

Notch or STING, which is in charge of CD4 T cell fate?
Abstract
Dysregulation of T cell apoptosis contributes to the pathogenesis of acute systemic inflammation–induced immunosuppression, as seen in sepsis and trauma. However, the regulatory mechanisms of T cell apoptosis are unclear. Activation of stimulator of interferon genes (STING) has been shown to induce T cell apoptosis. Notch was previously identified as the top negative regulator of STING in macrophages through a kinase inhibitor library screening. However, how Notch signaling regulates STING activation in T cells is unknown. Here, using a γ-secretase inhibitor to block Notch signaling, we found that Notch protected CD4 T cells from STING-mediated apoptosis during endotoxemia. Mechanistically, Notch intracellular domain (NICD) interacted with STING at the cyclic dinucleotide (CDN) binding domain and competed with CDN to inhibit STING activation. In conclusion, our data reveal a previously unidentified role of Notch in negative regulation of STING-mediated apoptosis in CD4 T cells.
INTRODUCTION
Loss of immune cells through apoptosis is one of the known mechanisms responsible for impaired immune defenses in acute critical illness as seen in sepsis, trauma, burns, and other causes of a dysregulated immune response (1, 2). CD4 T cells are particularly sensitive to apoptotic loss, and this is thought to contribute to increased susceptibility toward secondary infections and mortality in patients with sepsis (1, 3, 4). However, the mechanisms of CD4 T cell apoptosis in critically ill patients are unclear.
The Notch signaling pathway was originally identified as a regulator of embryonic development (5). It is now known to play roles in a variety of immune and nonimmune cells by regulating proliferation and cell fate decisions (6). The Notch intracellular signaling domain (NICD) acts as a transcription factor and positively regulates the production of lipopolysaccharide (LPS)–induced inflammatory cytokines in macrophage (7, 8). A recent study indicates that Notch activation suppresses T helper 1 (TH1)/TH17 differentiation and the cytotoxicity of peripheral CD8 T cells in septic patients (9). However, the mechanisms by which Notch regulates T cell fate are not understood.
We have previously shown that deletion of stimulator of interferon genes (STING) increases survival in a mouse polymicrobial intra-abdominal sepsis model (10). A targeted screen of known major signaling pathways identified Notch as a negative regular of STING signaling in macrophages (10). However, how Notch signaling regulates STING is not known. STING is an adaptor protein located in the endoplasmic reticulum (ER) that can be activated upon STING engagement of cyclic dinucleotides (CDNs) produced by cyclic GMP-AMP synthase (cGAS) or generated by bacteria. Upon the binding of CDN, STING translocates to the ER-Golgi intermediate compartment to initiate type I interferon responses (11). Recent studies have shown that activation of STING also induces apoptosis and necroptosis in a variety of immune cells, including T cells (12, 13). However, the roles of STING or Notch in regulating T cell survival during a systemic inflammatory response, as seen in sepsis, are not known.
In this study, we used a mouse endotoxemia model to investigate the roles of Notch and STING signaling pathways in splenic T cell apoptosis. We found that inhibition of Notch using a γ-secretase inhibitor (DAPT) substantially increased STING-dependent splenic CD4 T cell apoptosis during endotoxemia. Mechanistic studies showed that NICD blocks STING activation by preventing CDN binding to STING. These studies demonstrate the central roles of Notch and STING signaling in CD4 T cell apoptosis during acute systemic inflammation and reveal a previously unknown role for Notch as a negative regulator of STING.
RESULTS
Inhibition of Notch enhances mortality, inflammation, and apoptosis during endotoxemia
To assess the roles of Notch in regulating the response of T cells during a severe acute systemic inflammatory response, we administered the Notch inhibitor DAPT to mice before a bolus of LPS. Notch signaling, represented by increases in detection of NICD, was transiently up-regulated in the spleen after LPS treatment (Fig. 1A). NICD levels peaked at 8 hours and then gradually decreased to baseline by 24 hours (Fig. 1A). Hairy and enhancer of split 1 (HES1), a downstream transcriptional target of NICD, was also transiently up-regulated upon LPS stimulation and followed a similar time-dependent expression pattern as NICD (Fig. 1A). Splenocytes pretreated with DAPT 3 hours before LPS injection showed decreased levels of NICD and Hes1, compared to splenocytes treated only with LPS (Fig. 1A). Notch inhibition during endotoxemia was associated with more splenocyte apoptosis, as represented by increased cleaved (activated) caspase-3 in the spleen (Fig. 1A). To confirm this, we used in situ TMR red to stain for TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling)–positive nuclei in spleens from endotoxemia mice at 8 hours after LPS injection, a time point where Notch signaling is most activated. Consistent with the previous findings, we found significantly more TUNEL-positive nuclei (red) in spleens from mice treated with DAPT + LPS compared with mice receiving vehicle + LPS (Fig. 1B). Circulating inflammatory mediators, including interleukin-1α (IL-1α) and high mobility group box 1 (HMGB1), increased significantly in mice treated with DAPT compared with the group treated with vehicle at 8 hours after LPS treatment (Fig. 1, C and D). The mortality rate of mice treated with DAPT was significantly higher than that of mice treated with vehicle after a lethal dose of LPS (10 mg/kg) challenge (Fig. 1E). Together, these results indicated that the transient Notch activation during endotoxemia acts as a prosurvival signal to splenocytes.
Fig. 1. Inhibition of Notch enhances mortality, inflammation, and apoptosis during endotoxemia.

(A to C) WT mice were administrated LPS [5 mg/kg, intraperitoneally (ip)] with or without DAPT before treatment (100 mg/kg, 3 hours before LPS injection). (A) Representative Western blots and quantification for NICD, HES1, and cleaved caspase-3 (Cl-cas3) in splenocyte from WT mice at indicated time points after LPS ± DAPT treatment. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (B) Representative images of TMR red staining in spleen from WT mice at 8 hours after LPS ± DAPT treatment. Arrows indicate representative TUNEL-positive nuclei (red). Percentage of TMR+ nuclei number/total nuclei number was quantified. n = 4 per group. Scale bars, 50 μm. (C and D) Plasma levels of IL-1α and HMGB1 were detected by ELISA. Data are from two or three separate experiments. Symbols represent individual mouse. Data are mean ± SEM; statistical difference was tested using one-way ANOVA with Tukey’s correction; *P < 0.05 and **P < 0.01. (E) Seven-day survival. WT mice were administrated LPS (10 mg/kg, ip) with or without DAPT before treatment (100 mg/kg, 3 hours before LPS injection). n = 30 in LPS group; n = 28 in LPS + DAPT group. Data are from three separate experiments. Statistical difference was tested using the log-rank test; *P < 0.05.
