

The microbial community, mostly composed of bacterial species, residing in the human gut degrades and ferments polysaccharides derived from plants (dietary fiber) that would not otherwise be digested. In this way, the collective metabolic actions of community members extract additional energy from the human diet. While the variety of bacteria present in the microbial community is well known, the formation of bacterial consortia, and the consequent interactions that result in the digestion of dietary polysaccharides, has not been studied extensively. The importance of our work was the establishment, under laboratory conditions, of a consortium of gut bacteria that formed around a dietary constituent commonly present in cereals. This enabled the metabolic interplay between the bacterial species to be studied. This kind of knowledge is required to construct an interactive, metabolic blueprint of the microbial community that inhabits the human gut.
KEYWORDS: RNAseq, bacterial consortium, beta-glucan, gut microbiota, whole-transcriptome analysis
ABSTRACT
Whole-transcriptome analysis was used to investigate the molecular interplay between three bacterial species that are members of the human gut microbiota. Bacteroides ovatus, Subdoligranulum variabile, and Hungatella hathewayi formed associations in cocultures fed barley β-glucan, a constituent of dietary fiber. B. ovatus depolymerized β-glucan and released, but did not utilize, 3-O-β-cellobiosyl-d-glucose (DP3) and 3-O-β-cellotriosyl-d-glucose (DP4). These oligosaccharides provided growth substrates for S. variabile and H. hathewayi with a preference for DP4 in the case of the latter species. There was increased transcription of a B. ovatus mixed-linkage-β-glucan utilization locus, as well as carbohydrate transporters in S. variabile and H. hathewayi when in batch coculture. Increased transcription of the β-glucan utilization locus did not occur in continuous culture. Evidence for interactions relating to provision of cobalamin, alterations to signaling, and modulation of the “stringent response” (an adaptation to nutrient deprivation) were detected. Overall, we established a bacterial consortium based on barley β-glucan in vitro, which can be used to investigate aspects of the functional blueprint of the human gut microbiota.
IMPORTANCE The microbial community, mostly composed of bacterial species, residing in the human gut degrades and ferments polysaccharides derived from plants (dietary fiber) that would not otherwise be digested. In this way, the collective metabolic actions of community members extract additional energy from the human diet. While the variety of bacteria present in the microbial community is well known, the formation of bacterial consortia, and the consequent interactions that result in the digestion of dietary polysaccharides, has not been studied extensively. The importance of our work was the establishment, under laboratory conditions, of a consortium of gut bacteria that formed around a dietary constituent commonly present in cereals. This enabled the metabolic interplay between the bacterial species to be studied. This kind of knowledge is required to construct an interactive, metabolic blueprint of the microbial community that inhabits the human gut.
INTRODUCTION
The human gut microbiota is a complex community, in which competitive and cooperative interactions between bacterial species play an important role in its assemblage and maintenance (1–3). Plant polysaccharides (glycans) constitute chemically and structurally diverse sources of carbon and energy for gut bacteria. They are commonly present in the diet but are not degraded by human digestive processes (4, 5). Bacteria in the colon have the ability to depolymerize the glycans and ferment the mono- and oligosaccharides that then become available (6). In at least some cases, cross-feeding networks form around glycans, in which some species degrade particular substrates that are metabolically inaccessible to others, but leave extracellular hydrolytic products in the habitat that are then used by other species for growth (7–10). Plant polysaccharides can thus form the nucleus for the assembly of combinations of bacterial species (consortia) with special metabolic activities. However, this is a relatively unexplored aspect of gut microbiota research (3).
The purpose of our work was to develop an experimental approach to aid in disentangling the dynamics of the human gut microbiota through synthetic ecology (11). To do this, we built an in vitro consortium of gut bacteria based on barley β-glucan, which is a (1→3),(1→4)-β-d-glucan present in plant cell walls, as nutritive substrate (12). Three bacterial species commonly present in the human gut microbiota were selected through experimentation as bacterial constituents of the consortium (13, 14). Bacteroides ovatus is a member of one of the dominant phyla present in the human gut microbiota, Bacteroidetes (15). Members of this phylum have large genomes that encode many carbohydrate-active enzymes (16). They are considered to be important functionally as “generalist” bacteria in the human gut (5, 17). The metabolic capacity of Bacteroides species has been studied for decades, but the molecular systems involved in the degradation of complex glycans have been described more recently (9, 18–29). The remaining members of the bacterial consortium belong to the Firmicutes, another dominant phylum in the human gut microbiota. Subdoligranulum variabile is a member of the family Ruminococcaceae (30), and Hungatella hathewayi is a member of the family Lachnospiraceae (31, 32). Hence, the consortium contained species that are members of the three predominant families of the human gut microbiota (Bacteroidaceae, Ruminococcaceae, and Lachnospiraceae) (33).
Transcriptomics provides a powerful tool to study consortium interplay (7). It is referred to as a means of eavesdropping on bacterial “conversations” within the community and to identify interactions that sustain the assemblage of bacterial species (7, 34). Therefore, the aim of our study was to determine the transcriptional responses of the different bacteria with regard to their relationships within the consortium.
RESULTS
Choice of bacterial species.
B. ovatus ATCC 8483T, S. variabile DSM 15176T, and H. hathewayi DSM 13479T were chosen as prospective members of the consortium because of the results of screening experiments using 49 bacterial species (Table 1) commonly present as members of human gut microbiotas (13, 14). Highly significant increases in the amount of growth obtained in β-glucan medium compared to basal medium were evident for five of the species, Bacteroides cellulosilyticus, Bacteroides ovatus, Bacteroides uniformis, Prevotella copri, and Eubacterium rectale. B. ovatus was chosen as the potential β-glucan-degrading member of the consortium because of its ability to grow well in β-glucan medium and because the molecular biology of its degradative abilities is well-described (20, 21). We then tested 19 common members of the gut microbiota for the ability to grow in medium containing β-glucan-derived oligosaccharides produced during the growth of B. ovatus (Table 2). Two species (Hungatella hathewayi and Subdoligranulum variabile) had highly significantly increased growth in oligosaccharide medium compared to basal medium. These species could not grow in β-glucan medium to a greater extent than in basal medium. Therefore, these two species were chosen for further study because they could potentially benefit nutritionally from the hydrolytic activity of B. ovatus in the presence of β-glucan. It is noteworthy that some species had less growth in β-glucan medium than in basal medium (such as Bacteroides pectinophilus and Alistipes putredinis) or less growth in oligosaccharide medium than in basal medium (such as Bacteroides fragilis, Bacteroides massiliensis, Bacteroides thetaiotaomicron, and Bacteroides vulgatus). The physiological basis for these inhibitions is not known.
TABLE 1.
Bacterial species tested for their ability to grow in barley β-glucan mediuma
| Species | Mean A600 | SD | A600 ratio | P valueb |
|---|---|---|---|---|
| Bacteroides cellulosilyticus DSM 14838T | 0.117 | 0.00173205 | 1.513 | 7.6022e-06 |
| Bacteroides ovatus ATCC 8483T | 0.493 | 0.0057735 | 2.851 | 8.8174e-08 |
| Bacteroides uniformis ATCC 8492T | 0.189 | 0.00503322 | 1.586 | 3.8781e-05 |
| Prevotella copri DSM 18205T | 0.143 | 0.00585947 | 2.337 | 5.3198e-05 |
| Eubacterium rectale DSM 17629T | 0.121 | 0.00251661 | 3.772 | 1.9103e-06 |
| Bifidobacterium adolescentis DSM 20083T | 0.134 | 0.00493288 | 0.982 | |
| Bifidobacterium breve ATCC 15700T | 0.236 | 0.00984886 | 1.354 | 0.00192629 |
| Bifidobacterium dentium DSM 20436T | 0.203 | 0.00568624 | 1.340 | 0.00040374 |
| Bifidobacterium longum subsp. longum ATCC 15707T | 0.133 | 0.001 | 0.990 | |
| Bifidobacterium pseudocatenulatum DSM 20438T | 0.217 | 0.01322876 | 1.146 | 0.02529211 |
| Collinsella aerofaciens DSM 3979T | 0.156 | 0.00650641 | 0.923 | |
| Bacteroides caccae ATCC 43185T | 0.143 | 0.00351188 | 0.953 | |
| Bacteroides dorei DSM 17855T | 0.142 | 0.01078579 | 0.806 | |
| Bacteroides eggerthii ATCC 27754T | 0.116 | 0.00585947 | 1.600 | 0.00055184 |
| Bacteroides finegoldii DSM 17565T | 0.250 | 0.03 | 2.632 | 0.00107981 |
| Bacteroides fragilis ATCC 25285T | 0.151 | 0.0052915 | 0.998 | |
| Bacteroides intestinalis DSM 17393T | 0.073 | 0.0034641 | 1.244 | 0.00439986 |
| Bacteroides massiliensis DSM 17679T | 0.037 | 0.00404145 | 0.836 | |
| Bacteroides pectinophilus ATCC 43243T | 0.003 | 0.00152753 | 0.400 | |
| Bacteroides stercoris ATCC 43183T | 0.139 | 0.00288675 | 1.146 | 0.00816623 |
| Bacteroides thetaiotaomicron ATCC 29148T | 0.262 | 0.01184624 | 0.913 | |
| Bacteroides vulgatus ATCC 29327 | 0.056 | 0.00650641 | 0.928 | |
| Bacteroides xylanisolvens DSM 18836T | 0.637 | 0.06110101 | 3.141 | 0.00025133 |
| Parabacteroides distasonis DSM 20701T | 0.123 | 0.002 | 0.918 | |
| Parabacteroides goldsteinii DSM 19448T | 0.120 | 0.001 | 0.832 | |
| Paraprevotella clara DSM 19731T | 0.014 | 0.00057735 | 1.433 | 0.00289001 |
| Alistipes putredinis DSM 17216T | 0.006 | 0.00152753 | 0.395 | |
| Lactobacillus salivarius DSM 20555T | 0.236 | 0.00152753 | 0.968 | |
| Blautia hansenii DSM 20583T | 0.060 | 0.03865661 | 0.767 | |
| Clostridium asparagiforme DSM 15981T | 0.470 | 0.01464013 | 0.929 | |
| Hungatella hathewayi DSM 13479T | 0.333 | 0.0321455 | 1.020 | NS |
| Clostridium nexile DSM 1787T | 0.129 | 0.00208167 | 0.998 | |
| Clostridium scindens DSM 5676T | 0.065 | 0.00251661 | 0.920 | |
| Clostridium symbiosum DSM 934T | 0.340 | 0.03874274 | 0.899 | |
| Clostridium propionicum DSM 1682T | 0.010 | 0.00057735 | 1.526 | 0.02411011 |
| Coprococcus comes ATCC 27758T | 0.677 | 0.18175075 | 0.883 | |
| Dorea formicigenerans DSM 3992T | 0.026 | 0.00458258 | 1.300 | NS |
| Dorea longicatena DSM 13814T | 0.135 | 0.00550757 | 0.888 | |
| Eubacterium ventriosum DSM 3988T | 0.044 | 0.00550757 | 0.771 | |
| Ruminococcus gnavus ATCC 29149T | 0.112 | 0.00458258 | 1.222 | 0.00253049 |
| Ruminococcus torques ATCC 27756T | 0.055 | 0.003 | 0.855 | |
| Ruminococcus obeum ATCC 29174T | 0.031 | 0.00378594 | 1.022 | NS |
| Clostridium leptum DSM 753T | 0.069 | 0.0051316 | 2.286 | 0.00027607 |
| Faecalibacterium prausnitzii DSM 17677T | 0.018 | 0.0034448 | 1.494 | 0.00631048 |
| Flavonifractor plautii DSM 4000T | 0.176 | 0.00321455 | 1.111 | 0.02816079 |
| Pseudoflavonifractor capillosus DSM 23940T | 0.527 | 0.1569501 | 2.476 | 0.02622434 |
| Subdoligranulum variabile DSM 15176T | 0.012 | 0.00252982 | 0.847 | |
| Holdemania filiformis DSM 12042T | 0.062 | 0.005 | 0.964 | |
| Bilophila wadsworthia DSM 11045T | 0.073 | 0.00519615 | 1.237 | 0.01855356 |
Optical density (A600) was measured after 24 h of growth. Bacterial growth in barley β-glucan (0.2% [wt/vol]) medium is reported as mean A600 of bacterial cultures (n = 3) with standard deviation (SD). The ratio between the mean A600 of bacterial cultures grown in barley β-glucan medium and basal medium (n = 3) is given. P values of <0.05 were considered statistically significant. P values are shown only when the A600 ratio is >1.
