

Abstract
Neuromyelitis optica (NMO)/NMO spectrum disorder (NMOSD) is a chronic, recurrent, antibody-mediated, inflammatory demyelinating disease of the central nervous system, characterized by optic neuritis and transverse myelitis. The binding of NMO-IgG with astrocytic aquaporin-4 (AQP4) functions directly in the pathogenesis of >60% of NMOSD patients, and causes astrocyte loss, secondary inflammatory infiltration, demyelination, and neuron death, potentially leading to paralysis and blindness. Current treatment options, including immunosuppressive agents, plasma exchange, and B-cell depletion, are based on small retrospective case series and open-label studies. It is noteworthy that monoclonal antibody (mAb) therapy is a better option for autoimmune diseases due to its high efficacy and tolerability. Although the pathophysiological mechanisms of NMOSD remain unknown, increasingly, therapeutic studies have focused on mAbs, which target B cell depletion, complement and inflammation cascade inactivation, blood-brain-barrier protection, and blockade of NMO-IgG-AQP4 binding. Here, we review the targets, characteristics, mechanisms of action, development, and potential efficacy of mAb trials in NMOSD, including preclinical and experimental investigations.
Keywords: Neuromyelitis optica spectrum disorders, Monoclonal antibody, AQP4-IgG, Astrocyte, Central nervous system
Introduction
Neuromyelitis optica (NMO)/NMO spectrum disorder (NMOSD) is a devastating autoimmune inflammatory disease of the central nervous system (CNS), with a predilection for causing lesions in the optic nerve and spinal cord [1–3]. NMO was previously considered a subtype of multiple sclerosis (MS); however, the discovery of an autoantibody against aquaporin-4 (AQP4), the dominant water channel which is strongly expressed on astrocyte end-feet, distinguishes NMO from MS and helps to establish a diagnosis of NMO [1, 4]. It is pertinent to distinguish between MNO and MS at diagnosis, as some MS therapies, such as natalizumab and interferon-β, aggravate rather than treat NMO [5, 6]. Autoantibodies against AQP4 (AQP4–IgG), which are present in the large majority of NMO patients, function directly in the pathogenesis of disease [7, 8]. In 2015, the Wingerchuck criteria, an internationally recognized means of diagnosing NMO, were updated. AQP4-IgG plays a crucial role in AQP4-IgG-positive NMOSD patients. The main features include clinical evidence, or MRI findings, associated with the optic nerve, spinal cord, area postrema, and other brainstem, diencephalic, or cerebral presentations. In seronegative patients, a diagnosis of NMOSD requires more stringent clinical and MRI criteria [9]. Specific antibodies against myelin oligodendrocyte glycoprotein (MOG–IgG) on the outermost surface of myelin sheaths, are present in ~40% of all AQP4–IgG seronegative NMO patients [10–13], and a few NMO patients are positive for both antibodies [14, 15]. MOG syndrome is a disorder distinct from AQP4–IgG-positive NMOSD. It has been reported that the treatment response to mAb therapy such as rituximab in MOG syndrome is not as good as in AQP4–IgG-positive NMOSD [16]. Previous studies have shown that MOG–IgG also plays an important role in the pathogenesis of seronegative NMOSD, demonstrating that MOG–IgG-positive patients have a more favorable clinical outcome than patients with AQP4–IgG [17–21]. At present, evidence from the laboratory and clinic suggests that AQP4–IgG is pathogenic in NMO. Thus, research on NMO treatment is mainly focused on the series of pathological inflammatory reactions caused by the binding of AQP4-IgG to AQP4.
Female predominance in NMO is common, especially in AQP4-IgG-positive patients; the female-to-male ratio is 5–9:1 [22–24]. The median age of onset is 35–37 years, but the first attack may occur in early childhood or in the elderly [23, 25]. Furthermore, 80%–90% of NMO patients experience a relapse during disease progression rather than a monophasic course, and most patients follow a course of early incremental disability [26, 27]. One reason for these pathogenic events is that the AQP4-IgG titer has significant clinical and immunological implications, as a higher serum titer means there is more AQP4-IgG in the CNS [28]. Therefore, studies on NMO treatment have focused on blocking the binding of AQP4–IgG to AQP4, using medications such as aquaporumab [29–32]. At present, a few drugs are available for the treatment of NMO, including general immunosuppressive agents, plasma exchange, and B-cell depletion, targeting AQP4-IgG and inflammatory reactions [33–35]. Unfortunately, even with the application of current treatment options, NMO patients frequently suffer from paralysis and blindness, and even die [36, 37]. Therefore, improved therapies for NMO are needed to ameliorate acute attacks and prevent exacerbations.
Recently, many targeted therapies with monoclonal antibodies (mAbs) have been introduced as a treatment strategy for autoimmune diseases, demonstrating a high rate of efficacy and tolerability [38]. Some treatments are currently being investigated in clinical trials for the treatment of NMO. For example, rituximab, eculizumab, tocilizumab, and bevacizumab are all effective for the treatment of NMO [33, 39, 40]. Based on this evidence, we review specific mAbs for the treatment of NMO based on the pathogenesis of the disease.
Attempts at mAb Therapy for NMO
At present, >70 antibody drugs have obtained marketing approval for clinical use. In 1975, Kohler and Milstein created the hybridoma technology for lymphocytes by which specific binding of mAbs to an antigenic site can be obtained [41]. Many therapeutic antibodies have been obtained by mouse hybridoma technology. However, mouse antibodies are recognized as foreign proteins by humans and induce human anti-mouse antibody (HAMA), limiting their clinical application [42, 43]. To avoid this problem, mouse sequences have been partly exchanged with the homologous human sequences by genetic engineering technology (Fig. 1). The chimeric antibodies have mouse constant domains replaced by human constant domains, as in rituximab; the humanized antibodies have mouse variable region frameworks and constant domains that have been replaced by the homologous human sequences [44]. Only three complementarity-determining regions of humanized antibodies are mouse sequences; the mouse sequences have been further reduced compared to chimeric antibodies, as in eculizumab [43, 44]. These engineered mouse antibodies have occupied the major proportion of therapeutic antibody approvals over several decades. In fact, the first completely human mAb, adalimumab, was obtained by phage display in 1994 [45]. mAbs are a growing group of therapeutic proteins, mainly focused on the treatment of cancer and autoimmune diseases.