Notch signaling protects splenic CD4 T cells from apoptosis during endotoxemia
To understand which splenic cell types undergo apoptosis after Notch inhibition during endotoxemia, we isolated and cultured the CD19-positive subset (mainly B cells) and the CD19-negative subsets (non-B cells) from the total population of splenocytes. We found that non-B cells were the Notch-expressing cells, while the B cells showed no detectable NICD at 8 hours after LPS stimulation (Fig. 2A). We next exposed isolated splenic T cells to LPS in vitro. T cells showed increased NICD levels after LPS stimulation, while the non-T cells had no significant increase of NICD after LPS treatment (Fig. 2B). We next tested whether Notch is involved in CD4 or CD8 T cell survival. Splenocytes were isolated from mice treated with LPS for 8 hours ± DAPT pretreatment and analyzed for apoptosis using flow cytometry. There was a significant increase in the percentage of apoptotic CD4 T cells [both annexin V+/PI– (propidium iodide–) early apoptotic cells and annexin V+/PI+ late apoptotic cells] in mice treated with LPS + DAPT compared with LPS treatment alone. In contrast, we found that there was no significant increase in apoptotic CD8 T cells or B cells during endotoxemia after treatment with DAPT (Fig. 2C). Consistently, the levels of cleaved caspase-3 were significantly higher in CD4 T cells with LPS + DAPT treatment compared with LPS only treatment (Fig. 2D). Jurkat cells, an immortalized CD4 T lymphocyte cell line obtained from a patient with T cell leukemia, are known to overexpress Notch. Notably, inhibition of notch activation in cultured Jurkat cells substantially increased cleaved caspase-3 at baseline and after LPS stimulation (Fig. 2D). Together, these data reveal the importance of Notch activation in promoting CD4 T cell survival during acute inflammatory responses.
Fig. 2. Notch signaling protects splenic CD4 T cells from apoptosis during endotoxemia.
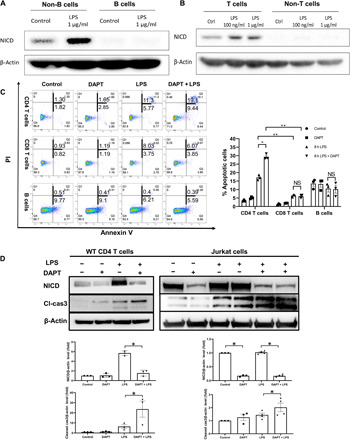
(A and B) Splenocytes were isolated from WT mice and treated with LPS ex vivo for 8 hours. (A) Representative Western blot for NICD expression in splenic B cells (CD19+ cells) versus non-B cells (CD19− cells) stimulated with LPS (1 μg/ml). (B) Representative Western blots of NICD expression in splenic T cells (CD3+) and non-T cells (CD3−) challenged with LPS (1 μg/ml or 100 ng/ml). (C) WT mice were administrated LPS (5 mg/kg, ip) with or without DAPT before treatment (100 mg/kg, 3 hours before LPS injection). Spleens were collected at 8 hours after LPS ± DAPT treatment. Percentage of early (annexin V+/PI−) and late (annexin V+/PI+) apoptotic cells was measured by flow cytometry. Data are from three separate experiments. Symbols represent individual mouse. Data are mean ± SEM. *P < 0.05 and **P < 0.01. NS, not significant. (D) Representative Western blots of NICD and cleaved caspase-3 expression in splenic CD4 T cells from WT and Jurkat cells at 8 hours after LPS (5 mg/kg or 1 μg/ml) ± DAPT (100 mg/kg or 10 μM, 3 hours before LPS injection) treatment. Data are from three separate experiments. Symbols represent individual mouse. Data are mean ± SEM. Statistical difference was tested using one-way ANOVA with Tukey’s correction; *P < 0.05.
Notch activation protects CD4 T cells from STING-mediated apoptosis during endotoxemia
STING was originally identified as a regulator of intracellular DNA sensing. STING activation induces a potent production of interferon in immune cells (11). However, recent studies revealed that STING activation is highly proapoptotic in immune cells (12). Our previous work showed that Notch may serve as a negative regulator of STING in bone marrow–derived monocytes (10). Therefore, we hypothesized that Notch modulates CD4 T cell homeostasis by regulating the STING signaling pathway.
To test our hypothesis, wild-type (WT) and Stinggt mice that have no STING expression were treated with LPS ± DAPT, and splenic CD4 T cells were isolated for NICD expression and apoptosis analysis. NICD levels increased in both WT and Stinggt mice upon 8 hours of LPS challenge (Fig. 3A). Pretreatment with DAPT suppressed NICD levels in cells from both strains (Fig. 3A). As expected, the levels of cleaved caspase-3 increased in WT mice treated with DAPT + LPS compared with LPS only. Inhibition of Notch activation resulted in increased STING and TANK binding kinase 1 (TBK1) phosphorylation in WT CD4 T cells, suggesting that the Notch signaling negatively regulated STING activation during endotoxemia. However, there were minimal increases in the levels of cleaved caspase-3 in LPS-treated Stinggt mice with DAPT before treatment compared with vehicle treatment (Fig. 3A). Furthermore, the percentage of apoptotic CD4 T cells from Stinggt mice exhibited no significant difference between groups with or without DAPT before treatment (Fig. 3B). Consistent with our previous findings (10), STING deficiency significantly reduced circulating IL-6 levels and mortality during endotoxemia. However, unlike WT mice (Fig. 1E), inhibition of Notch using DAPT had minimal effects on IL-6 levels and mortality in Stinggt mice after LPS treatment (Fig. 3, C and D). Together, our results reveal that the Notch signaling negatively regulates STING activation and suppresses STING-mediated apoptosis in CD4 T cell.
Fig. 3. Notch activation protects CD4 T from STING-mediated apoptosis during endotoxemia.
