

Abstract
Increased aortic stiffness may contribute to kidney damage by transferring excessive flow pulsatility to susceptible renal microvasculature, leading to constriction or vessel loss. We previously demonstrated that 5 weeks of dietary sodium restriction (DSR) reduces large-elastic artery stiffness as well as blood pressure in healthy middle-aged/older adults with moderately elevated systolic blood pressure (SBP) who are free from chronic kidney disease (CKD). We hypothesized that DSR in this cohort would also reduce urinary concentrations of renal tubular injury biomarkers, which predict incident CKD in the general population. We performed a post hoc analysis using stored 24 hours urine samples collected in 13 participants as part of a randomized, double-blind, crossover clinical trial of DSR (low sodium (LS) target: 50 mmol/day; normal sodium (NS) target: 150 mmol/day). Participants were 61±2 (mean±SEM) years (8 M/5 F) with a baseline blood pressure of 139±2/82±2 mm Hg and an estimated glomerular filtration rate of 79±3 mL/min/1.73 m2. Twenty-four hour urinary sodium excretion was reduced from 149±7 to 66±8 mmol/day during week 5. Despite having preserved kidney function, participants had a 31% reduction in urinary neutrophil gelatinase-associated lipocalin concentrations with just 5 weeks of DSR (LS: 2.8±0.6 vs NS: 4.2±0.8 ng/mL, p<0.05). Results were similar when normalized to urinary creatinine (urinary creatinine did not change between conditions). Concentrations of another kidney tubular injury biomarker, kidney injury molecule-1, were below the detectable limit in all but one sample. In conclusion, DSR reduces an established clinical biomarker of kidney tubular damage in adults with moderately elevated SBP who are free from prevalent kidney disease.
INTRODUCTION
Arterial stiffness increases with aging and is a major risk factor for cardiovascular diseases (CVD).1 Increased arterial stiffness may contribute to kidney damage by transferring excessive flow pulsatility to susceptible renal microvasculature, leading to constriction or vessel loss.2 It precedes the development of hypertension and contributes to pathogenesis of target organ damage, including the kidney.3,4 Using a randomized, placebo-controlled crossover study design, we recently demonstrated that DSR reduced arterial stiffness (measured by aortic pulse-wave velocity (aPWV)) and systolic blood pressure (SBP), in middle-aged and older adults with moderately elevated SBP who were otherwise healthy and free of kidney disease.5 Given these previous findings, we hypothesized that DSR may also reduce biomarkers of renal tubular injury, via reduced arterial stiffness and blood pressure, or other multifactorial effects of reduced dietary sodium.
High sodium intake promotes hypertension and also has detrimental effect on the kidney, including promoting proteinuria, hyperfiltration and reducing responsiveness to renin-angiotensin-aldosterone system blockade.6–10 Previous research on the effects of DSR on the kidneys has mainly focused on patients with CKD, with observed benefits including reduced BP, extracellular fluid volume and proteinuria.11–13 Urinary kidney injury molecule-1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) are well-developed biomarkers for acute and chronic renal injury.14–17 Higher levels of these biomarkers independently predict incident CKD in the general population,15,18 thus any reduction would be of high clinical significance in this cohort free from prevalent CKD.
Therefore, our objective was to examine the effect of DSR on these kidney injury markers in the aforementioned randomized double-blinded trial. We evaluated stored 24 hours urine samples collected during the trial.
MATERIALS AND METHODS
This was a post hoc analysis of a randomized, placebo-controlled crossover trial. The details of the parent study, conducted from February 2009 to January 2012, have been published previously.5 The trial was conducted at the University of Colorado Boulder Clinical and Translational Research Center.