NS, not significant.
TABLE 2.
Bacterial species that did not grow in barley β-glucan medium and therefore were tested for their ability to grow on barley β-glucan-derived oligosaccharidesa
| Species | Mean A600 | SD | A600 ratio | P valueb |
|---|---|---|---|---|
| Subdoligranulum variabile DSM 15176T | 0.284 | 0.01635441 | 23.058 | 2.293e-12 |
| Hungatella hathewayi DSM 13479T | 0.790 | 0.04195235 | 2.685 | 1.9604e-10 |
| Bifidobacterium adolescentis DSM 20083T | 0.133 | 0.00519615 | 1.025 | NS |
| Collinsella aerofaciens DSM 3979T | 0.152 | 0.00360555 | 0.896 | |
| Bacteroides dorei DSM 17855T | 0.108 | 0.00416333 | 0.806 | |
| Bacteroides fragilis ATCC 25285T | 0.053 | 0.00288675 | 0.377 | |
| Bacteroides massiliensis DSM 17679T | 0.011 | 0.00057735 | 0.101 | |
| Bacteroides thetaiotaomicron ATCC 29148T | 0.117 | 0.02083267 | 0.644 | |
| Bacteroides vulgatus ATCC 29327 | 0.013 | 0.00360555 | 0.140 | |
| Parabacteroides distasonis DSM 20701T | 0.130 | 0.00602771 | 0.620 | |
| Parabacteroides goldsteinii DSM 19448T | 0.248 | 0.04606951 | 0.996 | |
| Alistipes putredinis DSM 17216T | 0.046 | 0.0011547 | 1.631 | 0.00127325 |
| Lactobacillus salivarius DSM 20555T | 0.287 | 0.0085049 | 1.056 | NS |
| Blautia hansenii DSM 20583T | 0.141 | 0.02129476 | 1.615 | 0.00051887 |
| Clostridium nexile DSM 1787T | 0.175 | 0.07716389 | 1.314 | NS |
| Clostridium symbiosum DSM 934T | 0.048 | 0.00173205 | 0.873 | |
| Dorea longicatena DSM 13814T | 0.152 | 0.02404163 | 0.948 | |
| Eubacterium ventriosum DSM 3988T | 0.018 | 0.00556776 | 0.628 | |
| Ruminococcus torques ATCC 27756T | 0.046 | 0.00360555 | 0.789 |
Optical density of cultures (A600) was measured after 48 h of growth. Bacterial growth is reported as the mean A600 (n = 3) of bacterial cultures grown in medium containing barley β-glucan-derived oligosaccharides (0.05% [wt/vol]), with standard deviation (SD). The ratio between A600 of bacterial cultures grown in oligosaccharide medium and basal medium (n = 3) (A600 ratio) is given. P values of <0.05 were considered statistically significant. P values are shown only when the A600 ratio is >1.
NS, not significant.
Formation of a bacterial consortium based on the hydrolysis of barley β-glucan.
Growth curves of pure cultures of B. ovatus in β-glucan medium and of S. variabile and H. hathewayi in oligosaccharide medium are shown in Fig. 1. Barley β-glucan was hydrolyzed extracellularly (Fig. 2), and 3-O-β-cellobiosyl-d-glucose (DP3) and 3-O-β-cellotriosyl-d-glucose (DP4) oligosaccharides were released into the medium (Fig. 3A). These oligosaccharides were used by S. variabile and H. hathewayi for growth (Fig. 3B and C). The three species achieved consistent population levels in replicate batch and continuous (steady-state) cocultures, indicating that the species could reliably form a stable association in which oligosaccharides released as hydrolytic products by B. ovatus were used as growth substrates by the other two species (Fig. 4A and B). The S. variabile rate of utilization of DP3 was 13.3%/h, slightly faster than for DP4, which was 12.5%/h. In contrast, H. hathewayi showed a marked preference for DP4 (10.5%/h) compared to DP3 (2.8%/h). Prioritization of usage of different carbohydrates for growth may allow potential competitors to cohabit (5, 35), although in this case, S. variabile would be predicted to have the overall advantage because it used both oligosaccharides more rapidly than did H. hathewayi. However, H. hathewayi was numerically dominant over S. variabile in both batch and continuous cultures (Fig. 4A and B), indicating that the uptake of fermentable substrates must be complemented by the availability of other growth-limiting nutrients and/or that inhibitory substances are produced by H. hathewayi. Experiments would need to be conducted in chemically defined medium to identify the growth-limiting nutrients. This is a difficult and perhaps impossible task in the case of gut commensals that, to date, require rich medium for growth.
FIG 1.
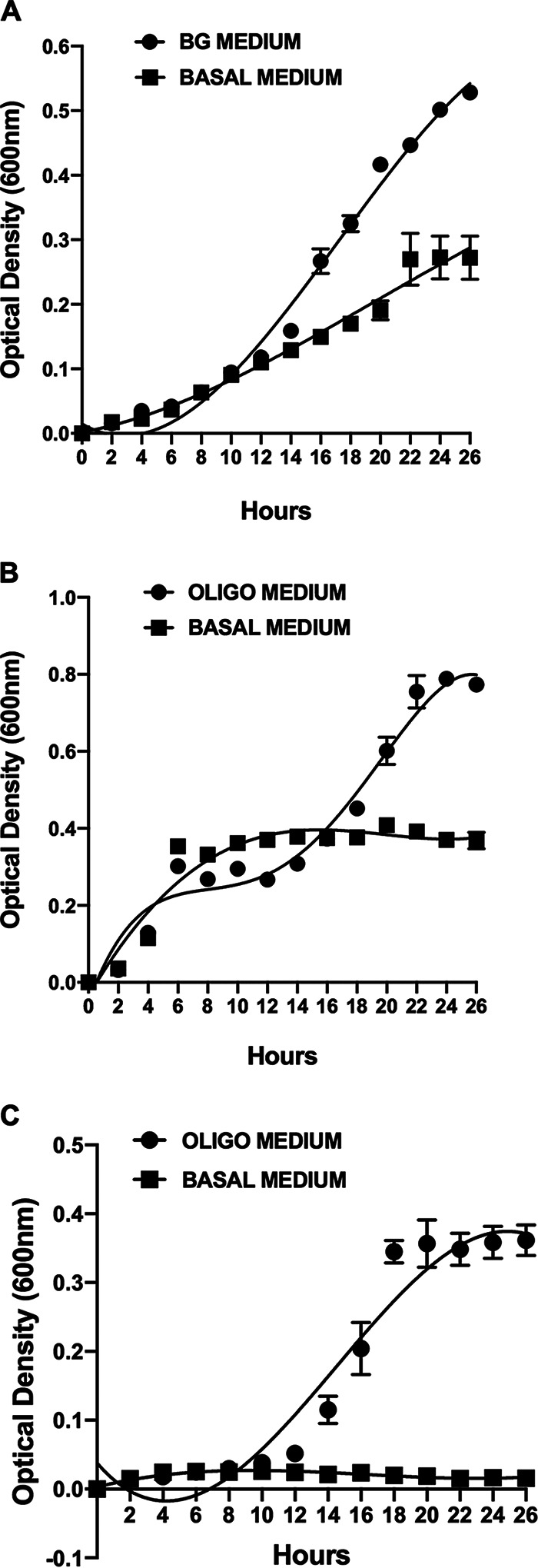
Growth of B. ovatus ATCC 8483T, H. hathewayi DSM 13479T, and S. variabile DSM 15176T in pure batch cultures during anaerobic incubation at 37°C. Mean (standard error of the mean [SEM]) optical densities at A600 of triplicate cultures are shown. (A) B. ovatus ATCC 8483T growth in barley β-glucan (BG) medium compared to basal medium. (B) H. hathewayi DSM 13479T growth in oligosaccharide (oligo) medium compared to basal medium. Note the diauxic growth profile in oligosaccharide medium where initial growth relied on constituents in basal medium, followed by utilization of oligosaccharides. (C) S. variabile DSM 15176T growth in oligosaccharide (oligo) medium compared to basal medium.
FIG 2.