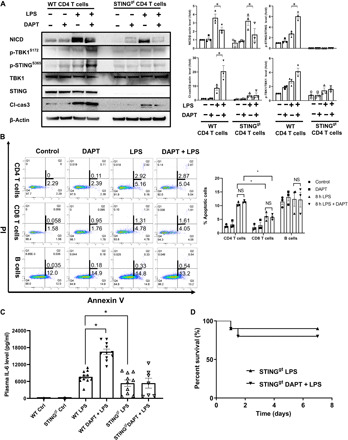
(A to C) WT and Stinggt mice were administrated LPS (5 mg/kg, ip) with or without DAPT before treatment (100 mg/kg, 3 hours before LPS injection). Spleens were collected at 8 hours after LPS ± DAPT treatment. (A) Representative Western blots of NICD, p-TBK1S172, p-STINGs365, TBK1, STING, and cleaved caspase-3 level in splenic CD4 T cells from indicated mice. Data are from three separate experiments. Symbols represent individual mouse. Data are mean ± SEM. *P < 0.05. (B) Percentage of early (annexin V+/PI−) and late (annexin V+/PI+) apoptotic cells was measured by flow cytometry in splenic CD4 T cells, CD8 T cells, and B cells from Stinggt mice after indicated treatments. (C) Plasma IL-6 level was determined by ELISA. Data are from two separate experiments. Symbols represent individual mouse. Data are mean ± SEM. Statistical difference was tested using one-way ANOVA with Tukey’s correction; *P < 0.05. (D) Seven-day survival. WT (Fig. 1E) and Stinggt mice were administrated LPS (10 mg/kg, ip) ± DAPT before treatment (100 mg/kg, 3 hours before LPS injection). n = 12 in Stinggt-LPS group; n = 12 in Stinggt-LPS + DAPT group. Data are from two separate experiments. Statistical difference was tested using the log-rank test.
NICD interacts with STING in the cytoplasm of CD4 T cells
To test whether Notch directly interacts with STING, we performed a coimmunoprecipitation analysis using a STING- or NICD-specific antibody to pull down STING or NICD in isolated splenic CD4 T cells from LPS-treated WT mice with or without DAPT before treatment. We found that coimmunoprecipitation of NICD with STING increased after 8 hours of LPS injection and that this increase was abrogated by DAPT (Fig. 4A). We also found that NICD was associated with STING at baseline and after LPS treatment in Jurkat cells (Fig. 4B). The association between NICD and STING in Jurkat cells was also attenuated after DAPT treatment (Fig. 4B). To confirm the interaction between NICD and STING, we cotransfected complementary DNA (cDNA) expressing hemagglutinin (HA)–tagged STING and V5-tagged NICD into human embryonic kidney (HEK) 293T cells. Notably, HEK293T cells have undetectable cGAS and STING protein expression at baseline (14). The transfected NICD could be coimmunoprecipitated with STING (Fig. 4C). Using immunofluorescence staining, we observed that transfected NICD-V5 colocalized with transfected-STING-HA in the cytoplasm of HEK293T cells (Fig. 4D). These data reveal an interaction between NICD and STING in the cytoplasm of CD4 T cells.
Fig. 4. NICD interacts with STING in the cytoplasm of CD4 T cells.
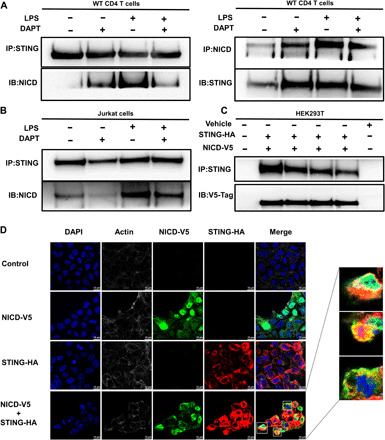
(A) WT mice were administrated LPS (5 mg/kg, ip) ± DAPT before treatment (100 mg/kg, 3 hours before LPS injection). Spleens were collected at 8 hours after LPS ± DAPT treatment. Splenic CD4 T cells were isolated, lysed, and immunoprecipitated with the rabbit anti-STING or rabbit anti-NICD antibody. The level of NICD (left) or STING (right) in immunoprecipitants was analyzed by Western blotting. (B) Jurkat cells were treated with LPS (1 μg/ml) ± DAPT (10 μM, 3 hours before LPS challenge). Cells were lysed and immunoprecipitated with the rabbit anti-STING antibody, and the level of NICD in immunoprecipitants was analyzed by Western blotting. (C and D) NICD-V5 plasmid (1000 ng) was cotransfected with STING-HA (1000 ng) plasmid into HEK293T cells for 48 hours. (C) Cells were lysed and immunoprecipitated with the rabbit anti-STING antibody. The level of transfected NICD-V5 in immunoprecipitants was analyzed by Western blotting. (D) Immunofluorescence staining for V5 tag (green), HA (red), actin (white), and nuclei (blue). The right panel indicates representative colocalization of V5 and HA (yellow). Data are from two separate experiments.
NICD interacts with STING at the CDN-binding domain
Next, we cotransfected NICD with various STING variants to identify the basis of the interaction between STING and NICD (Fig. 5A). The transfection efficiency of STING variants was verified using anti-STING antibodies that detect the different domains of STING (fig. S2). Full-length human STING consists of 379 amino acid residues, which are divided into three functional domains: the N-terminal transmembrane domain (NTD; 1 to 154 amino acids), the dimerization and CDN-binding domain (CBD; 155 to 342 amino acids), and the TBK-interacting C-terminal tail (CTT; 343 to 379 amino acids) (Fig. 5A). STING-R232H and STING-HAQ are natural variant alleles of full-length human STING. Of note, nonsynonymous variants of human STING-HAQ and STING-R232H did not affect NICD binding to STING (Fig. 5B). To test whether NICD interacted with STING at the NTD, STING-β (15), an alternatively spliced isoform of human STING lacking NTD (Fig. 5A), was cotransfected with NICD-V5 into HEK293T cells. Both full-length STING and STING-β were coimmunoprecipitated with NICD (Fig. 5B), indicating that NICD did not interact with STING at the NTD. To test whether NICD interacted with STING at the CTT site, STING-△342, STING-△354, and STING-△368, which CTT were partially or completely deleted (Fig. 5A), were cotransfected with NICD-V5 into HEK293T cells. NICD also coimmunoprecipitated with these STING variants (Fig. 5C), suggesting that STING did not interact with NICD at CTT. To test whether STING interacted with NICD at CBD, we coexpressed the NICD-V5 together with STING-MRP (Fig. 5A) (16), a variant of STING lacking the CDN binding and conserved TBK1 interaction domain. STING-MRP was not coimmunoprecipitated with NICD (Fig. 5D), indicating that NICD interacted with STING at CBD.
