

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease affecting motoneurons (MNs) in a motor-unit (MU)-dependent manner. Glial dysfunction contributes to numerous aspects of the disease. At the neuromuscular junction (NMJ), early alterations in perisynaptic Schwann cell (PSC), glial cells at this synapse, may impact their ability to regulate NMJ stability and repair. Indeed, muscarinic receptors (mAChRs) regulate the repair phenotype of PSCs and are overactivated at disease-resistant NMJs [soleus muscle (SOL)] in SOD1G37R mice. However, it remains unknown whether this is the case at disease-vulnerable NMJs and whether it translates into an impairment of PSC-dependent repair mechanisms. We used SOL and sternomastoid (STM) muscles from SOD1G37R mice and performed Ca2+-imaging to monitor PSC activity and used immunohistochemistry to analyze their repair and phagocytic properties. We show that PSC mAChR-dependent activity was transiently increased at disease-vulnerable NMJs (STM muscle). Furthermore, PSCs from both muscles extended disorganized processes from denervated NMJs and failed to initiate or guide nerve terminal sprouts at disease-vulnerable NMJs, a phenomenon essential for compensatory reinnervation. This was accompanied by a failure of numerous PSCs to upregulate galectin-3 (MAC-2), a marker of glial axonal debris phagocytosis, on NMJ denervation in SOD1 mice. Finally, differences in these PSC-dependent NMJ repair mechanisms were MU type dependent, thus reflecting MU vulnerability in ALS. Together, these results reveal that neuron-glia communication is ubiquitously altered at the NMJ in ALS. This appears to prevent PSCs from adopting a repair phenotype, resulting in a maladapted response to denervation at the NMJ in ALS.
SIGNIFICANCE STATEMENT Understanding how the complex interplay between neurons and glial cells ultimately lead to the degeneration of motor neurons and loss of motor function is a fundamental question to comprehend amyotrophic lateral sclerosis (ALS). An early and persistent alteration of glial cell activity takes place at the neuromuscular junction (NMJ), the output of motor neurons, but its impact on NMJ repair remains unknown. Here, we reveal that glial cells at disease-vulnerable NMJs often fail to guide compensatory nerve terminal sprouts and to adopt a phagocytic phenotype on denervated NMJs in SOD1G37R mice. These results show that glial cells at the NMJ elaborate an inappropriate response to NMJ degeneration in a manner that reflects motor-unit (MU) vulnerability and potentially impairs compensatory reinnervation.
Keywords: galectin-3, muscarinic receptors, neuron-glia interactions, perisynaptic Schwann cells, purinergic receptors, sprouting
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the progressive loss of motoneurons (MNs), with fast-fatigable (FF) motor-units (MUs) being more vulnerable than fast-fatigue-resistant (FR) and slow (S) MUs (Frey et al., 2000; Pun et al., 2006; Hegedus et al., 2007, 2008). Over the last decade, evidence of disease onset and progression being modulated by numerous non-cell autonomous mechanisms has highlighted the importance of glial cells in this disease (Boillée et al., 2006a; Ilieva et al., 2009; Kang et al., 2013; Ditsworth et al., 2017). Evidence from ALS patients and SOD1 mouse models also shows early loss of neuromuscular junctions (NMJ) before disease onset (Fischer et al., 2004; Pun et al., 2006; Hegedus et al., 2007, 2008; Martineau et al., 2018), supporting the notion that ALS has a long silent presymptomatic phase (Eisen et al., 2014). Interestingly, an early and persistent functional alteration of perisynaptic Schwann cells (PSCs), glial cells at the NMJ, was reported in the SOD1G37R mouse model (Arbour et al., 2015).
Early PSC dysfunction in ALS could be of particular importance owing to their ability to detect and modulate synaptic communication, regulate NMJ stability, and oversee NMJ repair. Importantly, these three functions are interdependent as PSC synaptic decoding regulates their ability to stabilize or repair NMJs, notably through muscarinic receptors (mAChRs). Indeed, activation of PSC mAChRs and purinergic receptors triggers a transient increase in cytoplasmic calcium (Ca2+ response), through which they modulate neurotransmitter release (Robitaille, 1998; Rochon et al., 2001; Rousse et al., 2010; Todd et al., 2010). Blockade of PSC mAChRs destabilizes NMJs (Wright et al., 2009), demonstrating that proper PSC synaptic decoding is necessary for NMJ maintenance.
Importantly, PSCs contribute to NMJ reinnervation following denervation of NMJs by adopting a “pro-regenerative” (repair) phenotype. For instance, PSCs extend long processes known to guide reinnervation through nerve terminal sprouting (Reynolds and Woolf, 1992; Son and Thompson, 1995a,b; Son et al., 1996; O'Malley et al., 1999). Similarly to axonal SCs (Reichert et al., 1994; Painter et al., 2014; Brosius Lutz et al., 2017), PSCs also actively phagocytose presynaptic debris following denervation (Duregotti et al., 2015; Cunningham et al., 2020), which is essential for efficient axonal regrowth and NMJ reinnervation (Kang and Lichtman, 2013). Finally, complete PSC endplate coverage is essential for reinnervation since parts of the endplate vacated by PSCs are forsaken during reinnervation (Kang et al., 2014). Importantly, mAChRs activation represses PSC's repair phenotype and can modulate NMJ repair by regulating PSC gene expression (Georgiou et al., 1994, 1999).
Interestingly, Arbour et al. (2015) reported an enhanced mAChR contribution to PSC Ca2+ responses at NMJs innervated by disease-resistant or moderately vulnerable MUs [S and FR MU, respectively, soleus (SOL) muscle] in SOD1G37R mice. As previously proposed (Ko and Robitaille, 2015; Arbour et al., 2017), we postulated that inappropriate mAChR signaling could prevent PSCs from adopting a repair phenotype, thus hindering NMJ reinnervation in ALS. However, this repair phenotype remains unevaluated in symptomatic animals. Furthermore, recent evidence revealed opposite changes in the synaptic properties of NMJs from different MU types in SOD1G37R mice (Tremblay et al., 2017). This raises the possibility that PSC properties, notably mAChR overactivation, may be different at NMJs innervated by vulnerable FF MUs.
In the present study, we sought to examine three main questions. First, we assessed if PSCs mAChR overactivation was also present at NMJs innervated by vulnerable FF MUs in the sternomastoid (STM) muscle at different stages of the disease. Second, we evaluated PSCs' phagocytic phenotype at vulnerable [STM or extensor digitorum longus (EDL)] or partially resistant (SOL) NMJs, in presymptomatic mice following experimentally-induced denervation or in symptomatic SOD1G37R mice. Finally, we evaluated the presence of extended PSC processes, nerve terminal sprouts in symptomatic SOD1G37R mice at vulnerable or partially resistant NMJs. Albeit through a different mechanism, PSCs in the STM also displayed an early, but transient, increase in mAChR-dependent decoding. Consistent with defects in PSC-dependent NMJ repair mechanisms in all these muscles, PSCs displayed abnormal process extension as well as paradoxical expression of galectin-3 (MAC-2), a marker of glial phagocytosis. Altogether, these results suggest that changes in PSCs are maladapted in ALS.
Materials and Methods
Animals
Male transgenic mice heterozygote for the human Sod1 gene carrying the G37R mutation (SOD1G37R, line 29; [B6.Cg-Tg(SOD1*G37R)29Dpr/J]; stock number 008229), or, in some experiments, the G93A mutation (SOD1G93A; [B6SJL-Tg(SOD1*G93A)1Gur/J]; stock number 002726), were obtained from The Jackson Laboratory. SOD1G37R mice were maintained on a C57BL/6 background and were genotyped by PCR for the hSOD1 gene performed on a tail sample taken at the time of the weaning.
For some experiments, transgenic mice expressing the yellow fluorescent protein (YFP) in all motor neurons (Feng et al., 2000) were used (homozygous Thy1-YFP, line 16; [B6.Cg-Tg(Thy1-YFP)16Jrs/J]; stock number 003709). These mice were also obtained from The Jackson Laboratory and were maintained on a C57BL/6 background.
Presymptomatic SOD1G37R mice were used at P119–P131 (P120) and P179–P194 (P180; Fig. 1A). Phenotypically matched late symptomatic SOD1G37R mice were used between P505 and P565. Age-matched wild-type (WT) brothers were used as controls. Animals were killed by exposition to a lethal dose of isoflurane (CDMV, Saint-Hyacinthe, Canada). All experiments were performed in accordance with the guidelines of the Canadian Council of Animal Care and the Comité de Déontologiede l'Expérimentation sur les Animaux of Université de Montréal.
Figure 1.

Disease progression in SOD1G37R mice and fiber type composition of the STM muscle. A, Kaplan–Meier plot of the ages at which disease onset (green; median age: 456 d) and early disease (blue; median age: 506 d) are reached in SOD1G37R mice under our conditions. All the symptomatic animals displayed similar motor phenotypes and were killed (red, median 527 d) after the early disease stage. Experiments on presymptomatic animals were carried at P120 and P180. B, C, Immunostaining of STM cross-sections for MHC Type IIb (FF, red) and IIa (FR, green; B) or all isotypes except IIx (all but IIx, green) and Type IIx (FF, red; C). Scale bars = 200 μm. D, Representative examples of presynaptic nerve terminals (green, SV2 and NF-M), postsynaptic nAChRs red (α-BTX), and glia (blue; S100β) labeling from the STM of P120 WT and SOD1G37R mice. Scale bars = 20 µm.
Phenotype evaluation
Starting at P290 or earlier, mice were weighted weekly to assess disease onset and progression as previously described (Lobsiger et al., 2009; Parone et al., 2013). Briefly, disease onset and early disease were defined as peak body weight and 10% loss from peak body weight, respectively. Consistent with the recent reduction in transgene copy number in this line (Zwiegers et al., 2014), the median age of onset of the symptomatic animals used in this study was 456 d, while the median age of early disease was 506 d (N = 19; Fig. 1A). Phenotypically matched symptomatic animals were killed for experimentation when three criteria were met (P505–P565) and are hereafter referred to as “symptomatic SOD1G37R mice”: (1) they passed the early disease stage (10% weight loss); (2) they displayed hindlimb paralysis as assessed by tail suspension; and (3) they showed a reduced grid hanging time (<10 s). The average duration of the disease progression (from age of onset to killing) was 78.26 ± 6.40 d.
Sciatic nerve crush surgery
Mice were anesthetized using isoflurane (2–3% in 98–97% O2) in an induction chamber and anesthesia was then maintained under a breathing mask. Lubricant (Optixcare) was applied on the eyes to prevent dryness. Mice were placed in prone position and an incision was made on the left mid-thigh. The gluteus maximus and the biceps femoris muscles were delicately separated to expose the sciatic nerve. The nerve was then crushed with Moria microserrated curved forceps (MC31; maximal pressure for 15 s). Muscles were then gently repositioned and the wound closed using 5–0 or 6–0 Vicryl suture (CDMV). Buprenorphine (3 μg/10 g of body weight; Temgesic) was subcutaneously administered three times during the following 24 h. At the end of the surgery, mice were also injected subcutaneously with 0.25 ml of warm 0.9% saline twice to prevent dehydration. For sham-operated animals, the same procedure was followed, but the sciatic nerve was only exposed and not crushed.
STM calcium imaging
Both STM muscles and their nerves were dissected from the same animal in oxygenated REES solution containing the following: 110 mm NaCl, 5 mm KCl, 25 mm NaHCO3, 2 mm CaCl2, 1 mm MgCl2, 11 mm glucose, 0.3 mm glutamic acid, 0.4 mm glutamine, 5 mm BES (N,N-Bis(2-hydroxyethyl)taurine), 0.036 mm choline chloride, and 4.34 × 10−4 mm thiamine pyrophosphate (all Sigma-Aldrich). Muscles were pinned in a Sylgard184-coated (Dow Corning) recording chamber, and the nerves were placed in two suction electrodes and were independently stimulated using a Pulsemaster A300 stimulator (square pulses: 20 mV to 2 V, 0.1 ms in duration; WPI). PSCs were loaded with the fluorescent Ca2+ indicator Rhod-3 (Life Technologies) by incubating muscles twice for 45 min in preoxygenated REES solution containing 5 μm Rhod-3 A.M. (Life Technologies), 0.02% pluronic acid (Life Technologies), and 0.15% dimethyl sulfoxide (DMSO) at 29 ± 2°C (Todd et al., 2010; Arbour et al., 2015). NMJs were located by labeling nAChRs before the start of the experiment with a closed-bath application of Alexa Fluor 488-conjugated α-bungarotoxin (BTX) (5 μg for 10 min; Life Technologies). PSCs were easily identified using transmitted light microscopy and their identity was further confirmed through their apposition over BTX staining. For experiments performed at P180, changes in fluorescence were monitored using a Zeiss LSM510 confocal microscope equipped with a 40× water-immersion lens (NA: 0.8; Zeiss). Rhod-3 was excited using a 543-nm HeNe laser and emission was filtered using a 560-nm-long pass filter. For experiments performed on symptomatic animals, changes in fluorescence were monitored using an Olympus FV-1000 confocal microscope equipped with a 60× water-immersion lens (NA: 0.9; Olympus). Rhod-3 was excited using a 559-nm diode laser and emission detected through a 570- to 625-nm spectral window. Changes in fluorescence were measured on the PSCs soma using the ImageJ software and were expressed as % ΔF/F0 = (F – F0)/F0. Recordings of PSC Ca2+ responses were not included when baseline fluorescence was unstable, focus changes occurred, spontaneous Ca2+ activity was observed or when Ca2+ levels did not return to the baseline after PSC activation. No direct comparisons between data obtained on different systems were made.
Ca2+ responses to endogenous neurotransmitter release were evoked by high-frequency stimulation of the nerve (50 Hz, 20 s). This pattern is known to elicit robust neurotransmitter release in this preparation (Wyatt and Balice-Gordon, 2008) and to efficiently trigger PSC Ca2+ responses in another fast-twitch muscle (Rousse et al., 2010). Alternatively, PSC mAChRs or purinergic receptors were activated by local application of agonists diluted in the extracellular Ringer REES solution, respectively, muscarine (10 μm; Sigma) or ATP (1 μm; Sigma). Local drug application was achieved by applying a brief pulse of positive pressure (20 PSI, 150 ms) using a Picospritzer II (Parker Instruments) through a glass micropipette (∼2 μm tip, 5 MΩ) positioned next to the NMJ. Because of the rundown of PSC Ca2+ responses following repeated activation (Jahromi et al., 1992; Rochon et al., 2001), local applications of agonists were performed before the high-frequency nerve stimulation was performed, each agonist was applied only once on each NMJ, and ATP and muscarine were applied at least 20 min apart on any given NMJ. Only a single high-frequency stimulation was executed on each muscle and PSC Ca2+ responses were imaged on a naive NMJ. NMJs where high-frequency nerve stimulation did not induce a presynaptic Ca2+ increase were discarded.
