebo. Pharmacokinetic data confirmed that IV vortioxetine facilitated reaching steady-state plasma concentration within 24 h. IV plus oral vortioxetine was well tolerated, with low levels of nausea as the most common adverse event.
Introduction
Major depressive disorder (MDD) is a disabling illness with symptoms spanning across emotional, cognitive and physical domains, and with a substantial negative impact on patients’ daily functioning and quality of life (American Psychiatric Association, 2016). The immediate goal of antidepressant treatment is to resolve acute emotional symptoms and obtain symptomatic remission. However, long-term treatment aims broader at achieving sustained functional recovery and help patients return to their usual family, psychosocial, and work functioning (Malhi and Mann, 2018).
There is evidence that the duration of the depressive episode is critical for treatment outcomes and long-term prognosis (Kraus et al., 2019). In addition to early and effective intervention, fast onset of therapeutic action may facilitate symptomatic relief at an early illness stage and shorten the time during which patients are emotionally distressed, potentially at risk of suicide, and functionally impaired.
While most of the currently available treatments may require several weeks to induce clinically relevant symptomatic improvement (Machado-Vieira et al., 2008), a number of new compounds with fast-acting properties are being developed, including the recently approved adjunctive intranasal esketamine for treatment-resistant depression (Popova et al., 2019) and intravenous (IV) brexanolone for postpartum depression (Meltzer-Brody et al., 2018), and oral SAGE-217 for major depression in phase II development (Gunduz-Bruce et al., 2019). However, these glutamatergic/GABAergic agents have significant side effects, primarily dissociation and sedation, and esketamine and brexanolone are available only through a Risk Evaluation and Mitigation Strategies (REMS) program. Consequently, a medical need remains for rapid-acting treatments with long-term efficacy and tolerability for broad MDD.
Vortioxetine is an efficacious antidepressant with a favorable risk–benefit profile (Katona et al., 2012; Thase et al., 2016; Baldwin et al., 2016a; Citrome, 2016) and potential for fast onset based on mechanism of action (Bétry et al., 2013). Specifically, 5-HT1A receptor agonism with vortioxetine may lead to rapid desensitizing of autoreceptors in the raphe nucleus (Bétry et al., 2013), and 5-HT3 receptor antagonism may reduce GABAergic input from hippocampal interneurons, which in turn may increase the serotonin-mediated activation of glutamatergic neurons in the frontal cortex (Dale et al., 2018). Thereby, vortioxetine may counteract the initial suppression of 5-HT neurotransmission following inhibition of the SERT, which is thought to account for the 2-to 3-week delay in the onset of effect typically seen with selective serotonin reuptake inhibitors (SSRIs) (Bétry et al., 2013; Artigas et al., 2018).
While the steady-state plasma concentration level with oral vortioxetine is reached after approximately 2 weeks (Areberg et al., 2012), an initial, single IV dose of 17 mg added to oral vortioxetine 10 mg/day has been demonstrated to facilitate a rapid increase (within 24 h) of vortioxetine exposure to the steady-state level (Vieta et al., 2019). In this previous study, however, no separation of 17 mg IV vortioxetine and 10 mg/day oral vortioxetine from IV placebo plus oral vortioxetine was seen at day 7 in the primary endpoint, the mean change from baseline in Montgomery–Åsberg Depression Rating Scale (MADRS) total score. The aim of the current study was to explore the early onset of efficacy and the safety and tolerability within the first week of treatment, of a single initial, IV dose of 25 mg vortioxetine added to 10 mg daily oral vortioxetine, using a study design aiming at mitigating potential limitations in the previous IV study.
Methods
Patients and study design
This multisite, parallel-group, fixed-dose study included patients aged 18–65 years with a DSM-5 diagnosis of recurrent MDD and a current major depressive episode (MDE; classification code 296.3x), who were admitted to hospital per clinical practice. Patients had to have had the current MDE for at least 3 months and less than 12 months, and experience severe depressive symptoms MADRS total score ≥30 at screening and at baseline] after one trial of SSRI/serotonin-norepinephrine reuptake inhibitor (SNRI) monotherapy (citalopram, escitalopram, paroxetine, duloxetine, venlafaxine, sertraline) at approved dose, for at least 6 weeks. Patients with any psychiatric comorbidity as assessed using the Mini International Neuropsychiatric Interview (Sheehan et al., 1998) – including alcohol or other substance abuse/dependence – or with a history of mania, hypomania, or any psychotic disorder, were excluded, as were patients who were pregnant or at the risk of suicide.