Fig. 5. NICD interacts with STING at the CBD.
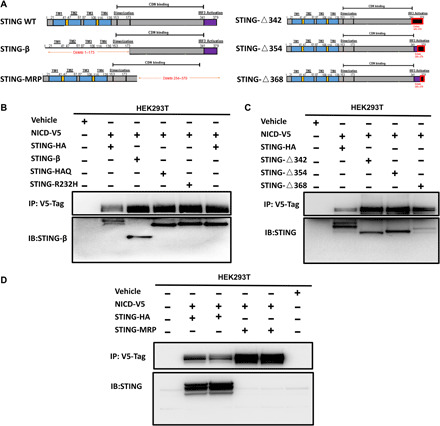
(A) Structure of STING-WT, STING-β, STING-MRP, STING-△342, STING-△354, and STING-△368. (B) NICD-V5 plasmid (1000 ng) was cotransfected with STING-HA (WT) (1000 ng) plasmid, STING-β plasmid (1000 ng), STING-HAQ plasmid (1000 ng), or STING-R232H plasmid (1000 ng) into HEK293T cells. After 48 hours, coimmunoprecipitation was carried out with rabbit anti-V5 Tag antibody. Immunoprecipitates were analyzed by Western blotting with rabbit anti-STING antibody. (C) NICD-V5 plasmid (1000 ng) plus STING-△342 (1000 ng) or NICD-V5 plasmid (1000 ng) plus STING-△354 (1000 ng) plasmids or NICD-V5 plasmid (1000 ng) plus STING-△368 (1000 ng) plasmids were cotransfected into HEK293T cells. Cells were harvested and lysed after 48 hours. Immunoprecipitation was carried out with rabbit anti-V5 antibody. Immunoprecipitates were probed with rabbit anti-STING antibody. (D) NICD-V5 plasmid (1000 ng) was cotransfected with STING-HA (1000 ng) plasmid or STING-MRP plasmid (1000 ng) into HEK293T cells. After 48 hours, cells were harvested and lysed. Coimmunoprecipitation was carried out with rabbit anti-V5 antibody. Immunoprecipitates were analyzed by Western blotting with rabbit anti-STING middle region antibody. Results in each panel are representative of three independent experiments.
NICD competes with cGAMP for the CDN-binding site at STING
Recent studies have reported that activation of cGAMP-STING signaling induces apoptosis in T cells and B cells (12, 13). Cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), synthesized by cGAS, binds to STING at the CDN domain and initiates downstream signaling. Our data indicated that NICD interacted with STING at the CDN domain. Therefore, we hypothesized that NICD negatively regulated STING-mediated apoptosis by competing the CDN-binding site with cGAMP. To test our hypothesis, Jurkat cells were treated with 2,3-cGAMP ± DAPT. As expected, treatment with 2,3-cGAMP activated the STING signaling, demonstrated by increased phospho-STING and phospho-TBK (Fig. 6A). Furthermore, activation of STING by 2,3-cGAMP increased apoptotic cell death in Jurkat cells, shown by increased cleaved caspase-3 (Fig. 6A). Inhibition of NICD using DAPT enhanced STING activation as well as apoptosis in Jurkat cells (Fig. 6A). To test whether the protection mediated by NICD depended on 2,3-cGAMP, splenic CD4 T cells were isolated from cGAS−/− mice. Deletion of cGAS did not alter the expression of NICD or STING in CD4 T cells (fig. S3). As expected, the percentage of apoptotic CD4 T cells increased after LPS treatment (Fig. 6B). However, there was no significant difference in the percentage of apoptotic CD4 T cells between the groups with or without DAPT treatment (Fig. 6B). Addition of 2,3-cGAMP significantly increased the percentage of apoptotic T cells after LPS treatment (Fig. 6B). Notably, inhibition of Notch using DAPT further increased the number of apoptotic T cells in cGAS−/− mice after LPS treatment in the presence of 2,3-cGAMP (Fig. 6B). There was minimal difference in the percentage of apoptotic splenic CD4 T cells between the groups treated with or without DAPT in cGAS−/− mice in vivo (Fig. 6C). Furthermore, cGAS deficiency significantly reduced circulating IL-6 levels and mortality during endotoxemia (Fig. 6, D and E). However, unlike WT mice (Fig. 1E), inhibition of Notch using DAPT had minimal effect on IL-6 levels and mortality in cGAS−/− mice after LPS treatment (Fig. 6, D and E). These data supported the idea that NICD protected CD4 T cells from STING-mediated apoptosis by competing with cGAMP.
Fig. 6. NICD competes with cGAMP for the CDN-binding site at STING.
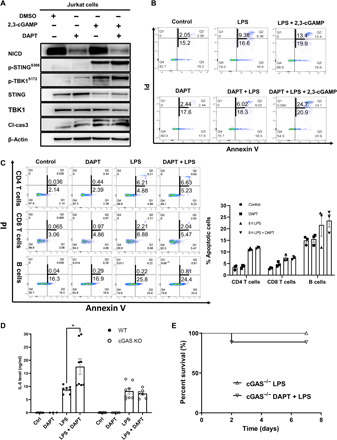
(A) Representative Western blots of NICD, p-STINGs366, STING, p-TBK1s172, TBK1, and cleaved caspase-3 in 2,3-cGAMP–treated (2.5 mg/ml) Jurkat cells ± DAPT before treatment (10 μM, 3 hours before 2,3-cGAMP treatment). (B) Splenic CD4 T cells were isolated from cGAS−/− mice; cells were treated with LPS (100 ng/ml) ± 2,3-cGAMP (2.5 mg/ml) or DAPT (10 μM, 3 hours before LPS ± 2,3-cGAMP challenge). Percentage of early (annexin V+/PI−) and late (annexin V+/PI+) apoptotic cells was measured by flow cytometry at 8 hours after indicated treatments. (C and D) cGAS−/− mice were administrated LPS (5 mg/kg, ip) ± DAPT before treatment (100 mg/kg, 3 hours before LPS injection). Spleens were collected at 8 hours after LPS ± DAPT treatment. Percentage of early and late apoptotic cells was measured by flow cytometry in splenic CD4 T cells, CD8 T cells, and B cells from cGAS−/− mice after indicated treatments. (D) Plasma IL-6 level was determined by ELISA in WT versus cGAS−/− mice at 8 hours after indicated treatments. Data are from two separate experiments. Symbols represent individual mouse. Data are mean ± SEM. Statistical difference was tested using one-way ANOVA with Tukey’s correction; *P < 0.05. (E) Seven-day survival. WT (Fig. 1E) and cGAS−/− mice were administrated LPS (10 mg/kg, ip) with or without DAPT before treatment (100 mg/kg, 3 hours before LPS injection). n = 12 in cGAS−/− LPS group; n = 12 in cGAS−/− LPS + DAPT group. Data are from two separate experiments. Statistical difference was tested using the log-rank test.