In all experiments, muscles were perfused (60–80 ml/h) with heated (28 ± 1°C) REES solution containing 2.3 μg/ml of D-tubocurarine chloride (Sigma) to prevent muscle contraction during nerve stimulation. In some experiments, mAChRs were blocked by adding the non-selective antagonist atropine (10 μm; Sigma) to the extracellular REES solution. To ensure efficient blocking of mAChRs, muscles were perfused at least 45 min before the start of the experiment. PSCs responding to local application of muscarine after atropine blockade were discarded.
Whole-mount muscle preparations and immunohistochemistry
The STM muscle is divided in two easily distinguishable parts on its ventral side: a lateral part composed exclusively of FF MUs on the surface (the “white” part), and a medial part composed of a mixture of FF and FR MUs (the “red” part) as inferred by their fiber type composition (Fig. 1B,C; Brichta et al., 1987). To exclusively evaluate vulnerable FF MUs in all experiments, we restricted our analysis to the white part of the STM which is easily observable using transmitted light. All surface NMJs analyzed in the EDL were associated with vulnerable FF MUs as previously shown (Tremblay et al., 2017). All NMJs analyzed in the soleus (SOL) were associated with either FR or S MUs as previously described (Pun et al., 2006; Arbour et al., 2015; Tremblay et al., 2017).
Immunohistochemical labeling was performed as described elsewhere (Todd et al., 2010; Darabid et al., 2013; Arbour et al., 2015). STM and SOL muscles were dissected in oxygenated (95% O2, 5% CO2) REES physiological solution and were pinned in a Sylgard-coated (Dow Corning) 10 mm Petri dish and fixed for 10 min in 4% formaldehyde (Mecalab) diluted in PBS (137 mm NaCl, 10 mm Na2HPO4, 2.7 mm KCl, and 2 mm KH2PO4; all Sigma-Aldrich) at room temperature (RT). Then, muscles were permeabilized in 100% ice-cold methanol for 6 min at −20°C and non-specific labeling was reduced using 10% normal donkey serum (NDS) diluted in PBS containing 0.01% Triton X-100 (20-min incubation). Muscles were incubated overnight at 4°C with a rat IgG2a anti-MAC-2 (galectin-3) antibody (1:250; clone M3/38; Cedarlane, catalog #CL8942AP), then 2 h at RT with a rabbit IgG anti-S100β antibody (1:250; Dako-Agilent, catalog #Z031101-2 or between 1:4 and 1:5, Dako-Agilent, catalog #IR50461-2) followed by 2 h at RT with the chicken IgY anti-neurofilament medium chain (NF-M; 1:2000; Rockland Immunochemicals; catalog #212-901-D84) and the mouse IgG1 anti-synaptic vesicular protein 2 (SV2; 1:2000; Developmental Studies Hybridoma bank; concentrate) antibodies. Secondary antibodies donkey anti-rabbit IgG Alexa Fluor 594, donkey anti-rat IgG Alexa Fluor 647, goat anti-mouse IgG1 Alexa Fluor 488 and donkey anti-chicken Alexa Fluor 488 were incubated together for 1 h at RT (all 1:500; Jackson ImmunoResearch). All antibody dilutions were made in PBS buffer containing 0.01% Triton X-100 and 2% NDS. Finally, postsynaptic nicotinic receptors (nAChRs) were labeled using α-BTX conjugated with CF405 (4 μg/ml; Biotium; catalog #00002) for 45 min in PBS. In some cases (nerve crush experiment at P90); the anti-SV2 antibody was replaced with goat anti-synaptotagmin (SyT; 1:250). In these cases, goat anti-mouse IgG1 Alexa Fluor 405, donkey anti-chicken Alexa Fluor 405 (both 1:250; Jackson ImmunoResearch), donkey anti-rabbit IgG Alexa 488 (1:500; Jackson ImmunoResearch) and α-BTX conjugated to Alexa Fluor 594 (1.33 μg/ml; Life Technologies; catalog #B13423) were used instead. After each incubation (except NDS blocking), muscles were rinsed three times for 5 min in PBS containing 0.01% Triton X-100. Muscles were mounted in ProlongGold anti-fade reagent (Life Technologies). Images were acquired on an Olympus FV1000 confocal microscope equipped with a 60× oil-immersion objective (N.A: 1.4; Olympus). Fluorescence was excited using a 488-nm (Alexa Fluor 488) argon laser and a 405-, a 559-, and a 633-nm laser diode. Fluorescence emission was filtered using appropriate custom spectral windows (425–475, 500–545, 570–630, and 650–750 nm). For figure representation, γ was adjusted to enhance visibility of certain NMJ structures using Adobe Photoshop. No quantitative measurements were performed on the adjusted images.
For some experiments, muscles were fixed and immunolabeled after calcium imaging experiments to determine their innervations status post hoc. In these experiments, confocal stacks of the BTX labeling pattern of the imaged and neighboring NMJs were acquired live and were used to reidentify the same NMJs following immunolabeling as previously described (Arbour et al., 2015).
Gal-3 labeling on non-permeabilized whole-mount muscle tissue
To label extracellular Gal-3, muscles from Thy1-YFP animals were fixed in 4% formaldehyde and non-specific labeling was blocked using 10% NDS. Muscles were then incubated overnight at 4°C with the rat IgG2a anti-MAC-2 (galectin-3) antibody (1:100), which was then revealed using a donkey anti-rat IgG Alexa Fluor 647 antibody (1:500; 60 min). Postsynaptic nAChRs were labeled with BTX conjugated to Alexa Fluor 594 (5 μg/ml; 45 min). Muscles were rinsed three times for 5 min each between incubations and all dilutions and rinses were made in PBS buffer without Triton X-100. Muscles were then mounted in Prolong Diamond anti-fade reagent containing DAPI (Life Technologies). Using this approach, no labeling of the presynaptic markers SV2 and NF-M could be observed, confirming that cells were not inadvertently permeabilized during the procedure (data not shown).
Analysis of NMJ morphology
Analysis of NMJ morphology was performed using the seven criteria previously described and illustrated by Tremblay et al. (2017). Briefly, NMJ innervation and postsynaptic receptor organization (faint clustered or ectopic nAChRs) were analyzed, and the presence of nerve terminal sprouting, polyinnervation and PSC process extensions was quantified. Although PSCs downregulate the S100β marker following denervation (Magill et al., 2007), PSC somata on fully denervated endplates were still discernible, allowing quantification of galectin-3-expressing PSCs. NMJs without any detectable S100β staining (presumably denervated for a long time) were not included in this study. Notwithstanding the limitations of S100β for the quantification of PSC process extensions in these conditions (Son and Thompson, 1995a), a large number of processes were labeled allowing quantification based on their presence on an NMJ rather than their number. Furthermore, we never observed a galectin-3-positive S100β-negative process or a presynaptic sprout in absence of S100β staining, showing that S100β is reliable for this analysis in these conditions.
Muscle cross-section immunohistochemistry
Immunostaining was performed similarly as previously described (Tremblay et al., 2017). Briefly, the STM muscle was dissected and frozen in cold optimal cutting medium compound (OCT; TissueTek) using isopentane at −80°C. Transverse cryosections (10 μm) were made and incubated in blocking solution (10% NDS in PBS). Sections were incubated with either mouse IgG1 anti-myosin heavy chain (MHC) Type IIa (SC-71, 1:200), mouse IgG2b anti-MHC Type I (BA-D5, 1:100), and mouse IgM anti-MHC Type IIb (BF-F3, 1:200) or mouse IgM anti-MHC Type IIx (6H1; 1:10) and mouse IgG1 anti-MHC all but IIx (BF-35, 1:200) for 1 h at RT (all from Developmental Studies Hybridoma Bank; concentrates; Schiaffino et al., 1989; Lucas et al., 2000). They were revealed with appropriate secondary antibodies (1:500, Jackson ImmunoResearch) and mounted in Prolong Gold antifade reagent (Life Technologies).
Experimental design and statistical analyses
For Ca2+ imaging experiments, the number of animals used (N; biological replicates) and the number of PSCs analyzed (n; statistical replicates) are indicated in the text. Unpaired t tests were performed to compare two different conditions from different experiments. Two-way ANOVAs tests were used to evaluate the effect of two independent variables on Ca2+ responses, using Tukey's multiple comparisons (Tukey's test) as a post hoc test. To analyze the heterogeneity of Ca2+ responses belonging to the same NMJs, the amplitude of the response of each cell was expressed as a percentage of the average response of all imaged cells on that NMJ. Then, the SD of the resulting distribution for each group were compared using the F test for unequal variance, using Holm–Sidak's method to correct for multiple comparisons. Statistical tests were performed in GraphPad Prism 7 software. Unless otherwise stated, all results are presented as mean ± SEM. All tests used a confidence interval of 95% (α = 0.05).
For the morphologic analysis, all visible surface NMJs from left and right STM and SOL muscles were included. Number of animals used (N; representing both the biological and statistical replicates) and the number of NMJs or PSCs analyzed (n; number of observations) in each condition are indicated in the text. Because of the nature of the analysis (count data), the data does not meet the assumptions of following a Gaussian distribution or having a variance equal between groups and symmetric (Crawley, 2007). Hence, generalized linear models (GLM) were created, using a binomial error structure and a logit link function (logistic distribution), to analyze the results as previously described (Tremblay et al., 2017). For all criteria, the effect of the genotype (WT vs SOD1G37R) and the muscle (STM vs SOL) were evaluated. Furthermore, the effect of the innervation status of NMJs (fully innervated vs completely denervated) on PSCs' repair response, identified by process extensions and Gal-3 expression, was also evaluated. The muscle and the innervation status were defined as within-subject factors. The effects of all two-way and three-way interactions were also evaluated. Finally, biologically relevant pairwise comparisons are illustrated in the relevant section and were made using the estimated marginal means (EM means), using Holm–Sidak's method to correct for multiple comparisons (hereafter referenced by GLM post hoc test). All analyses were made using the SPSS software (v.24.0.0.0; IBM). All tests used a confidence interval of 95% (α = 0.05).
Results
We have previously shown an increased activation of PSC mAChR during synaptic communication at S and FR NMJs in presymptomatic SOD1G37R mice (P120 and P380; Arbour et al., 2015). However, NMJs innervated by different MU types display opposite changes in their synaptic properties in SOD1G37R mice, i.e., neurotransmitter release is increased at S NMJs and decreased at FF NMJs (Tremblay et al., 2017). Knowing that alterations in synaptic activity can induce plastic changes in PSC activity (Bélair et al., 2010), PSC excitability may be different at FF NMJs in SOD1G37R mice. In the present study, we first tested whether PSCs at vulnerable FF NMJs also displayed an enhance mAChR activation during synaptic communication (mAChR contribution) in SOD1G37R mice. For most experiments, the STM (FF MUs on its ventro-lateral face; Fig. 1B,C) and the SOL (FR and S MUs; Arbour et al., 2015) nerve muscle preparations were used because of their similar rate of denervation in SOD1 mice despite their different MU type composition (Valdez et al., 2012). Their similar disease course allowed the analysis of the impact of the MU type on PSC repair properties under similar levels of endogenous denervation (symptomatic animals). FF NMJs in the STM of SOD1G37R mice also showed a similar decrease in synaptic activity (our unpublished observations) to FF NMJs in the EDL at P180 (Tremblay et al., 2017), indicating that, despite denervation occurring at later stages than in hindlimb fast-twitch muscles, early synaptic alterations are also present in the STM.
Reduced PSC Ca2+ activity in the STM of presymptomatic SOD1G37R mice
To address the first question, we examined the ability of PSCs to detect endogenous neurotransmitter release elicited by a motor nerve stimulation (50 Hz, 20 s) in the STM. PSC activity was monitored by imaging transient intracellular Ca2+ changes, a well-established reporter of their decoding activity (Rochon et al., 2001; Rousse et al., 2010). Experiments were performed at P120, age where the first alterations were observed in the SOL of SOD1G37R mice (Arbour et al., 2015). At this stage, no signs of denervation were observed in the STM of SOD1G37R mice (50 NMJs, N = 2; Fig. 1D).
At P120, all PSCs of WT (18 PSCs; 9 NMJs; N = 6) and SOD1G37R mice (19 PSCs; 7 NMJs; N = 4) responded to nerve stimulation-induced neurotransmitter release in the STM. PSC Ca2+-response amplitude was similar between WT and SOD1G37R mice (333.7 ± 31.1% ΔF/F0, n = 18 vs 279.2 ± 27.4% ΔF/F0, n = 19, respectively; t(35) = 1.320; p = 0.1954 unpaired t test). This is consistent with the different progression of limb and trunk muscles and the observation that PSC Ca2+ responses were unchanged in the SOL two months earlier (P60; Arbour et al., 2015).
We next examined PSC properties in the STM at P180 to test whether changes in PSC activity were delayed in the STM compared with the SOL. Again, PSCs responsiveness to nerve stimulation was similar between SOD1G37R and WT mice (SOD1G37R: 31 out of 33 PSCs; 12 NMJs; N = 8; WT: 20 out 22 PSCs; 11 NMJs; N = 8). However, unlike at P120, PSC Ca2+ responses at P180 were significantly smaller in SOD1G37R mice (WT-Ctrl: 403.2 ± 22.8% ΔF/F0, n = 20 vs SOD1-Ctrl: 270.6 ± 24.3% ΔF/F0, n = 31; effect of genotype: F(1,77) = 25.13; p < 0.0001, two-way ANOVA; p = 0.0005, Tukey's test on two-way ANOVA; Fig. 2A,D). Hence, PSC properties were also altered in the STM of SOD1G37R mice at an early presymptomatic stage, but in an opposite manner to what was observed in the SOL (Arbour et al., 2015).
Figure 2.