Eligible patients were randomly switched with a ratio of 1:1 directly, without a washout period, from current SSRI/SNRI to double-blind treatment with either a single dose of IV vortioxetine 25 mg (250 ml saline infused over 2 hours) followed by 1 week of daily oral vortioxetine 10 mg (vortioxetine group), or to a single initial dose of IV placebo saline infusion followed by 1 week of daily oral placebo (placebo group). Patients were hospitalized per clinical practice throughout the 1-week treatment period, with a safety follow-up approximately 4 weeks after the end of treatment. To minimize the placebo response induced by the IV procedure during the randomized treatment period, all eligible patients received a single-blind IV placebo infusion followed by a placebo tablet on the day before randomization. An internal, blinded safety evaluation was conducted after randomization of the first eight patients before continuing the study. A safety follow-up was conducted approximately 1 month after completion of the randomized treatment period.
The IV dose of 25 mg vortioxetine was selected based on a pharmacokinetic model evaluating plasma exposure with a 25 mg IV dose combined with an oral dose of 10 mg/day (recommended starting dose). The pharmacokinetic model predicted that this regimen within 24 h would be comparable to steady-state levels of 15 mg/day oral vortioxetine dosing.
The study was conducted between December 2018 and July 2019 at 13 sites in Bulgaria, Estonia, and Latvia. The study protocol was approved by the independent ethics committee of each study site. The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practices guidelines and with the ethical principles of the Declaration of Helsinki. The trial is registered at ClinicalTrials.gov (NCT03766867).
Assessments
Efficacy data were collected at day 0 (baseline), and at days 1, 3, and 7, except for the patient-reported Patient Global Impression of Change (PGI-C, see below), which was assessed daily from days 1 through 7; safety data in addition on day 1 (i.e., in the placebo lead-in) and at 2 h after the IV infusion (given in the morning of day 0). Pharmacokinetic samples were collected at 2 h, at bedtime and at 24 h post-IV infusion, and at day 7 (sampling time unspecified).
Depressive symptom severity was rated independently and blindly by clinicians using the MADRS (Montgomery and Åsberg, 1979), modified to assess symptoms within the past 24 h at each visit. The primary efficacy scale was the six-item MADRS-6 subscale score (possible score range 0–36), calculated as the sum of the following items: Apparent sadness, reported sadness, inner tension, lassitude, inability to feel, and pessimistic thoughts (item numbers 1, 2, 3, 7, 8, and 9). Developed to obtain a more unidimensional measure of the core symptoms of depression compared with the full scale, the MADRS-6 subscale has shown sensitivity to detect treatment effects and early improvement with antidepressant treatment (Bech et al., 2002; Bech, 2006; Thase et al., 2012; Vieta et al., 2019). The total score (possible score range 0–60) was calculated as the sum of scores on the 10 items of the full MADRS. Higher MADRS scores indicate worse symptom severity.
Illness severity and improvement was recorded by clinicians using the Clinical Global Impressions – Severity of Illness (CGI-S) and CGI – Improvement (CGI-I; not assessed at randomization baseline) (Guy, 1976). Both scales range from 1 to 7, with higher CGI scores indicating worse status/worsening of illness, a CGI-I score of 4 reflecting neither improvement nor worsening relative to baseline. A patient-version of the CGI, the PGI-C (Hurst and Bolton, 2004), was used to record patient-reported improvement in overall illness status relative to a baseline, with higher scores indicating larger improvement and a score of 1 indicating no change. In addition, patient-reported symptoms of depression and anxiety were assessed using the Hospital Anxiety and Depression Scale subscales for depression (HADS-D) and anxiety (HADS-A) (Zigmond and Snaith, 1983). Scores for each of the 7-item subscales (possible range 0-21) were calculated as the sum of item scores, higher scores indicating worse symptom severity.
Safety and tolerability assessments included adverse events, vital signs, clinical safety laboratory tests, electrocardiograms (ECGs), and physical examinations.
Statistical analyses
Safety analyses comprised all patients who received the IV infusion or took at least one tablet in the randomized period (treated patients). Treatment-emergent adverse events (TEAEs) were summarized using descriptive statistics. Efficacy analyses comprised all randomized patients who received the IV infusion and took at least one tablet, and who had valid MADRS-6 subscale score assessments at baseline and at least once postbaseline [full-analysis set (FAS)].
The primary efficacy endpoint assessing the change from baseline at day 1 in the MADRS-6 subscale score was analyzed using a restricted maximum likelihood-based mixed model for repeated measurements (MMRM), including study site, visit, and treatment as fixed effects, baseline MADRS-6 subscale score as a covariate, and interactions for treatment-by-visit and baseline MADRS-6 subscale score-by-visit.
P values for the treatment differences were estimated based on the least squares mean for the treatment-by-visit interaction in the MMRM. Other endpoints were analyzed using MMRM models similar to that specified for the primary endpoint.
To account for multiplicity, a hierarchical testing procedure was applied, testing first the mean treatment difference for the primary endpoint (the change from baseline to day 1 in MADRS-6 subscale score), then for the key secondary endpoint (change from baseline to day 3 in MADRS-6 subscale score), provided the primary endpoint met statistical significance at a threshold of 0.05. If not, testing according to the strategy was stopped, and subsequent P values considered nominal.