DISCUSSION
The goal of this work was to determine whether Notch regulates T cell apoptosis during acute systemic inflammatory responses. We show that Notch suppresses STING-mediated CD4 T cell apoptosis when NICD interacts with the CDN-binding domain within STING. These findings have implications for the regulation of CD4 T cell fate in settings associated with pathologic apoptosis of T cells during systemic inflammation as seen during sepsis.
Notch signaling modulation of apoptosis is multifactorial, involving intricate cross-talk with many pathways that regulate cell cycle, growth, and survival. Thus, like many other signaling pathways in a biological system, the phenotypic outcome of Notch activity in regard to apoptosis is highly cell type and context dependent (17). Lymphocytes, including T and B cells, are known to express Notch (18, 19), and Notch signaling plays critical roles in controlling lymphocyte function and cell fate in lymphocyte malignancies (20, 21). Activating Notch mutations are found in about 60% of patients with T cell acute lymphoblastic leukemia (T-ALL) and across all T-ALL subtypes (22). Decades of evidence proved that Notch enhances antiapoptotic signals and protects CD4+ T cells against apoptosis in the inflammation model (23). Notch signaling has also been shown to enhance T cell function during immunotherapy (24) and to mediate T cell metabolism (23). Furthermore, Notch signaling is involved in the generation and activation of both CD4 and CD8 T cells during adaptive immune responses (25, 26). Inhibition of Notch signaling enhances CD4 T cell differentiation and CD8 T cell cytotoxicity (9, 20) in peripheral T cells from septic patients. Using a model of in vivo endotoxemia, we found that Notch was activated only in CD4 T cells and not in CD8 T cells or B cells in the spleen. CD4 T cells are reported to be more vulnerable to apoptosis compared to CD8 T cells in models of sepsis (3, 27). We confirmed the higher sensitivity of CD4 T cells to apoptosis during endotoxemia and demonstrate the role of the STING pathway as a major driver of the early T cell apoptosis in the spleen. The selective Notch activation in CD4 T cells appears to be part of a cell survival pathway aimed to protect CD4 T cells.
NICD functions as a transcription factor regulating embryo development and cell fate decision by coordinating transcriptional programs by binding to DNA binding protein RBP-J (recombining binding protein suppressor of hairless) (28). Intranuclear Notch signaling is implicated in the expression of inflammatory mediators in macrophages and HUVECs (human umbilical vein endothelial cells) (29) in response to LPS. Despite the intensive investigations on NICD as a transcription factor, recent studies have suggested that it also acts as a cytoplasmic mediator of cellular activities. Cytoplasmic NICD is involved in regulating the AKT/GSK-3 (glycogen synthase kinase–3) pathway through mechanisms independent of the transcriptional function of NICD (30, 31). Furthermore, NICD has been shown to form a complex with LC3, P62, and β-catenin to facilitate protein degradation (32). We extend the list of nontranscriptional activities of NICD by discovering that NICD inhibits STING activation through a protein-protein interaction mechanism. STING is an ER-resident transmembrane adaptor protein (33). After binding to CDNs, STING translocates to ER-Golgi intermediate compartments and elicits an innate immune response (11). While our findings firmly establish that NICD blocks STING-dependent apoptosis, further studies are required to elucidate whether NICD binding to STING can impede STING translocation to the ER-Golgi intermediate compartment (ERGIC). We also found that Notch activation in splenocytes leads to up-regulation of the transcription factor HES, and this suggests that Notch also regulates other pathways in lymphocytes during endotoxemia.
STING drives type I interferon response in innate immune cells, such as macrophages and dendritic cells (12). Activation of STING also induces apoptosis in lymphocytes (12) in multiple pathogenic conditions, such as SAVI (STING-associated vasculopathy with onset in infancy) patients (34). The differences in STING-mediated response between innate immune cells and lymphocytes may be due to the fact that degradation of STING is less efficient in lymphocytes than in other immune cell types (35). Whether Notch also inhibits STING activation in other immune cells and the consequence of Notch-STING interaction in these cells is unclear. What is clear from our previous work is that STING contributes to the immune dysfunction of sepsis (10), and the current work provides further rationale for the targeting of STING to prevent lymphocyte apoptosis during acute critical illness.
CDNs are the only ligands known to activate STING. CDNs are synthesized from adenosine triphosphate (ATP) and/or guanosine triphosphate (GTP), and three molecular combinations have been found: cyclic diguanylic acid (c-di-GMP), cyclic diadenylic acid (c-di-AMP), and cyclic GMP-AMP (cGAMP). c-di-GMP and c-di-AMP are mainly pathogen-derived CDNs (36, 37), while cGAMP is synthesized endogenously by cGAS upon DNA sensing in the cytosol (38). All three CDNs have been shown to interact with STING directly and to activate STING signaling (36, 38, 39). Our data demonstrate that NICD interacts with STING at the CDN-binding site and suggest that NICD competes with cGAMP to inhibit STING activation in CD4 T cells. Because the CDN-binding domain of STING is conserved for all three CDN (40), it is possible that NICD also competes with pathogen-derived CDNs (c-di-AMP and c-di-GMP) to inhibit STING activation in CD4 T cells. However, further studies are required to elucidate this point.