Relative increase of mAChR-dependent PSC activity in the STM of SOD1G37R mice at a presymptomatic stage. A, Average PSC Ca2+ responses ± SEM (pale area) in SOD1G37R mice (right) and WT controls (left) induced by high-frequency nerve stimulation at P180 in absence of atropine (Ctrl). Top, Localization of the PSC on an endplate (left, apposed over BTX staining) and representative false color images of the changes in Rhod-3 fluorescence representing changes in intracellular Ca2+ levels before, during, and after motor nerve stimulation. B, Example of PSC Ca2+ responses in WT controls evoked by local application of muscarine (left) or nerve stimulation (right) in presence of 10 μm atropine. Note the blockade of muscarine-induced Ca2+ response and the reduced amplitude of nerve stimulation-evoked Ca2+ responses (left, compared with A). C, Average PSC Ca2+ responses ± SEM (pale area) in SOD1G37R mice (right) and WT controls (left) induced by high-frequency nerve stimulation at P180 in presence of 10 μm atropine. Note the reduced amplitude of nerve stimulation-evoked Ca2+ responses compared with the average peak amplitude without atropine (dashed lines) Top, Localization of the PSC on an endplate (left) and representative false color images of the changes in Rhod-3 fluorescence representing changes in Ca2+ levels before, during, and after motor nerve stimulation. D, Histogram of the amplitude of PSC Ca2+ responses in all conditions P180. Scale bar = 5 μm; *p <0.05, **p < 0.01, ***p < 0.005.
Relative increase of mAChR-dependent PSC activity in STM at a presymptomatic stage
Next, we tested whether the contribution of mAChR activation to PSC Ca2+ activity (mAChR contribution) was increased in the STM, despite the smaller PSC Ca2+ responses.
For this, Ca2+ imaging was performed in presence of the general mAChR antagonist atropine (10 μm) at P180. As shown in Figure 2B, blockade of PSC mAChRs was confirmed before the nerve stimulation by the inability of local muscarine applications (10 μm) to elicit Ca2+ responses. Out of the 32 cells analyzed (14 in WTs and 18 in SOD1s), only two PSCs from WT mice exhibited a small response to local application of muscarine in presence of atropine and were discarded. In the presence of atropine, all PSCs responded to nerve stimulation-induced neurotransmitter release in both WT (12 PSCs; 7 NMJs; N = 4) and SOD1G37R mice (18 PSCs; 8 NMJs; N = 4). As expected, the amplitude of PSC Ca2+ responses was significantly decreased in both groups (effect of atropine: F(1,77) = 22.63, p < 0.0001; two-way ANOVA; interaction: F(1,77) = 0.01446, p = 0.9046). In WT mice, it was reduced from 403.2 ± 22.8% ΔF/F0 (n = 20) to 277.5 ± 18.4% ΔF/F0 in presence of atropine (n = 12; p = 0.0194; Tukey's test on two-way ANOVA). This represents a 31% contribution of mAChRs to PSC Ca2+ responses that is similar to our observation in the adult SOL (Arbour et al., 2015). Likewise, PSC Ca2+ responses in SOD1G37R mice had a significantly smaller amplitude in presence of atropine (SOD1-Ctrl: 270.6 ± 24.3% ΔF/F0, n = 31 vs SOD1-atropine: 138.5 ± 27.6% ΔF/F0, n = 18; p = 0.0013; Tukey's test on two-way ANOVA; Fig. 2D). However, this represents a 49% mAChR contribution in SOD1G37R mice, which is 1.6-fold larger than in WT animals. Hence, the contribution of mAChR activation to PSC Ca2+-activity is also increased in the STM of SOD1G37R mice.
Furthermore, PSC Ca2+ responses in SOD1G37R mice remained significantly smaller than in WT mice in presence of atropine (p = 0.0094; Tukey's test on two-way ANOVA). As such, the reduction of the amplitude of PSC Ca2+ responses in the STM of SOD1G37R seem to be attributable to the reduced activation of non-muscarinic PSC receptors. The other major type of receptors by which PSCs detect synaptic activity at the mammalian NMJ are purinergic receptors (Rochon et al., 2001). However, their specific contribution to nerve-evoked PSC Ca2+ response was not evaluated as the underlying set of receptors remain undetermined at the adult mammalian NMJs (Rochon et al., 2001; Arbour et al., 2015).
As a whole, these results suggest that a relative increase in the contribution of mAChR to PSC activity is a common pathologic feature between both fast-twitch and slow-twitch muscles in SOD1G37R mice.
Heterogeneous nerve-induced PSC activity at individual NMJs in the STM at a presymptomatic stage
Interestingly, PSCs belonging to the same NMJ in the STM of SOD1G37R mice displayed heterogeneous responses to nerve stimulation, an unseen characteristic in WT mice (Fig. 3A). When only considering NMJs where multiple PSCs were imaged, PSC Ca2+ responses were all within ±20% of the average response on their NMJ in WT animals (SD = 11.17%; N = 8; n = 20; Fig. 3B). However, in SOD1G37R mice, PSC Ca2+ responses ranged from −99% up to +85% of the average response on their NMJ (SD = 43.65%; N = 8; n = 31; Fig. 3B), which is significantly more than in WT mice (F(30,19) = 15.27; p = 0.0004; F test). This suggests an uneven alteration of PSCs excitability at presymptomatic stage.
Figure 3.
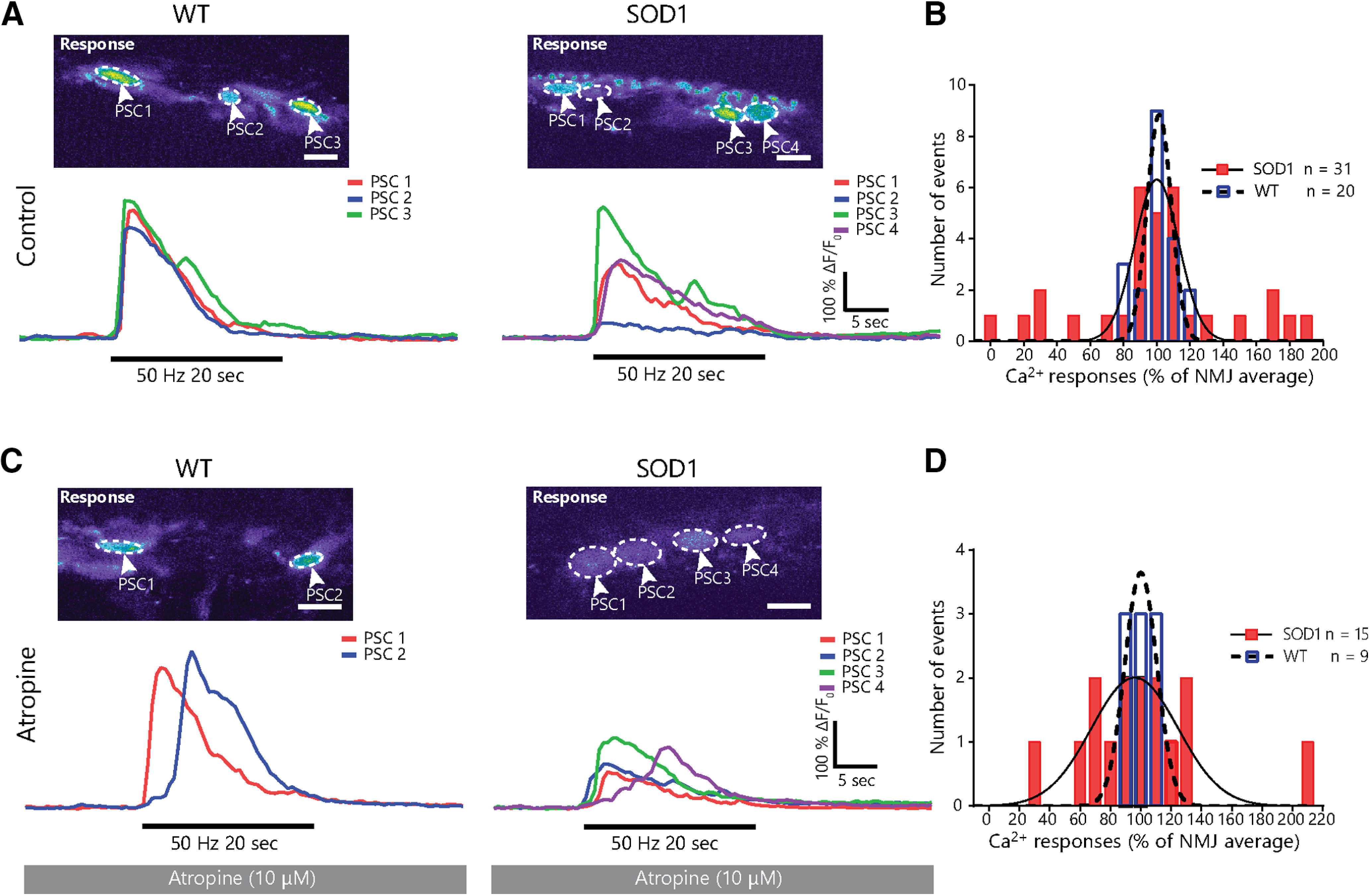
Muscarinic receptor-independent heterogeneity in PSC activity at individual NMJs in the STM of presymptomatic SOD1G37R animals. A, Examples of PSC Ca2+ responses from a single NMJ in SOD1G37R mice (right) and WT controls (left) in absence of atropine. Note the greater heterogeneity in Ca2+ responses in SOD1G37R mice. B, Frequency distribution of PSC Ca2+ in WT (blue, dashed line) and SOD1G37R (red, solid line) mice normalized to the average response of all PSCs on their NMJ. C, Example of individual PSC Ca2+ responses from an NMJ in SOD1G37R mice (right) and WT controls (left) in presence of atropine. D, Frequency distribution of PSC Ca2+ in WT (blue, dashed line) and SOD1G37R (red, solid line) mice normalized to the average response of all PSCs on their NMJ in presence of atropine. Lines in B, C represent best-fit Gaussian curves for each sample (WT: dashed line; SOD1G37R: solid line) and are used for representation purposes only. Scale bar = 10 μm.
These results could imply that the increased mAChR-dependent PSC activity is also uneven in the STM of SOD1G37R mice and therefore may not be a common feature to all PSCs. If this were the case, blockade of PSC mAChRs would restore homogeneity in Ca2+ responses over a single NMJ. However, heterogeneity in PSC Ca2+ responses was maintained following blockade of mAChRs in SOD1G37R mice (Fig. 3C). In presence of atropine, PSC Ca2+ responses in WT animals varied between −20% and +30% of each other on a given NMJ (N = 4; n = 9; SD = 15.12%; Fig. 3D), while PSC Ca2+ responses in SOD1G37R mice varied between −71% and +113% (N = 3; n = 15; SD = 47.51%; Fig. 3D), thus maintaining the difference observed in control condition (F(14,8) = 9.877; p = 0.0087; F test). Indeed, presence of atropine did not significantly affect the variability of Ca2+ responses on single NMJs in WT (WT-Ctrl vs WT-atropine: F(8,19) = 1.832; p = 0.4610; F test) or in SOD1G37R mice (SOD1-Ctrl vs SOD1-atropine: F(14,30) = 1.185; p = 0.6702; F test). These results reveal that heterogeneity in PSC Ca2+ responses in SOD1G37R mice is independent of mAChR activation. Hence, increased relative mAChR contribution to PSC synaptic decoding appears to be present in all PSCs in the STM of SOD1G37R mice.
Reduced PSC detection of purinergic signals in STM of presymptomatic SOD1G37R mice
PSC Ca2+ activity heavily depends on their intrinsic properties (Bélair et al., 2010; Rousse et al., 2010; Darabid et al., 2013; Arbour et al., 2015), such as the presence and sensitivity of their muscarinic and purinergic receptors. To further investigate PSC properties in the STM of SOD1G37R mice, we first tested PSCs sensitivity to cholinergic signals by monitoring PSC Ca2+ responses elicited by local application of the mAChR agonist muscarine (10 μm). As shown in Figure 4A, PSCs in both groups exhibited similar responsiveness (WT: 28 out 32; 17 NMJs; N = 10 vs SOD1: 32 out of 33; 16 NMJs; N = 8) and Ca2+ responses of similar amplitude following muscarine application (191.6 ± 23.1% ΔF/F0, n = 28 vs 228.0 ± 23.0% ΔF/F0, n = 32, respectively; t(58) = 1.127; p = 0.2644; unpaired t test; Fig. 4B). Thus, these results are consistent with the suggested reduced activation of another type of PSC receptors during synaptic communication in the STM of SOD1G37R mice.
Figure 4.

PSC Ca2+ responses elicited by local application of muscarine or ATP in the STM of presymptomatic SOD1G37R mice. A, Average PSC Ca2+ responses ± SEM (pale area) in SOD1G37R mice (right) and WT controls (left) induced by local application of muscarine at P180. Top, Localization of the PSC on an endplate (left, apposed over BTX staining) and representative false color images of the changes in Rhod-3 fluorescence representing changes in intracellular Ca2+ levels before, during, and after the response. B, Histogram of the amplitude of PSC Ca2+ responses induced by muscarine application at P180. C, Average PSC Ca2+ responses ± SEM (pale area) in SOD1G37R mice (right) and WT controls (left) induced by local application of ATP at P180. Top, Localization of the PSC on an endplate and representative false color images of the changes in Rhod-3 fluorescence representing changes in Ca2+ levels before, during, and after the response. D, Histogram of the amplitude of PSC Ca2+ responses induced by ATP application at P180. Scale bar = 5 μm; *p < 0.05.
Next, PSCs' sensitivity to purinergic agonists was examined by monitoring PSC Ca2+ responses after local application of the general purinergic agonist ATP (1 μm). Local application of ATP robustly triggered a Ca2+ response in 100% of PSC in both groups (WT: 32 out of 32; 15 NMJs; N = 9 vs SOD1: 21 out 21; 13 NMJs; N = 7). However, as shown in Figure 4C, ATP-induced PSC Ca2+ responses in the STM of SOD1G37R mice were smaller than in WT mice (315.2 ± 35.6% ΔF/F0, n = 21 vs 394.3 ± 20.2% ΔF/F0, n = 32, respectively; t(51) = 2.078; p = 0.0429; unpaired t test; Fig. 4D). These results suggest that PSCs are less sensitive to purinergic signals in the STM of SOD1G37R mice.
Altogether, our results obtained by imaging PSC Ca2+ activity show that the relative contribution of mAChR to PSC activity is increased, while PSCs' sensitivity to purinergic signals is decreased at FF NMJs in SOD1G37R mice. Despite having slightly different properties depending on the MU type they are associated with (Arbour et al., 2015), these results show that the contribution of mAChR to PSCs' activity increases at all NMJ types in SOD1G37R mice.
Altered PSC Ca2+ activity does not persist until the symptomatic stage in the STM of SOD1G37R mice
We previously showed that PSC muscarinic receptor activation remained elevated until the preonset stage (P380) in the SOL (Arbour et al., 2015). Therefore, we asked whether the alteration of PSCs properties at FF NMJs would also persist until the symptomatic stage. To address this question, we first examined PSC Ca2+ activity in the STM of phenotypically matched late symptomatic SOD1G37R mice and age-matched WT mice. PSC Ca2+ activity was induced by high-frequency nerve stimulation (50 Hz 20 s) or by local application of muscarine (10 µm) or ATP (1 µm).