Mean changes from baseline and mean treatment differences with standard errors (SEs) are reported, unless otherwise stated. All statistical tests are two-sided. Data were analyzed using SAS, Version 9.4 statistical software.
Sample size determination
Assuming an effect size at day 1 of 3 MADRS-6 points, no missing data, and a standard deviation (SD) of 4.5, a total of 80 patients (40 per treatment group) would provide a power of at least 80% at a two-sided alpha level of 5%.
Population pharmacokinetic analysis
The sampled plasma concentrations of vortioxetine were analyzed and modeled using non-linear mixed effect methods, based on a previously developed model for vortioxetine (Areberg et al., 2014). The population pharmacokinetic model was used to simulate full individual plasma concentration-time profiles.
Results
All 81 patients who were screened entered the placebo lead-in, and 80 (98.8%) were randomized to the double-blind treatment period (Fig. 1). Of those randomized, 78 patients (97.5%) completed the study.
Fig. 1.
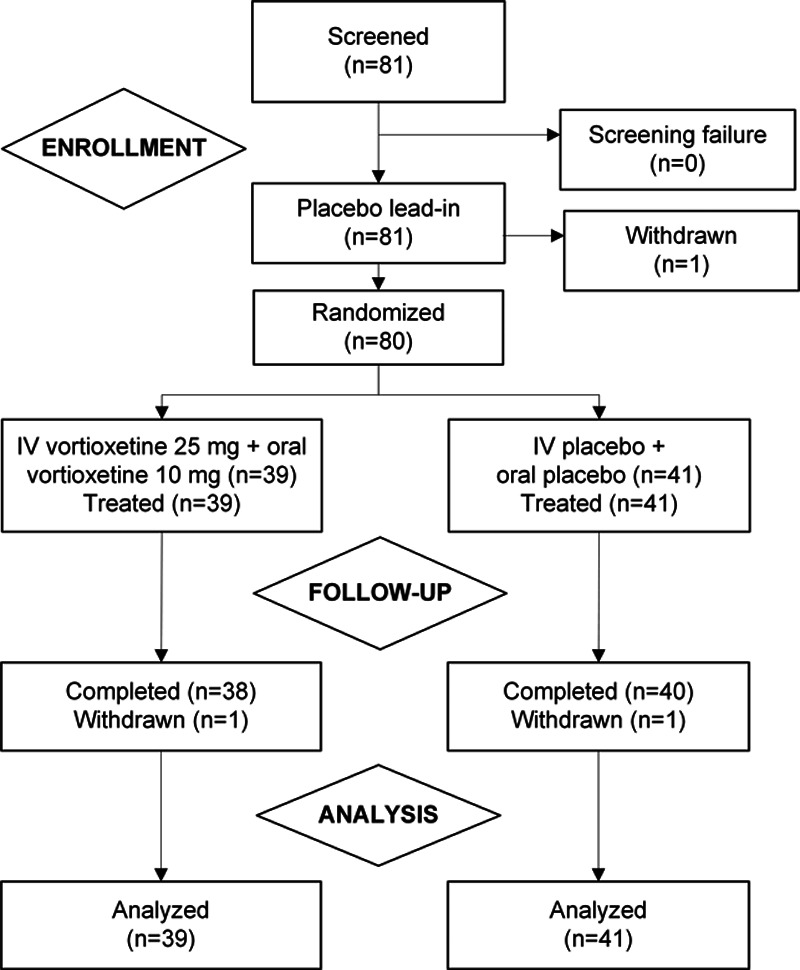
Patient disposition.
The mean age was 47 years and most (73%) patients were females; the mean MADRS total score at baseline was 35, indicating severe depression (Table 1). The treatment groups were comparable in demographic and clinical characteristics.
Table 1.
Demographic and baseline clinical characteristics
| Vortioxetine (n = 39) | Placebo (n = 41) | |
|---|---|---|
| Demographic and clinical characteristics | ||
| Women, n (%) | 28 (71.8) | 30 (73.2) |
| Age (years) | 47.3 (11.0) | 46.9 (13.7) |
| Ethnicity, n (%) | ||
| Caucasian | 39 (100) | 40 (97.6) |
| Black or African-American | 0 | 1 (2.4) |
| BMI (kg/m2) | 25.0 (3.7) | 24.6 (3.5) |
| Duration of current episode (days) | 150.6 (50.2) | 165.5 (77.1) |
| Median (days) | 140 | 131 |
| Number of previous episodes | 3.4 (2.0) | 2.8 (1.7) |
| Range | 1-10 | 1-8 |
| Treatment at study entry | ||
| SSRI, n (%) | 26 (66.7) | 29 (70.7) |
| SNRI, n (%) | 13 (23.3) | 12 (29.3) |
| Baseline clinical assessments | ||
| MADRS total score | 35.6 (3.4) | 34.7 (2.7) |
| MADRS-6 subscale score | 23.4 (2.3) | 22.9 (2.2) |
| CGI-S score | 5.1 (0.6) | 5.1 (0.5) |
| HADS-D score | 16.5 (3.2) | 15.7 (3.2) |
| HADS-A score | 12.1 (3.0) | 11.8 (3.6) |
| PGIC score | 1.4 (0.6) | 1.5 (0.6) |
Mean (SD) reported unless otherwise specified. Demographic and clinical characteristics based on all treated patients; clinical assessments based on the full-analysis set.