In conclusion, we have provided compelling evidence that Notch signaling protects CD4 T cells from STING-mediated apoptosis during endotoxemia. The mechanisms underlying the protective effect of Notch on CD4 T cells is via the interaction between Notch and STING at the CDN-binding site to inhibit STING activation. Dysregulation of CD4 T cell apoptosis is involved in a variety of pathological conditions, such as immunosuppression in acute critical illness and leukemia. Therefore, our newly identified Notch-STING mechanism in controlling CD4 T cell apoptosis will provide opportunities for devising therapeutic strategies for sepsis and other T cell–mediated diseases, such as leukemia.
MATERIALS AND METHODS
Reagents
DAPT was purchased from Selleck Chemicals and dissolved in 4% dimethyl sulfoxide [DMSO; American Type Culture Collection (ATCC)] for in vitro studies. DAPT was dissolved in coil oil (Selleck Chemicals) for intraperitoneal injection. Ultrapure LPS from Escherichia coli O111:B4 was purchased from Sigma-Aldrich and dissolved in endotoxin-free ultrapure water (Sigma-Aldrich). 2′3′-cGAMP was obtained from InvivoGen.
Animal
Male C57BL/6J (WT) mice were purchased from The Jackson Laboratory. Stinggt mice and cGAS−/− mice on a C57BL/6J background were obtained from The Jackson Laboratory and bred in our facility. Mice aged 8 to 12 weeks, weighing 21 to 30 g, were used in our experiments.
Endotoxemia model
Endotoxemia was induced by intraperitoneal injection of ultrapure LPS (5 mg/kg; Sigma-Aldrich) at a sublethal level. For survival experiments, LPS (10 mg/kg) was used. All experimental protocols were approved by the Institutional Animal Use and Care Committee of the University of Pittsburgh.
Inhibition of Notch signaling in vitro and in vivo
To inhibit Notch signaling in vitro, cells were pretreated with the γ-secretase inhibitors: N- [N-(3,5-difluorophenacetyl)-l-alanyl]-(S)-phenylglycine-t-butyl ester (DAPT; Selleck Chemicals) (10 μM, dissolved in 4% DMSO) at 3 hours before LPS stimulation. To block Notch activation in vivo, DAPT (100 mg/kg, dissolved in corn oil) was administered intraperitoneally at 3 hours before LPS challenge.
Cell culture and primary cell isolation
HEK293T and Jurkat cells were obtained from the ATCC. HEK293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Lonza). Jurkat cells were cultured in RPMI 1640 (Lonza) at 37°C. CD19+ B cells, CD19- cells, CD3+ pan-T cells, or non-T cells were isolated from mouse spleen using positive selection with CD19 or CD3 microbeads (STEMCELL Technologies) according to the manufacturer’s instructions. CD4 T cells were obtained by negative selection using reagents from STEMCELL Technologies, and the purification was verified by flow cytometry (fig. S1). All primary cells were cultured in RPMI 1640 (Lonza). All cells are cultured at 37°C with 5% CO2 (v/v). All media were supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 U/ml), streptomycin (100 U/ml), and 1× GlutaMAX Supplement (Thermo Fisher Scientific).
Plasmid transfection
V5-tagged NICD, STING△342, STING△354, and STING△368 were purchased from Addgene. Recommended protocol from Addgene was followed. HA-tagged STING, STING-R232H, STING-HAQ, STING-MRP, and STING-β were ordered from InvivoGen. Plasmids (1 μg) were transfected into cells using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. The transfection efficiency was validated using Western blot for the expression levels of the target protein.
cGAMP stimulation of cells
2′3′-cGAMP, a STING ligand, was obtained from InvivoGen. 2′3′-cGAMP was delivered into splenic CD4 T cells and Jurkat cells using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Flow cytometry analysis of apoptosis
Cells were blocked for Fc receptors with anti-mouse CD16/32 (BD Biosciences) for 15 min and then were stained with fluorochrome-conjugated antibody (table S1) for 30 min at 4°C in the dark. Data were acquired with a Cytek Aurora flow cytometer (Cytek Biosciences) and analyzed with FlowJo analytical software (Tree Star).
Western blot
Cells were lysed in radioimmunoprecipitation assay lysis buffer (Sigma-Aldrich) supplemented with 1× Protease/Phosphatase Inhibitor Cocktail (Cell Signaling Technology). Whole-cell lysates were denatured for 5 min at 99°C. A total of 20 μg of reduced samples was separated by 4 to 20% precast gradient gels (GenScript). Each gel was run for 65 min at 150 V in Mops buffer and then transferred onto a polyvinylidene difluoride membrane (Thermo Fisher Scientific) at 200 mA for 65 min. The membrane was blocked in 5% milk (Bio-Rad) for 1 hour at room temperature and then incubated at 4°C overnight with primary antibody (table S1) in 1% milk overnight. Membranes were washed three times in tris-buffered saline Tween (TBS-T) for 10 min, incubated with horseradish peroxidase–conjugated secondary antibody for 1 hour, and then washed three times for 10 min in TBS-T before being developed for chemiluminescence (Bio-Rad). Images were quantified by volume analysis using ImageLab software (Bio-Rad) and presented as a ratio of loading controls.
Confocal and immunofluorescence
HEK293T cells were plated on poly-lysine–coated coverslips (Corning) before transfection with plasmids. Two days after transfection of NICD-V5 with or without STING-HA, cells were fixed with 2% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 15 min, blocked with 20% normal goat serum, and then washed with cold PBS and 0.5% bovine serum albumin (BSA) repeatedly. Cells were incubated with the fluorescence-labeled primary antibodies (table S1). Samples were then incubated with F-actin phalloidin for 1 hour. A Hoechst nuclear stain was applied for 30 s followed by a single rinse of PBS to remove excess dye. Spleen tissue was removed after perfusion with cold PBS, and 2% paraformaldehyde was incubated for an additional 2 hours to complete tissue fixation and then incubated for 24 hours in 30% sucrose, followed by cryopreservation in liquid nitrogen–cooled 2-methylbutane. Tissue sections of 6 μm were permeabilized with 0.3% Triton X-100 for 20 min, followed by staining according to the manufacturer’s protocol of the In Situ Cell Death Detection Kit, TMR red (Roche). All immunofluorescent experiment staining sets involved staining F-actin and nuclear stain for use in quantitation. Imaging conditions were maintained at identical settings within each antibody-labeling experiment, with original gating performed using the primary depletion control. Large-area images equivalent to nine unique fields were taken in X and Y with a Nikon A1 confocal microscope (purchased with 1S10OD019973-01 awarded to S. C. Watkins). Quantification was performed using NIS-Elements (Nikon).