Subjects
In this post hoc analysis, total of 13 of the 17 subjects from the parent study who had sufficient stored urine samples remaining were included. The inclusion and exclusion criteria for the parent study have been described previously.5 Briefly, all subjects had a resting SBP within 130–159 mm Hg, that is, high normal or stage I systolic hypertension, and diastolic blood pressure <99 mm Hg, verified on a minimum of two occasions,19 but were otherwise free of CVD, diabetes, kidney disease and other clinical disorders as assessed by medical history, physical examination, ankle-brachial index (≤0.9), blood chemistries and resting and exercise ECG. All subjects were non-smokers, had a body mass index <40 kg/m2 and were not taking dietary supplements known to influence vascular function, including those with antioxidant properties. Women (n=5) were postmenopausal and were not taking hormone replacement therapy. Subjects were either sedentary or recreationally active, but none were performing regular, vigorous exercise.
Experimental design and dietary sodium restriction
The trial used a double-blind, placebo-controlled, randomized, crossover design, as described previously.5 Briefly, a LS intake of ~1500 mg/day (65 mmol/day) was compared with an NS intake of 3600 mg/day (150 mmol/day). During the entire 10-week intervention period, subjects reduced dietary sodium (target was 50 mmol/day) and were instructed to take a total of 10 tablets spread across the day with meals. For five of the weeks the tablets were placebo pills, whereas, for the other 5 weeks the tablets were slow-release NaCl tablets (10 mmol (0.23 g) per tablet) (HK Pharma, UK), in a randomized order. The slow-release NaCl tablets aimed to return sodium intake to the ~150 mmol/day target. Subjects were provided with comprehensive dietary education and weekly counseling by Clinical and Translational Research Center bionutritionists in order to reduce dietary sodium intake without changing caloric intake, dietary composition or potassium intake (these data have all been published previously).5 The investigators were blinded to sodium condition in the analysis and recording of all variables (including blood pressure). There was no washout period between conditions and no evidence of a carryover effect.5
Blood pressure, urine assays and aortic pulse-wave velocity
Resting blood pressure assessment and 24 hours urine collections were performed at baseline (twice) and then weekly throughout the 10-week dietary intervention (twice during the final week of each condition), for a total of 14 measurements per subject, as previously described in detail for this study.5 Urine NGAL (uNGAL) and KIM-1 (uKIM-1) levels were assessed using ELISA (R&D Systems) using stored samples collected during the final week of each sodium condition. The sensitivity for assays is 0.04 ng/mL for uNGAL and 0.045 ng/mL for uKIM-1. The intra-assay precisions are 3.7% and 4.2%; the interassay precisions are 6.5% and 6.4% for uNGAL and uKIM-1, respectively. aPVW was measured during the final week (week 5) of each sodium condition and methods have been described previously.19
Statistics
Differences in subject characteristics and outcome variables were assessed using paired t-tests between dietary sodium conditions. The influence of SBP on sodium-related differences in uNGAL was analyzed using analysis of covariance. Bivariate associations were determined using the Pearson’s correlation coefficient. All data are reported as means±SEM. Analyses were performed using SPSS V.24 and statistical significance for all analyses was set at p<0.05.
RESULTS
Effectiveness of dietary sodium restriction and baseline clinical characteristics
Participants were 61±2 years (8 M/5 F) with a baseline SBP of 139±2, diastolic BP (DBP) of 82±2 mm Hg and a baseline estimated glomerular filtration rate (eGFR) of 79±3 mL/min/1.73 m2 (modification of diet in renal disease study equation). DSR successfully reduced sodium excretion, SBP (and aPVW, as reported previously).5,20 It also significantly reduced DBP, although to a lesser degree. Other clinical characteristics, including eGFR and dietary components were not changed (table 1 and previous publication).5
Table 1.
Clinical characteristics
| Variable | Low sodium (n=13) | Normal sodium (n=13) | P value |
|---|---|---|---|
| Sex (female/male) | (5/8) | – | – |
| Age (years) | 61±2 | – | |
| Race (% (n) Caucasian, Asian) | 85% (11), 15% (2) | – | – |
| MDRD eGFR (mL/min/1.73 m2) | 80±4 | 83±5 | 0.28 |
| Urine sodium (mmol/24 hour) | 66±8 | 150±7 | <0.0001 |
| Urine creatinine (mg/L) | 645±69 | 586±75 | 0.42 |
| Urine potassium (mmol/24 hour) | 71 ±6 | 77±7 | 0.32 |
| SBP (mm Hg) | 126±2 | 138±4 | <0.001 |
| DBP (mm Hg) | 77±2 | 80±2 | 0.01 |
| aPWV (cm/s) | 747±42 | 884±32 | <0.0001 |
Data are mean±SEM.
aPWV, aortic pulse-wave velocity; DBP, diastolic blood pressure; eGFR, estimated glomerular filtration rate; MDRD, modification of diet in renal disease; SBP, systolic blood pressure.