Depolymerization of barley β-glucan by B. ovatus ATCC 8483T in pure batch culture at 37°C. Size exclusion chromatography (SEC) profiles of uninoculated barley β-glucan medium, barley β-glucan standard, and supernatant from B. ovatus ATCC 8483T culture in barley β-glucan medium incubated for 24 h.
FIG 3.

High-performance anion-exchange chromatography (HPAEC) profiles of supernatants collected during the growth of B. ovatus ATCC 8483T, S. variabile DSM 15176T, and H. hathewayi DSM 13479T in pure culture during anaerobic incubation at 37°C. Peaks corresponding to DP2 (probably laminaribiose), DP3 (3-O-β-cellobiosyl-d-glucose), and DP4 (3-O-β-cellotriosyl-d-glucose) oligosaccharides are indicated by reference to standards. Sampling times are indicated, and chromatographic profiles of samples that were analyzed with whole-transcriptomics analysis (RNA-seq) are colored red/orange. (A) B. ovatus ATCC 8483T supernatants from barley β-glucan medium. (B) H. hathewayi DSM 13479T supernatants from oligosaccharide medium. (C) S. variabile DSM 15176T supernatants from oligosaccharide medium.
FIG 4.

Populations of B. ovatus ATCC 8483T, S. variabile DSM 15176T, and H. hathewayi DSM 13479T in batch (24 h of incubation) or continuous coculture and utilization of DP3 and DP4 oligosaccharides in continuous coculture. (A) CFU/ml of B. ovatus ATCC 8483T, S. variabile DSM 15176T, and H. hathewayi DSM 13479T in combinations of batch coculture. Means (SEMs) of duplicate cultures are shown. (B) CFU/ml of B. ovatus ATCC 8483T, S. variabile DSM 15176T, and H. hathewayi DSM 13479T in continuous coculture. Means (SEMs) of three chemostat runs are shown. (C) High-performance anion-exchange chromatography (HPAEC) profiles of supernatants collected from continuous coculture of B. ovatus ATCC 8483T, S. variabile DSM 15176T, and H. hathewayi DSM 13479T. Profiles from three chemostat runs are shown. BO, B. ovatus ATCC 8483T in pure culture; BO+SV+HH, the three species in coculture. Peaks corresponding to DP3 (3-O-β-cellobiosyl-d-glucose) and DP4 (3-O-β-cellotriosyl-d-glucose) oligosaccharides are indicated.
Differential transcription of genes.
The transcription levels of genes in the genomes of the three species in pure culture were determined and compared using whole-transcriptome analysis of cells collected at different stages of the growth curve. These comparisons were aimed at determining gene transcription during degradation and uptake of nutrients sourced from β-glucan by actively growing cells (for example, B. ovatus in pure culture sampled at 16, 20, and 26 h of growth; intraculture comparisons). Transcriptional comparisons of cocultures were made using cells collected after 24 h of growth because all three species were in the same growth phase by then (for example, S. variabile in pure culture at 24 h compared to B. ovatus/S. variabile at 24 h). Although the population levels of the three species were not the same within the coculture at the sampling time, they were similar (no more than 10-fold difference) for a given species between pure culture and coculture, and therefore sufficiently standardized comparisons could be made. S. variabile and H. hathewayi transcriptomes were compared in pure culture and coculture under batch conditions. Continuous coculture comparisons were not made, due to logistical difficulties in large-scale preparation of oligosaccharide medium required for comparative chemostat experiments. General data relating to RNA sequencing and whole transcriptome analysis are summarized in Table S1 in the supplemental material. Differential transcription of genes in Cluster of Orthologous Genes (COG) categories from the perspective of individual members of cocultures and enrichment of Gene Ontology (GO) terms are summarized in Fig. S1 and Table S2, respectively. The numbers of differentially transcribed genes common or unique to specific combinations of the three bacterial species are summarized in Venn diagrams (Fig. S2).
Particularly noteworthy, transcriptomics analysis revealed the increased transcription of B. ovatus genes involved in barley β-glucan degradation (Table 3) during growth in pure culture and coculture under batch conditions. These genes have been recently described as part of a mixed-linkage-β-glucan utilization locus in B. ovatus (20, 21). In particular, glycoside hydrolase GH16 is an outer membrane-anchored endo-glucanase responsible for the breakdown of barley β-glucan molecules into oligosaccharides, the smallest released being mixed-linkage tri- and tetrasaccharides (21). Consistent with our results, a recent study showed that cell surface glycan-binding proteins belonging to the B. ovatus mixed-linkage-β-glucan utilization locus preferentially bind oligosaccharides with a degree of polymerization >6 and do not bind mixed-linkage tri- and tetrasaccharides (20). The transcription of the β-glucan utilization genes in batch coculture was mainly influenced by the presence of H. hathaweyi, because coculture with S. variabile did not affect differential transcription of the locus to any extent compared to pure culture. Similarly, transcription of the genes was the same in B. ovatus cells obtained from pure culture and coculture under continuous conditions (Table 3). Nevertheless, β-glucan was hydrolyzed in the chemostat, and DP3 and DP4 oligosaccharides were utilized by the coculture as observed for batch conditions (Fig. 3A and 4C).
TABLE 3.
Differential transcription of genes involved in degradation of barley β-glucan in B. ovatus ATCC 8483T in pure culture and coculturea
| Gene name | Product (EC no.) | Pure culture (batch)b
|
Coculture (batch)b
|
Coculture (continuous)b
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Log2FC BO16 vs BO20 | Log2FC BO16 vs BO26 | FDR | Log2FC BOHH24 vs BO24 | Log2FC BOSV24 vs BO24 | Log2FCBOSVHH24 vs BO24 | FDR | Log2FC BOSVHH vs BO | FDR | ||
| Bovatus_RS15290 | Glycoside hydrolase family 16 (GH16; EC 3.2.1.-) | 3.786 | 7.074 | 1.55e-04 | 6.695 | –c | 5.395 | 1.26e-03 | –0.037 | 0.955 |
| Bovatus_RS15295 | SusC/RagA family TonB-linked outer membrane protein | 2.819 | 6.775 | 3.69e-04 | 6.413 | 1.180 | 5.398 | 9.49e-04 | –0.428 | 0.358 |
| Bovatus_RS15300 | SusC/RagB family nutrient-binding outer membrane lipoprotein | 2.734 | 6.676 | 2.86e-04 | 6.113 | 1.043 | 5.364 | 1.52e-03 | –0.696 | 0.083 |
| Bovatus_RS15305 | Hypothetical protein | 2.507 | 7.055 | 1.23e-04 | 7.252 | 1.912 | 6.557 | 2.73e-04 | –0.769 | 0.053 |
| Bovatus_RS15310 | β-Glucosidase (EC 3.2.1.-) | 2.542 | 6.996 | 5.05e-04 | 6.862 | 1.345 | 6.159 | 1.12e-03 | –0.686 | 0.080 |
Gene name, product, Log2FC (fold change) of each comparison, and FDR (false discovery rate) are reported. Genes showing an FDR of <0.05 and absolute value of Log2FC of >1 were considered statistically differentially transcribed.
BO, B. ovatus; BOHH, coculture of B. ovatus and H. hathewayi; BOSV, coculture of B. ovatus and S. variabile; BOSVHH, coculture of B. ovatus, S. variabile, and H. hathewayi. Comparisons in pure culture are between transcription at 16 h and 20 or 26 h of incubation. Comparisons in coculture are between pure culture and coculture, both at 24 h of incubation.
–, absolute value Log2FC < 1.
S. variabile had increased transcription of genes encoding carbohydrate transporters during pure growth but decreased transcription of most of these in coculture, which may be related to competition with H. hathewayi, which was numerically dominant (Fig. 4A). H. hathewayi had increased transcription of genes encoding carbohydrate receptors and permease transporters in both pure culture and coculture. These gene products are likely to be involved in the uptake of the oligosaccharides (Table 4).
TABLE 4.
Differential transcription of exemplar genes encoding carbohydrate receptors and transporters in S. variabile DSM 15176T and H. hathewayi DSM 13479T in pure and coculturea
| Species and gene name | Product | Pure cultureb
,c
|
Cocultureb
|
|||||
|---|---|---|---|---|---|---|---|---|
| Log2FC SV12 vs SV14 |
Log2FC SV12 vs SV16 | FDR |
Log2FC BOSV vs SV24 | Log2FC BOSVHH vs SV24 | FDR | |||
| S. variabile | ||||||||
| SUBVAR_RS01140 | Carbohydrate ABC transporter permease | 1.914 | 1.805 | 9.45e-03 | –6.402 | –3.233 | 2.98e-32 | |
| SUBVAR_RS01150 | Carbohydrate ABC transporter substrate-binding protein | 1.553 | 1.271 | 4.91e-02 | –6.100 | –5.316 | 5.21e-58 | |
| SUBVAR_RS12285 | Carbohydrate ABC transporter permease | 1.487 | 1.109 | 3.72e-03 | –2.595 | –2.442 | 7.40e-32 | |
| SUBVAR_RS12505 | PTS sugar transporter subunit IIBCA | 2.126 | 2.497 | 2.23e-03 | –7.116 | –5.950 | 2.95e-87 | |
| SUBVAR_RS14155 | Carbohydrate ABC transporter substrate-binding protein | – | – | NSd | 1.549 | 1.911 | 4.13e-19 | |
| Log2FC HH06 vs HH16 | Log2FC HH06 vs HH18 | Log2FC HH06 vs HH20 | FDR | Log2FC BOHH vs HH24 | Log2FC BOSVHH vs HH24 | FDR | ||
| H. hathewayi | ||||||||
| CLOSTHATH_RS02755 | Carbohydrate ABC transporter permease | 3.186 | 4.197 | 3.932 | 2.22e-07 | 2.209 | 2.352 | 1.78e-02 |
| CLOSTHATH_RS02760 | Sugar ABC transporter permease | 3.520 | 4.275 | 4.275 | 8.15e-08 | 3.949 | 3.575 | 4.95e-05 |
| CLOSTHATH_RS02765 | Sugar ABC transporter substrate-binding protein | 3.820 | 4.728 | 4.754 | 5.50e-06 | 3.566 | 3.317 | 2.13e-04 |
Gene name, product, and Log2FC (fold change) of each comparison and FDR (false-discovery rate) are reported. Genes showing an FDR of <0.05 and absolute value of Log2FC >1 were considered statistically differentially transcribed.