Measurement of cytokines
Plasma was collected for the measurement of cytokines. Concentrations of IL-1α, IL-6 (R&D Systems), or high-mobility group box 1 (Tecan) in samples were determined using enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions.
Coimmunoprecipitation
WT splenic CD4 T cells, Jurkat cells, and HEK293T cells stimulated with LPS ± DAPT or transfected with the specified plasmids encoding V5-tagged NICD, HA-tagged STING, STING-β, STING-MRP, STING-Δ342, STING-Δ354, or STING-Δ368 were lysed in Pierce IP Lysis/Wash Buffer (Thermo Fisher Scientific) complemented with 1× Protease/Phosphatase Inhibitor Cocktail (Cell Signaling Technology). The cell lysates were then subjected to immunoprecipitation using antibodies against V5 (Cell Signaling Technology) for transfected proteins or using anti-STING and anti-NICD (Cell Signaling Technology) antibody for endogenous STING or NICD proteins. The antibody-antigen complex was precipitated using Pierce Protein A/G Magnetic Beads (Thermo Fisher Scientific). Immunoprecipitated complexes were eluted from the magnetic beads with an elution buffer and a neutralization buffer (Thermo Fisher Scientific). The sample was resolved on a 4 to 20% SurePAGE bis-tris precast gradient gel and blotted for the respective proteins.
Statistical analysis
All data were analyzed using GraphPad Prism 8.0 software (GraphPad, San Diego, CA). Statistical analysis was performed using the two-tailed Student’s t test for calculating the statistical significance of two experimental groups. One-way analysis of variance (ANOVA) was used for calculating the statistically significant differences between the means of three or more independent groups. A P value of <0.05 was considered statistically significant. All values are presented as the mean ± SEM.
Ethics statement
All experimental protocols were approved by the Institutional Animal Use and Care Committee of the University of Pittsburgh. Experimental procedures were carried out in accordance with all regulations regarding the care and use of experimental animals (National Institutes of Health). Male C57BL/6 mice on arrival were housed in groups of five in standard cages. Animals were maintained under controlled temperature (22 ± 2°C) on a 12-hour dark/12-hour light cycle with free access to food and water.
Supplementary Material
Acknowledgments
We thank D. Reiser for managing the mouse colonies. Funding: We received no financial support for the research, authorship, and publication of this article. Author contributions: J.L. conceived the project, designed the project, performed the experiments, extracted and analyzed the data, drafted the manuscript, and approved the final manuscript. C.Y. conceived the project, designed the project, performed the experiments, extracted and analyzed the data, and approved the final manuscript. Y.Z. performed the animal experiments and analyzed the data. P.L., F.G., H.L., and Y.L. performed the experiments. M.J.S. and D.T. provided conceptual insights. T.R.B. and M.D. provided conceptual insights and edited the manuscript. M.D. supervised the project. All authors provided feedback on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The data that support the findings of this study are available from the corresponding author upon reasonable request through a material transfer agreement.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/39/eabc5447/DC1
REFERENCES AND NOTES
- 1.Delano M. J., Ward P. A., Sepsis-induced immune dysfunction: Can immune therapies reduce mortality? J. Clin. Invest. 126, 23–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hotchkiss R. S., Nicholson D. W., Apoptosis and caspases regulate death and inflammation in sepsis. Nat. Rev. Immunol. 6, 813–822 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Boomer J. S., To K., Chang K. C., Takasu O., Osborne D. F., Walton A. H., Bricker T. L., Jarman S. D. II, Kreisel D., Krupnick A. S., Srivastava A., Swanson P. E., Green J. M., Hotchkiss R. S., Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306, 2594–2605 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabrera-Perez J., Condotta S. A., Badovinac V. P., Griffith T. S., Impact of sepsis on CD4 T cell immunity. J. Leukoc. Biol. 96, 767–777 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Appel B., Givan L. A., Eisen J. S., Delta-Notch signaling and lateral inhibition in zebrafish spinal cord development. BMC Dev. Biol. 1, 13 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radtke F., Fasnacht N., Macdonald H. R., Notch signaling in the immune system. Immunity 32, 14–27 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Fazio C., Ricciardiello L., Inflammation and Notch signaling: A crosstalk with opposite effects on tumorigenesis. Cell Death Dis. 7, e2515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monsalve E., Ruiz-García A., Baladrón V., Ruiz-Hidalgo M. J., Sánchez-Solana B., Rivero S., García-Ramírez J. J., Rubio A., Laborda J., Díaz-Guerra M. J., Notch1 upregulates LPS-induced macrophage activation by increasing NF-κB activity. Eur. J. Immunol. 39, 2556–2570 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Jin B., Liang Y., Liu Y., Zhang L.-X., Xi F.-Y., Wu W.-J., Li Y., Liu G.-H., Notch signaling pathway regulates T cell dysfunction in septic patients. Int. Immunopharmacol. 76, 105907 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Zeng L., Kang R., Zhu S., Wang X., Cao L., Wang H., Billiar T. R., Jiang J., Tang D., ALK is a therapeutic target for lethal sepsis. Sci. Transl. Med. 9, eaan5689 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Motwani M., Pesiridis S., Fitzgerald K. A., DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 20, 657–674 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Gulen M. F., Koch U., Haag S. M., Schuler F., Apetoh L., Villunger A., Radtke F., Ablasser A., Signalling strength determines proapoptotic functions of STING. Nat. Commun. 8, 427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang C.-H., Zundell J. A., Ranatunga S., Lin C., Nefedova Y., Del Valle J. R., Hu C.-C. A., Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 76, 2137–2152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L., Wu J., Du F., Chen X., Chen Z. J., Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang P.-H., Fung S.-Y., Gao W.-W., Deng J.