Fig. 1.
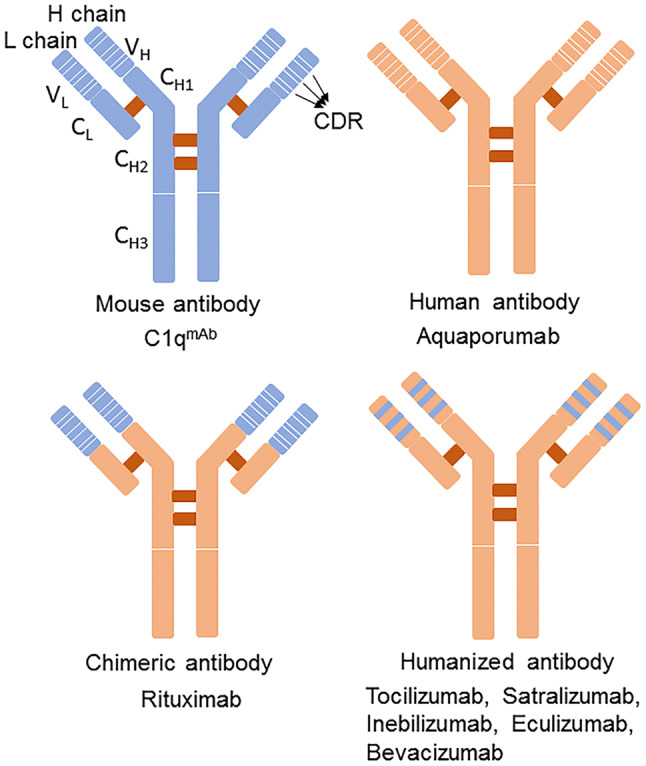
Components and classification of monoclonal antibodies. Based on the mouse-derived to human-derived ratio, mAbs can be divided into four categories. The mouse sequences are partly exchanged with the homologous human sequences through genetic engineering. In chimeric antibodies, mouse constant domains are replaced with human constant domains; the humanized antibodies have mouse variable-region frameworks and the constant domains are replaced by homologous human sequences, further reducing the mouse sequences. The higher the proportion of human-derived components, the lower the risk of HAMA reactions. H chain, heavy chain; L chain, light chain; CDR, complementarity determining region; CH, heavy chain constant region; CL, light chain constant region; VH, heavy chain variable region; VL, light chain variable region; C1qmAb, C1q-targeted monoclonal antibody.
Though there have yet to be any approved mAbs for NMO, several that are under development are described in this review. For example, rituximab is widely used as a preventive therapy in NMO [46]; ublituximab, another anti-CD20+ mAb, exhibits greater antibody-dependent cell-mediated cytotoxicity (ADCC) activity than rituximab [47]; and eculizumab significantly reduced the annualized relapse rate (ARR), and improved the neurological disability in an NMO open-label trial [48]. Tocilizumab also reduces the ARR, and gradually ameliorates intractable pain and general fatigue in NMO patients [49], while satralizumab, an extended version of tocilizumab, had a greater duration of action than tocilizumab in a phase III clinical trial. Lastly, bevacizumab [anti-VEGF (vascular endothelial growth factor)-A] is a safe add-on therapy for NMO patients with an acute relapse [50]; while non-pathogenic aquaporumab may also be a promising treatment for NMO, though there has been no clinical report as yet [40]. A schematic illustration of the mechanisms of action of these drugs is shown in Fig. 2 and information on these drugs for NMO is listed in Table 1 (according to www.clinicaltrials.gov).
Fig. 2.

Pharmacological targets of antibodies in the mechanisms of NMO pathogenesis. The entry of AQP4-IgG produced by plasma cells into the CNS through the BBB plays an important role in the pathogenesis of NMO. Tocilizumab and satralizumab specifically bind to interleukin (IL)-6 receptors and block the IL-6 signaling pathway and reduce plasma cells derived from B cells. Inebilizumab specifically binds to CD19 and rituximab specifically binds to CD20, deleting B cells that inhibit the B/T-cell interaction, decreasing pro-inflammatory cytokines, increasing regulatory T cells, and modulating the T-cell compartment. Aquaporumab specifically binds to astrocyte AQP4 to block the binding of AQP4-IgG and AQP4, thus decreasing CDC and ADCC; C1qmAb specifically binds to complement C1q to block the classical complement pathway. Eculizumab specifically binds to complement C5 and blocks all terminal pathways of complement activation (classical, lectin, and alternative) and prevents the formation of MAC. Bevacizumab specifically binds to VEGF-A, which is beneficial for the BBB and decreases the entry of AQP4–IgG and inflammatory cells into the CNS, especially in acute NMO exacerbations. mAbs specifically block the pathological processes of NMO, thereby reducing astrocyte lysis, oligodendrocyte loss, demyelination, and neuron loss. IL-6, interleukin-6; IL-6R, IL-6 receptor; VEGF-A, vascular endothelial growth factor A; VEGFR2, VEGF receptor 2; AQP4, aquaporin-4; CDC, complement-dependent cytotoxicity; MAC, membrane attack complex; NMO, neuromyelitis optica; BBB, blood brain barrier; CNS, central nervous system.
Table 1.