CGI-I, Clinical Global Impressions – Improvement; CGI-S, Hospital Anxiety and Depression Scale – Depression; HADS-A, Hospital Anxiety and Depression Scale – Anxiety; MADRS, Montgomery–Åsberg Depression Scale; PGI-C, patient global impression of change; SNRI, serotonin-norepinephrine reuptake inhibitors; SSRI, selective serotonin reuptake inhibitors.
Efficacy
Improvement during the single-blind lead-in was −0.6 MADRS-6 point; at day 1 after the IV infusion with either vortioxetine or placebo, improvements were seen of −3.6 (SE = 0.6) and −2.8 (SE = 0.6) MADRS-6 points for vortioxetine and placebo, respectively, with a nonsignificant difference of −0.8 (SE = 0.7, P = 0.263) in favor of vortioxetine (Fig. 2a). As the primary endpoint was not met, testing according to the strategy was stopped, and subsequent P values considered nominal. The corresponding changes from baseline in MADRS total score were −4.8 (SE = 0.8) for vortioxetine and −4.2 (SE = 0.8) for placebo (P = 0.514; Fig. 2b). Similar results were seen at days 3 and 7, with a slightly larger numerical improvement in the placebo group at the end of treatment (Fig. 2).
Fig. 2.
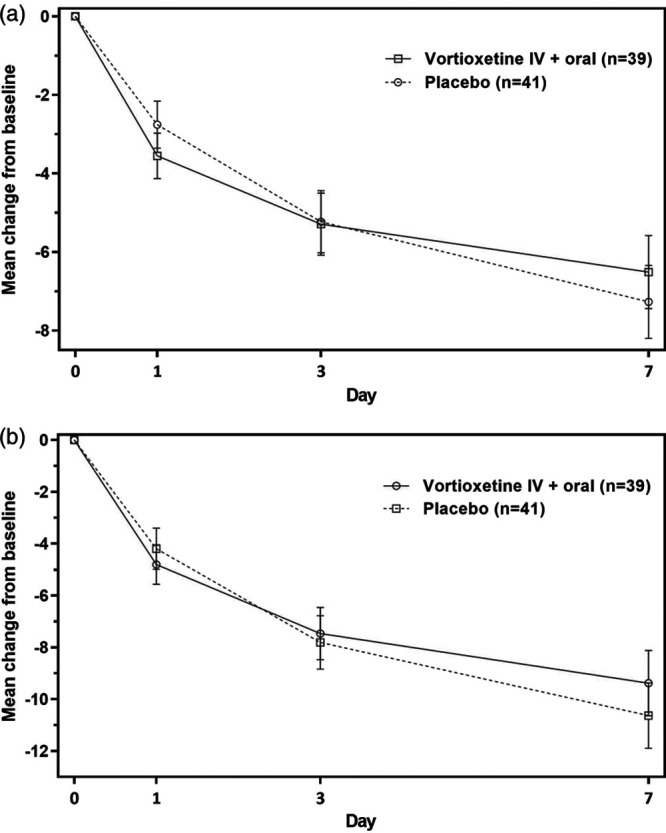
MADRS-6 subscale score (a) and MADRS total score (b) across study period (FAS, MMRM). Treatment differences are based on the least squares mean; error bars represent standard errors. FAS, full-analysis set; MADRS, Montgomery–Åsberg Depression Scale; MMRM, mixed model for repeated measurements.
Other endpoints showed comparable results (Table 2) except the HADS-A subscale score, for which numerically larger improvements were seen with vortioxetine than with placebo, most pronouncedly at the earlier timepoints [mean difference vs placebo = −1.3 (P = 0.064) and −1.4 (P = 0.063) at days 1 and 3, respectively (Table 2)].
Table 2.