SV, S. variabile; BO, B. ovatus; BOSV, coculture of B. ovatus and S. variabile; BOSVHH, coculture of B. ovatus, S. variabile, and H. hathewayi; HH, H. hathewayi; BOHH, B. ovatus and H. hathewayi in coculture; BOSVHH, B. ovatus, S. variabile, and H. hathewayi in coculture. Comparisons in S. variabile pure culture are between transcription at 12 h and 14 or 16 h. Comparisons in H. hathewayi pure culture are between transcription at 6 h and 16, 18, or 20 h of incubation. Comparisons in coculture are between pure culture and coculture, both at 24 h of incubation.
–, absolute value Log2FC < 1.
NS, not significant.
Having obtained evidence of a nutritional relationship based on barley β-glucan oligosaccharides within the coculture of three species, we searched transcriptomics data for indications of other adaptations by the species. Three phenomena of interest were detected, which we report here for future reference. They are, at this stage, observational and thus speculative and require biochemical validation. Cobalamin (vitamin B12) is an essential cofactor for the growth of many bacteria. Species that do not possess the biosynthetic pathway for cobalamin synthesis have mechanisms to acquire it from the environment (36–39). Cobalamin availability may thus represent an important factor in the establishment of microbial networks (36). A search of S. variabile and H. hathewayi genome sequences (NCBI RefSeq accession assembly numbers GCF_000157955.1 and GCF_000160095.1, respectively) revealed the presence of genes representative of cobalamin biosynthetic pathways. The B. ovatus genome sequence (NCBI RefSeq accession assembly number GCF_001314995.1) did not contain evidence of a cobalamin biosynthetic pathway, but a gene encoding a cobalamin-binding protein was present. The transcription of the cobalamin biosynthetic operon of S. variabile was unchanged during pure growth or in coculture with B. ovatus but had decreased transcription in coculture with H. hathewayi (Table 5). This suggests that H. hathewayi was the principal source of cobalamin in coculture.
TABLE 5.
Transcription of genes belonging to the cobalamin biosynthetic operon of S. variabile DSM 15176Ta
| Gene name | Product (EC no.) | Pure cultureb,c
|
Cocultureb,c
|
||||
|---|---|---|---|---|---|---|---|
| Log2FC SV12 vs SV14 | Log2FC SV12 vs SV16 | FDRd | Log2FC BOSV vs SV24 | Log2FC BOSVHH vs SV24 | FDR | ||
| SUBVAR_RS06700 | Iron ABC transporter permease | – | – | NSd | – | –2.055 | 6.55e-04 |
| SUBVAR_RS06705 | Precorrin-8X methylmutase (EC 5.4.99.60) | – | – | NS | – | –1.744 | 2.62e-03 |
| SUBVAR_RS06720 | Cobalamin biosynthesis protein CobD | – | – | NS | – | –1.187 | 3.60e-03 |
| SUBVAR_RS06735 | Adenosylcobinamide-GDP Ribazoletransferase (EC 2.7.8.26) | – | – | NS | – | –1.051 | 4.98e-02 |
| SUBVAR_RS06750 | Cobyrinate a,c-diamide synthase (EC 6.3.5.11) | – | – | NS | – | –1.636 | 1.30e-02 |
| SUBVAR_RS06775 | Precorrin-4 C(11)-methyltransferase (EC 2.1.1.271) | – | – | NS | – | –2.200 | 1.35e-05 |
| SUBVAR_RS06780 | Precorrin-2 C(20)-methyltransferase (EC 2.1.1.151) | – | – | NS | – | –2.206 | 6.64e-03 |
| SUBVAR_RS06785 | Cobalamin biosynthesis protein CbiD | – | – | NS | – | –1.969 | 4.39e-05 |
| SUBVAR_RS06790 | Cobalt chelatase (EC 4.99.1.3) | – | – | NS | – | –2.449 | 1.97e-06 |
| SUBVAR_RS06805 | ABC transporter substrate-binding protein | – | – | NS | – | –2.312 | 1.09e-06 |
aGene name, product, and Log2FC (fold change) of each comparison and FDR (false discovery rate) are reported. Genes showing an FDR of <0.05 and absolute value of Log2FC >1 were considered statistically differentially transcribed.
bSV, S. variabile; BO, B. ovatus; BOSV, coculture of B. ovatus and S. variabile; BOSVHH, coculture of B. ovatus, S. variabile, and H. hathewayi. Comparisons in pure culture are between transcription at 12 h and 14 or 16 h of incubation. Comparisons in coculture are between pure culture and coculture, both at 24 h of incubation.
–, absolute value Log2FC < 1.
NS, not statistically significant.
Another possible adaptation detected was in relation to the “stringent response” (40, 41), where the key regulators are the intracellular molecules guanosine-5′-3′-bispyrophosphate (ppGpp) and guanosine pentaphosphate (pppGpp), together denoted by (p)ppGpp (42). In the presence of nutrient starvation conditions, increased levels of (p)ppGpp cause reduced transcription of genes involved in translation machinery and cell growth and division and increased transcription of genes involved in stress response (41, 43). We noted that the bifunctional-(p)ppGpp synthetase/guanosine-3′-5′-bis(diphosphate)-3′-pyrophosphohydrolase (SUBVAR_RS12040) gene had decreased transcription when S. variabile was in coculture with B. ovatus and in coculture with B. ovatus and H. hathewayi (Table 6). Moreover, ObgE (SUBVAR_RS07640), an essential GTPase that acts as a negative regulator of the stringent response preventing activation in a nutrient-rich environment (40, 44), had increased transcription in S. variabile in cocultures (Table 6). Together with increased transcription of ribosomal protein and aminoacyl-tRNA ligase genes in S. variabile (Table 6 and Table S2), these observations indicate a suppression of the stringent response when in coculture and hence indicate that the species was not suffering from nutrient starvation. In the case of H. hathewayi, however, the bifunctional-(p)ppGpp-synthetase/guanosine-3′-5′-bis(diphosphate)-3′-pyrophosphohydrolase gene (CLOSTHATH_RS21480) had increased transcription in coculture both with B. ovatus and with B. ovatus and S. variabile. The GTPase OgbE (CLOSTHATH_RS30045) gene had increased transcription when H. hathewayi was in coculture with B. ovatus and S. variabile, indicating some nutritional stress (Table 6), but genes encoding ribosomal structural proteins nevertheless had increased transcription in coculture.
TABLE 6.
Transcription of exemplar genes encoding ribosome structural proteins, aminoacyl-tRNA ligases, and regulators of stringent response in S. variabile DSM 15176T and H. hathewayi DSM 13479Ta
| Species and gene name | Product (EC no.) | Pure cultureb
,c
|
Cocultureb
|
|||||
|---|---|---|---|---|---|---|---|---|
| Log2FC SV12 vs SV14 | Log2FC SV12 vs SV16 | FDRd | Log2FC BOSV vs SV24 | Log2FC BOSVHH vs SV24 | FDR | |||
| S. variabile | ||||||||
| SUBVAR_RS01425 | 30S ribosomal protein S9 | – | – | NS | 4.291 | 3.961 | 1.89e-54 | |
| SUBVAR_RS02945 | 30S ribosomal protein S18 | – | – | NS | 3.387 | 3.033 | 1.59e-41 | |
| SUBVAR_RS04660 | 50S ribosomal protein L19 | – | – | NS | 3.291 | 2.966 | 1.57e-42 | |
| SUBVAR_RS05155 | 50S ribosomal protein L31 | – | – | NS | 4.744 | 4.195 | 4.26e-60 | |
| SUBVAR_RS05835 | 50S ribosomal protein L10 | – | – | NS | 4.011 | 3.423 | 2.19e-51 | |
| SUBVAR_RS07650 | 50S ribosomal protein L21 | – | – | NS | 2.640 | 2.428 | 1.73e-30 | |
| SUBVAR_RS04130 | Alanine-tRNA ligase (EC 6.1.1.7) | – | – | NS | 2.332 | 2.945 | 1.13e-31 | |
| SUBVAR_RS04425 | Tyrosine-tRNA ligase (EC 6.1.1.1) | – | – | NS | 2.301 | 2.408 | 3.05e-23 | |
| SUBVAR_RS05990 | Asparagine-tRNA ligase (EC 6.1.1.22) | – | – | NS | 2.185 | 2.207 | 3.84e-23 | |
| SUBVAR_RS06090 | Phenylalanine-tRNA ligase subunit beta (EC 6.1.1.20) | – | – | NS | 1.385 | 1.356 | 3.14e-11 | |
| SUBVAR_RS09185 | Lysine-tRNA ligase (EC 6.1.1.6) | – | – | NS | 2.172 | 1.630 | 1.97e-21 | |
| SUBVAR_RS09715 | Threonine-tRNA ligase (EC 6.1.1.3) | – | – | NS | 1.619 | 2.114 | 5.55e-18 | |
| SUBVAR_RS12040 | Bifunctional (p)ppGpp synthetase/guanosine-3, 5′-bis(diphosphate) 3′-pyrophosphohydrolase (EC 3.1.7.2) | – | – | NS | –1.456 | –11.357 | 1.48e-09 | |
| SUBVAR_RS07640 | GTPase ObgE | – | – | NS | 1.232 | 1.013 | 1.91e-08 | |
| Log2FC HH06 vs HH16 | Log2FC HH06 vs HH18 | Log2FC HH06 vs HH20 | FDR | Log2FC BOHH vs HH24 | Log2FC BOSVHH vs HH24 | FDR | ||
| H. hathewayi | ||||||||
| CLOSTHATH_RS11480 | 50S ribosomal protein L31 | –2.080 | –2.217 | –1.890 | 6.50e-05 | 3.550 | 3.486 | 8.79e-05 |
| CLOSTHATH_RS18880 | 30S ribosomal protein S2 | –1.550 | –1.583 | –1.338 | 5.84e-04 | 2.598 | 2.353 | 2.49e-04 |
| CLOSTHATH_RS18945 | 30S ribosomal protein S21 | –2.511 | –2.503 | –2.416 | 4.68e-04 | 3.912 | 3.597 | 4.24e-05 |
| CLOSTHATH_RS20325 | 50S ribosomal protein L28 | –2.543 | –2.716 | –2.587 | 1.10e-04 | 4.578 | 4.351 | 2.39e-05 |
| CLOSTHATH_RS28340 | 50S ribosomal protein L35 | –1.908 | –1.956 | –1.821 | 1.82e-04 | 2.545 | 2.716 | 2.65e-03 |
| CLOSTHATH_RS30700 | 30S ribosomal protein S20 | –2.869 | –2.588 | –2.106 | 2.44e-05 | 3.198 | 2.701 | 1.23e-04 |
| CLOSTHATH_RS07400 | Glutamine-tRNA ligase/YqeY domain fusion protein (EC 6.1.1.18) | –1.646 | –1.838 | –1.653 | 2.91e-04 | 2.536 | 2.572 | 3.20e-04 |
| CLOSTHATH_RS12900 | Glycine-tRNA ligase (EC 6.1.1.14) | –1.202 | –1.213 | –1.306 | 2.29e-04 | 1.131 | 1.208 | 1.90e-02 |
| CLOSTHATH_RS21545 | Histidine-tRNA ligase (EC 6.1.1.21) | – | –1.031 | – | 2.32e-03 | 1.857 | 2.400 | 6.22e-04 |
| CLOSTHATH_RS27790 | Arginine-tRNA ligase (EC 6.1.1.19) | –1.110 | –1.476 | –1.347 | 2.89e-04 | 1.268 | 1.598 | 3.80e-03 |
| CLOSTHATH_RS28080 | Lysine-tRNA ligase (EC 6.1.1.6) | –1.161 | –1.382 | – | 1.66e-03 | 2.201 | 2.302 | 3.73e-04 |
| CLOSTHATH_RS21480 | Bifunctional (p)ppGpp synthetase/guanosine-3′,5′-bis(diphosphate) 3′-pyrophosphohydrolase (EC 3.1.7.2) | –1.052 | –1.239 | –1.189 | 3.80e-03 | 1.997 | 1.866 | 7.53e-04 |
| CLOSTHATH_RS30045 | GTPase ObgE | – | – | – | NS | – | 1.531 | 3.28e-02 |
Gene name, product, and Log2FC (fold change) of each comparison and FDR (false discovery rate) are reported. Genes showing an FDR of <0.05 and absolute value of Log2FC >1 were considered statistically differentially transcribed.