All clinical trials on the treatment of NMO with monoclonal antibodies (www.clinicaltrials.gov).
| Intervention | Other name | Mode of action | NCT number | Patients | Type of trial | Status | Purpose of trial | Adverse events | |
|---|---|---|---|---|---|---|---|---|---|
| Chimeric antibody | Rituximab | Rituxan | Depletes CD20+ B cells | 00501748 | 14 | I | Completed | Safety and tolerability in NMO | Immunosuppression; immunogenicity; PML; hepatitis B virus |
| Chimeric antibody | Rituximab | RediTux® | Depletes CD20+ B cells | 03002038 | 76 | II, III | Completed | Comparison of efficacy and safety between azathioprine and rituximab in NMOSD | |
| Humanized antibody | Ublituximab | LFB-R603 | Depletes CD20+ B cells | 02276963 | 5 | I | Active, not recruiting | Safety and adverse events as add-on therapy in acute NMO/NMOSD; | Immunosuppression; immunogenicity; diarrhea, constipation, fatigue; neutropenia |
| Humanized antibody | Inebilizumab | MEDI-551 | Depletes CD19+ B cells | 02200770 | 231 | II, III | Completed | Efficacy and safety in relapsing NMO/NMOSD | Urinary tract infection, arthralgia, headache, hypoesthesia, and eye pain |
| Humanized antibody | Eculizumab | Soliris | Inhibits C5 | 02003144 | 119 | III | Active, not recruiting | Long-term safety in relapsing NMO/NMOSD | Headache; immunosuppression; immunogenicity; infection (meningococcus) |
| Humanized antibody | Eculizumab | Soliris | Inhibits C5 | 01892345 | 143 | III | Completed | Efficacy and safety in relapsing NMO/NMOSD | |
| Humanized antibody | Eculizumab | Soliris | Inhibits C5 | 00904826 | 14 | I, II | Completed | Long-term safety, efficacy; pharmacodynamics and pharmacokinetic in NMO/NMOSD | |
| Humanized antibody | Tocilizumab | ACTEMRA® | Anti-IL-6 receptor | 03350633 | 118 | II, III | Active, not recruiting | Efficacy and safety in relapsing NMO/NMOSD | Immunosuppression; immunogenicity; cholesterol elevation; leukocytopenia; lymphocytopenia; anemia; deep venous thrombosis; autoimmune neurological disorders |
| Humanized antibody | Tocilizumab | ACTEMRA® | Anti-IL-6 receptor | 03062579 | 10 | I, II | Active, not recruiting | Efficacy and safety as monotherapy in relapsing NMO/NMOSD | |
| Humanized antibody | Satralizumab | SA237 | Anti-IL-6 receptor | 02028884 | 70 | III | Completed | Efficacy, safety, pharmacodynamic, pharmacokinetic and immunogenic profiles as add-on therapy in NMO/NMOSD | No clinical trials or case reports on adverse events |
| Humanized antibody | Satralizumab | SA237 | Anti-IL-6 receptor | 02073279 | 95 | III | Active, not recruiting | Efficacy, safety, pharmacodynamic, pharmacokinetic and immunogenic profiles as monotherapy in NMO/NMOSD | |
| Humanized antibody | Bevacizumab | Avastin | VEGF inhibitor | 01777412 | 10 | Ib | Completed | Tolerability, safety and efficacy as add-on therapy in acute NMO/NMOSD | Thromboembolic disease; gastrointestinal hemorrhage |
NMO, neuromyelitis optica; NMOSD, NMO spectrum disorder; IL-6, interleukin-6; VEGF, vascular endothelial growth factor; PML, progressive multifocal leukoencephalopathy; NCT number, Clinical trial registry number.
Current Clinical Trials of mAb Therapy for NMO
Rituximab
Rituximab, a chimeric monoclonal IgG1 that specifically binds to CD20 on B cells, has mouse constant domains replaced by human constant domains. Rituximab has been used as preventive therapy in NMO for years [46, 51]. B cells are essential in the pathogenesis of NMO [52, 53]. In most NMO patients, AQP4-IgG is produced by plasmablasts, a subpopulation of B cells [8]. In the CNS, with AQP4-IgG binding, AQP4 initiates an inflammatory cascade that ultimately leads to demyelination and neuron death [54, 55]. Rituximab effectively depletes CD20+ B cells in peripheral blood, while AQP4-IgG is not consistently decreased after repeated courses of rituximab [56, 57]. Other mechanisms beyond antibody reduction may contribute to the clinical stabilization, including inhibition of B/T-cell interactions, decreasing pro-inflammatory cytokines, increasing regulatory T cells, and modulating the T-cell compartment [58–60].
Rituximab is usually administered intravenously at 375 mg/m2 once weekly for 4 weeks and 1000 mg twice at a 2-week interval. Most patients remain B-cell-deficient for 6 months, so an additional dose can be given 6–9 months after the initial treatment. [33]. In a trial in China, Yang et al. first reported that low-dose rituximab (100 mg intravenously infused once weekly for 3 weeks and re-infused at the same dose when CD19+B cells exceed 1%) depletes B cells and maintains low B-cell counts. In their study, of the five treated NMO patients, none experienced a relapse during the 1-year follow-up [61]. During the past decade, rituximab has been used to treat immune-mediated neurological disorders over a long trial period and is well tolerated [62–65]. In 2005, Cree et al. first reported a prospective open-label rituximab study of eight patients with severe NMO. The results showed that treatment was well tolerated with a significant ARR reduction (2.6 to 0) and disability improvement [66].
There have been, however, several trials of rituximab for NMO in which the mean ARR significantly declined [67, 68]. Rituximab is an effective and well-tolerated treatment for refractory NMO with the ARR defined by at least one relapse during immunosuppressive therapy [69, 70]. In 2013, a 5-year follow-up study of rituximab in NMO patients showed that 87% of patients exhibited a clear reduction in ARR from 2.4 to 0.3, and 60% of overall patients were relapse-free after treatment [69]. Similarly, in an open-label clinical trial for the treatment of NMO from September 2015 to December 2016 (NCT03002038, phase II, III), rituximab was shown to be more effective (ARR decreased from 1.30 to 0.21) than azathioprine (ARR decreased from 1 to 0.51) [71].