DSR and uNGAL and KIM-1 excretion
uNGAL excretion was reduced by 31% following 5 weeks of LS compared with 5 weeks of high sodium (LS: 2.8±0.6 ng/mL vs NS: 4.2±0.8 ng/mL, p<0.05, figure 1A). Individual changes in uNGAL with DSR are also shown (figure 1B), with all but two subjects reducing uNGAL concentrations with LS versus NS. Results were similar when normalized to urinary creatinine (LS: 5.0±1.1 vs NS: 8.5±2. μg/g, p<0.05) (urinary creatinine did not change between conditions). The group difference in NGAL was attenuated when adjusting for SBP (p=0.09), but not for aPWV (p<0.01). uKim-1 concentrations, however, were below the detectable limit in all but one sample. There were no significant correlations between uNGAL, SBP, aPWV or sodium excretion, or among changes in these variables.
Figure 1.
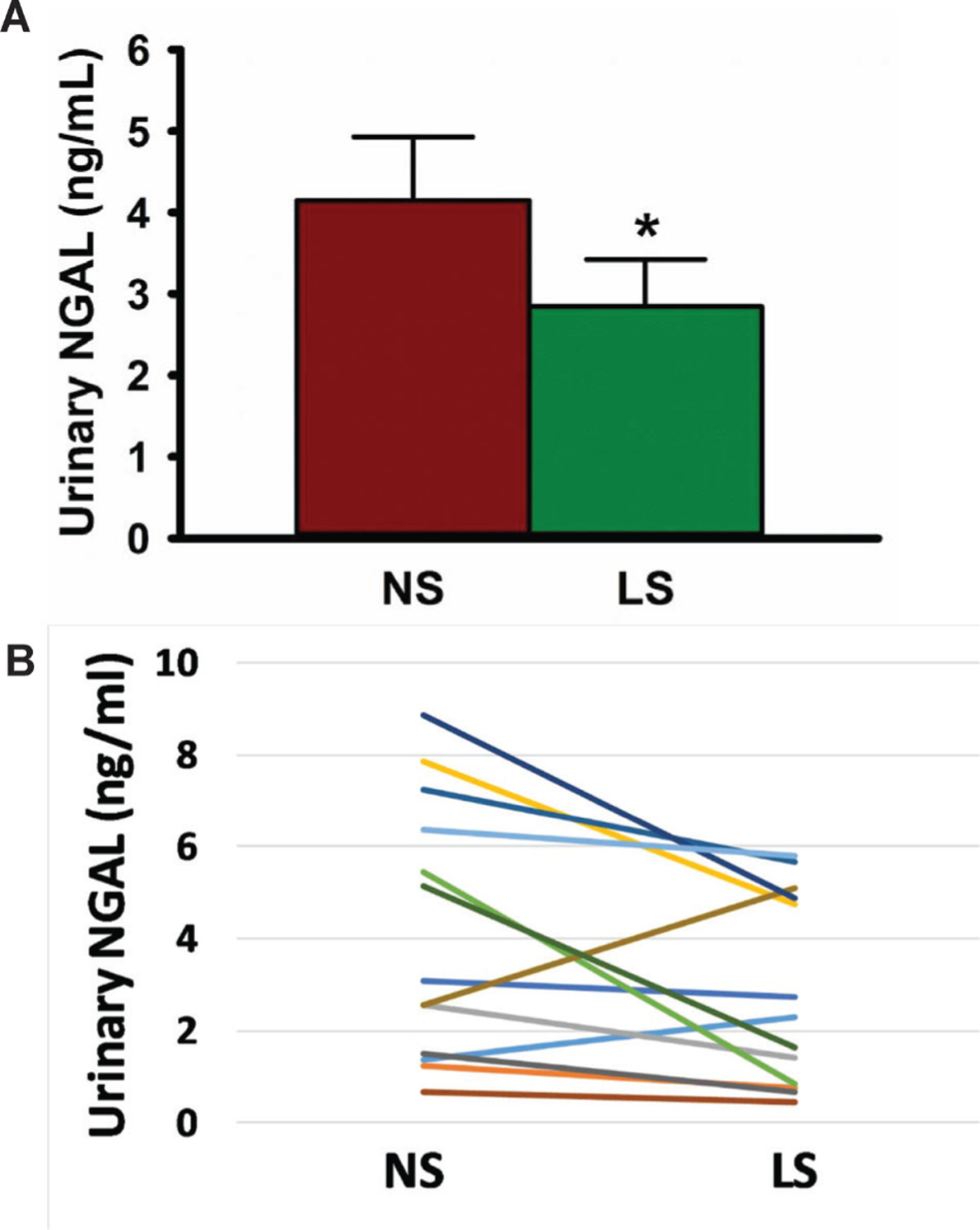
Dietary sodium restriction reduces urinary NGAL concentrations. (A) Tweny-four hour urinary NGAL concentrations as measured during the final week (week 5) of each sodium condition; (B) individual changes of urinary NGAL concentrations as measured during the final week (week 5) of each sodium condition. LS, low sodium; NGAL, neutrophil gelatinase-associated lipocalin; NS, normal sodium. Data are mean±SEM. *P<0.05.
DISCUSSION
This is the first evidence that DSR reduces the kidney injury marker NGAL in individuals with moderately elevated SBP but otherwise in good health. Our results provide initial support to the hypothesis that DSR may be an effective strategy to reduce kidney tubular injury. As markers of kidney tubular injury predict incident CKD in the general population,15,18 this finding is of great clinical relevance.
Our initial hypothesis was that DSR would reduce evidence of kidney tubular injury via reduced arterial stiffness (less transfer of excessive flow pulsatility to susceptible renal microvasculature). However, we did not observe any association between uNGAL, aPWV, SBP, urinary sodium excretion or among changes in any of these variables, limiting direct support for this mechanistic explanation of the observed change in NGAL with DSR. This is very likely due to the very small sample size and a lack of power. Notably, the observed reduction in uNGAL with sodium restriction was no longer statistically significant when it was adjusted for SBP. Thus, reduced