Among PSCs that met our inclusion criteria (see Materials and Methods), responsiveness to nerve stimulation, local application of muscarine, or local application of ATP was similar between groups (nerve stimulation: SOD1G37R 12 out of 12 PSCs, N = 5 vs WT 13 out of 13 PSCs N = 6; muscarine: SOD1G37R 43 out of 52 PSCs, N = 8 vs WT 33 out of 39 PSCs, N = 6; ATP: SOD1G37R 49 out of 49 PSCs, N = 8 vs WT 39 out of 39 PSCs, N = 6). Although some PSC Ca2+ responses to endogenous transmitter release in SOD1G37R mice tended to be slightly smaller than in WT mice, the difference was not statistically significant (294.8 ± 35.7% ΔF/F0, n = 12 vs 353.4 ± 31.2% ΔF/F0, n = 13, respectively; t(23) = 1.227; p = 0.2324 unpaired t test; Fig. 5A,B). Similarly, the amplitude of PSC Ca2+ responses to local application of muscarine and ATP were similar between WT and symptomatic SOD1G37R mice (muscarine: 230.6 ± 18.8% ΔF/F0, n = 33 vs 233.3 ± 19.1% ΔF/F0, n = 43, respectively; t(74) = 1.007; p = 0.9200 unpaired t test; ATP: 261.8 ± 16.9% ΔF/F0, n = 39 vs 288.9 ± 17.2% ΔF/F0, n = 49, respectively; t(86) = 1.106; p = 0.2718 unpaired t test; Fig. 5C,D). Hence, PSC Ca2+ properties seem to have evolved during disease progression in the STM, with Ca2+ responses to endogenous transmitter release or local ATP and muscarine applications no longer being reduced in symptomatic SOD1G37R mice.
Figure 5.
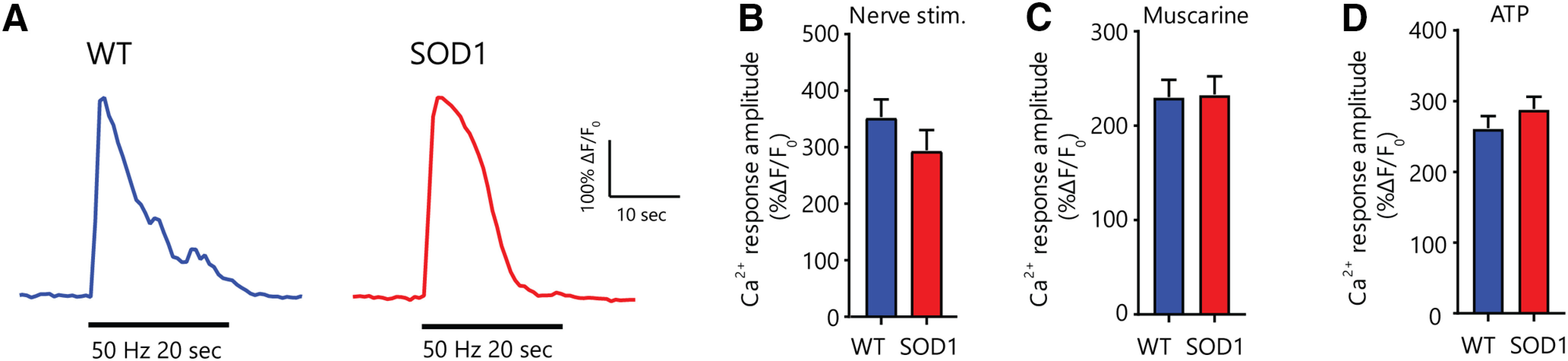
Unchanged PSC properties in the STM of phenotypically-matched symptomatic SOD1-G37R mice. A, Representative examples of PSC Ca2+ responses induced by high-frequency nerve stimulation in WT (blue) and SOD1G37R (red) mice. B–D, Histograms of the amplitude of PSC Ca2+ responses induced by high-frequency nerve stimulation (B) and local application of 10 μm muscarine (C) or 1 μm ATP (D).
PSCs express and secrete the phagocytic marker Gal-3 following denervation in WT mice
Our second goal was to examine PSCs' ability to contribute to debris clearance. Knowing that PSC mAChR activation regulates gene expression and represses their repair phenotype, we postulated that altered muscarinic properties, even if only transient, could be associated with defects in PSC-dependent NMJ repair mechanisms. Notably, PSCs become phagocytic and participate in the clearance of nerve terminal debris following denervation (Duregotti et al., 2015) similarly to axonal SCs (Reichert et al., 1994; Brosius Lutz et al., 2017). Axonal debris clearance is essential for efficient NMJ reinnervation, cellular debris hindering axonal growth in the endoneural tube (Kang and Lichtman, 2013). Hence, to address the second main question, a series of experiments were design to test whether PSCs could adopt a phagocytic phenotype in presymptomatic and symptomatic SOD1G37R mice.
Interestingly, phagocytic axonal SCs express the β-galactoside-binding lectin galectin-3 (Gal-3; also known as MAC-2; Reichert et al., 1994; Rotshenker et al., 2008), similarly to other phagocytic glial cells (Rotshenker, 2009; Nguyen et al., 2011; Morizawa et al., 2017), suggesting that its expression could also reflect PSC phagocytic activity. However, Gal-3 expression has never been evaluated in PSCs. Thus, we first investigated whether PSCs expressed Gal-3 at denervated NMJs in WT animals by performing an IHC for the three synaptic components (presynaptic, postsynaptic, and glia) and Gal-3. We induced the complete denervation of SOL NMJs in adult WT animals by sciatic nerve crush and monitored the presence of Gal-3 2 d later (nerve crush: N = 3, n = 43; sham: N = 3, n = 42; Fig. 6A). As expected, we observed a robust expression of Gal-3 in the soma and processes of PSCs following sciatic nerve crush (85.66 ± 2.02% of PSCs, n = 134; Fig. 6A,E) compared with sham surgery (2.883 ± 1.92% of PSCs, n = 105; p < 0.001; GLM post hoc test). Interestingly, we found that PSC Gal-3 expression increased 1 d after nerve crush and remained stable from 2 to 5 d after nerve crush (data not shown), in line with findings in axonal SCs showing expression from 2 d to at least 12 d after nerve crush (Rotshenker, 2011) These results confirm that PSCs express Gal-3 following NMJ denervation as part of their normal response to injury in WT animals.
Figure 6.
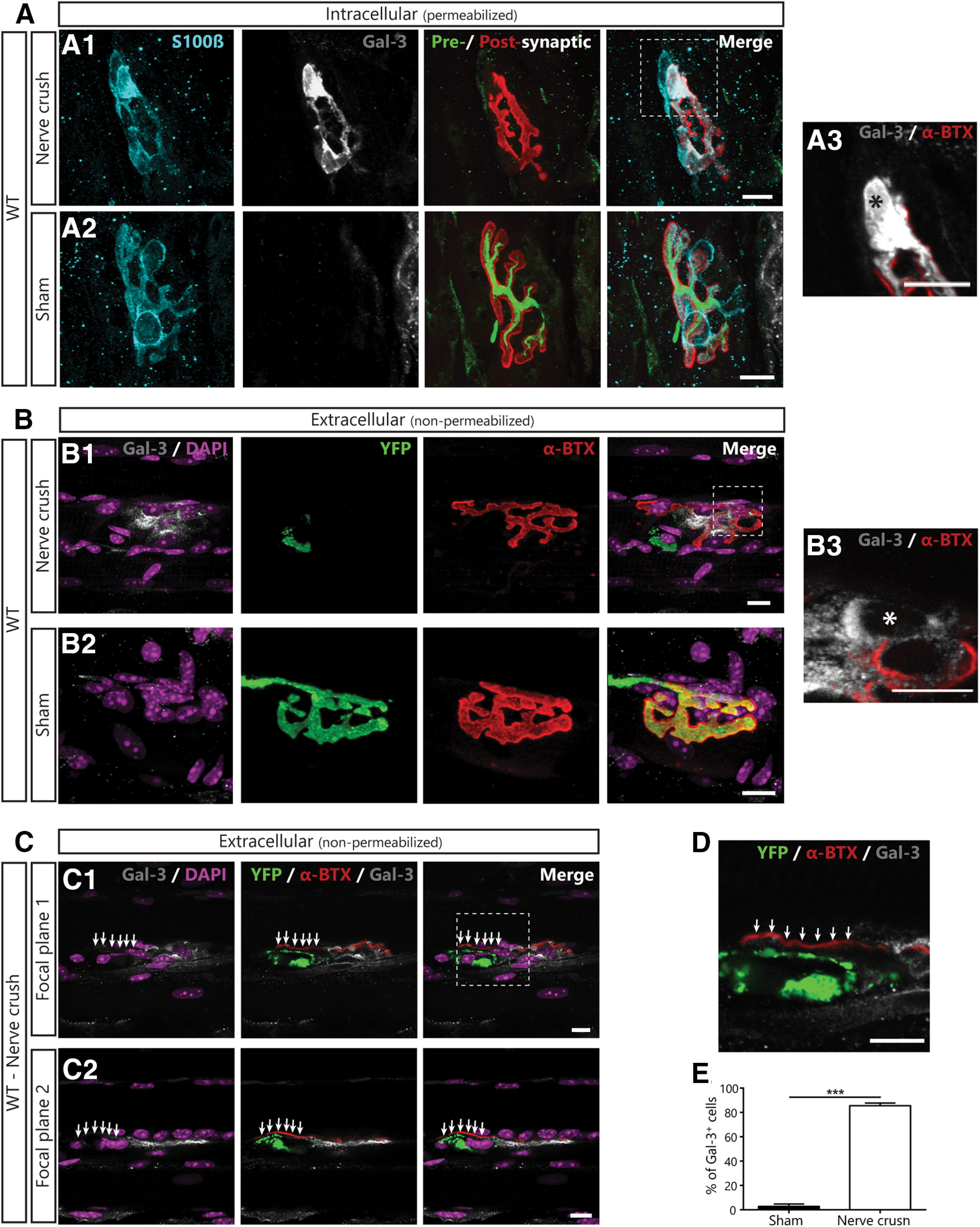
PSCs express the phagocytic marker Gal-3 following denervation in WT mice. A, Representative confocal images of immunolabeling of glial cells (blue; S100β), Gal-3 (white), presynaptic nerve terminals and postsynaptic nAChRs (merged: respectively, green, SyT; red, α-BTX) from the SOL of WT mice following a sciatic nerve crush (A1), or in sham operated animal (A2). A3, High-magnification image of the region identified in A1 showing that Gal-3 is present inside the soma (*). B, Representative images of the extracellular labeling of postsynaptic nAChRs (red), Gal-3 (white) and nuclei (DAPI; purple) from the SOL of Thy1-YFP mice following sciatic nerve crush (B1) or sham surgery (B2). B3, High-magnification image of the region identified in B1 showing that Gal-3 is present around the soma (*). Note that Gal-3 appears to be both intracellular and extracellular following NMJ denervation. C, Two single confocal planes of a denervated NMJ showing that YFP+-debris accumulation on the endplate (arrows) correlates with the lack of extracellular Gal-3 following sciatic nerve crush. D, High magnification of the region in C1 showing that Gal-3 is absent next to YFP+ debris. E, Percentage of PSCs expressing Gal-3. Quantification is based on permeabilized immunolabeling. Scale bar = 10 μm; ***p < 0.005.
Gal-3 can be present intracellularly, secreted or associated with transmembrane proteins (Dumic et al., 2006; Yang et al., 2008). Previous studies on axonal SCs suggested that Gal-3 may exert its function in the extracellular space (Reichert et al., 1994). To further characterize its role at the NMJ, we examined whether Gal-3 was present in the extracellular space 2 d after sciatic nerve crush by performing an IHC in absence of membrane permeabilization. Since our presynaptic markers are intracellular, the efficacy of the denervation was validated by performing experiments on animals expressing the YFP in all motor neurons (homozygous Thy1-YFP16 mice). As shown in Figure 6B, Gal-3 was present in the extracellular space following sciatic nerve crush (N = 3, n = 45; Fig. 6B1) but not sham surgery (N = 2, n = 27; Fig. 6B2). Extracellular Gal-3 seemed membrane bound as it was present all around PSCs' somata (Fig. 6B3). However, close comparison of Gal-3 staining in permeabilized and non-permeabilized tissue (Fig. 6A3,B3, respectively) reveals that part of the staining was missing in non-permeabilized tissues (asterisk). This difference shows that Gal-3 was also present in the cytoplasm and possibly in the nucleus. Interestingly, accumulation of YFP+ axonal debris on the endplate often correlated with the lack of extracellular Gal-3 on neighboring PSCs (six out of seven observations; Fig. 6C,D, arrows). Altogether, these results show that Gal-3 is expressed by PSCs following denervation, is present in the extracellular space at the NMJ and correlates with efficient clearance of presynaptic debris on the endplate.
Some PSCs fail to upregulate Gal-3 following denervation in presymptomatic SOD1G37R mice
Based on our Ca2+-imaging results, we hypothesized that altered PSCs' decoding properties would be associated with an inadequate phagocytic phenotype on denervated NMJs in presymptomatic SOD1G37R mice. Thus, we tested whether PSCs upregulated Gal-3 following an experimentally-induced denervation in presymptomatic SOD1G37R mice (sciatic nerve crush at ∼P180). We evaluated PSCs Gal-3 expression in the EDL (FF NMJs on its surface; Tremblay et al., 2017) and the SOL (FR and S NMJs) to assess any MU type-dependent differences. The EDL was used instead of the STM for nerve-crush experiments, since the denervation of SOL and EDL NMJs can be induced using the same surgical procedure (sciatic nerve crush), thereby reducing interanimal and interprocedure variability. Furthermore, the surgery to induce the denervation of STM NMJs is much more invasive, causing a strong inflammatory response in the muscle (data not shown) and possibly altering PSC properties. Based on our hypothesis, we predict that PSC Gal-3 expression would be reduced at denervated NMJs of both types in SOD1G37R mice.