Efficacy endpoints (full-analysis set, mixed model for repeated measurements)
| Mean (SE) change from baseline | Mean (SE) difference vs placebo | P value | |
|---|---|---|---|
| Day 1 | |||
| MADRS-6 subscale score | −3.6 (0.6) | −0.8 (0.7) | 0.263 |
| MADRS total score | −4.8 (0.8) | −0.6 (0.9) | 0.514 |
| CGI-S score | −0.3 (0.1) | 0.1 (0.1) | 0.624 |
| CGI-I scorea | 3.3 (0.1) | −0.1 (0.2) | 0.759 |
| HADS-D subscale score | −2.6 (0.6) | 0.1 (0.7) | 0.874 |
| HADS-A subscale score | −3.4 (0.5) | −1.3 (0.7) | 0.064 |
| PGI-C scorea | 2.9 (0.3) | −0.2 (0.3) | 0.465 |
| Day 3 | |||
| MADRS-6 subscale score | −5.3 (0.8) | −0.1 (1.0) | 0.956 |
| MADRS total score | −7.5 (1.0) | 0.3 (1.3) | 0.798 |
| CGI-S score | −0.7 (0.1) | 0.0 (0.2) | 0.944 |
| CGI-I scorea | 3.1 (0.1) | −0.0 (0.2) | 0.830 |
| HADS-D subscale score | −3.6 (0.7) | 0.2 (0.9) | 0.864 |
| HADS-A subscale score | −4.1 (0.6) | −1.4 (0.7) | 0.063 |
| PGI-C scorea | 3.2 (0.3) | −0.3 (0.3) | 0.353 |
| Day 7 | |||
| MADRS-6 subscale score | −6.5 (0.9) | 0.8 (1.2) | 0.542 |
| MADRS total score | −9.4 (1.3) | 1.2 (1.7) | 0.461 |
| CGI-S score | −0.9 (0.2) | 0.1 (0.2) | 0.563 |
| CGI-I scorea | 2.8 (0.2) | −0.1 (0.2) | 0.749 |
| HADS-D subscale score | −5.1 (0.8) | 0.8 (1.1) | 0.464 |
| HADS-A subscale score | −4.4 (0.6) | −0.5 (0.7) | 0.518 |
| PGI-C scorea | 3.8 (0.3) | −0.2 (0.4) | 0.545 |
Vortioxetine, n = 39; placebo, n = 41.
CGI-S, Clinical Global Impressions – Severity of Illness; CGI-I, Clinical Global Impressions – Improvement; FAS, full-analysis set; HADS-D, Hospital Anxiety and Depression Scale – Depression; HADS-A, Hospital Anxiety and Depression Scale – Anxiety; MADRS, Montgomery–Åsberg Depression Scale; MMRM, mixed model for repeated measurements; PGI-C, patient global impression of change.
Absolute score. Treatment differences are based on the least squares mean.
Pharmacokinetics
Plasma concentrations were within the lower expected range based on simulations (see Supplementary Fig., Supplemental digital content 1, http://links.lww.com/ICP/A78 which shows the estimated vs prior expected vortioxetine plasma concentration), and steady-state was reached within 24 h after dosing.
Safety
In the placebo lead-in, a total of five patients (6.2%) reported an adverse event. In the randomized treatment period, adverse events were reported by 21 patients (53.8%) treated with vortioxetine and 15 (36.6%) treated with placebo (Table 3); the overall most common TEAEs (≥10% in either group) were nausea, dizziness, and erythema. More patients treated with vortioxetine reported erythema, insomnia, anxiety, and increased blood pressure, whereas events of sedation and somnolence were more common among those receiving placebo. Two patients, both in the vortioxetine group, reported mild infusion site skin reactions.
Table 3.
Summary of treatment-emergent adverse events and treatment-emergent adverse events occurring in ≥5% in either treatment group during the 1-week treatment period
| Vortioxetine, n (%) | Placebo, n (%) | |
|---|---|---|
| Number of treated patients | 39 | 41 |
| Patients with TEAEs | 21 (53.8) | 15 (36.6) |
| TEAEs occurring in ≥5% in either group | ||
| Nauseaa | 5 (12.8) | 7 (17.1) |
| Dizziness | 4 (10.3) | 3 (7.3) |
| Erythema | 4 (10.3) | 0 |
| Insomnia | 3 (7.7) | 1 (2.4) |
| Headache | 2 (5.1) | 1 (2.4) |
| Diarrhea | 2 (5.1) | 1 (2.4) |
| Anxiety | 2 (5.1) | 0 |
| Blood pressure increased | 2 (5.1) | 0 |
| Sedation | 1 (2.6) | 3 (7.3) |
| Somnolence | 1 (2.6) | 4 (9.8) |
No serious adverse events or deaths occurred during the study. No patients withdrew from the study due to adverse events.
TEAE, treatment-emergent adverse event.
One patient in the vortioxetine group reported vomiting.
More patients in the placebo group reported nausea [7 (17.1%) vs 5 (12.8%) in the vortioxetine group]. Most of the patients (four of five) in the vortioxetine group who reported nausea did so within 24 h postinfusion, with no new incidences after day 3, whereas in the placebo group, nausea occurred relatively later, with five of seven patients reporting nausea on day 2 or later. Nausea was mostly mild (four of six events for vortioxetine and four of seven for placebo) and otherwise of moderate intensity. One patient in the vortioxetine group reported vomiting of mild intensity. The median duration of nausea within the 7-day treatment period was 3 days among patients treated with vortioxetine, and 1 day among those receiving placebo.