SBP by DSR could also be a factor mediating the beneficial effects of DSR on uNAGL. However, the modest change in the p value following adjustment for SBP suggests that the beneficial effect of DSR on uNGAL likely goes beyond reduced SBP. This could be explained by the multifactorial effects of DSR beyond these mechanisms. For example, in the setting of DSR, the resulting relative volume contraction could lead to increased proximal tubular reabsorption of NGAL and thus less excretion. Our original observation that DSR trended to increase (although statistically insignificant) serum levels of renin, angiotensin II and aldosterone supports this notion.5
DSR may also have other direct effects on the kidneys. Prior evidence suggests that sodium intake modifies both proteinuria and kidney function in the setting of kidney disease. High salt intake blunts the antiproteinuric effects of ACE inhibitors (ACEI), independent of BP control, and increases the risk of progression to end-stage renal disease in patients without diabetes but with CKD.6 Short-term high salt intake also increases microalbuminuria in patients with type II diabetes.10 Similarly, angiotensin receptor blockers (ARBs) are more efficacious for slowing renal disease progression in patients with diabetes consuming a low compared with high sodium diet.21 Consistent with epidemiological evidence, dietary sodium intake modifies the antiproteinuric effects of ACEI/ARB in clinical trials of proteinuric patients.22,23 In the a large number of participants with prevalent CKD in the Chronic Renal Insufficiency Cohort study, higher 24 hours urinary sodium excretion was associated with increased risk of CKD progression.24
Mechanisms underlying the direct effect of DSR on kidney injury remain unclear. A high sodium diet has direct effects on the kidney via induction of transforming growth factor-β, oxidative stress25 and compensatory renal growth.26 A beneficial effect of DSR on kidney injury could be attributed to its ability to potentiate the effect of the renin-angiotensin-aldosterone system blockade.22,27 We have previously shown that DSR reduces endothelial cell NADPH oxidase expression and vascular oxidative stress20 supporting the hypothesis that the beneficial effect of DSR on the kidney may be due in part to a reduction in oxidative stress.