Interestingly, fewer PSCs upregulated Gal-3 in the EDL and the SOL of P180 SOD1G37R mice than of littermate controls 2 d after sciatic nerve crush (Fig. 7A,B, arrows; Fig. 7C, WT EDL: N = 4, 83 NMJs, n = 246 PSCs; WT SOL: N = 4, 108 NMJs, n = 288 PSCs; SOD1 EDL: N = 4, 90 NMJs, n = 252 PSCs; SOD1 SOL: N = 4, 89 NMJs, 205 PSCs, Effect of genotype: p < 0.001, GLM). Importantly, PSC Gal-3 expression was also dependent on the muscle type (effect of muscle: p < 0.001, GLM; interaction between genotype and muscle: p = 0.028, GLM; Fig. 7C) such that significantly fewer PSCs expressed Gal-3 following denervation in the EDL than in the SOL in WT mice (74.65 ± 4.73% vs 88.25 ± 1.65%, respectively, p = 0.022, GLM post hoc test; Fig. 7C) and in SOD1G37R mice (42.00 ± 3.85% vs 81.25 ± 3.01%, respectively; p < 0.001, GLM post hoc test; Fig. 7C). Although more PSCs failed to upregulate Gal-3 in both muscles in SOD1G37R mice compared with WT mice (EDL: WT vs SOD1, p < 0.001; SOL: WT vs SOD1 p = 0.022, GLM post hoc test; Fig. 7C), this effect was much more pronounced in the EDL than in the SOL. Hence, PSCs seem less likely to adopt a phagocytic phenotype at denervated NMJs in presymptomatic SOD1G37R mice in a manner consistent with NMJ vulnerability in ALS. These results suggest that PSCs may not adequately promote NMJ repair on the endogenous denervation of NMJs during the symptomatic phase in SOD1G37R mice.
Figure 7.
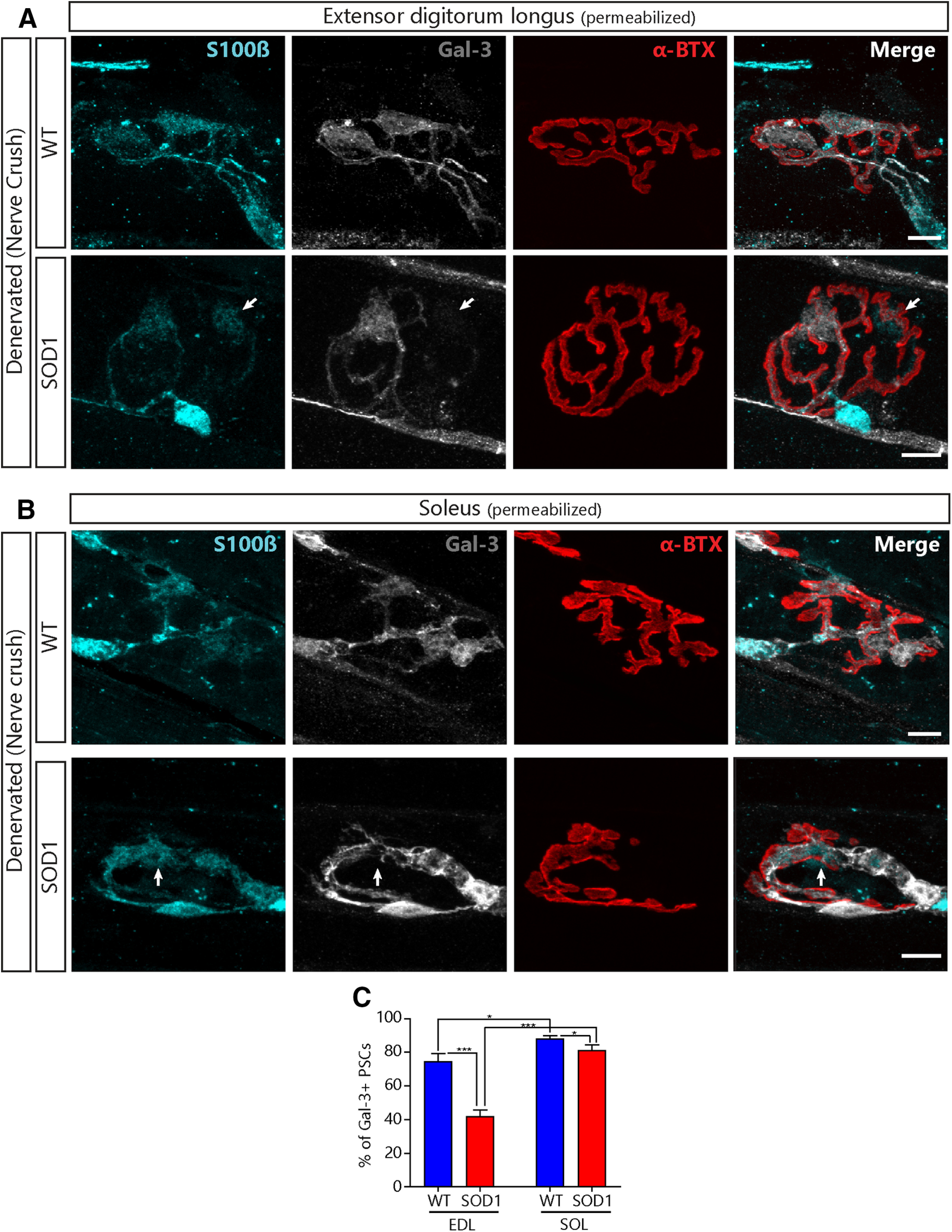
Lower expression of Gal-3 in PSCs of presymptomatic SOD1G37R mice following denervation. Representative confocal images of the immunolabeling of glial cells (blue; S100β), Gal-3 (white), and postsynaptic nAChRs (red, α-BTX) from the EDL (A) or the SOL (B) of WT (top) and SOD1G37R mice (bottom) 2 after a sciatic nerve crush. Note the absence of Gal-3 expression in some of the PSCs from SOD1 mice (arrows). C, Quantification of the percentage of PSCs expressing Gal-3 2 d following a sciatic nerve crush as a function of the genotype and the muscle type. Scale bar = 10 μm; *p < 0.05, ***p < 0.001.
Similar level of denervation in STM and SOL muscles of symptomatic SOD1G37R mice
We next wanted to evaluate whether PSC's ability to upregulate Gal-3 or to promote NMJ repair was still impaired in symptomatic SOD1G37R mice. Using IHC, PSCs' repair properties (Gal-3 expression, PSC process extensions and nerve terminal sprouting) were evaluated at all NMJ types under similar levels of denervation, by comparing the STM and the SOL of phenotypically matched late symptomatic SOD1G37R animals (P505–P565; Fig. 1A). This experimental design allowed us to isolate the effect of the muscle type on the repair properties since, unlike the STM, denervation levels in leg fast-twitch muscles, such as the EDL, are drastically higher than in the SOL at this stage, with only few NMJs being fully innervated (our unpublished observations; Tremblay et al., 2017). Observations were analyzed as a function of the innervation state of the NMJs (innervated vs denervated) in addition to the muscle type (STM vs SOL) and the genotype (WT vs SOD1G37R) as illustrated in Figure 8A.
Figure 8.
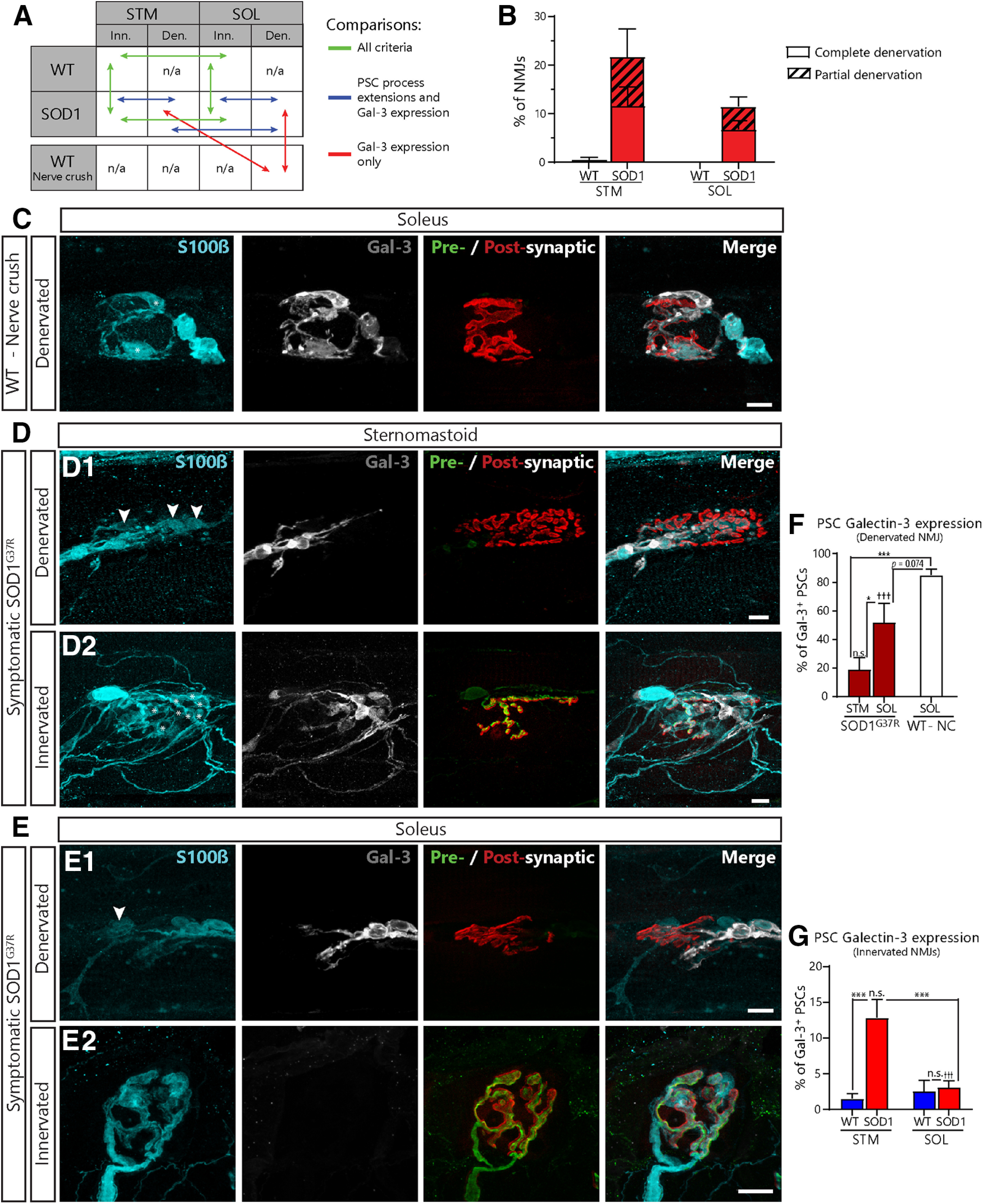
Paradoxical expression of Gal-3 in the STM and SOL of symptomatic SOD1G37R mice. A, Schematic of the biologically relevant pairwise comparisons to assess the effect of genotype (WT vs SOD1G37R), muscle type (STM vs SOL), and innervation status (innervated vs denervated). B, Histogram of the percentage of completely and partially denervated NMJs across all groups. Den., denervated; Inn., innervated. C–E, Immunolabeling of glia (blue; S100β), Gal-3 (white), and presynaptic nerve terminals and postsynaptic nAChRs (merged: respectively, green, SV2 and NF-M; red, α-BTX). C, Representative confocal images of NMJs from the SOL of aged WT mice following nerve-crush induced denervation. D, Representative confocal images of denervated (D1) and innervated (D2) NMJs from the STM of symptomatic SOD1G37R mice. Note the presence of Gal-3 in PSCs (*) on an innervated NMJ in the STM of SOD1G37R mice. Also note the disorganization of PSC processes. E, Representative confocal images of denervated (E1) and innervated (E2) NMJs from the SOL of SOD1G37R mice. Note the presence of Gal-3 in axonal SCs and its absence from PSCs (B, C, arrowheads). Percentage of PSCs expressing Gal-3 on denervated NMJs (F) and innervated NMJs in all groups (G). Symbols on the top of histogram bars represent the difference between innervated and denervated NMJs (D vs E). Scale bars = 10 μm. ns, nonsignificant; ***p < 0.005; †††p < 0.005.
First, we needed to confirm that the level of NMJ denervation in each muscle were comparable. Consistent with previous observations in the SOD1G93A mice (Schaefer et al., 2005; Valdez et al., 2012), we confirmed that the extent of denervation (partial or complete) was comparable between the STM and the SOL in symptomatic SOD1G37R mice, not being significantly higher in the STM than in the SOL (STM: 21.71 ± 5.76%, N = 9 vs SOL: 11.51 ± 1.98%, N = 7; p = 0.116; GLM post hoc test; Fig. 8B). Indeed, we found that 11.63 ± 3.91% of NMJs in the STM were completely denervated (31 out of 266, N = 9) compared with 6.80 ± 1.84%; in the SOL (20 out of 282, N = 7). Similar results were obtained for partially denervated NMJs (STM, 10.08 ± 3.36%; 29 out of 266 NMJs, N = 9 vs SOL, 4.11 ± 1.97%; 15 out of 282, N = 7). Of note, only one out of the 156 NMJs analyzed in the STM of WT animals (0.54 ± 0.54%; N = 6) was completely denervated and none were partially denervated. In the SOL, none of the 152 NMJs analyzed in WT animals (N = 5) showed any sign of denervation. A partially denervated NMJ could be either undergoing denervation or reinnervation, which are two very distinct physiological states (Martineau et al., 2018). Because of this uncertainty, partially denervated NMJs were excluded from the following analyses.
As intended with this experimental design, these observations confirm that a possible difference in the level of denervation will not bias the analysis of Gal-3 expression or PSC repair properties at symptomatic stage.
Reduced Gal-3 expression at denervated NMJs in symptomatic SOD1 animals
We examined Gal-3 expression in PSCs NMJs in the STM and the SOL of symptomatic SOD1G37R mice. To rule out age-related changes in the debris clearance pathway (Kang and Lichtman, 2013), we also performed a sciatic nerve crush in aged WT mice (P450–P500). PSCs in the SOL of aged WT mice robustly expressed Gal-3 following sciatic nerve crush (85.03 ± 4.07%, N = 4, n = 120; Fig. 8C) and at similar levels than in P180 mice (Fig. 6). Hence, PSCs maintain their ability to upregulate Gal-3 in 14- to 16-month-old WT mice.