SV, S. variabile; BO, B. ovatus; BOSV, coculture of B. ovatus and S. variabile; BOSVHH, coculture of B. ovatus, S. variabile, and H. hathewayi; HH, H. hathewayi; BOHH, B. ovatus and H. hathewayi in coculture; BOSVHH, B. ovatus, S. variabile, and H. hathewayi in coculture. Comparisons in S. variabile pure culture are between 12 h and 14 or 16 h of incubation. Comparisons in H. hathewayi pure culture are between transcription at 6 h and 16, 18 or 20 h of incubation. Comparisons in coculture are between pure culture and coculture, both at 24 h of incubation.
–, absolute value Log2FC < 1.
NS, not statistically significant.
Genes belonging to the COG category “signal transduction mechanisms” were differentially transcribed in the three species (Fig. S1). Several of these genes encode sensor-histidine kinases known to be important for sensing and responding to environmental signals (45). Of particular interest was the increased transcription of the gene encoding S-ribosylhomocysteine lyase (SUBVAR_RS06625) of S. variabile when in coculture with B. ovatus but not with B. ovatus and H. hathewayi (Table 7). S-ribosylhomocysteine lyase (LuxS) catalyzes the synthesis of autoinducer-2 (AI-2), a molecule considered a universal signal for interspecies communication (46, 47).
TABLE 7.
Differential transcription of exemplar genes involved in molecular signaling in S. variabile DSM 15176T and H. hathewayi DSM 13479T in pure culture and coculturea
| Species and gene name | Product (EC no.) | Pure cultureb
,c
|
Coculture |
|||||
|---|---|---|---|---|---|---|---|---|
| Log2FC SV12 vs SV14 | Log2FC SV12 vs SV16 | FDRd | Log2FC BOSV vs SV24 | Log2FC BOSVHH vs SV24 | FDR | |||
| S. variabile | ||||||||
| SUBVAR_RS00065 | HAMP domain-containing histidine kinase (EC 2.7.13.3) | – | – | NS | –2.538 | –10.576 | 4.19e-12 | |
| SUBVAR_RS05565 | HAMP domain-containing protein | – | – | NS | –2.518 | –9.765 | 5.16e-08 | |
| SUBVAR_RS06880 | HAMP domain-containing histidine kinase (EC 2.7.13.3) | – | – | NS | –3.041 | –11.549 | 6.32e-22 | |
| SUBVAR_RS10870 | Sensor histidine kinase (EC 2.7.13.3) | – | – | NS | –1.986 | –3.518 | 8.78e-18 | |
| SUBVAR_RS14765 | Two-component sensor histidine kinase (EC 2.7.13.3) | – | – | NS | –1.584 | –1.732 | 7.14e-12 | |
| SUBVAR_RS15315 | HAMP domain-containing histidine kinase (EC 2.7.13.3) | – | – | NS | –3.479 | –11.767 | 1.68e-26 | |
| SUBVAR_RS06625 | S-ribosylhomocysteine lyase (EC 4.4.1.21) | – | – | NS | 1.446 | – | 1.52e-09 | |
| Log2FC HH06 vs HH16 | Log2FC HH06 vs HH18 | Log2FC HH06 vs HH20 | FDR | Log2FC BOHH vs HH24 | Log2FC BOSVHH vs HH24 | FDR | ||
| H. hathewayi | ||||||||
| CLOSTHATH_RS01390 | Sensor histidine kinase (EC 2.7.13.3) | – | – | – | NS | 1.815 | 1.745 | 1.08e-02 |
| CLOSTHATH_RS14145 | Sensor histidine kinase (EC 2.7.13.3) | –1.206 | –1.567 | –1.550 | 1.72e-04 | 1.719 | 1.214 | 7.46e-03 |
| CLOSTHATH_RS30795 | HAMP domain-containing protein | –1.206 | –1.315 | –1.390 | 6.01e-04 | 1.650 | 1.582 | 2.04e-02 |
| CLOSTHATH_RS00055 | Diguanylate cyclase (EC 2.7.7.65) | 1.070 | 1.589 | 1.460 | 2.99e-04 | –1.270 | –1.837 | 6.34e-03 |
| CLOSTHATH_RS07675 | Diguanylate cyclase (EC 2.7.7.65) | – | – | – | NS | 5.132 | – | 4.24e-04 |
| CLOSTHATH_RS08820 | GGDEF domain-containing protein | –2.119 | –1.877 | –1.993 | 7.40e-05 | 4.138 | 1.411 | 2.79e-02 |
| CLOSTHATH_RS17865 | Diguanylate cyclase (EC 2.7.7.65) | – | –1.065 | –1.089 | 3.54e-03 | 1.367 | 1.308 | 2.17e-02 |
| CLOSTHATH_RS23095 | EAL domain-containing protein | –1.034 | –1.008 | –1.338 | 2.58e-02 | 1.617 | 1.819 | 2.23e-02 |
| CLOSTHATH_RS26500 | EAL domain-containing protein | – | – | – | NS | 1.801 | 1.686 | 1.43e-03 |
| CLOSTHATH_RS27510 | GGDEF domain-containing protein | – | – | – | NS | –1.461 | –1.831 | 4.49e-02 |
| CLOSTHATH_RS29240 | Diguanylate cyclase (EC 2.7.7.65) | – | – | – | NS | –1.040 | – | 4.91e-02 |
| CLOSTHATH_RS30530 | Sensor domain-containing diguanylate cyclase (EC 2.7.7.65) | – | – | – | NS | 2.112 | 2.085 | 1.08e-02 |
| CLOSTHATH_RS13885 | Flagellin | – | 1.067 | – | 3.60e-03 | –1.474 | –1.415 | 1.72e-02 |
Gene name, product, and Log2FC (fold change) of each comparison and FDR (false discovery rate) are reported. Genes showing an FDR of <0.05 and absolute value of Log2FC >1 were considered statistically differentially transcribed.
SV, S. variabile; BOSV, coculture of B. ovatus and S. variabile; HH, H. hathewayi; BOHH, B. ovatus and H. hathewayi in coculture. Comparisons in B. ovatus pure culture are between transcription at 16 h and 20 or 26 h of incubation. Comparisons in S. variabile pure culture are between 12 h and 14 or 16 h of incubation. Comparisons in H. hathewayi pure culture are between transcription at 6 h and 16, 18 or 20 h of incubation Comparisons in coculture are between pure culture and coculture, both at 24 h of incubation.
–, absolute value Log2FC < 1.
NS, not statistically significant.
DISCUSSION
While there have been major increases in knowledge about the composition of microbial communities during recent decades, details of the functional interactions that occur between bacterial species within communities are much less advanced (3). As a result, Widder and colleagues (11) advocated the development of integrated model systems with which carefully chosen species can be studied in vitro. These defined model systems could be used to gain fundamental knowledge of how communities function. Although synthetic communities do not reproduce the complex diversity of natural communities, elucidation of basic principles of the operation of microbial communities is considered to be possible (11). In accordance with these views, we assembled a consortium of bacterial species that are commonly present in the human gut microbiota. Based on the hydrolysis of a common dietary fiber, barley β-glucan, we investigated the functional interplay between members of the consortium using whole-transcriptome sequencing and analysis. Coculture of bacteria produced transcriptional changes of genes contained in 16 COG categories in all three bacterial species, as well as enrichment or depletion of many GO terms, indicating that the bacteria responded to life in a more complex biological environment. Of particular interest was the increased transcription of genes associated with the hydrolysis of barley β-glucan (B. ovatus) and the increased transcription of carbohydrate receptors and transporters when oligosaccharides resulting from β-glucan hydrolysis by B. ovatus were available in the medium (S. variabile and H. hathewayi). The increased transcription of β-glucan utilization genes in B. ovatus in batch but not in continuous culture raises the interesting question as to which culture condition is most appropriate in simulating the human gut. Probably, the colon provides conditions that are intermediate between batch and continuous systems. Food travels to the colon in boluses (which could be likened to batches). Movement of digesta into the colon is controlled by the ileo-cecal valve. Thus, the flow of nutrients is semicontinuous, not continuous as is the situation in a chemostat. Technically sophisticated models of the human gut may be required to take into account intermittent addition of digesta to the colon.