One patient allocated to placebo reported a serious adverse event (exacerbation of MDD) 15 days after first dose (safety follow-up period). No deaths occurred during the study, and no patients withdrew due to adverse events. There were no notable findings in the clinical safety laboratory tests, vital signs, or ECG parameters.
Discussion
Although this small-scale, exploratory study confirmed previous findings that a single initial dose of vortioxetine facilitated plasma-concentration at the steady-state level within 24 h after dosing (Vieta et al., 2019), no difference vs placebo was seen in the early antidepressant effect of a single initial IV dose of vortioxetine added to oral daily treatment at any timepoint during a 7-day treatment period. Thus, the study did not meet its primary endpoint.
Albeit statistically nonsignificant, the numerically larger improvement in patient-rated symptoms of anxiety (HADS-A) among patients treated with vortioxetine compared with placebo is consistent with an early anxiolytic effect of vortioxetine seen in previous studies showing reduced levels of anxiety in anxious depression (Alvarez et al., 2012; Baldwin et al., 2016b) as well as generalized anxiety (Bidzan et al., 2012; Liebowitz et al., 2017). In contrast, transient anxiogenic effects are seen in some patients after the initial administration of SSRIs, potentially due to 5-HT1A-mediated reductions in serotonergic firing (Piñeyro and Blier, 1999; Zienowicz et al., 2006; Akimova et al., 2009). In this study, the numerical reductions in symptoms of anxiety with vortioxetine vs placebo were most pronounced at earlier timepoints, consistent with the findings from the previous study with IV vortioxetine (Vieta et al., 2019). Such early improvements in anxiety symptoms with vortioxetine may hypothetically be related to modulation of serotonin receptors in addition to SERT blockade, specifically 5-HT1A agonism leading to desensitization of autoreceptors and increased serotonergic neurotransmission (Bétry et al., 2013).
In the previous exploratory study of the efficacy of a single IV infusion of vortioxetine compared with IV placebo, in which both treatments were followed by daily oral vortioxetine, all patients improved fast and substantially (at approximately 14 points in MADRS total score at day 7), with no separation between the treatments for the primary endpoint at day 7; however, numerically larger improvements with IV vortioxetine at days 1 and 3 were seen consistently across outcomes (Vieta et al., 2019). Based on these findings, the current study design applied features aiming at mitigating limitations related to assay sensitivity in the previous IV study, foremost the high response rate among patients who received IV placebo plus oral vortioxetine. Thus, the current study used a placebo-only (IV and oral) comparator, hence omitting any effects from oral vortioxetine in the placebo arm, and counteracting expectation bias by reducing the probability of receiving active treatment. Likewise, the single-blind placebo lead-in in this study aimed at reducing the impact from expectation bias by masking the time when patients could potentially receive active treatment. Finally, patients in this study were required to be hospitalized per standard of care to reduce improvement caused by per-protocol hospital care and environment.
Keeping in mind the study design differences between this and the previous IV study and the consequent limitations in their direct comparison, overall, the response in the placebo arm in this study was smaller compared to Vieta et al. (2019) (−4.2 vs −5.9 MADRS total score points at day 1) and the lead-in period may thus have had some impact in attenuating the placebo response, as may the blinding of raters to patients’ previous status. However, the response in the vortioxetine arm in this study was also smaller, resulting in a net effect of comparable magnitude to that observed by Vieta et al. (2019).
This study confirmed the favorable tolerability profile that has previously been shown with vortioxetine, also as a switch therapy (Baldwin et al., 2016a; Thase et al., 2017). In this study, switching patients directly and rapidly from an SSRI/SNRI to a single high dose of vortioxetine did not compromise tolerability, with lower levels of nausea compared both with the previous examination of 17 mg IV vortioxetine in a nonswitch population (Vieta et al., 2019) and with oral doses of vortioxetine (5–20 mg) (Baldwin et al., 2016a), likely reflecting tolerance to the side effects elicited by SERT inhibition developed with the previous SSRI/SNRI therapy.
Limitations
The single-blind placebo lead-in may have succeeded only partly in reducing the placebo response, as indicated by the limited improvement after the IV placebo infusion in the lead-in period relative to the substantial response seen after IV placebo in the randomized treatment period. Because investigators were not blinded to the time of randomization, this might suggest that the clinician-rated assessments (MADRS; CGI scales) may have been subject to some level of bias related to their awareness of when improvements could be expected. Although the patients were blinded to the placebo lead-in study feature, a similar pattern was observed for the patient-reported assessments (HADS scales; PGI), which might hypothetically be explained by ‘socially transmitted’ placebo effects (Chen et al., 2019), that is, investigators’ inadvertent and subconscious communication of their own expectations to the patient, and/or by patients not expecting an effect immediately after the first (placebo-) IV based on their previous experience or knowledge about antidepressant (SSRI) treatment. Altogether, such limitations may have led to reduced assay sensitivity, hence attenuating any treatment differences between vortioxetine and placebo, in this already small study.