As expected, we found that PSC Gal-3 expression varied according to the state of innervation and the muscle in SOD1G37R mice and in age-matched controls (effect of genotype: p < 0.001, effect of innervation: p < 0.001, genotype × innervation interaction: p < 0.001; muscle × innervation interaction: p < 0.001, GLM). Interestingly however, numerous PSCs did not express Gal-3 on completely denervated NMJs in SOD1G37R mice in both muscles (Fig. 8D1,E1, arrowheads). Consistent with our finding in presymptomatic mice (Fig. 7), significantly less PSCs expressed Gal-3 in the STM of SOD1G37R animals when compared with nerve crush-induced denervated NMJs in WT mice (18.94 ± 8.32% of PSCs; n = 136 vs 85.03 ± 4.07% of PSCs n = 120; p < 0.001; GLM post hoc test; Fig. 8F). Furthermore, presence of Gal-3 was highly variable in the SOL, ranging from 0.00% to 77,78% of PSCs (51.81 ± 13.56%, n = 49), and an overall trend toward lower levels was observed when compared with nerve crush-induced denervation (SOD1G37R SOL vs nerve crush WT SOL; p = 0.074; GLM post hoc test; Fig. 8F). Interestingly, PSCs Gal-3 expression on denervated NMJs was significantly higher in the SOL than in the STM (SOD1G37R SOL vs STM; p = 0.020; GLM post hoc test; Fig. 8F), again reflecting the higher vulnerability of FF NMJs. Given the time course of Gal-3 expression in SCs (Rotshenker, 2011), one could expect not to observe PSC Gal-3 expression on very recently denervated NMJs (<1 d after denervation) or on chronically denervated NMJs (>12 d) in SOD1 mice. However, we do not believe that the majority of PSCs in our sample fit into these scenarios as all analyzed PSCs still expressed S100β, arguing against chronic denervation, and axonal SCs associated with denervated NMJs robustly expressed Gal-3 in both muscles (Fig. 8D,E). Worthy of note, the latter finding also suggests that this alteration is specific to PSCs. Altogether these results show that PSCs unreliably upregulate Gal-3 on denervated NMJs in symptomatic SOD1G37R mice, especially on vulnerable NMJs.
Gal-3 is expressed on some innervated and partially innervated NMJs in the STM of symptomatic SOD1 mice
Paradoxically, while Gal-3 was absent from PSCs on denervated STM NMJs in SOD1G37R mice, it was expressed in some PSCs on fully innervated NMJs in the STM (Fig. 8D2, asterisks, G, SOD1G37R: 12.84 ± 2.54% of PSCs, n = 866 vs WT: 1.55 ± 1.68% of PSCs, n = 618; p < 0.001; GLM post hoc test). More importantly, a similar proportion of PSCs expressed Gal-3 whether the NMJ was innervated or denervated in the STM of SOD1G37R mice (12.84 ± 2.54% vs 18.94 ± 8.32%; p = 0.132; GLM post hoc test; Fig. 8F,G) suggesting that Gal-3 expression in PSCs was independent of the state of innervation in this ALS model. This upregulation of Gal-3 on innervated NMJs was not observed in the SOL of SOD1G37R animals (SOD1G37R SOL innervated: 3.12 ± 0.92%, n = 708 vs WT SOL innervated: 2.59 ± 1.50, n = 435; p = 0.231; GLM post hoc test; Fig. 8E2), where its expression was specific to denervated NMJs (innervated: 3.12 ± 0.92% vs denervated: 51.81 ± 13.56%; p < 0.001; GLM post hoc test). Gal-3 expression was also not observed at innervated NMJs in presymptomatic (P120–P180) and preonset (P350) SOD1G37R mice (data not shown).
In line with this paradoxical observation, a number of PSCs on partially innervated NMJs also expressed Gal-3 and were usually associated with the nerve terminal in the STM of symptomatic SOD1G37R mice (Fig. 9A1). Again, this was not observed in the SOL (Fig. 9A2). Indeed, 20 out of 29 (69.97%) partially innervated NMJs had at least one Gal-3+-PSC in the STM compared with only two out 15 (13.33%) in the SOL. In 80% of these cases in the STM, Gal-3+-PSCs were preferentially associated with the presynaptic nerve terminal. Altogether, these results are suggestive of active PSC phagocytosis of presynaptic elements on denervating or reinnervating NMJs.
Figure 9.

Gal-3 in PSCs associated with nerve terminals on partially innervated NMJs in the STM of symptomatic SOD1G37R mice. Immunolabeling of glia (blue; S100β), Gal-3 (white), and presynaptic nerve terminals and postsynaptic nAChRs (merged: respectively, green, SV2 and NF-M; red, α-BTX). A, Representative confocal images of partially innervated NMJs from the STM and (B) the SOL of symptomatic SOD1G37R mice. Note that Gal-3+-PSCs are preferentially associated with the innervated part of the endplate rather than the denervated part. Scale bars = 10 μm.
PSCs extend disorganized processes in both the STM and the SOL of symptomatic SOD1 mice
Finally, we addressed the third main question and sought to characterize another fundamental PSC mechanism contributing to NMJ repair: the extension of glial processes and the guidance of terminal sprouting. Importantly, this aspect of PSC repair properties could not be evaluated in presymptomatic animals following a complete nerve crush, since this procedure only poorly induces the extension of PSC processes (Love and Thompson, 1999). We monitored the presence and targeting of PSC process extensions in symptomatic SOD1G37R animals. These extensions are normally observed from denervated NMJs toward nearby innervated NMJs, a phenomenon essential in the initiation and guidance of nerve terminal sprouting (Reynolds and Woolf, 1992; Son and Thompson, 1995a,b; Son et al., 1996; O'Malley et al., 1999).
In general, we found significantly more NMJs associated with extended PSC processes in SOD1G37R than in WT mice (effect of genotype: p < 0.001, GLM; Fig. 10A,B), but this effect varied slightly according to the muscle type (interaction genotype × muscle: p = 0.044; GLM). Denervated NMJs were more likely to be associated with extended PSC processes than innervated NMJs (effect of innervation: p < 0.001, GLM), but this effect also varied depending on the muscle (interaction innervation × muscle: p = 0.013, GLM).
Figure 10.

Abnormal and disorganized nerve terminal sprouting and PSC process extension in the STM and the SOL of symptomatic SOD1G37R mice. Immunolabeling of glial cells (blue; S100β), presynaptic nerve terminals (green; NF-M and SV2) and postsynaptic nAChRs (red; α-BTX). A, Representative confocal images of NMJs from the STM of age-matched WT controls (A1) and of symptomatic SOD1G37R mice (A2, denervated; A3, innervated). B, Representative confocal images of NMJs from the SOL of WT animals (B1) and of symptomatic SOD1G37R mice (B2: denervated; B3: innervated). Note the nerve terminal sprouting avoiding the nearby denervated NMJ (B2; arrowheads). C, The percentage of NMJs associated with PSC processes in the STM or the SOL according to the state of innervation. D, Percentage of innervated NMJs displaying nerve terminal sprouting. Scale bars = 10 μm. ns, nonsignificant; *p < 0.05, ***p < 0.005.
Indeed, in the STM, completely denervated NMJs were not significantly more likely to be associated with extended PSC processes than innervated NMJs in SOD1G37R mice (51.05 ± 14.99%, n = 31 vs 38.48 ± 7.11%, n = 206, respectively; p = 0.858; GLM post hoc test; Fig. 10A3, arrowheads, C). Conversely, innervated NMJs in the STM of SOD1G37R mice were more likely to be associated with extended glial processes than innervated NMJs in WT mice (13.73 ± 4.05% n = 155; p = 0.047; GLM post hoc test; Fig. 10A,C). Hence, PSCs in the STM of SOD1G37R extended their processes but this did not seem to be dependent on the innervation state of NMJs. These results contrast with those obtained in the SOL. Indeed, PSCs on denervated NMJs in the SOL of SOD1G37R mice robustly extended their processes compared with PSCs on innervated NMJs (71.67 ± 13.76, n = 36 vs 15.65 ± 3.68, n = 247; p < 0.001; GLM post hoc test; Fig. 10B,C). Hence, while PSC process extensions from denervated NMJs occurred as often in both muscles (p = 0.858; GLM post hoc test), they were abnormally more likely to be associated with innervated NMJs in the STM than in the SOL (p = 0.047; GLM post hoc test).
Importantly though, these results also show that 49% of denervated NMJs in the STM and 28% in the SOL lacked PSC process extensions (Fig. 6A2). These proportions are higher than the expected 1% based on studies using partial denervation (Son and Thompson, 1995b), and, in most cases, only one process per NMJ was observed (1.13 ± 0.67 processes on average; Son and Thompson, 1995b; O'Malley et al., 1999). Furthermore, PSC processes were often highly disorganized in the STM and SOL of SOD1G37R mice (47.09 ± 9.87% and 31.02 ± 18.62% of processes on average, respectively; Fig. 10A3,B2), often changing direction, not following muscle fibers and frequently making loops around themselves. Hence, although present, PSC process extensions were often disorganized and less abundant than expected on denervated NMJs regardless of the muscle type. These results further suggest that PSC properties are heterogeneous, which is reminiscent of the heterogeneity in PSC decoding properties observed at a presymptomatic stage.
Nerve terminal sprouting is absent in the STM and disorganized in the SOL of symptomatic SOD1 mice
Extended PSC processes contacting nearby innervated NMJs can initiate and guide nerve terminal sprouting back to denervated NMJs (Son and Thompson, 1995b; Son et al., 1996; O'Malley et al., 1999). However, previous studies reported conflicting results whereby nerve terminal sprouting is absent (Schaefer et al., 2005; Tallon et al., 2016) or abundant in SOD1G93A mice (Valdez et al., 2012). Hence, we next tested whether nerve terminal sprouting would be present, albeit disorganized, on innervated NMJs in SOD1 animals. Based on the lower, but not null, ability of FF NMJs to elaborate terminal sprouts (O'Malley et al., 1999; Frey et al., 2000; Wright et al., 2009), we expected fewer terminals sprouts in the STM compared with the SOL.
In general, we found significantly more NMJs associated with nerve terminal sprouts in the SOL of SOD1G37R animals than any other groups (effect of genotype: p = 0.001; effect of muscle: p < 0.001; interaction genotype × muscle: p = 0.015; GLM; Fig. 10A–C). Indeed, 30.26 ± 6.96% of innervated NMJs in the SOL of SOD1G37R mice (n = 259; Fig. 10B3) formed sprouts compared with 2.89 ± 1.46% of SOL NMJs in WT mice and 5.26 ± 1.37% of STM NMJs in SOD1G37R mice (n = 152 and n = 206, p < 0.001 and p < 0.001, respectively; GLM post hoc test; Fig. 10A3,B1,D), which suggests an active reinnervation effort in the SOL. However, consistent with previous observations made on FF NMJs (Martineau et al., 2018), we did not observe significantly more nerve terminal sprouts at STM NMJs in SOD1G37R mice than in WT mice (5.26 ± 1.37%, n = 206 vs 2.56 ± 1.64%, n = 155; p = 0.311). This finding was nevertheless surprising considering that over a third of them were being contacted by extended PSC processes (Fig. 10A3). Therefore, initiation of nerve terminal sprouting in symptomatic SOD1 mice appears deficient in the STM, but not the SOL, resulting in “empty” extended PSCs processes contacting both innervated and denervated endplates (Fig. 10C).
Importantly though, nerve terminal sprouts in the SOL were often disorganized, going in many directions and even “avoiding” nearby denervated NMJs (Fig. 10B2, arrowheads). Indeed, 33.46 ± 6.50% of sprouting NMJs had disorganized sprouts in the SOL of SOD1G37R mice, which is consistent with the muddled PSC process extensions. As a whole, these results show that nerve terminal sprouting was almost completely absent in the STM and disorganized in the SOL of symptomatic SOD1G37R animals.
In summary, these results show that PSCs display a paradoxical Gal-3 expression in symptomatic SOD1G37R animals, being inconsistent on denervated NMJs of both muscles and unexpectedly present on innervated and partially innervated NMJs in the STM. This suggests that PSCs do not adopt a phagocytic phenotype on denervation of vulnerable NMJs, and also to a lesser extent of partially resistant NMJs. Along with the disorganized processes and the abnormal nerve terminal sprouting, these results suggest that PSCs do not adopt an appropriate phenotype for reinnervation in ALS. Importantly, these main findings were replicated in the STM of a small number of preonset (P70) SOD1G93A animals. Namely, PSCs on denervated NMJs failed to upregulate Gal-3 and to extend processes, resulting in the absence of nerve terminal sprouting (data not shown). Altogether, these results show that alteration PSC repair properties are not specific to a single SOD1 model.
Discussion
Our results reveal inappropriate glial repair properties at the NMJs of a fast-twitch and a slow-twitch muscle in symptomatic SOD1 animals. These changes are associated with an altered PSC excitability, long before disease onset. Despite multiple differences in PSC properties (decoding and repair) between muscles and the heterogeneity in PSC properties, our data converges toward the same end results whereby PSCs on denervated NMJs inconsistently adopted a phagocytic phenotype, formed muddled processes, failed to guide nerve terminal sprouting appropriately and presented an increased mAChR-dependent activity, albeit transiently in a fast twitch muscle. Thus, PSC excitability and repair properties are incompatible with NMJ reinnervation across all MU types in SOD1 models.
NMJ repair defects in ALS
Here, we show that numerous PSC-dependent NMJ repair mechanisms are abnormal in SOD1 animals, suggesting that NMJ reinnervation may be deficient during the progression of ALS. Although compensatory reinnervation is known to occur during the disease (Schaefer et al., 2005; Martineau et al., 2018), this process may be suboptimal, resulting in inefficient or unstable NMJ repair. Such bungled compensation could contribute to the loss of motor function in ALS by exacerbating loss of NMJs. Consistent with this hypothesis, numerous studies documented delays in NMJ reinnervation following axonal injury in ALS models (Sharp et al., 2005; Pun et al., 2006; Henriques et al., 2011; Swarup et al., 2012; Mesnard et al., 2013; Carrasco et al., 2016b). These observations suggest that a number of core NMJ repair mechanisms are altered in these models. For instance, an intrinsically slower axonal growth or cellular debris accumulation in the endoneural tube and at the NMJ that would deter NMJ reformation after denervation (Kang and Lichtman, 2013). Our observation that PSCs fail to adopt a phagocytic phenotype on denervated NMJs (Fig. 8) is consistent with these mechanisms. However, since the inadequate Gal-3 expression was specific to PSCs, and was not observed for axonal SCs, our results suggest that other mechanisms besides debris accumulation in the endoneural tube could contribute to the delay in NMJ reinnervation after nerve injury.