Decreased eGFR (increased serum creatinine) has been previously observed with DSR.11,13 This was likely caused by the correction of hyperfiltration, a sign of poor renal prognosis.7,28 In our study, baseline eGFR was normal and not changed by DSR, indicating minimal renal hemodynamic change in this population. Notably, renal tubular injury biomarkers are sensitive to change is response to an intervention, with reductions observed response to intensive blood pressure lowering in the Systolic Blood Pressure Intervention Trial, despite loss of eGFR thought to reflect hemodynamic changes.29,30 Of note, there was also no difference in the current study between the two groups in urinary potassium excretion, another potential confounding factor that might affect renal outcomes.24
Even though eGFR of our hypertensive subjects was normal, the likelihood of early renal structural damage related to aging is quite plausible.31 The observed 30% reduction in uNGAL with DSR supports this notion. Traditional markers of kidney injury, such as serum creatinine, are neither sensitive nor specific for the diagnosis of acute kidney injury.32,33 NGAL is a small secreted protein initially identified in mature neutrophil granules,34 but has since been found to be expressed in many cells including renal cells.35 It is synthesized in the distal nephron and secreted freely into the urine in response to structural kidney injury.36 uNGAL is stable and resistant to proteases, making it a good candidate for clinical use.36 Both urinary and circulating levels increase markedly in various acute and chronic kidney injuries.37 We have observed that although uNGAL concentrations were relatively low in our healthy cohort, they were significantly reduced with DSR, which could have clinically significant implications long-term. However, the low concentrations of uNGAL even in the NS group, may potentially limit the clinical relevance of the observed reduction with DSR. The relatively low concentrations compared with other studies15,38 could be partly due to different detection methods (eg, immunoblot38 vs ELISA).
It is also important to highlight that the predictive value of uNGAL for CKD progression is not consistently reported, particularly after adjustment for other variables such as urinary creatinine and albumin levels.15,39,40 However, in a subset of patients with CKD with primarily inflammatory renal diseases, urinary biomarkers including uNGAL appear to be favorable candidates.40 Thus, the clinical relevance of urinary NGAL as a biomarker might be dependent on type and stage of renal disease.41 Additionally, the utility of NGAL as a biomarker in acute kidney injury is inconsistent and complicated by the fact that it is a complex molecule produced by multiple tissues is different molecular forms41. Of note, although the current study cannot distinguish between homodimeric (mainly by neutrophils) and monomeric (by stressed kidney) forms of NGAL, it is unlikely that neutrophils would be a major source of urinary NGAL in this cohort, due to the nature of the study population (hypertensive otherwise healthy normal adults).
uKIM-1 is a marker for proximal tubular injury and its concentrations are strongly related to its tubular expression in acute kidney injury.17,42 uKIM-1 concentrations, however, were not detectable in our cohort of relatively healthy middle-aged and older adults. It is unlikely that uKIM-1 was degraded since it is very stable even at ambient temperature.43 The absence of uKIM-1 in the study could be a sign of minimal proximal tubular injury. Plasma KIM-1 (pKIM-1) was recently found to be a biomarker for acute and chronic kidney injury.44 It predicts progression to ESRD in type I diabetes,44 and predicts future decline of eGFR and risk of CKD in a healthy middle-aged population.18 While we did not have sufficient remaining sample to measure pKIM-1 in the current study, pKIM-1 should be investigated as an alternate biomarker in future research.