Conclusion
This small-scale, exploratory study showed no difference in the early antidepressant effect of a single initial IV dose of vortioxetine added to daily oral treatment versus placebo at any timepoint during a 7-day treatment period. Except a numerically larger improvement in anxiety symptoms with vortioxetine vs placebo, similar results were seen for other endpoints. Pharmacokinetic data confirmed that IV vortioxetine facilitated reaching steady-state plasma concentration within 24 h. IV plus oral vortioxetine was well tolerated, with low levels of nausea as the most common adverse event, and no new safety signals compared to previous studies with vortioxetine administered as IV or oral formulations.
Acknowledgements
The authors thank all participants in the study, as well as the investigators and sites involved in conducting the trial, and Simon N. Schmidt, biostatistician at H. Lundbeck A/S, for statistical support. Medical writing support was provided by Hanne-Lise F. Eriksen, PhD, employee of H. Lundbeck A/S. This study was supported by H. Lundbeck A/S.
Conflicts of interest
E.R. has received research grants from Gedeon Richter and Lundbeck, and speaker honoraria from and is a member of advisory panels for Abbvie, Gedeon Richter, Grindex, Janssen Cilag, Lundbeck, Servier, and Zentiva. J.Z., M.D., C.B., J.A., A.E., and I.F. are employees of H. Lundbeck A/S.
Supplementary Material
Footnotes
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.intclinpsychopharm.com
References
- Akimova E, Lanzenberger R, Kasper S. The serotonin-1A receptor in anxiety disorders. Biol Psychiatry. 2009; 66:627–635 [DOI] [PubMed] [Google Scholar]
- Alvarez E, Perez V, Dragheim M, Loft H, Artigas F. A double-blind, randomized, placebo-controlled, active reference study of Lu AA21004 in patients with major depressive disorder. Int J Neuropsychopharmacol. 2012; 15:589–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders. 20165th ed, Washington, DC: American Psychiatric Association [Google Scholar]
- Areberg J, Petersen KB, Chen G, Naik H. Population pharmacokinetic meta-analysis of vortioxetine in healthy individuals. Basic Clin Pharmacol Toxicol. 2014; 115:552–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Areberg J, Søgaard B, Højer AM. The clinical pharmacokinetics of Lu AA21004 and its major metabolite in healthy young volunteers. Basic Clin Pharmacol Toxicol. 2012; 111:198–205 [DOI] [PubMed] [Google Scholar]
- Artigas F, Bortolozzi A, Celada P. Can we increase speed and efficacy of antidepressant treatments? Part I: general aspects and monoamine-based strategies. Eur Neuropsychopharmacol. 2018; 28:445–456 [DOI] [PubMed] [Google Scholar]
- Baldwin DS, Chrones L, Florea I, Nielsen R, Nomikos GG, Palo W, Reines E. The safety and tolerability of vortioxetine: analysis of data from randomized placebo-controlled trials and open-label extension studies. J Psychopharmacol. 2016; 30:242–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin DS, Florea I, Jacobsen PL, Zhong W, Nomikos GG. A meta-analysis of the efficacy of vortioxetine in patients with major depressive disorder (MDD) and high levels of anxiety symptoms. J Affect Disord. 2016; 206:140–150 [DOI] [PubMed] [Google Scholar]
- Bech P. Rating scales in depression: limitations and pitfalls. Dialogues Clin Neurosci. 2006; 8:207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bech P, Tanghøj P, Andersen HF, Overø K. Citalopram dose-response revisited using an alternative psychometric approach to evaluate clinical effects of four fixed citalopram doses compared to placebo in patients with major depression. Psychopharmacology (Berl). 2002; 163:20–25 [DOI] [PubMed] [Google Scholar]
- Bétry C, Pehrson AL, Etiévant A, Ebert B, Sánchez C, Haddjeri N. The rapid recovery of 5-HT cell firing induced by the antidepressant vortioxetine involves 5-HT3 receptor antagonism. Int J Neuropsychopharmacol. 2013; 16:1115–1127 [DOI] [PubMed] [Google Scholar]
- Bidzan L, Mahableshwarkar AR, Jacobsen P, Yan M, Sheehan DV. Vortioxetine (Lu AA21004) in generalized anxiety disorder: results of an 8-week, multinational, randomized, double-blind, placebo-controlled clinical trial. Eur Neuropsychopharmacol. 2012; 22:847–857 [DOI] [PubMed] [Google Scholar]