Importantly, global nerve injuries do not recapitulate the progressive denervation occurring in ALS. Hence, evaluating NMJ repair following partial denervation would be more instructive as it would allow compensatory reinnervation to take place through nodal (axonal) and nerve terminal sprouting of intact motor axons (Thompson and Jansen, 1977; Son and Thompson, 1995b). Since PSCs unreliably extended muddled processes (Fig. 10), we predict that reinnervation based on these sprouting events would also be drastically impaired. Indeed, despite being more abundant than in controls, PSC processes were less abundant than expected (Son and Thompson, 1995b; O'Malley et al., 1999), which could underlie the reduced terminal sprouting observed following blockade of neurotransmission in SOD1 mice (Frey et al., 2000). Interestingly, Schaefer et al. (2005) and Martineau et al. (2018) observed substantial MU enlargement in the STM during disease progression, but these events appeared to be mostly because of nodal sprouts. This is consistent with the absence or disorganization of nerve terminal sprouts (Fig. 10) and the apparent sparing of axonal SCs documented here (Fig. 8). As a whole, these results suggest that the absence of terminal sprouting from FF NMJs is because of altered NMJ repair mechanisms rather than a general incapacity to form neuronal sprouts.
MU type dependent and independent diversity in PSC properties
Results presented here and previously (Arbour et al., 2015) show (1) that increased mAChR-dependent PSC activity is present long before disease onset, (2) that this alteration is associated with an inability to adopt a phagocytic phenotype on denervation in presymptomatic animals, and (3) that PSCs' phenotype is incompatible with NMJ repair in symptomatic animals, regardless of the MU type they are associated with. However, excitability and repair properties of PSCs associated with vulnerable FF MUs (STM) contrasted with those observed on PSCs associated with less vulnerable FR and S MUs (SOL).
Indeed, PSC Ca2+ signaling is affected differentially depending on the MU type in SOD1G37R mice, revealing a unique set of complementary changes in PSC activity at FF NMJs in contrast with those reported at FR and S MUs (Arbour et al., 2015). For instance, while maintaining a higher relative mAChR contribution, nerve-induced PSC Ca2+ responses on FF MUs were smaller, possibly because of a reduced sensitivity of the purinergic system (Figs. 2, 4). In contrast, PSC Ca2+ responses on FR and S MUs were larger because of an increased activation of mAChRs (FR and S) and an increased sensitivity of the purinergic system (FR, preonset stage only; Arbour et al., 2015). This highlights the importance of the balance regulation of PSCs activity between muscarinic and purinergic receptors. A possible explanation for these opposite changes may be linked to the differential MU-specific changes in NMJ activity occurring in SOD1 mice (Tremblay et al., 2017). Indeed, chronic changes in NMJ activity have been shown to induce plastic changes in PSC properties (Bélair et al., 2010).
Furthermore, our results suggest that changes in PSC excitability may only be transient on FF NMJs while they seem to become more pronounced at the preonset stage on S and FR MUs (Arbour et al., 2015). These results suggest that the altered PSC excitability may also confer some benefits on NMJ stability (i.e., before it denervates). Alternatively, but not exclusively, transient changes in PSC excitability may reflect longer-lasting changes in PSC function. Further examination of PSC excitability on denervated NMJs in symptomatic SOD1 mice could shed light on this question. Unfortunately, nerve-muscle preparations from symptomatic SOD1 animals are more fragile and PSCs on denervated NMJs seem more vulnerable to the AM loading procedure (Arbour et al., 2015). Future investigation of PSC properties using another approach, such as GCaMP expression (Heredia et al., 2018), may potentially circumvent those limitations.
Moreover, PSC repair properties varied according to the MU type in symptomatic SOD1G37R animals, where PSC properties on FF NMJs were out of phase with the innervation status while PSCs on FR or S NMJs adopted a less pronounced phenotype. Similarly, the expression of the chemorepellent Semaphorin 3A was selectively increased in FF NMJs following denervation or blockage of neurotransmitter release (De Winter et al., 2006). All these results are consistent with the known vulnerability of FF MUs compared with FR and S MUs in the disease (Frey et al., 2000; Atkin et al., 2005; Pun et al., 2006). Importantly, these differences were not because of STM being further along the progression of the disease than the SOL considering that denervation levels were comparable between the STM and the SOL at this stage. Hence, PSC repair properties reflect MU vulnerability at presymptomatic and symptomatic stages of the disease. Furthermore, the lack of PSC-dependent synaptic repair at FF NMJs could contribute to their increased vulnerability in ALS.
An additional level of complexity arises from the increased heterogeneity in PSC activity (nerve-induced Ca2+ responses) and repair properties (Gal-3 expression and process extensions) at individual NMJs in SOD1 animals. Heterogeneity in PSC responses to synaptic activity could arise from two scenarios. First, a mAChR-independent component of PSC Ca2+ responses could be inconsistently decreased among PSCs. Second, knowing that PSCs cover exclusive synaptic territories (Brill et al., 2011) and that synaptic activity is altered at the NMJ in ALS (Armstrong and Drapeau, 2013a,b; Rocha et al., 2013; Shahidullah et al., 2013; Arbour et al., 2015; Tremblay et al., 2017), neurotransmitter release could be more heterogeneous between active zones, thus inducing variable levels of Ca2+ activity in PSCs depending on the active zones they cover.
Altogether, common pathologic alterations in PSC properties are complemented by numerous context-dependent differences that could contribute to, or at least reflect, NMJ dysfunction and MU vulnerability in ALS.
Non-cell autonomy at the NMJ and impact of PSC dysfunction on disease progression
Similarly to glial cells in the CNS (Boillée et al., 2006a,b; Yamanaka et al., 2008; Ilieva et al., 2009; Haidet-Phillips et al., 2011; Kang et al., 2013; Meyer et al., 2014; Ditsworth et al., 2017), results presented here and in recent studies (Carrasco et al., 2016a; Van Dyke et al., 2016) suggest that alterations in PSC properties could contribute to ALS onset and progression. Direct manipulation of mutant SOD1 levels in astrocytes, microglia or oligodendrocyte progenitors directly affected their function and revealed their contribution to the disease. These results raise the question of whether changes in PSCs are directly caused by the expression of the mutant protein or reflect a maladaptive response to the diseased MN. Previous studies evaluating the contribution of mutant SOD1 expression in SCs (both PSCs and axonal SCs) to the disease provided disparate results depending on the dismutase competency of the mutated SOD1 protein (Lobsiger et al., 2009; Turner et al., 2010; Wang et al., 2012). However, the P0 promoter used in these studies poorly targets PSCs (Lobsiger et al., 2009). Furthermore, we show here that axonal SCs robustly adopted a phagocytic phenotype following denervation in symptomatic SOD1 mice, while PSCs failed to do so (Fig. 8), suggesting that different types of SCs may be affected differently in the disease. Unfortunately, lack of a known specific PSC promoter has so far prevented a direct manipulation of mutant SOD1 expression in PSCs. Interestingly, Castro et al. (2020) recently suggested a genetic strategy to identify PSCs which could open the possibility to develop new tools to target them selectively. Hence, the contribution of mutant SOD1 expression in PSCs to their dysfunction and to ALS onset and progression remains to be determined.
Role of Gal-3 in ALS
Numerous studies have evaluated the role of galectin-3 (MAC-2) in ALS and its usefulness as a potential biomarker for the disease (Zhou et al., 2010). For instance, Gal-3 expression is increased early in the spinal cord of SOD1G93A mice and ALS patients, in skeletal muscles of SOD1G86R mice (possibly in axonal SCs based on our results), and rises throughout disease progression (Ferraiuolo et al., 2007; Gonzalez de Aguilar et al., 2008; Zhou et al., 2010; Lerman et al., 2012; Baker et al., 2015). Its expression correlates with microglial activation (Yamanaka et al., 2008; Lerman et al., 2012) and is also inexplicably upregulated in MNs during disease progression (Ferraiuolo et al., 2007). These observations have been interpreted as indications that Gal-3 expression could play a detrimental role in disease progression or would at least reflect increasing levels of neuroinflamation.
However, Gal-3 has been implicated in numerous cellular functions which could be protective in ALS (Dumic et al., 2006; Yang et al., 2008) such as phagocytosis in astrocytes (Nguyen et al., 2011; Baker et al., 2015; Morizawa et al., 2017) and myelin phagocytosis in axonal SCs (Reichert et al., 1994). Although still debated, evidence suggest it could play a similar role in microglia (Rotshenker, 2009; Lalancette-Hébert et al., 2012). These results suggest that its upregulation may be linked to increased cellular debris clearance in ALS. Our results suggest that extracellular Gal-3 at the NMJ and in peripheral nerves would exert a beneficial effect on disease progression, being inversely correlated to the accumulation of axonal debris. However, a role of intracellular Gal-3 cannot be cast out as both fractions could not be labeled simultaneously. Lack of debris clearance would delay NMJ reinnervation (Kang and Lichtman, 2013) causing PSCs to gradually abandon parts of the endplate, thus leading to incomplete NMJ reinnervation (Kang et al., 2014). In agreement with this possibility, crossing SOD1G93A mice to Gal-3 knock-out (lgal3−/−) mice significantly accelerated disease progression (Lerman et al., 2012), further supporting a protective role of Gal-3 in ALS. As a whole, these results suggest that the effect of Gal-3 in ALS is context-specific, exerting different adaptive or maladaptive roles in different cell types.
Conclusion
We show that PSC excitability and repair properties are incompatible with NMJ repair across all MU types, in two SOD1 ALS mice models, in a manner which reflects their selective vulnerability. These results suggest that compensatory NMJ reinnervation may be defective in ALS because of misguided nerve terminal sprouting and inadequate endplate debris clearance. Further studies aimed at understanding the molecular basis of the diversity and the changes in PSCs in ALS could unravel novel therapeutic targets. Restoring PSCs' repair capabilities could enhance synaptic reestablishment which could improve motor function in ALS patients.
Footnotes
This work was supported by grants from the Canadian Institutes for Health Research (MOP-111070; to R.R.), Robert Packard Center for ALS Research (R.R.), Canadian Foundation for Innovation (R.R), and an infrastructure grant from the Fonds Recherche Québec-Santé Leader Opportunity Fund to the Groupe de Recherche sur le Système Nerveux Central (Université de Montréal). É.M. held a doctoral studentship from the ALS Society of Canada and a master's degree studentship from the Fonds Recherche Québec-Santé. We thank Félix-Antoine Robert for his help regarding the statistical analysis.
The authors declare no competing financial interests.
References
- Arbour D, Tremblay E, Martineau E, Julien JP, Robitaille R (2015) Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J Neurosci 35:688–706. 10.1523/JNEUROSCI.1379-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour D, Vande Velde C, Robitaille R (2017) New perspectives on amyotrophic lateral sclerosis: the role of glial cells at the neuromuscular junction. J Physiol 595:647–661. 10.1113/JP270213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong GA, Drapeau P (2013a) Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum Mol Genet 22:4282–4292. 10.1093/hmg/ddt278 [DOI] [PubMed] [Google Scholar]
- Armstrong GA, Drapeau P (2013b) Calcium channel agonists protect against neuromuscular dysfunction in a genetic model of TDP-43 mutation in ALS. J Neurosci 33:1741–1752. 10.1523/JNEUROSCI.4003-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkin JD, Scott RL, West JM, Lopes E, Quah AK, Cheema SS (2005) Properties of slow- and fast-twitch muscle fibres in a mouse model of amyotrophic lateral sclerosis. Neuromuscul Disord 15:377–388. 10.1016/j.nmd.2005.02.005 [DOI] [PubMed] [Google Scholar]
- Baker DJ, Blackburn DJ, Keatinge M, Sokhi D, Viskaitis P, Heath PR, Ferraiuolo L, Kirby J, Shaw PJ (2015) Lysosomal and phagocytic activity is increased in astrocytes during disease progression in the SOD1 (G93A) mouse model of amyotrophic lateral sclerosis. Front Cell Neurosci 9:410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélair EL, Vallée J, Robitaille R (2010) In vivo long-term synaptic plasticity of glial cells. J Physiol 588:1039–1056. 10.1113/jphysiol.2009.178988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée S, Vande Velde C, Cleveland DW (2006a) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52:39–59. 10.1016/j.neuron.2006.09.018 [DOI] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW (2006b) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392. 10.1126/science.1123511 [DOI] [PubMed] [Google Scholar]
- Brichta AM, Callister RJ, Peterson EH (1987) Quantitative analysis of cervical musculature in rats: histochemical composition and motor pool organization. I. Muscles of the spinal accessory complex. J Comp Neurol 255:351–368. 10.1002/cne.902550304 [DOI] [PubMed] [Google Scholar]
- Brill MS, Lichtman JW, Thompson W, Zuo Y, Misgeld T (2011) Spatial constraints dictate glial territories at murine neuromuscular junctions. J Cell Biol 195:293–305. 10.1083/jcb.201108005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosius Lutz A, Chung WS, Sloan SA, Carson GA, Zhou L, Lovelett E, Posada S, Zuchero JB, Barres BA (2017) Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc Natl Acad Sci USA 114:E8072–E8080. 10.1073/pnas.1710566114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco DI, Seburn KL, Pinter MJ (2016a) Altered terminal Schwann cell morphology precedes denervation in SOD1 mice. Exp Neurol 275:172–181. 10.1016/j.expneurol.2015.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco DI, Bahr BA, Seburn KL, Pinter MJ (2016b) Abnormal response of distal Schwann cells to denervation in a mouse model of motor neuron disease. Exp Neurol 278:116–126. 10.1016/j.expneurol.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro R, Taetzsch T, Vaughan SK, Godbe K, Chappell J, Settlage R, Valdez G (2020) Identification of a molecular fingerprint for synaptic glia. eLife 9:e56935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley MJ. (2007) The R book. Chichester; Hobokens: Wiley. [Google Scholar]
- Cunningham ME, Meehan GR, Robinson S, Yao D, McGonigal R, Willison HJ (2020) Perisynaptic Schwann cells phagocytose nerve terminal debris in a mouse model of Guillain-Barré syndrome. J Peripher Nerv Syst 25:143–151. 10.1111/jns.12373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabid H, Arbour D, Robitaille R (2013) Glial cells decipher synaptic competition at the mammalian neuromuscular junction. J Neurosci 33:1297–1313. 10.1523/JNEUROSCI.2935-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Winter F, Vo T, Stam FJ, Wisman LA, Bär PR, Niclou SP, van Muiswinkel FL, Verhaagen J (2006) The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol Cell Neurosci 32:102–117. 10.1016/j.mcn.2006.03.002 [DOI] [PubMed] [Google Scholar]