The most serious adverse events with rituximab are immunosuppressive in nature, including infections and HAMA reactions [72]. Progressive multifocal leukoencephalopathy (PML) has also been reported in rituximab-treated patients, although the pathophysiology of PML after rituximab therapy remains uncertain. Regardless, the effect of rituximab on T-cell function is recognized as a potential mechanism of JC virus reactivation [73, 74]. To date, there have been no reports of PML with rituximab in NMO patients. Rituximab treatment, however, may reactivate hepatitis B virus [75]. Therefore, it is desirable to check for the hepatitis B virus antigen before rituximab treatment.
Ublituximab (LFB-R603, TGT-1101, TGTX-1101)
Ublituximab is a glycol-engineered chimeric IgG1 targeting CD20 with a low fucose content of oligosaccharides [76]. ADCC activity is dependent on the fucose content [77]. Ublituximab designed with low fucose content exhibits high-affinity binding to FcγRIIIa that increases ADCC activity 100 times more than rituximab [47]. A recent clinical trial (NCT02276963, phase I) evaluated the safety of ublituximab as an add-on therapy in acute NMO/NMOSD. Patients were intravenously infused with 450 mg ublituximab once on day 1, plus 1000 mg glucocorticoids daily on days 1-5. The primary outcome measure was the number of participants with adverse events over a 90-day period. The major adverse events of ublituximab were mostly immunosuppressive, while common side-effects included diarrhea, constipation, fatigue, and neutropenia.
Inebilizumab (MEDI-551)
Inebilizumab, a humanized IgG1 mAb against CD19, is expressed in a wide range of B cells at various stages [78]. While rituximab depletes mature naïve and memory B cells [51], the pre-B cell developmental stages and plasma cells do not express CD20 on the surface. CD19 has broader expression during B-cell development than CD20 in that it is expressed at the pre-B cell stage and in a proportion of plasma cells [58, 79]. Plasmablasts are responsible for producing AQP4-IgG. In addition, CD19 is selectively expressed on B cells, but CD20 is also expressed on T cells [80, 81]. Anti-CD19 therapy is a promising treatment for autoimmune diseases [82]. In sum, inebilizumab is expected to more efficiently deplete circulating plasmablasts than other B-cell-targeting mAbs, and is believed to be effective for B-cell-related autoimmune diseases [83]. The results from a phase II/III trial (NCT02200770) of inebilizumab in NMO showed that 21 (12%) of 174 participants with inebilizumab treatment had an attack versus 22 (39%) of 56 participants with placebo treatment. Inebilizumab reduced the risk of an NMOSD attack compared with placebo [84].
The major serious adverse events of inebilizumab are likely immune suppression. The depletion of CD19+ B cells offers potential advantages in efficacy, but a potentially high risk of infectious complications [85]. Urinary tract infection, arthralgia, headache, hypoesthesia, and eye pain are nominally more frequent with inebilizumab [84].
Eculizumab (Soliris)
Eculizumab is a recombinant, humanized, monoclonal IgG2 antibody in the complement system [86]. The complement system is essential in the pathogenesis of NMO, as AQP4-IgG binding to AQP4 activates complement, which then amplifies the inflammatory response and is formative in the membrane attack complex (MAC) in astrocyte membranes, cell lysis, BBB disruption, demyelination, and neuron death [7, 54, 87–89]. C5 can be cleaved into C5a and C5b in all pathways of complement activation (classical, lectin, and alternative), which initiates the terminal complement cascade [90]. Therefore, C5 is a crucial target in NMO treatment [91]. Currently, eculizumab is approved for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome [86, 92]. Though still at an experimental stage, eculizumab is also expected to be applied to NMO.
In an open-label trial (NCT00904826, phase I, II), 14 seropositive NMO patients were intravenously infused with 600 mg eculizumab weekly for four weeks, 900 mg in the fifth week, and then 900 mg every two weeks for 48 weeks. Serum complement activation decreased significantly during the treatment. Twelve (85%) patients were relapse-free during the first year of treatment, and demonstrated improved neurological disability including visual acuity. However, one patient had meningococcal sepsis two months after the first eculizumab infusion [48]. Recently, the PREVENT double-blind clinical trial (NCT01892345, phase III) showed that 97.9% of AQP4-positive NMOSD patients treated with eculizumab did not relapse within 48 weeks compared with 63.2% who received placebo [93]. Another placebo-controlled clinical trial (NCT02003144, phase III) is actively investigating the long-term safety and efficacy of eculizumab in relapsing NMO over a 4-year period.
The most frequent adverse event related to eculizumab is headache. The other major adverse events are immunosuppressive and immunogenic, including the HAMA reaction and infections, as seen with other mAbs. Although eculizumab is a humanized monoclonal IgG in which the mouse sequence is further reduced, long-term use in patients may continue to HAMA, therefore limiting its clinical application. Because eculizumab inhibits the terminal complex C5b-9 (MAC), the risk of infection by meningococcus (polysaccharide-encapsulated bacteria) increases. As described previously, there have been reports of NMO patients presenting with meningococcal sepsis when treated with eculizumab. Thus, it is desirable that patients receive meningococcal vaccination 2 weeks prior to eculizumab therapy.
Tocilizumab (ACTEMRA®)
Tocilizumab, a humanized mAb against the IL-6R, has demonstrated efficacy in the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, and Castleman disease [94]. The IL-6/IL-6R signaling pathway plays a variety of roles in the pathogenesis of NMO. IL-6 promotes the survival of plasmablasts and the production of AQP4-IgG, while serum and cerebrospinal fluid IL-6 and the soluble IL-6R level are particularly elevated during NMO relapse [95–97]. Plasmablasts increase in the peripheral blood of NMO, and anti-IL6R antibody reduces the survival of plasmablasts. Tocilizumab is intravenously administered 8 mg/kg once every 4 weeks, or 6 weeks if possible [95].