- Chen PA, Cheong JH, Jolly E, Elhence H, Wager TD, Chang LJ. Socially transmitted placebo effects. Nat Hum Behav. 2019; 3:1295–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citrome L. Vortioxetine for major depressive disorder: an indirect comparison with duloxetine, escitalopram, levomilnacipran, sertraline, venlafaxine, and vilazodone, using number needed to treat, number needed to harm, and likelihood to be helped or harmed. J Affect Disord. 2016; 196:225–233 [DOI] [PubMed] [Google Scholar]
- Dale E, Grunnet M, Pehrson AL, Frederiksen K, Larsen PH, Nielsen J, et al. The multimodal antidepressant vortioxetine may facilitate pyramidal cell firing by inhibition of 5-HT3 receptor expressing interneurons: An in vitro study in rat hippocampus slices. Brain Res. 2018; 1689:1–11 [DOI] [PubMed] [Google Scholar]
- Gunduz-Bruce H, Silber C, Kaul I, Rothschild AJ, Riesenberg R, Sankoh AJ, et al. Trial of SAGE-217 in patients with major depressive disorder. N Engl J Med. 2019; 381:903–911 [DOI] [PubMed] [Google Scholar]
- Guy W. Guy W. Clinical global impressions. ECDEU Assessment Manual for Psychopharmacology. Revised ed. 1976, Rockville, MD: National Institute of Mental Health [Google Scholar]
- Hurst H, Bolton J. Assessing the clinical significance of change scores recorded on subjective outcome measures. J Manipulative Physiol Ther. 2004; 27:26–35 [DOI] [PubMed] [Google Scholar]
- Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Int Clin Psychopharmacol. 2012; 27:215–223 [DOI] [PubMed] [Google Scholar]
- Kraus C, Kadriu B, Lanzenberger R, Zarate CA, Jr, Kasper S. Prognosis and improved outcomes in major depression: a review. Transl Psychiatry. 2019; 9:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebowitz MR, Careri J, Blatt K, Draine A, Morita J, Moran M, Hanover R. Vortioxetine versus placebo in major depressive disorder comorbid with social anxiety disorder. Depress Anxiety. 2017; 34:1164–1172 [DOI] [PubMed] [Google Scholar]
- Machado-Vieira R, Salvadore G, Luckenbaugh DA, Manji HK, Zarate CA., Jr Rapid onset of antidepressant action: a new paradigm in the research and treatment of major depressive disorder. J Clin Psychiatry. 2008; 69:946–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi GS, Mann JJ. Depression. Lancet. 2018; 392:2299–2312 [DOI] [PubMed] [Google Scholar]
- Meltzer-Brody S, Colquhoun H, Riesenberg R, Epperson CN, Deligiannidis KM, Rubinow DR, et al. Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet. 2018; 392:1058–1070 [DOI] [PubMed] [Google Scholar]
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979; 134:382–389 [DOI] [PubMed] [Google Scholar]
- Piñeyro G, Blier P. Autoregulation of serotonin neurons: role in antidepressant drug action. Pharmacol Rev. 1999; 51:533–591 [PubMed] [Google Scholar]
- Popova V, Daly EJ, Trivedi M, Cooper K, Lane R, Lim P, et al. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. Am J Psychiatry. 2019; 176:428–438 [DOI] [PubMed] [Google Scholar]
- Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini-International Neuropsychiatric Interview (MINI): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998; 59:22–33 [PubMed] [Google Scholar]
- Thase ME, Bowden CL, Nashat M, Eudicone JM, Marcus R, McQuade RD, Carlson BX. Aripiprazole in bipolar depression: a pooled, post-hoc analysis by severity of core depressive symptoms. Int J Psychiatry Clin Pract. 2012; 16:121–131 [DOI] [PubMed] [Google Scholar]
- Thase ME, Danchenko N, Brignone M, Florea I, Diamand F, Jacobsen PL, Vieta E. Comparative evaluation of vortioxetine as a switch therapy in patients with major depressive disorder. Eur Neuropsychopharmacol. 2017; 27:773–781 [DOI] [PubMed] [Google Scholar]
- Thase ME, Mahableshwarkar AR, Dragheim M, Loft H, Vieta E. A meta-analysis of randomized, placebo-controlled trials of vortioxetine for the treatment of major depressive disorder in adults. Eur Neuropsychopharmacol. 2016; 26:979–993 [DOI] [PubMed] [Google Scholar]
- Vieta E, Florea I, Schmidt SN, Areberg J, Ettrup A. Intravenous vortioxetine to accelerate onset of effect in major depressive disorder: a 2-week, randomized, double-blind, placebo-controlled study. Int Clin Psychopharmacol. 2019; 34:153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zienowicz M, Wisłowska-Stanek A, Lehner M, Taracha E, Maciejak P, Sobolewska A, et al. Fluoxetine-induced anxiety and nervousness. Pharmacol Rep. 2006; 58:115–119 [PubMed] [Google Scholar]
- Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983; 67:361–370 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.