- Ditsworth D, Maldonado M, McAlonis-Downes M, Sun S, Seelman A, Drenner K, Arnold E, Ling SC, Pizzo D, Ravits J, Cleveland DW, Da Cruz S (2017) Mutant TDP-43 within motor neurons drives disease onset but not progression in amyotrophic lateral sclerosis. Acta Neuropathol 133:907–922. 10.1007/s00401-017-1698-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumic J, Dabelic S, Flögel M (2006) Galectin-3: an open-ended story. Biochim Biophys Acta 1760:616–635. 10.1016/j.bbagen.2005.12.020 [DOI] [PubMed] [Google Scholar]
- Duregotti E, Negro S, Scorzeto M, Zornetta I, Dickinson BC, Chang CJ, Montecucco C, Rigoni M (2015) Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc Natl Acad Sci USA 112:E497–E505. 10.1073/pnas.1417108112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen A, Kiernan M, Mitsumoto H, Swash M (2014) Amyotrophic lateral sclerosis: a long preclinical period? J Neurol Neurosurg Psychiatry 85:1232–1238. 10.1136/jnnp-2013-307135 [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28:41–51. 10.1016/S0896-6273(00)00084-2 [DOI] [PubMed] [Google Scholar]
- Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ (2007) Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci 27:9201–9219. 10.1523/JNEUROSCI.1470-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185:232–240. 10.1016/j.expneurol.2003.10.004 [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P (2000) Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 20:2534–2542. 10.1523/JNEUROSCI.20-07-02534.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou J, Robitaille R, Trimble WS, Charlton MP (1994) Synaptic regulation of glial protein expression in vivo. Neuron 12:443–455. 10.1016/0896-6273(94)90284-4 [DOI] [PubMed] [Google Scholar]
- Georgiou J, Robitaille R, Charlton MP (1999) Muscarinic control of cytoskeleton in perisynaptic glia. J Neurosci 19:3836–3846. 10.1523/JNEUROSCI.19-10-03836.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez de Aguilar JL, Niederhauser-Wiederkehr C, Halter B, De Tapia M, Di Scala F, Demougin P, Dupuis L, Primig M, Meininger V, Loeffler JP (2008) Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model. Physiol Genomics 32:207–218. 10.1152/physiolgenomics.00017.2007 [DOI] [PubMed] [Google Scholar]
- Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, Kaspar BK (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29:824–828. 10.1038/nbt.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T (2007) Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 28:154–164. 10.1016/j.nbd.2007.07.003 [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Tyreman N, Gordon T (2008) Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol 586:3337–3351. 10.1113/jphysiol.2007.149286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques A, Pitzer C, Dittgen T, Klugmann M, Dupuis L, Schneider A (2011) CNS-targeted viral delivery of G-CSF in an animal model for ALS: improved efficacy and preservation of the neuromuscular unit. Mol Ther 19:284–292. 10.1038/mt.2010.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia DJ, Feng CY, Hennig GW, Renden RB, Gould TW (2018) Activity-induced Ca(2+) signaling in perisynaptic Schwann cells of the early postnatal mouse is mediated by P2Y1 receptors and regulates muscle fatigue. Elife 7:e30839 10.7554/eLife.30839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW (2009) Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187:761–772. 10.1083/jcb.200908164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahromi BS, Robitaille R, Charlton MP (1992) Transmitter release increases intracellular calcium in perisynaptic Schwann cells in situ. Neuron 8:1069–1077. 10.1016/0896-6273(92)90128-z [DOI] [PubMed] [Google Scholar]
- Kang H, Lichtman JW (2013) Motor axon regeneration and muscle reinnervation in young adult and aged animals. J Neurosci 33:19480–19491. 10.1523/JNEUROSCI.4067-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Tian L, Mikesh M, Lichtman JW, Thompson WJ (2014) Terminal Schwann cells participate in neuromuscular synapse remodeling during reinnervation following nerve injury. J Neurosci 34:6323–6333. 10.1523/JNEUROSCI.4673-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE (2013) Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 16:571–579. 10.1038/nn.3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko CP, Robitaille R (2015) Perisynaptic Schwann cells at the neuromuscular synapse: adaptable, multitasking glial cells. Cold Spring Harb Perspect Biol 7:a020503. 10.1101/cshperspect.a020503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J (2012) Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci 32:10383–10395. 10.1523/JNEUROSCI.1498-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman BJ, Hoffman EP, Sutherland ML, Bouri K, Hsu DK, Liu FT, Rothstein JD, Knoblach SM (2012) Deletion of galectin-3 exacerbates microglial activation and accelerates disease progression and demise in a SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Brain Behav 2:563–575. 10.1002/brb3.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobsiger CS, Boillee S, McAlonis-Downes M, Khan AM, Feltri ML, Yamanaka K, Cleveland DW (2009) Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc Natl Acad Sci USA 106:4465–4470. 10.1073/pnas.0813339106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love FM, Thompson WJ (1999) Glial cells promote muscle reinnervation by responding to activity-dependent postsynaptic signals. J Neurosci 19:10390–10396. 10.1523/JNEUROSCI.19-23-10390.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas CA, Kang LH, Hoh JF (2000) Monospecific antibodies against the three mammalian fast limb myosin heavy chains. Biochem Biophys Res Commun 272:303–308. 10.1006/bbrc.2000.2768 [DOI] [PubMed] [Google Scholar]
- Magill CK, Tong A, Kawamura D, Hayashi A, Hunter DA, Parsadanian A, Mackinnon SE, Myckatyn TM (2007) Reinnervation of the tibialis anterior following sciatic nerve crush injury: a confocal microscopic study in transgenic mice. Exp Neurol 207:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau E, Di Polo A, Vande Velde C, Robitaille R (2018) Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. Elife 7:e41973 10.7554/eLife.41973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesnard NA, Haulcomb MM, Tanzer L, Sanders VM, Jones KJ (2013) Delayed functional recovery in presymptomatic mSOD1 mice following facial nerve crush axotomy. J Neurodegener Regen 4:21–25. [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Ferraiuolo L, Miranda CJ, Likhite S, McElroy S, Renusch S, Ditsworth D, Lagier-Tourenne C, Smith RA, Ravits J, Burghes AH, Shaw PJ, Cleveland DW, Kolb SJ, Kaspar BK (2014) Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci USA 111:829–832. 10.1073/pnas.1314085111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizawa YM, Hirayama Y, Ohno N, Shibata S, Shigetomi E, Sui Y, Nabekura J, Sato K, Okajima F, Takebayashi H, Okano H, Koizumi S (2017) Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat Commun 8:28. 10.1038/s41467-017-00037-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JV, Soto I, Kim KY, Bushong EA, Oglesby E, Valiente-Soriano FJ, Yang Z, Davis CH, Bedont JL, Son JL, Wei JO, Buchman VL, Zack DJ, Vidal-Sanz M, Ellisman MH, Marsh-Armstrong N (2011) Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci USA 108:1176–1181. 10.1073/pnas.1013965108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley JP, Waran MT, Balice-Gordon RJ (1999) In vivo observations of terminal Schwann cells at normal, denervated, and reinnervated mouse neuromuscular junctions. J Neurobiol 38:270–286. [DOI] [PubMed] [Google Scholar]
- Painter MW, Brosius Lutz A, Cheng YC, Latremoliere A, Duong K, Miller CM, Posada S, Cobos EJ, Zhang AX, Wagers AJ, Havton LA, Barres B, Omura T, Woolf CJ (2014) Diminished Schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron 83:331–343. 10.1016/j.neuron.2014.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E, Cleveland DW (2013) Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci 33:4657–4671. 10.1523/JNEUROSCI.1119-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P (2006) Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9:408–419. 10.1038/nn1653 [DOI] [PubMed] [Google Scholar]
- Reichert F, Saada A, Rotshenker S (1994) Peripheral nerve injury induces Schwann cells to express two macrophage phenotypes: phagocytosis and the galactose-specific lectin MAC-2. J Neurosci 14:3231–3245. 10.1523/JNEUROSCI.14-05-03231.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds ML, Woolf CJ (1992) Terminal Schwann cells elaborate extensive processes following denervation of the motor endplate. J Neurocytol 21:50–66. 10.1007/BF01206897 [DOI] [PubMed] [Google Scholar]
- Robitaille R. (1998) Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron 21:847–855. 10.1016/s0896-6273(00)80600-5 [DOI] [PubMed] [Google Scholar]
- Rocha MC, Pousinha PA, Correia AM, Sebastião AM, Ribeiro JA (2013) Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS One 8:e73846. 10.1371/journal.pone.0073846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochon D, Rousse I, Robitaille R (2001) Synapse-glia interactions at the mammalian neuromuscular junction. J Neurosci 21:3819–3829. 10.1523/JNEUROSCI.21-11-03819.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotshenker S. (2009) The role of galectin-3/MAC-2 in the activation of the innate-immune function of phagocytosis in microglia in injury and disease. J Mol Neurosci 39:99–103. 10.1007/s12031-009-9186-7 [DOI] [PubMed] [Google Scholar]
- Rotshenker S. (2011) Wallerian degeneration: the innate-immune response to traumatic nerve injury. J Neuroinflammation 8:109. 10.1186/1742-2094-8-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotshenker S, Reichert F, Gitik M, Haklai R, Elad-Sfadia G, Kloog Y (2008) Galectin-3/MAC-2, Ras and PI3K activate complement receptor-3 and scavenger receptor-AI/II mediated myelin phagocytosis in microglia. Glia 56:1607–1613. 10.1002/glia.20713 [DOI] [PubMed] [Google Scholar]
- Rousse I, St-Amour A, Darabid H, Robitaille R (2010) Synapse-glia interactions are governed by synaptic and intrinsic glial properties. Neuroscience 167:621–632. 10.1016/j.neuroscience.2010.02.036 [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Sanes JR, Lichtman JW (2005) A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol 490:209–219. 10.1002/cne.20620 [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Gorza L, Sartore S, Saggin L, Ausoni S, Vianello M, Gundersen K, Lømo T (1989) Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. J Muscle Res Cell Motil 10:197–205. 10.1007/BF01739810 [DOI] [PubMed] [Google Scholar]
- Shahidullah M, Le Marchand SJ, Fei H, Zhang J, Pandey UB, Dalva MB, Pasinelli P, Levitan IB (2013) Defects in synapse structure and function precede motor neuron degeneration in Drosophila models of FUS-related ALS. J Neurosci 33:19590–19598. 10.1523/JNEUROSCI.3396-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PS, Dick JR, Greensmith L (2005) The effect of peripheral nerve injury on disease progression in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neuroscience 130:897–910. 10.1016/j.neuroscience.2004.09.069 [DOI] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ (1995a) Schwann cell processes guide regeneration of peripheral axons. Neuron 14:125–132. 10.1016/0896-6273(95)90246-5 [DOI] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ (1995b) Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron 14:133–141. 10.1016/0896-6273(95)90247-3 [DOI] [PubMed] [Google Scholar]
- Son YJ, Trachtenberg JT, Thompson WJ (1996) Schwann cells induce and guide sprouting and reinnervation of neuromuscular junctions. Trends Neurosci 19:280–285. 10.1016/S0166-2236(96)10032-1 [DOI] [PubMed] [Google Scholar]
- Swarup V, Audet JN, Phaneuf D, Kriz J, Julien JP (2012) Abnormal regenerative responses and impaired axonal outgrowth after nerve crush in TDP-43 transgenic mouse models of amyotrophic lateral sclerosis. J Neurosci 32:18186–18195. 10.1523/JNEUROSCI.2267-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallon C, Russell KA, Sakhalkar S, Andrapallayal N, Farah MH (2016) Length-dependent axo-terminal degeneration at the neuromuscular synapses of type II muscle in SOD1 mice. Neuroscience 312:179–189. 10.1016/j.neuroscience.2015.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson W, Jansen JK (1977) The extent of sprouting of remaining motor units in partly denervated immature and adult rat soleus muscle. Neuroscience 2:523–535. 10.1016/0306-4522(77)90049-5 [DOI] [PubMed] [Google Scholar]
- Todd KJ, Darabid H, Robitaille R (2010) Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci 30:11870–11882. 10.1523/JNEUROSCI.3165-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay E, Martineau E, Robitaille R (2017) Opposite synaptic alterations at the neuromuscular junction in an ALS mouse model: when motor units matter. J Neurosci 37:8901–8918. 10.1523/JNEUROSCI.3090-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BJ, Ackerley S, Davies KE, Talbot K (2010) Dismutase-competent SOD1 mutant accumulation in myelinating Schwann cells is not detrimental to normal or transgenic ALS model mice. Hum Mol Genet 19:815–824. 10.1093/hmg/ddp550 [DOI] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR (2012) Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 7:e34640. 10.1371/journal.pone.0034640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke JM, Smit-Oistad IM, Macrander C, Krakora D, Meyer MG, Suzuki M (2016) Macrophage-mediated inflammation and glial response in the skeletal muscle of a rat model of familial amyotrophic lateral sclerosis (ALS). Exp Neurol 277:275–282. 10.1016/j.expneurol.2016.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Pytel P, Feltri ML, Wrabetz L, Roos RP (2012) Selective knockdown of mutant SOD1 in Schwann cells ameliorates disease in G85R mutant SOD1 transgenic mice. Neurobiol Dis 48:52–57. 10.1016/j.nbd.2012.05.014 [DOI] [PubMed] [Google Scholar]
- Wright MC, Potluri S, Wang X, Dentcheva E, Gautam D, Tessler A, Wess J, Rich MM, Son YJ (2009) Distinct muscarinic acetylcholine receptor subtypes contribute to stability and growth, but not compensatory plasticity, of neuromuscular synapses. J Neurosci 29:14942–14955. 10.1523/JNEUROSCI.2276-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt RM, Balice-Gordon RJ (2008) Heterogeneity in synaptic vesicle release at neuromuscular synapses of mice expressing synaptopHluorin. J Neurosci 28:325–335. 10.1523/JNEUROSCI.3544-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253. 10.1038/nn2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RY, Rabinovich GA, Liu FT (2008) Galectins: structure, function and therapeutic potential. Expert Rev Mol Med 10:e17. 10.1017/S1462399408000719 [DOI] [PubMed] [Google Scholar]
- Zhou JY, Afjehi-Sadat L, Asress S, Duong DM, Cudkowicz M, Glass JD, Peng J (2010) Galectin-3 is a candidate biomarker for amyotrophic lateral sclerosis: discovery by a proteomics approach. J Proteome Res 9:5133–5141. 10.1021/pr100409r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwiegers P, Lee G, Shaw CA (2014) Reduction in hSOD1 copy number significantly impacts ALS phenotype presentation in G37R (line 29) mice: implications for the assessment of putative therapeutic agents. J Negat Results Biomed 13:14. 10.1186/1477-5751-13-14 [DOI] [PMC free article] [PubMed] [Google Scholar]