Several case reports have shown that monthly intravenous infusions of tocilizumab in NMO patients cause a rapid reduction in the number of plasmablasts and the AQP4-IgG titer, and significantly reduce the ARR [39, 49, 98–100]. Conversely, in a Japanese trial of tocilizumab for AQP4-IgG-seropositive NMO, the ARR of patients with immunotherapy including azathioprine and corticosteroids still averaged 2.9. Yet, when tocilizumab was implemented as an add-on therapy in seven patients, the ARR dropped to 0.4 and five of the seven patients remained relapse-free during treatment [39].
In a long-term retrospective observational study between December 2010 and February 2015, patients with highly active AQP4-IgG-seropositive NMO in whom prior medications including rituximab had failed, were intravenously infused with tocilizumab as monotherapy for four years. The ARR significantly decreased from 4.0 before tocilizumab to 0.4 during treatment [101]. Another trial also reported that the drug significantly decreased the ARR in three patients with aggressive NMO after failure of anti-CD20 therapy [102]. Specifically, the majority of patients with NMO experienced severe and intractable pain, even when taking pain medications [103]. Current studies show that tocilizumab ameliorates gradually intractable pain and general fatigue: 50% of patients were completely free of pain. It is also possible that epidural administration of tocilizumab reduces radicular pain in patients with lumbar spinal stenosis [104]. Tocilizumab may be a promising second-line alternative treatment for NMO. A clinical trial (NCT03062579, Phase I, II) of NMO/NMOSD patients compared the ARR before and one year after initial tocilizumab infusion. Another clinical trial is active (NCT03350633, Phase II) to compare the safety and efficacy of tocilizumab and azathioprine in preventing NMO relapse.
The major adverse events relative to tocilizumab treatment are immunosuppressive and immunogenic. Other events include infection, HAMA reaction, moderate cholesterol elevation, leukocytopenia and/or lymphocytopenia, anemia, and deep venous thrombosis. However, most events are not considered severe. It should be noted that tocilizumab may cause autoimmune neurological disorders [105–108]. The complementarity-determining regions of tocilizumab are mouse sequences and patients may experience HAMA after long-term use. Hence, the benefits and risks should be determined in future studies and latent infection and adverse events should be monitored.
Satralizumab (SA237)
Satralizumab (SA237) is another humanized anti-IL-6R mAb [109, 110]. The drug was previously used as an extended version of tocilizumab in clinical trials. A placebo-controlled, double-blind trial (NCT02028884, phase III) was conducted to evaluate the efficacy, safety, pharmacodynamics, pharmacokinetics, and immunogenic profile of SA237 as add-on therapy for NMO/NMOSD. Satralizumab added to an immunosuppressant led to a lower risk of relapse (20%) than placebo (43%) [111]. Another double-blinded trial evaluated SA237 monotherapy in NMO/NMOSD (NCT02073279, phase III). The European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) 2019 announced the complete results of the phase III clinical study of monotherapy for NMOSD: 55% reduction in the risk of relapse for SA237 monotherapy versus placebo, and 74% reduction in the risk of relapse for SA237 monotherapy versus placebo in AQP4-IgG seropositive patients. Satralizumab was approved by the US FDA for the treatment of NMOSD in 2019.
Overall, the serious adverse events of satralizumab are similar between the satralizumab monotherapy and placebo groups (NCT02073279), and between satralizumab as add-on therapy and placebo added to baseline therapy groups (NCT02028884).
Bevacizumab (Avastin)
Bevacizumab, a humanized mAb that specifically binds to vascular endothelial growth factor A (VEGF-A), inhibits the formation of new blood vessels [112]. The rationale for its use in the treatment of acute NMO exacerbations is that VEGF induces the breakdown of the BBB, although the mechanisms are unclear. Serum from AQP4-IgG-seropositive NMO patients increases the permeability of human astrocyte and endothelial cell co-cultures and reduces the expression of tight-junction proteins in cultured brain microvascular endothelial cells. VEGF thus exacerbates disruption of the BBB in CNS inflammatory disease [113–116]. Disruption of the BBB is considered to be the first step of NMO; when peripheral AQP4-IgG reaches the CNS through the disrupted BBB it leads to a series of inflammatory responses [113, 117].
A single-center, open label study of bevacizumab involved ten patients with AQP4-IgG seropositive NMO who presented with an acute attack of transverse myelitis, optic neuritis, or brainstem inflammation (NCT01777412). These patients were treated with 1 g of intravenous methylprednisolone daily, in addition to 10 mg/kg of intravenously infused bevacizumab on day 1. Bevacizumab is safe as an add-on therapy with high-dose corticosteroids for NMO patients with an acute relapse [50]. The main purpose of this clinical trial was to evaluate the safety of bevacizumab as an add-on therapy for acute NMO relapse. The primary outcome measure was based on the expanded disability status score.
The major adverse events of bevacizumab are thromboembolic disease or gastrointestinal hemorrhage, which have not been seen in NMO patients. At present, there are few clinical reports on the treatment of NMO with bevacizumab, and the numbers are not sufficient to show common adverse events. A large and long-term clinical trial is needed to determine the safety and efficacy of bevacizumab as an add-on therapy for NMO treatment.
Preclinical mAb Drugs for NMO
Novel therapeutic targets have been found on the basis of understanding NMO pathogenesis. In this section, we discuss potential mAb drugs under development for NMO treatment.