β-Glucans are a major component of grains and are therefore common in human foods. The consumption of β-glucans has been reported to reduce postprandial glycemia in type 2 diabetic patients and to reduce hypercholesterolemia and hyperlipidemia (48–52). Although the amount of β-glucan varies in grains according to plant variety and growth conditions, the glycans can be purified to provide products that, when ingested, may therefore support human health. Specific modulation of the gut microbiota through the consumption of β-glucans may also be feasible because, in our screen of 49 common gut commensals, relatively few (mostly Bacteroides species) had greatly augmented growth in barley β-glucan medium relative to control medium. The opportunities for syntrophic relationships based on the hydrolysis of barley β-glucan by B. ovatus were found to be limited on the basis of the screen of 19 common commensals that could grow in oligosaccharide medium but not in β-glucan medium, of which H. hathewayi and S. variabile were by far the most competent. Lactobacillus ruminis has also been reported to utilize oligosaccharides derived from barley β-glucan hydrolysis (53, 54). This trophic information points to the possibility that highly specific changes to microbiota taxa and associated function could be achieved by dietary intervention using barley β-glucan. Investigation of other plant polysaccharides common in human diets might reveal further clusters of bacterial species that cooperatively degrade particular dietary fibers (2, 3, 55). It would also be interesting to develop this work further by determining the hydrolytic products in culture supernatants that might have been released from barley β-glucan by species other than B. ovatus (such as Bacteroides cellulosilytcus, Bacteroides uniformis, Prevotella copri, and Eubacterium rectale). Fundamental aspects of microbiota ecology could also be advanced through the development of more complex consortia of gut commensals in vitro using trophic information.
The establishment of an in vitro consortium of anaerobic bacterial species common in the gut microbiota is consistent with limiting experimental systems to a manageable number of constituent species, making it easier in turn to measure interactions within the coculture (11, 56, 57). For example, augmented propionate production as a proportion of total short-chain fatty acids resulting from the fermentation of a mixture of plant glycans by a synthetic community (Bacteroides ovatus, Bifidobacterium longum subspecies longum, Megasphaera elsdenii, Ruminococcus gnavus, and Veillonella parvula) was recently reported (56). Increased propionate production was due to greater succinate production by B. ovatus from galactan fermentation and conversion of succinate to propionate by V. parvula. We recognize, nevertheless, that the need to use rich medium for the culture of the fastidious bacterial species in our experiments limits the amount of detailed nutritional information that can be obtained. Missing from our model, too, is a spatial perspective; bacterial cells are associated with plant particulate material in the digesta, and these bacteria doubtless have a role in the hydrolysis and fermentation of plant polysaccharides in the gut (33, 58). Recent work with Bacteroides species, including B. ovatus, showed that these bacteria have the capacity to bind to beads coated with polysaccharides, characteristic of dietary fiber, in the gut of gnotobiotic mice (59). Spatial associations on food particles of hydrolytic bacteria (producers) with other bacterial species that benefit from leakage of hydrolytic products that are potential growth substrates can be envisioned in the development of consortia in the gut ecosystem (2, 3, 7). However, we feel that overall, transcriptomics studies as well as functional studies of consortia, such as we have developed, have the potential to dissect important mechanisms and interactions that take place among bacteria sharing the same meal at the same table (commensalism) (7, 9–11).
MATERIALS AND METHODS
Screening bacterial species for growth on barley β-glucan and β-glucan-derived oligosaccharides.
Purified (1→3),(1→4)-β-d-glucan (β-glucan) was prepared from Glucagel (DKSH, Italy) (60). A total of 49 bacterial species (Table 1) commonly present as members of human gut microbiotas (13, 14) were tested for their ability to grow in medium containing barley β-glucan. Individual bacterial strains were cultured under anaerobic conditions for 18 h at 37°C following DSMZ (https://www.dsmz.de/) and ATCC (https://www.atcc.org/) protocols. The screening assay was performed in basal medium (55) containing 2 g/liter of β-glucan. Media were sterilized by autoclaving (121°C for 15 min), prereduced in an anaerobic glove box, and inoculated (1% [vol/vol]) with individual bacterial cultures. Following our previously published procedure (55), optical density (A600) of cultures was measured after 24 h of anaerobic incubation at 37°C, when supernatants from the cultures were collected and stored at −80°C (55). Unpaired t test with Welch’s correction (GraphPad Prism version 7.0b) was used to compare optical density values of bacterial cultures grown in the presence or absence of β-glucan.
Oligosaccharide medium was prepared from β-glucan medium inoculated (1% [vol/vol]) with Bacteroides ovatus ATCC 8483T culture. After anaerobic incubation at 37°C for 24 h, supernatant was collected by centrifugation at 4,720 × g for 20 min at 4°C, pH was adjusted to 6.8 (to match that of basal medium), sterilized by filtration (0.22 μm pore size), and stored at −20°C. The tri- and tetrasaccharides in oligosaccharide medium were present at 0.45 to 0.5 mg/ml, as estimated by comparison of the peak areas of tri- and tetrasaccharides of a standard lichenase digest of barley β-glucan. Bacterial species that did not grow in β-glucan medium were tested for growth after 48 h in oligosaccharide medium following the procedure described above (Table 2).
Pure culture experiments.
The temporal pattern of degradation of β-glucan by B. ovatus and utilization of β-glucan-derived oligosaccharides by Subdoligranulum variabile DSM 15176T and Hungatella hathewayi DSM 13479T in batch cultures was followed over a 26-h period after inoculation of the medium. B. ovatus, S. variabile, and H. hathewayi cultures were grown for 18 h at 37°C and used to inoculate (1% [vol/vol]) prereduced and prewarmed medium. Sample collection and optical density measurements (A600) were performed every 2 h. Then, 1-ml aliquots of cultures were centrifuged at 14,500 × g for 5 min at 4°C to collect bacterial cells and supernatant. Bacterial cell pellets were immediately resuspended in 1 ml of RNAprotect Bacteria Reagent (Qiagen) and stored at −80°C (7, 61, 62), and supernatants were also stored at −80°C. Two technical replicates and three biological replicates were carried out for each time course experiment.
Coculture experiments.
Batch cocultures containing combinations of B. ovatus, S. variabile, and H. hathewayi were used to assess the ability of the species to grow together. B. ovatus, S. variabile, and H. hathewayi cultures were grown separately, anaerobically for 18 h at 37°C. The individual bacterial cultures were used to inoculate (1% [vol/vol]; equal proportions of each strain) prereduced and prewarmed (37°C) β-glucan medium in the following combinations: B. ovatus and S. variabile; B. ovatus and H. hathewayi; and B. ovatus, S. variabile, and H. hathewayi. The cocultures were sampled over a 26-h period of incubation, and 1-ml aliquots were centrifuged to collect cells and supernatant as described above. Two technical replicates and two biological replicates were carried out for each experiment.
Continuous (steady-state) culture.
Continuous culture experiments were carried out in chemostats using cocultures of B. ovatus, H. hathewayi, and S. variabile. β-glucan medium was used in chemostat experiments which were run as described previously (7). This was in order to further demonstrate the persistence of the consortium in the longer term. In brief, the reactor vessel (chemostat) contained 30 ml of medium, and flow rate of 3 ml/h was controlled by peristaltic pumps. The chemostat was inoculated with 200 μl of a mixed culture of B. ovatus, H. hathewayi, and S. variabile. The mixed culture inoculum consisted of a batch culture of the three bacterial species grown together for 18 h at 37°C in anaerobiosis in β-glucan medium. Chemostat cultures were sampled after steady-state was reached (five complete changes of chemostat volume; 7). Chemostat experiments were run three times. Measurement of optical density of cultures and harvest of cells and supernatants was as described above.
Determining CFU/ml.
To determine CFU/ml of each bacterial species in cocultures, samples were serially diluted in 10-fold steps to 10−6 in prereduced basal medium. Aliquots were plated on brain heart infusion (Difco) agar supplemented with yeast extract (5 g/liter), l-cysteine (1 g/liter), vitamin K (0.5 ml/liter), hemin (5 mg/liter), resazurin (1 mg/liter), and Tween 80 (0.1% [vol/vol]) (BHISA) for B. ovatus and H. hathewayi enumeration according to their different colony morphologies. BHISA supplemented with tetracycline (16 μg/ml) was used to determine the CFU/ml of S. variabile. Plates were incubated for 48 h, or 5 days for BHISA plates supplemented with tetracycline, at 37°C in anaerobiosis before colony enumeration and CFU/ml calculation.
Carbohydrate analysis of culture supernatants.
Oligosaccharides that accumulated in culture supernatants were detected by high-performance anion-exchange chromatography (HPAEC) as described previously (55). Identification of DP3 and DP4 oligosaccharides was by reference to standards as described previously (53). The rate of utilization of DP3 and DP4 was calculated by plotting the amount (%) of oligosaccharide remaining at each time interval, relative to uninoculated medium, in pure cultures of S. variabile and H. hathewayi and then by determining the slope of the regression curve (percentage/h).
Whole-transcriptomics analysis (RNAseq).
Procedures were the same as those used in our previous studies (7, 61, 62). In summary, RNA was extracted from bacterial cells collected from the biological replicates from each culture. Total RNA purification was performed as described previously, standardized by using total RNA at 10 μg/μl from each sample for sequencing (7, 61, 62). RNA samples were sequenced at the Otago Genomics Facility (Dunedin, New Zealand) using the Illumina HiSeq 2500 platform. For RNA-seq analysis, reads were mapped with Rockhopper version 2.0.3 (61, 62) to the reference genomes B. ovatus ATCC 8483T (NCBI RefSeq accession assembly number GCF_001314995.1, complete genome), S. variabile DSM 15176T (NCBI RefSeq accession assembly number GCF_000157955.1, 11 scaffolds), and H. hathewayi DSM 13479T (NCBI RefSeq accession assembly number GCF_000160095.1, 714 scaffolds). Reads from coculture samples were mapped onto the individual reference genomes using a highly stringent cutoff (no mismatches allowed). Reads mapping on rRNA and tRNA were excluded from subsequent analysis to avoid cross-mapping between genomes (63). The number of mapped reads for each replicate is shown in Table S1. Pearson’s correlation coefficient was calculated between biological replicates using the R package Hmisc (http://biostat.mc.vanderbilt.edu/Hmisc). EdgeR package version 3.16.5 (64, 65) was used to normalize the raw reads and determine differentially transcribed (DT) genes using the quasi-likelihood F-test. Multiple testing correction was performed using the Benjamini-Hochberg method. Transcripts with counts per million (CPM) less than 0.5 in at least 2 samples were discarded. Genes were considered significantly differentially expressed when the absolute log2 fold change value in the comparisons was >1 and the false-discovery rate (FDR) value was <0.05. In pure cultures, comparisons were performed between all time points. In coculture, pairwise comparisons between coculture and pure culture at 24 h were carried out.