-J., Cheng Y., Chaudhary V., Yuen K.-S., Ho T.-H., Chan C.-P., Zhang Y., Kok K.-H., Yang W., Chan C.-P., Jin D.-Y., A novel transcript isoform of STING that sequesters cGAMP and dominantly inhibits innate nucleic acid sensing. Nucleic Acids Res. 46, 4054–4071 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H., Pei R., Zhu W., Zeng R., Wang Y., Wang Y., Lu M., Chen X., An alternative splicing isoform of MITA antagonizes MITA-mediated induction of type I IFNs. J. Immunol. 192, 1162–1170 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Dang T. P., Notch, apoptosis and cancer. Adv. Exp. Med. Biol. 727, 199–209 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Bailis W., Yashiro-Ohtani Y., Fang T. C., Hatton R. D., Weaver C. T., Artis D., Pear W. S., Notch simultaneously orchestrates multiple helper T cell programs independently of cytokine signals. Immunity 39, 148–159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radtke F., Wilson A., Mancini S. J. C., MacDonald H. R., Notch regulation of lymphocyte development and function. Nat. Immunol. 5, 247–253 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Amsen D., Helbig C., Backer R. A., Notch in T cell differentiation: All things considered. Trends Immunol. 36, 802–814 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Takam Kamga P., Dal Collo G., Midolo M., Adamo A., Delfino P., Mercuri A., Cesaro S., Mimiola E., Bonifacio M., Andreini A., Chilosi M., Krampera M., Inhibition of notch signaling enhances chemosensitivity in B-cell precursor acute lymphoblastic leukemia. Cancer Res. 79, 639–649 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Habets R. A., de Bock C. E., Serneels L., Lodewijckx I., Verbeke D., Nittner D., Narlawar R., Demeyer S., Dooley J., Liston A., Taghon T., Cools J., de Strooper B., Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci. Transl. Med. 11, eaau6246 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Maekawa Y., Ishifune C., Tsukumo S.-i., Hozumi K., Yagita H., Yasutomo K., Notch controls the survival of memory CD4+ T cells by regulating glucose uptake. Nat. Med. 21, 55–61 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Sierra R. A., Thevenot P., Raber P. L., Cui Y., Parsons C., Ochoa A. C., Trillo-Tinoco J., Del Valle L., Rodriguez P. C., Rescue of Notch-1 signaling in antigen-specific CD8+ T cells overcomes tumor-induced T-cell suppression and enhances immunotherapy in cancer. Cancer Immunol. Res. 2, 800–811 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuijk L. M., Verstege M. I., Rekers N. V., Bruijns S. C., Hooijberg E., Roep B. O., de Gruijl T. D., van Kooyk Y., Unger W. W. J., Notch controls generation and function of human effector CD8+ T cells. Blood 121, 2638–2646 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Charbonnier L.-M., Wang S., Georgiev P., Sefik E., Chatila T. A., Control of peripheral tolerance by regulatory T cell-intrinsic Notch signaling. Nat. Immunol. 16, 1162–1173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma A., Yang W.-L., Matsuo S., Wang P., Differential alterations of tissue T-cell subsets after sepsis. Immunol. Lett. 168, 41–50 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neves A., Priess J. R., The REF-1 family of bHLH transcription factors pattern C. elegans embryos through Notch-dependent and Notch-independent pathways. Dev Cell 8, 867–879 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Ge Y., Huang M., Ma Y.-F., The effects of microRNA-34a regulating Notch-1/NF-κB signaling pathway on lipopolysaccharide-induced human umbilical vein endothelial cells. World J. Emerg. Med. 8, 292–296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy A., Basak N. P., Banerjee S., Notch1 intracellular domain increases cytoplasmic EZH2 levels during early megakaryopoiesis. Cell Death Dis. 3, e380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sieiro D., Rios A. C., Hirst C. E., Marcelle C., Cytoplasmic NOTCH and membrane-derived β-catenin link cell fate choice to epithelial-mesenchymal transition during myogenesis. eLife 5, e14847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jia Z., Wang J., Wang W., Tian Y., XiangWei W., Chen P., Ma K., Zhou C., Autophagy eliminates cytoplasmic β-catenin and NICD to promote the cardiac differentiation of P19CL6 cells. Cell. Signal. 26, 2299–2305 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa H., Ma Z., Barber G. N., STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y., Jesus A. A., Marrero B., Yang D., Ramsey S. E., Sanchez G. A. M., Tenbrock K., Wittkowski H., Jones O. Y., Kuehn H. S., Lee C. C. R., Di Mattia M. A., Cowen E. W., Gonzalez B., Palmer I., Di Giovanna J. J., Biancotto A., Kim H., Tsai W. L., Trier A. M., Huang Y., Stone D. L., Hill S., Kim H. J., St. Hilaire C., Gurprasad S., Plass N., Chapelle D., Horkayne-Szakaly I., Foell D., Barysenka A., Candotti F., Holland S. M., Hughes J. D., Mehmet H., Issekutz A. C., Raffeld M., McElwee J., Fontana J. R., Minniti C. P., Moir S., Kastner D. L., Gadina M., Steven A. C., Wingfield P. T., Brooks S. R., Rosenzweig S. D., Fleisher T. A., Deng Z., Boehm M., Paller A., Goldbach-Mansky R., Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 371, 507–518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ablasser A., Hur S., Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol. 21, 17–29 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Burdette D. L., Monroe K. M., Sotelo-Troha K., Iwig J. S., Eckert B., Hyodo M., Hayakawa Y., Vance R. E., STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamegaya T., Kuroda K., Hayakawa Y., Identification of a Streptococcus pyogenes SF370 gene involved in production of c-di-AMP. Nagoya J. Med. Sci. 73, 49–57 (2011). [PMC free article] [PubMed] [Google Scholar]
- 38.Wu J., Sun L., Chen X., Du F., Shi H., Chen C., Chen Z. J., Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin L., Hill K. K., Filak H., Mogan J., Knowles H., Zhang B., Perraud A.-L., Cambier J. C., Lenz L. L., MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J. Immunol. 187, 2595–2601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kranzusch P. J., Wilson S. C., Lee A. S. Y., Berger J. M., Doudna J. A., Vance R. E., Ancient origin of cGAS-STING reveals mechanism of universal 2′,3′ cGAMP signaling. Mol. Cell 59, 891–903 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/39/eabc5447/DC1