Aquaporumab, a non-pathogenic human mAb, was generated from a recombinant pathogenic monoclonal AQP4-IgG. The Fc (fragment crystallizable) region of aquaporumab has been artificially mutated to eliminate its complement-dependent cytotoxicity (CDC) and ADCC effects. As noted above, AQP4-IgG is essential in the pathogenesis of NMO. Several potential therapeutic approaches have been developed to block the binding of AQP4-IgG to AQP4, thereby obstructing downstream inflammation [29–31]. Non-pathogenic aquaporumab greatly reduces NMO lesions in an animal model of NMO [40]. However, there are no clinical studies of aquaporumab. This mAb binding AQP4 is highly specific with minimal toxicity and immunogenicity. Another mAb against complement components is also being studied. mAbs targeted to complement C1q (C1qmAb) have been shown to be efficient in the mouse model of NMO [118]. C1q is the initiating protein in the classical complement pathway. C1q inhibition prevents formation of the MAC, CDCC, and amplification of the classical complement pathway. In addition, C1qmAb, unlike C5-targeted therapy, does not interfere with defense against bacteria involving the lectin and alternative activation pathways. Theoretically, the drug has a lower risk of meningococcal meningitis infection than eculizumab. C1qmAb may be useful during acute NMO exacerbation and chronic progression. This mAb is still at an experimental stage. Future studies should exclude off-target effects and immunogenicity, with a focus on randomized active-control trials.
At present, increasing numbers of mAbs are being studied for NMO treatment. However, their adverse events in humans are unknown because these drugs are still under development. Additional laboratory and clinical trials are needed to further determine their efficacy and safety.
mAb Treatment for NMO Patients during Pregnancy
Women are the vast majority of patients with NMO, so the management of therapy before and during pregnancy is a challenge for neurologists. The frequency of acute relapse increases in the postpartum period, with disability progression significantly worsening one year after delivery [119, 120]. Azathioprine, mycophenolate, and methotrexate, which are often administered to decrease NMO relapse, should not be continued during pregnancy [35]. Although mAbs are not contraindicated in pregnant women with autoimmune diseases, and a few anecdotal reports have shown that it may be safe to use rituximab during pregnancy, further studies are needed [121–123].
One study has reported that an anti-AQP4 antibody-seropositive patient received a low dose of rituximab (100 mg) 7 months before pregnancy and 100 mg rituximab was restarted two days after delivery. The mother and new-born baby showed no complications during the pregnancy. While IgG antibodies can enter the fetus through the placental barrier during the third trimester of pregnancy, the new-born showed reduced B cells in umbilical cord blood, which returned to normal 3 months later [124, 125]. In a recent study, no major safety issue was found with rituximab use within 6 months of conception in patients with NMOSD or MS, and the mother and baby were healthy [126]. Rituximab might be compatible with conception and prevent NMO exacerbation postpartum. A large sample size and long-term follow-up are needed to explore the safety of rituximab during pregnancy with NMO.
Currently, available treatments including clinical trials for pregnancy are very limited. Preclinical and clinical studies are needed to assess the potential benefits and risks of other mAbs for NMO patients during pregnancy.
Conclusions
In this review, we outline the safety and efficacy of mAbs for NMO treatment. At present, the main mAbs for NMO are engineered mouse antibodies (chimeric and humanized antibodies), which are recognized by humans and induce immunogenicity. With the development of genetic technology, fully human mAbs such as aquaporumab have been used in preclinical experiments. mAbs are a promising strategy for the treatment of NMO. Currently, several mAbs are under evaluation. Preclinical experiments and clinical trials present substantial challenges. In preclinical trials, the mouse model of brain injection with AQP4-IgG and complement has been widely used in the animal studies, but this model does not fully represent NMO [88, 127, 128]. In clinical trials, the number of participants is limited, and larger sample sizes are needed to determine the safety of NMO treatment. There are other concerns involving placebo-controlled trials because untreated NMO has irreversible neurological deficits, even death. Another challenge is the treatment of AQP4-IgG-seronegative patients, because their pathogenesis is unclear. Most of the current clinical trials have selected AQP4-IgG-seropositive patients. Further research is needed for seronegative NMO and to explore the effect of monoclonal antibodies on these patients. Most of the mAbs described above are highly effective and well-tolerated by NMO patients. The development of mAb drugs requires further clinical evaluation to define the risks and benefits. The development of effective, highly selective drug therapy is the central goal of NMO therapeutics.
Acknowledgements
This review was supported by the National Natural Science Foundation of China (81571596 and 81601044), the National Key R&D Program of China (2017YFC1701300), and Fundamental Research Funds for the Central Universities, China (GK201701009).