Gene Ontology (GO) term enrichment analysis for each bacterial species was carried out using GOseq (7, 66). Custom-made category mapping files were prepared to link B. ovatus, S. variabile, and H. hathewayi gene names to the associated GO terms (7). Additional functional enrichment analyses were carried out using eggNOG-mapper (67, 68) based on eggNOG 4.5 orthology data (69, 70). Enzyme nomenclature (EC) numbers were retrieved from the Carbohydrate-Active enZYmes Database (http://www.cazy.org/) or from the ExplorEnz database (https://www.enzyme-database.org/).
Reverse transcription quantitative PCR (RT-qPCR) was performed to validate transcription levels of selected target genes. Primer design and optimization (Table S3), reverse transcriptase reaction, and qPCR were carried out as described previously (7, 61). Two biological replicates and six technical replicates were assessed for each condition. The transcription levels of target genes were calculated by comparative CT method (71). Transcription levels of selected target genes for each strain were confirmed by RT-qPCR analysis (Table S4).
Data availability.
Transcriptomics data are available from NCBI BioProject accession number PRJNA531520.
Supplementary Material
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Plichta DR, Juncker AS, Bertalan M, Rettedal E, Gautier L, Varela E, Manichanh C, Fouqueray C, Levenez F, Nielsen T, Doré J, Machado AMD, de Evgrafov MCR, Hansen T, Jørgensen T, Bork P, Guarner F, Pedersen O, Sommer MOA, Ehrlich SD, Sicheritz-Pontén T, Brunak S, Nielsen HB, Nielsen HB, Metagenomics of the Human Intestinal Tract (MetaHIT) Consortium. 2016. Transcriptional interactions suggest niche segregation among microorganisms in the human gut. Nat Microbiol 1:16152. doi: 10.1038/nmicrobiol.2016.152. [DOI] [PubMed] [Google Scholar]
- 2.Tannock GW, Taylor MW. 2017. Embracing the co-operative society to better understand assembly of the gut microbiota. Environ Microbiol 19:2924–2925. doi: 10.1111/1462-2920.13752. [DOI] [PubMed] [Google Scholar]
- 3.Tannock GW. 2017. Understanding the gut microbiota. John Wiley & Sons, Inc, Hoboken, NJ. [Google Scholar]
- 4.Koropatkin NM, Cameron E, Martens EC. 2012. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol 10:323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cockburn DW, Koropatkin NM. 2016. Polysaccharide degradation by the intestinal microbiota and its influence on human health and disease. J Mol Biol 428:3230–3252. doi: 10.1016/j.jmb.2016.06.021. [DOI] [PubMed] [Google Scholar]
- 6.El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. 2013. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol 11:497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- 7.Centanni M, Ferguson SA, Sims IM, Biswas A, Tannock GW. 2019. Bifidobacterium bifidum ATCC 15696 and Bifidobacterium breve 24b metabolic interaction based on 2′-O-fucosyl-lactose studied in steady-state cultures in a Freter-style chemostat. Appl Environ Microbiol 85:e02783-18. doi: 10.1128/AEM.02783-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rivière A, Gagnon M, Weckx S, Roy D, De Vuyst L. 2015. Mutual cross-feeding interactions between Bifidobacterium longum NCC2705 and Eubacterium rectale ATCC 33656 explain the bifidogenic and butyrogenic effects of arabinoxylan-oligosaccharides. Appl Environ Microbiol 81:7767–7781. doi: 10.1128/AEM.02089-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luis AS, Briggs J, Zhang X, Farnell B, Ndeh D, Labourel A, Baslé A, Cartmell A, Terrapon N, Stott K, Lowe EC, McLean R, Shearer K, Schückel J, Venditto I, Ralet M-C, Henrissat B, Martens EC, Mosimann SC, Abbott DW, Gilbert HJ. 2018. Dietary pectic glycans are degraded by coordinated enzyme pathways in human colonic Bacteroides. Nat Microbiol 3:210–219. doi: 10.1038/s41564-017-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turroni F, Milani C, Duranti S, Mahony J, van Sinderen D, Ventura M. 2018. Glycan utilization and cross-feeding activities by bifidobacteria. Trends Microbiol 26:339–350. doi: 10.1016/j.tim.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 11.Widder S, Allen RJ, Pfeiffer T, Curtis TP, Wiuf C, Sloan WT, Cordero OX, Brown SP, Momeni B, Shou W, Kettle H, Flint HJ, Haas AF, Laroche B, Kreft JU, Rainey PB, Freilich S, Schuster S, Milferstedt K, Van Der Meer JR, Grobkopf T, Huisman J, Free A, Picioreanu C, Quince C, Klapper I, Labarthe S, Smets BF, Wang H, Soyer OS, Isaac Newton Institute Fellows. 2016. Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J 10:2557–2568. doi: 10.1038/ismej.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burton RA, Fincher GB. 2014. Evolution and development of cell walls in cereal grains. Front Plant Sci 5:456. doi: 10.3389/fpls.2014.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich SD, Wang J, MetaHIT Consortium. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI. 2011. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 108:6252–6257. doi: 10.1073/pnas.1102938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cato EP, Johnson JL. 1976. Reinstatement of species rank for Bacteroides fragilis, B. ovatus, B. distasonis, B. thetaiotaomicron, and B. vulgatus: designation of neotype strains for Bacteroides fragilis (Veillon and Zuber) Castellani and Chalmers and Bacteroides thetaiotaomicron (Distaso) Castellani and Chalmers. Int J Syst Bacteriol 26:230–237. doi: 10.1099/00207713-26-2-230. [DOI] [Google Scholar]
- 16.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wexler AG, Goodman AL. 2017. An insider’s perspective: Bacteroides as a window into the microbiome. Nat Microbiol 2:17026. doi: 10.1038/nmicrobiol.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salyers AA, Vercellotti JR, West SE, Wilkins TD. 1977. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl Environ Microbiol 33:319–322. doi: 10.1128/AEM.33.2.319-322.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grondin JM, Tamura K, Déjean G, Abbott DW, Brumer H. 2017. Polysaccharide utilization loci: fueling microbial communities. J Bacteriol 199:e00860-16. doi: 10.1128/JB.00860-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura K, Foley MH, Gardill BR, Dejean G, Schnizlein M, Bahr CME, Creagh AL, van Petegem F, Koropatkin NM, Brumer H. 2019. Surface glycan-binding proteins are essential for cereal beta-glucan utilization by the human gut symbiont Bacteroides ovatus. Cell Mol Life Sci 76:4319–4340. doi: 10.1007/s00018-019-03115-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamura K, Hemsworth GR, Déjean G, Rogers TE, Pudlo NA, Urs K, Jain N, Davies GJ, Martens EC, Brumer H. 2017. Molecular mechanism by which prominent human gut Bacteroidetes utilize mixed-linkage beta-glucans, major health-promoting cereal polysaccharides. Cell Rep 21:417–430. doi: 10.1016/j.celrep.2017.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salyers AA, West SE, Vercellotti JR, Wilkins TD. 1977. Fermentation of mucins and plant polysaccharides by anaerobic bacteria from the human colon. Appl Environ Microbiol 34:529–533. doi: 10.1128/AEM.34.5.529-533.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, Abbott DW, Henrissat B, Gilbert HJ, Bolam DN, Gordon JI. 2011. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol 9:e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larsbrink J, Rogers TE, Hemsworth GR, McKee LS, Tauzin AS, Spadiut O, Klinter S, Pudlo NA, Urs K, Koropatkin NM, Creagh AL, Haynes CA, Kelly AG, Cederholm SN, Davies GJ, Martens EC, Brumer H. 2014. A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506:498–502. doi: 10.1038/nature12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang M, Chekan JR, Dodd D, Hong P-Y, Radlinski L, Revindran V, Nair SK, Mackie RI, Cann I. 2014. Xylan utilization in human gut commensal bacteria is orchestrated by unique modular organization of polysaccharide-degrading enzymes. Proc Natl Acad Sci U S A 111:E3708–E3717. doi: 10.1073/pnas.1406156111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuskin F, Lowe EC, Temple MJ, Zhu Y, Cameron E, Pudlo N, Porter NT, Urs K, Thompson AJ, Cartmell A, Rogowski A, Hamilton BS, Chen R, Tolbert TJ, Piens K, Bracke D, Vervecken W, Hakki Z, Speciale G, Munōz-Munōz JL, Day A, Peña MJ, McLean R, Suits MD, Boraston AB, Atherly T, Ziemer CJ, Williams SJ, Davies GJ, Abbott DW, Martens EC, Gilbert HJ. 2015. Human gut Bacteroidetes can utilize yeast mannan through a selfish mechanism. Nature 517:165–169. doi: 10.1038/nature13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raghavan V, Groisman EA. 2015. Species-specific dynamic responses of gut bacteria to a mammalian glycan. J Bacteriol 197:1538–1548. doi: 10.1128/JB.00010-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rogowski A, Briggs JA, Mortimer JC, Tryfona T, Terrapon N, Lowe EC, Baslé A, Morland C, Day AM, Zheng H, Rogers TE, Thompson P, Hawkins AR, Yadav MP, Henrissat B, Martens EC, Dupree P, Gilbert HJ, Bolam DN. 2015. Glycan complexity dictates microbial resource allocation in the large intestine. Nat Commun 6:7481. doi: 10.1038/ncomms8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rakoff-Nahoum S, Coyne MJ, Comstock LE. 2014. An ecological network of polysaccharide utilization among human intestinal symbionts. Curr Biol 24:40–49. doi: 10.1016/j.cub.2013.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmstrøm K, Collins MD, Møller T, Falsen E, Lawson PA. 2004. Subdoligranulum variabile gen. nov., sp. nov. from human feces. Anaerobe 10:197–203. doi: 10.1016/j.anaerobe.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Steer T, Collins MD, Gibson GR, Hippe H, Lawson PA. 2001. Clostridium hathewayi sp. nov., from human faeces. Syst Appl Microbiol 24:353–357. doi: 10.1078/0723-2020-00044. [DOI] [PubMed] [Google Scholar]