The major limitation of the study is its small sample size and short follow-up. With a limited sample size, uNGAL did not correlate with aPWV, limiting our ability to support the hypothesis that dietary sodium may promote end-organ kidney damage via increased arterial stiffness. We also did not investigate other potential contributing mechanisms beyond arterial stiffness and blood pressure. Our population was primarily Caucasian adults, limited generalizability. Urinary protein was not measured, although this population was known to be free from CKD. Additionally, we did not measure uNGAL at baseline in participants; thus, it is possible that dietary changes across both conditions lowered uNGAL as compared with usual diet. However, our crossover design comparing the LS and NS allowed for isolation of sodium as the sole variable differing between the two conditions when uNGAL was measured. Our study also has notable strengths. Importantly, the application of measuring markers of kidney tubule damage in the setting of an intervention in a non-CKD population is innovative. Additionally, our crossover design allowed with isolation of dietary sodium as the sole manipulated dietary factor with outcomes assessed in the same participants under each sodium condition.
In conclusion, this is the first trial to demonstrate that DSR reduces a sensitive kidney injury marker, uNGAL, in middle-aged and older adults with moderately elevated SBP and preserved kidney function. Future research should further evaluate DSR as a potential strategy to prevent incident CKD, as well as slow progression of prevalent CKD. A randomized controlled trial involving large number of participants and longer duration of follow-up is warranted to fully understand the effects of DSR on kidney tubule damage and the effects on renal function.
Significance of this study.
What is already known about this subject?
Dietary sodium restriction (DSR) lowers blood pressure.
DSR reduces arterial stiffness.
Renal tubular injury biomarkers predict incident chronic kidney disease (CKD) in the general population.
What are the new findings?
DSR reduces urinary neutrophil gelatinase-associated lipocalin (NGAL) concentrations, a biomarker of kidney tubular damage in healthy middle-aged and older adults with moderately elevated blood pressure.
Concentrations of another kidney tubular injury biomarker, kidney injury molecule-1, were below the detectable limit in all but one sample.
Changes in urinary NGAL did not correlate with previously observed reductions in arterial stiffness or blood pressure; however, the sample size was limited.
How might these results change the focus of research or clinical practice?
Future research should further evaluate DSR as a potential strategy to prevent incident CKD, as well as slow progression of prevalent CKD.
A randomized controlled trial involving large number of participants and longer duration of follow-up is warranted to fully understand the effects of DSR on kidney tubule damage and the effects on renal function in this population.
The effects of DSR on kidney tubule damage should also be evaluated in a population with prevalent CKD.
Acknowledgements
The authors would like to thank the staff of the University of Colorado Boulder Clinical and Translational Research Center, particularly the bionutritionists for their technical assistance.
Funding This work was supported by National Institutes of Health awards NIH AG013038, AG006537, AG031141, AG033994, AG031617 and UL1 TR0002535.
Footnotes
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All procedures were approved by the Institutional Review Board of the University of Colorado Boulder (0608.12) and conformed with the Declaration of Helsinki. The nature, benefits and risks of the study were explained to the volunteers and their written informed consent was obtained prior to participation.
Data availability statement Data are available on reasonable request.