References
- 1.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- 2.Jacob A, McKeon A, Nakashima I, Sato DK, Elsone L, Fujihara K, et al. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2013;84:922–930. doi: 10.1136/jnnp-2012-302310. [DOI] [PubMed] [Google Scholar]
- 3.Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R, et al. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. 2008;4:202–214. doi: 10.1038/ncpneuro0764. [DOI] [PubMed] [Google Scholar]
- 4.Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 5.Kleiter I. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol. 2012;69:239. doi: 10.1001/archneurol.2011.216. [DOI] [PubMed] [Google Scholar]
- 6.Palace J, Leite MI, Nairne A, Vincent A. Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol. 2010;67:1016–1017. doi: 10.1001/archneurol.2010.188. [DOI] [PubMed] [Google Scholar]
- 7.Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221–2231. doi: 10.1212/01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- 8.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177–189. doi: 10.1212/WNL.0000000000001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bradl M, Reindl M, Lassmann H. Mechanisms for lesion localization in neuromyelitis optica spectrum disorders. Curr Opin Neurol. 2018;31:325. doi: 10.1097/WCO.0000000000000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamid SHM, Whittam D, Mutch K, Linaker S, Solomon T, Das K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017;264:2088–2094. doi: 10.1007/s00415-017-8596-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato DK. Distinction between MOG antibody- positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82:474–481. doi: 10.1212/WNL.0000000000000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sepúlveda M, Aldea M, Escudero D, Llufriu S, Arrambide G, Otero-Romero S, et al. Epidemiology of NMOSD in Catalonia: Influence of the new 2015 criteria in incidence and prevalence estimates. Mult Scler J. 2017;24:1843–1851. doi: 10.1177/1352458517735191. [DOI] [PubMed] [Google Scholar]
- 14.Yan Y, Li Y, Fu Y, Yang L, Su L, Shi K, et al. Autoantibody to MOG suggests two distinct clinical subtypes of NMOSD. Sci China Life Sci. 2016;59:1270–1281. doi: 10.1007/s11427-015-4997-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alves Do Rego C, Collongues N. Neuromyelitis optica spectrum disorders: Features of aquaporin-4, myelin oligodendrocyte glycoprotein and double-seronegative-mediated subtypes. Rev Neurol. 2018;174:458–470. doi: 10.1016/j.neurol.2018.02.084. [DOI] [PubMed] [Google Scholar]
- 16.Durozard P, Rico A, Boutiere C, Maarouf A, Lacroix R, Cointe S, et al. Comparison of the response to rituximab between myelin oligodendrocyte glycoprotein and aquaporin-4 antibody diseases. Ann Neurol. 2020;87:256–266. doi: 10.1002/ana.25648. [DOI] [PubMed] [Google Scholar]
- 17.Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. 2012;79:1273–1277. doi: 10.1212/WNL.0b013e31826aac4e. [DOI] [PubMed] [Google Scholar]
- 18.Kezuka T, Usui Y, Yamakawa N, Matsunaga Y, Matsuda R, Masuda M, et al. Relationship between NMO-antibody and anti–MOG antibody in optic neuritis. J Neuroophthalmol. 2012;32:107–110. doi: 10.1097/WNO.0b013e31823c9b6c. [DOI] [PubMed] [Google Scholar]
- 19.Hamid SHM, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, et al. Seizures and encephalitis in myelin oligodendrocyte glycoprotein IgG disease vs aquaporin 4 IgG disease. JAMA Neurol. 2018;75:65–71. doi: 10.1001/jamaneurol.2017.3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ungureanu A, de Seze J, Ahle G, Sellal F. Myelin oligodendrocyte glycoprotein antibodies in neuromyelitis optica spectrum disorder. Rev Neurol. 2018;01:378–383. doi: 10.1016/j.neurol.2018.01.378. [DOI] [PubMed] [Google Scholar]
- 21.Fang L. Myelin oligodendrocyte glycoprotein-IgG contributes to oligodendrocytopathy in the presence of complement, distinct from astrocytopathy induced by AQP4-IgG. Neurosci Bull. 2019;35:853–866. doi: 10.1007/s12264-019-00375-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bove R, Elsone L, Alvarez E, Borisow N, Cortez MM, Mateen FJ, et al. Female hormonal exposures and neuromyelitis optica symptom onset in a multicenter study. Neuroinflammation. 2017;4:e339. doi: 10.1212/NXI.0000000000000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flanagan EP, Cabre P, Weinshenker BG, Sauver JS, Jacobson DJ, Majed M, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum: Aquaporin-4-IgG Seroprevalence. Ann Neurol. 2016;79:775–783. doi: 10.1002/ana.24617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J Neuroinflammation. 2012;9:14. doi: 10.1186/1742-2094-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banwell B, Tenembaum S, Lennon VA, Ursell E, Kennedy J, Bar-Or A, et al. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology. 2008;70:344–352. doi: 10.1212/01.wnl.0000284600.80782.d5. [DOI] [PubMed] [Google Scholar]
- 26.Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome) Neurology. 1999;53:1107–1114. doi: 10.1212/wnl.53.5.1107. [DOI] [PubMed] [Google Scholar]
- 27.Wingerchuk DM, Weinshenker BG. Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology. 2003;60:848–853. doi: 10.1212/01.wnl.0000049912.02954.2c. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain. 2007;130:1235–1243. doi: 10.1093/brain/awm062. [DOI] [PubMed] [Google Scholar]
- 29.Miyazaki K, Abe Y, Iwanari H, Suzuki Y, Kikuchi T, Ito T, et al. Establishment of monoclonal antibodies against the extracellular domain that block binding of NMO-IgG to AQP4. J Neuroimmunol. 2013;260:107–116. doi: 10.1016/j.jneuroim.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 30.Phuan PW, Anderson MO, Tradtrantip L, Zhang H, Tan J, Lam C, et al. A small-molecule screen yields idiotype-specific blockers of neuromyelitis optica immunoglobulin G binding to aquaporin-4. J Biol Chem. 2012;287:36837–36844. doi: 10.1074/jbc.M112.408716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tradtrantip L, Zhang H, Anderson MO, Saadoun S, Phuan PW, Papadopoulos MC, et al. Small-molecule inhibitors of NMO-IgG binding to aquaporin-4 reduce astrocyte cytotoxicity in neuromyelitis optica. FASEB J. 2012;26:2197–2208. doi: 10.1096/fj.11-201608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tradtrantip L, Asavapanumas N, Verkman AS. Therapeutic cleavage of anti-aquaporin-4 autoantibody in neuromyelitis optica by an IgG-selective proteinase. Mol Pharmacol. 2013;83:1268–1275. doi: 10.1124/mol.113.086470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leavitt J. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Yearb Neurol Neurosurg. 2009;2009:112–113. [Google Scholar]