- 32.Kaur S, Yawar M, Kumar PA, Suresh K. 2014. Hungatella effluvii gen. nov., sp. nov. an obligately anaerobic bacterium isolated from an effluent treatment plant, and reclassification of Clostridium hathewayi as Hungatella hathewayi gen nov., comb. nov. Int J Syst Evol Microbiol 64:710–718. doi: 10.1099/ijs.0.056986-0. [DOI] [PubMed] [Google Scholar]
- 33.White BA, Lamed R, Bayer EA, Flint HJ. 2014. Biomass utilization by gut microbiomes. Annu Rev Microbiol 68:279–296. doi: 10.1146/annurev-micro-092412-155618. [DOI] [PubMed] [Google Scholar]
- 34.Moran MA, Satinsky B, Gifford SM, Luo H, Rivers A, Chan L-K, Meng J, Durham BP, Shen C, Varaljay VA, Smith CB, Yager PL, Hopkinson BM. 2013. Sizing up metatranscriptomics. ISME J 7:237–243. doi: 10.1038/ismej.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leth ML, Ejby M, Workman C, Ewald DA, Pedersen SS, Sternberg C, Bahl MI, Licht TR, Aachmann FL, Westereng B, Abou Hachem M. 2018. Differential bacterial capture and transport preferences facilitate co-growth on dietary xylan in the human gut. Nat Microbiol 3:570–580. doi: 10.1038/s41564-018-0132-8. [DOI] [PubMed] [Google Scholar]
- 36.Degnan PH, Taga ME, Goodman AL. 2014. Vitamin B12 as a modulator of gut microbial ecology. Cell Metab 20:769–778. doi: 10.1016/j.cmet.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wexler AG, Schofield WB, Degnan PH, Folta-Stogniew E, Barry NA, Goodman AL. 2018. Human gut Bacteroides capture vitamin B12 via cell surface-exposed lipoproteins. Elife 7:e37138. doi: 10.7554/eLife.37138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Degnan PH, Barry NA, Mok KC, Taga ME, Goodman AL. 2014. Human gut microbes use multiple transporters to distinguish vitamin B12 analogs and compete in the gut. Cell Host Microbe 15:47–57. doi: 10.1016/j.chom.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zafar H, Saier MH. 2018. Comparative genomics of transport proteins in seven Bacteroides species. PLoS One 13:e0208151. doi: 10.1371/journal.pone.0208151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Starosta AL, Lassak J, Jung K, Wilson DN. 2014. The bacterial translation stress response. FEMS Microbiol Rev 38:1172–1201. doi: 10.1111/1574-6976.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boutte CC, Crosson S. 2013. Bacterial lifestyle shapes stringent response activation. Trends Microbiol 21:174–180. doi: 10.1016/j.tim.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Atkinson GC, Tenson T, Hauryliuk V. 2011. The RelA/SpoT homolog (RSH) superfamily: distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLoS One 6:e23479. doi: 10.1371/journal.pone.0023479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu Rev Microbiol 62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- 44.Kint C, Verstraeten N, Hofkens J, Fauvart M, Michiels J. 2014. Bacterial Obg proteins: GTPases at the nexus of protein and DNA synthesis. Crit Rev Microbiol 40:207–224. doi: 10.3109/1040841X.2013.776510. [DOI] [PubMed] [Google Scholar]
- 45.Krell T, Lacal J, Busch A, Silva-Jiménez H, Guazzaroni M-E, Ramos JL. 2010. Bacterial sensor kinases: diversity in the recognition of environmental signals. Annu Rev Microbiol 64:539–559. doi: 10.1146/annurev.micro.112408.134054. [DOI] [PubMed] [Google Scholar]
- 46.Xavier KB, Bassler BL. 2003. LuxS quorum sensing: more than just a numbers game. Curr Opin Microbiol 6:191–197. doi: 10.1016/s1369-5274(03)00028-6. [DOI] [PubMed] [Google Scholar]
- 47.Pereira CS, Thompson JA, Xavier KB. 2013. AI-2-mediated signalling in bacteria. FEMS Microbiol Rev 37:156–181. doi: 10.1111/j.1574-6976.2012.00345.x. [DOI] [PubMed] [Google Scholar]
- 48.Braaten JT, Wood PJ, Scott FW, Wolynetz MS, Lowe MK, Bradley-White P, Collins MW. 1994. Oat β-glucan reduces blood cholesterol concentration in hypercholesterolemic subjects. Eur J Clin Nutr 48:465–474. [PubMed] [Google Scholar]
- 49.Braaten JT, Scott FW, Wood PJ, Riedel KD, Wolynetz MS, Brulé D, Collins MW. 1994. High beta-glucan oat bran and oat gum reduce postprandial blood glucose and insulin in subjects with and without type 2 diabetes. Diabet Med 11:312–318. doi: 10.1111/j.1464-5491.1994.tb00277.x. [DOI] [PubMed] [Google Scholar]
- 50.Davidson MH, Dugan LD, Burns JH, Bova J, Story K, Drennan KB. 1991. The hypocholesterolemic effects of β-glucan in oatmeal and oat bran: a dose-controlled study. JAMA 265:1833–1839. doi: 10.1001/jama.1991.03460140061027. [DOI] [PubMed] [Google Scholar]
- 51.Jenkins DJA, Kendall CWC, Vuksan V. 2000. Viscous fibers, health claims, and strategies to reduce cardiovascular disease risk. Am J Clin Nutr 71:401–402. doi: 10.1093/ajcn/71.2.401. [DOI] [PubMed] [Google Scholar]
- 52.Tappy L, Gügolz E, Würsch P. 1996. Effects of breakfast cereals containing various amounts of beta-glucan fibers on plasma glucose and insulin responses in NIDDM subjects. Diabetes Care 19:831–834. doi: 10.2337/diacare.19.8.831. [DOI] [PubMed] [Google Scholar]
- 53.Lawley B, Sims IM, Tannock GW. 2013. Whole-transcriptome shotgun sequencing (RNA-seq) screen reveals upregulation of cellobiose and motility operons of Lactobacillus ruminis L5 during growth on tetrasaccharides derived from barley β-glucan. Appl Environ Microbiol 79:5661–5669. doi: 10.1128/AEM.01887-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Snart J, Bibiloni R, Grayson T, Lay C, Zhang H, Allison GE, Laverdiere JK, Temelli F, Vasanthan T, Bell R, Tannock GW. 2006. Supplementation of the diet with high-viscosity beta-glucan results in enrichment for lactobacilli in the rat cecum. Appl Environ Microbiol 72:1925–1931. doi: 10.1128/AEM.72.3.1925-1931.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Centanni M, Hutchison JC, Carnachan SM, Daines AM, Kelly WJ, Tannock GW, Sims IM. 2017. Differential growth of bowel commensal Bacteroides species on plant xylans of differing structural complexity. Carbohydr Polym 157:1374–1382. doi: 10.1016/j.carbpol.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 56.Liu Y, Heath A-L, Galland B, Rehrer N, Drummond L, Wu X-Y, Bell TJ, Lawley B, Sims IM, Tannock GW. 2019. Prioritization of substrate use by a co-culture of five species of gut bacteria fed mixtures of arabinoxylan, xyloglucan, β-glucan, and pectin. Appl Environ Microbiol 86:e01905-19. doi: 10.1128/AEM.01905-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vrancken G, Gregory AC, Huys GRB, Faust K, Raes J. 2019. Synthetic ecology of the human gut microbiota. Nat Rev Microbiol 17:754–763. doi: 10.1038/s41579-019-0264-8. [DOI] [PubMed] [Google Scholar]
- 58.Walker AW, Duncan SH, Harmsen HJM, Holtrop G, Welling GW, Flint HJ. 2008. The species composition of the human intestinal microbiota differs between particle-associated and liquid phase communities. Environ Microbiol 10:3275–3283. doi: 10.1111/j.1462-2920.2008.01717.x. [DOI] [PubMed] [Google Scholar]
- 59.Patnode ML, Beller ZW, Han ND, Cheng J, Peters SL, Terrapon N, Henrissat B, Le Gall S, Saulnier L, Hayashi DK, Meynier A, Vinoy S, Giannone RJ, Hettich RL, Gordon JI. 2019. Interspecies competition impacts targeted manipulation of human gut bacteria by fiber-derived glycans. Cell 179:59–73.e13. doi: 10.1016/j.cell.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morgan KR, Ofman DJ. 1998. Glucagel, a gelling β-glucan from barley. Cereal Chem J 75:879–881. doi: 10.1094/CCHEM.1998.75.6.879. [DOI] [Google Scholar]
- 61.Centanni M, Lawley B, Butts CA, Roy NC, Lee J, Kelly WJ, Tannock GW. 2018. Bifidobacterium pseudolongum in the ceca of rats fed Hi-Maize starch has characteristics of a keystone species in bifidobacterial blooms. Appl Environ Microbiol 84:e00547-18. doi: 10.1128/AEM.00547-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawley B, Centanni M, Watanabe J, Sims I, Carnachan S, Broadbent R, Lee PS, Wong KH, Tannock GW. 2018. Tuf gene sequence variation in Bifidobacterium longum subspecies infantis detected in the fecal microbiota of Chinese infants. Appl Environ Microbiol 84:e00336-18. doi: 10.1128/AEM.00336-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tjaden B. 2015. De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol 16:1. doi: 10.1186/s13059-014-0572-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK, Tjaden B. 2013. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res 41:e140. doi: 10.1093/nar/gkt444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li B, Ibrahim M, Ge M, Cui Z, Sun G, Xu F, Kube M. 2014. Transcriptome analysis of Acidovorax avenae subsp. avenae cultivated in vivo and co-culture with Burkholderia seminalis. Sci Rep 4:5698. doi: 10.1038/srep05698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCarthy DJ, Chen Y, Smyth GK. 2012. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Young MD, Wakefield MJ, Smyth GK, Oshlack A. 2010. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, Bork P. 2017. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol Evol 34:2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, Jensen LJ, von Mering C, Bork P. 2016. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptomics data are available from NCBI BioProject accession number PRJNA531520.