REFERENCES
- 1.Mitchell GF. Aortic stiffness, pressure and flow pulsatility, and target organ damage. J Appl Physiol 2018;125:1871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitchell GF. Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end-organ damage. J Appl Physiol 2008;105:1652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitchell GF, Hwang S-J, Vasan RS, et al. Arterial stiffness and cardiovascular events: the Framingham heart study. Circulation 2010;121:505–1 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Najjar SS, Scuteri A, Shetty V, et al. Pulse wave velocity is an independent predictor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore longitudinal study of aging. J Am Coll Cardiol 2008;51:1377–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jablonski KL, Racine ML, Geolfos CJ, et al. Dietary sodium restriction reverses vascular endothelial dysfunction in middle-aged/older adults with moderately elevated systolic blood pressure. J Am Coll Cardiol 2013;61:335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vegter S, Perna A, Postma MJ, et al. Sodium intake, ACE inhibition, and progression to ESRD. J Am Soc Nephrol 2012;23:165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weir MR, Dengel DR, Behrens MT, et al. Salt-Induced increases in systolic blood pressure affect renal hemodynamics and proteinuria. Hypertension 1995;25:1339–44. [DOI] [PubMed] [Google Scholar]
- 8.Jones-Burton C, Mishra SI, Fink JC, et al. An in-depth review of the evidence linking dietary salt intake and progression of chronic kidney disease. Am J Nephrol 2006;26:268–75. [DOI] [PubMed] [Google Scholar]
- 9.Krikken JA, Laverman GD, Navis G. Benefits of dietary sodium restriction in the management of chronic kidney disease. Curr Opin Nephrol Hypertens 2009;18:531–8. [DOI] [PubMed] [Google Scholar]
- 10.Vedovato M, Lepore G, Coracina A, et al. Effect of sodium intake on blood pressure and albuminuria in type 2 diabetic patients: the role of insulin resistance. Diabetologia 2004;47:300–3. [DOI] [PubMed] [Google Scholar]
- 11.Campbell KL, Johnson DW, Bauer JD, et al. A randomized trial of sodium-restriction on kidney function, fluid volume and adipokines in CKD patients. BMC Nephrol 2014;15:57–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMahon EJ, Bauer JD, Hawley CM, et al. A randomized trial of dietary sodium restriction in CKD. J Am Soc Nephrol 2013;24:2096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saran R, Padilla RL, Gillespie BW, et al. A randomized crossover trial of dietary sodium restriction in stage 3–4 CKD. Clin J Am Soc Nephrol 2017;12:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singer E, Elger A, Elitok S, et al. Urinary neutrophil gelatinase-associated lipocalin distinguishes pre-renal from intrinsic renal failure and predicts outcomes. Kidney Int 2011;80:405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhavsar NA, Köttgen A, Coresh J, et al. Neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury molecule 1 (KIM-1) as predictors of incident CKD stage 3: the Atherosclerosis risk in communities (ARIC) study. Am J Kidney Dis 2012;60:233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han WK, Bailly V, Abichandani R, et al. Kidney injury molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int 2002;62:237–44. [DOI] [PubMed] [Google Scholar]
- 17.Waanders F, van Timmeren MM, Stegeman CA, et al. Kidney injury molecule-1 in renal disease. J Pathol 2010;220:7–16. [DOI] [PubMed] [Google Scholar]
- 18.Schulz C-A, Engström G, Nilsson J, et al. Plasma kidney injury molecule-1 (p-KIM-1) levels and deterioration of kidney function over 16 years. Nephrol Dial Transplant 2020;35:265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seals DR, Tanaka H, Clevenger CM, et al. Blood pressure reductions with exercise and sodium restriction in postmenopausal women with elevated systolic pressure: role of arterial stiffness. J Am Coll Cardiol 2001;38:506–13. [DOI] [PubMed] [Google Scholar]
- 20.Jablonski KL, Fedorova OV, Racine ML, et al. Dietary sodium restriction and association with urinary marinobufagenin, blood pressure, and aortic stiffness. Clin J Am Soc Nephrol 2013;8:1952–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambers Heerspink HJ, Holtkamp FA, Parving H-H, et al. Moderation of dietary sodium potentiates the renal and cardiovascular protective effects of angiotensin receptor blockers. Kidney Int 2012;82:330–7. [DOI] [PubMed] [Google Scholar]
- 22.Vogt L, Waanders F, Boomsma F, et al. Effects of dietary sodium and hydrochlorothiazide on the antiproteinuric efficacy of losartan. J Am Soc Nephrol 2008;19:999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D’Elia L, Rossi G, Schiano di Cola M, et al. Meta-Analysis of the effect of dietary sodium restriction with or without concomitant renin-angiotensin-aldosterone System-Inhibiting treatment on albuminuria. Clin J Am Soc Nephrol 2015;10:1542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He J, Mills KT, Appel LJ, et al. Urinary sodium and potassium excretion and CKD progression. J Am Soc Nephrol 2016;27:1202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng S, Roberts LJ, Cason GW, et al. Superoxide dismutase and oxidative stress in Dahl salt-sensitive and -resistant rats. Am J Physiol Regul Integr Comp Physiol 2002;283:R732–8. [DOI] [PubMed] [Google Scholar]
- 26.Lax DS, Benstein JA, Tolbert E, et al. Effects of salt restriction on renal growth and glomerular injury in rats with remnant kidneys. Kidney Int 1992;41:1527–34. [DOI] [PubMed] [Google Scholar]
- 27.Esnault VLM, Ekhlas A, Delcroix C, et al. Diuretic and enhanced sodium restriction results in improved antiproteinuric response to Ras blocking agents. J Am Soc Nephrol 2005;16:474–81. [DOI] [PubMed] [Google Scholar]
- 28.Bigazzi R, Bianchi S, Baldari D, et al. Microalbuminuria in salt-sensitive patients. A marker for renal and cardiovascular risk factors. Hypertension 1994;23:195–9. [DOI] [PubMed] [Google Scholar]
- 29.Malhotra R, Craven T, Ambrosius WT, et al. Effects of intensive blood pressure lowering on kidney tubule injury in CKD: a longitudinal subgroup analysis in sprint. Am J Kidney Dis 2019;73:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang WR, Craven TE, Malhotra R, et al. Kidney damage biomarkers and incident chronic kidney disease during blood pressure reduction: a case-control study. Ann Intern Med 2018;169:610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension 2004;44:595–601. [DOI] [PubMed] [Google Scholar]
- 32.Beker BM, Corleto MG, Fieiras C, et al. Novel acute kidney injury biomarkers: their characteristics, utility and concerns. Int Urol Nephrol 2018;50:705–13. [DOI] [PubMed] [Google Scholar]
- 33.Vaidya VS, Ferguson MA, Bonventre JV. Biomarkers of acute kidney injury.Annu Rev Pharmacol Toxicol 2008;48:463–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kjeldsen L, Johnsen AH, Sengeløv H, et al. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem 1993;268:10425–32. [PubMed] [Google Scholar]
- 35.Liu F, Yang H, Chen H, et al. High expression of neutrophil gelatinase-associated lipocalin (NGAL) in the kidney proximal tubules of diabetic rats. Adv Med Sci 2015;60:133–8. [DOI] [PubMed] [Google Scholar]
- 36.Mishra J, Ma Q, Prada A, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 2003;14:2534–43. [DOI] [PubMed] [Google Scholar]
- 37.Buonafine M, Martinez-Martinez E, Jaisser F. More than a simple biomarker: the role of NGAL in cardiovascular and renal diseases. Clin Sci 2018;132:909–23. [DOI] [PubMed] [Google Scholar]
- 38.Nickolas TL, O’Rourke MJ, Yang J, et al. Sensitivity and specificity of a single emergency department measurement of urinary neutrophil gelatinase-associated lipocalin for diagnosing acute kidney injury. Ann Intern Med 2008;148:810–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu C-Y, Xie D, Waikar SS, et al. Urine biomarkers of tubular injury do not improve on the clinical model predicting chronic kidney disease progression. Kidney Int 2017;91:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seibert FS, Sitz M, Passfall J, et al. Prognostic value of urinary calprotectin, NGAL and KIM-1 in chronic kidney disease. Kidney Blood Press Res 2018;43:1255–62. [DOI] [PubMed] [Google Scholar]
- 41.Mårtensson J, Bellomo R. The rise and fall of NGAL in acute kidney injury. Blood Purif 2014;37:304–10. [DOI] [PubMed] [Google Scholar]
- 42.van Timmeren MM, van den Heuvel MC, Bailly V, et al. Tubular kidney injury molecule-1 (KIM-1) in human renal disease. J Pathol 2007;212:209–17. [DOI] [PubMed] [Google Scholar]
- 43.Bonventre JV. Kidney injury molecule-1 (KIM-1): a urinary biomarker and much more. Nephrol Dial Transplant 2009;24:3265–8. [DOI] [PubMed] [Google Scholar]
- 44.Sabbisetti VS, Waikar SS, Antoine DJ, et al. Blood kidney injury molecule-1 is a biomarker of acute and chronic kidney injury and predicts progression to ESRD in type I diabetes. J Am Soc Nephrol 2014;25:2177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]