- 34.Miyamoto K, Kusunoki S. Intermittent plasmapheresis prevents recurrence in neuromyelitis optica. Ther Apher Dial. 2009;13:505–508. doi: 10.1111/j.1744-9987.2009.00780.x. [DOI] [PubMed] [Google Scholar]
- 35.Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol. 2014;10:493–506. doi: 10.1038/nrneurol.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim SH, Kim W, Huh SY, Lee KY, Jung IJ, Kim HJ. Clinical efficacy of plasmapheresis in patients with neuromyelitis optica spectrum disorder and effects on circulating anti-aquaporin-4 antibody levels. J Clin Neurol. 2013;9:36–42. doi: 10.3988/jcn.2013.9.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Herle K, Behne JM, Van Herle A, Blaschke TF, Smith TJ, Yeaman MR. Integrative continuum: accelerating therapeutic advances in rare autoimmune diseases. Annu Rev Pharmacol Toxicol. 2012;52:523–547. doi: 10.1146/annurev-pharmtox-010611-134628. [DOI] [PubMed] [Google Scholar]
- 38.Klotz L, Wiendl H. Monoclonal antibodies in neuroinflammatory diseases. Expert Opin Biol Ther. 2013;13:831–846. doi: 10.1517/14712598.2013.767329. [DOI] [PubMed] [Google Scholar]
- 39.Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: A pilot study. Neurology. 2014;82:1302–1306. doi: 10.1212/WNL.0000000000000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC, et al. Anti-Aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol. 2012;71:314–322. doi: 10.1002/ana.22657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Biotechnology. 1975;24:524. [PubMed] [Google Scholar]
- 42.Tjandra JJ, Ramadi L, McKenzie IFC. Development of human anti-murine antibody (HAMA) response in patients. Immunol Cell Biol. 1990;68:367–376. doi: 10.1038/icb.1990.50. [DOI] [PubMed] [Google Scholar]
- 43.Hwang WYK, Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36:3–10. doi: 10.1016/j.ymeth.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Presta LG. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function. Adv Drug Deliv Rev. 2006;58:640–656. doi: 10.1016/j.addr.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 45.Jespers LS, Roberts A, Mahler SM, Winter G, Hoogenboom HR. Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen. Biotechnology. 1994;12:899. doi: 10.1038/nbt0994-899. [DOI] [PubMed] [Google Scholar]
- 46.Zéphir H, Bernard-Valnet R, Lebrun C, Outteryck O, Audoin B, Bourre B, et al. Rituximab as first-line therapy in neuromyelitis optica: efficiency and tolerability. J Neurol. 2015;262:2329–2335. doi: 10.1007/s00415-015-7852-y. [DOI] [PubMed] [Google Scholar]
- 47.Le GTM, Herbi L, De RC, Nguyen-Khac F, Davi F, Grelier A, et al. Antibody-dependent cellular cytotoxicity of the optimized anti-CD20 monoclonal antibody ublituximab on chronic lymphocytic leukemia cells with the 17p deletion. Leukemia. 2014;28:230–233. doi: 10.1038/leu.2013.240. [DOI] [PubMed] [Google Scholar]
- 48.Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12:554–562. doi: 10.1016/S1474-4422(13)70076-0. [DOI] [PubMed] [Google Scholar]
- 49.Araki M, Aranami T, Matsuoka T, Nakamura M, Miyake S, Yamamura T. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod Rheumatol. 2013;23:827–831. doi: 10.1007/s10165-012-0715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mealy MA, Shin K, John G, Levy M. Bevacizumab is safe in acute relapses of neuromyelitis optica. Clin Exp Neuroimmunol. 2015;6:413–418. doi: 10.1111/cen3.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taylor RP, Lindorfer MA. Immunotherapeutic mechanisms of anti-CD20 monoclonal antibodies. Curr Opin Immunol. 2008;20:444–449. doi: 10.1016/j.coi.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, et al. Intrathecal pathogenic anti–aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66:617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hohlfeld R. B-cells as therapeutic targets in neuro-inflammatory diseases. Clin Immunol. 2018;186:51–53. doi: 10.1016/j.clim.2017.07.013. [DOI] [PubMed] [Google Scholar]
- 54.Hinson SR, McKeon A, Fryer JP, Apiwattanakul M, Lennon VA, Pittock SJ. Prediction of neuromyelitis optica attack severity by quantitation of complement-mediated injury to aquaporin-4–expressing cells. Arch Neurol. 2009;66:1164–1167. doi: 10.1001/archneurol.2009.188. [DOI] [PubMed] [Google Scholar]
- 55.Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123:861–872. doi: 10.1007/s00401-012-0986-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monson NL, Cravens PD, Frohman EM, Hawker K, Racke MK. Effect of rituximab on the peripheral blood and cerebrospinal fluid B Cells in patients with primary progressive multiple sclerosis. Arch Neurol. 2005;62:258–264. doi: 10.1001/archneur.62.2.258. [DOI] [PubMed] [Google Scholar]
- 57.Rastetter W, Molina A, White CA. Rituximab: Expanding role in therapy for lymphomas and autoimmune diseases. Annu Rev Med. 2004;55:477–503. doi: 10.1146/annurev.med.55.091902.104249. [DOI] [PubMed] [Google Scholar]
- 58.Dalakas MC. B cells as therapeutic targets in autoimmune neurological disorders. Nat Clin Pract Neurol. 2008;4:557–567. doi: 10.1038/ncpneuro0901. [DOI] [PubMed] [Google Scholar]
- 59.Graves J, Vinayagasundaram U, Mowry EM, Matthews IR, Marino JA, Cheng J, et al. Effects of rituximab on lymphocytes in multiple sclerosis and neuromyelitis optica. Mult Scler Relat Disord. 2014;3:244–252. doi: 10.1016/j.msard.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 60.Wilk E, Witte T, Marquardt N, Horvath T, Kalippke K, Scholz K, et al. Depletion of functionally active CD20+ T cells by rituximab treatment. Arthritis Rheum. 2009;60:3563–3571. doi: 10.1002/art.24998. [DOI] [PubMed] [Google Scholar]
- 61.Yang CS. Responsiveness to reduced dosage of rituximab in Chinese patients with neuromyelitis. Optica. 2013;81:710–713. doi: 10.1212/WNL.0b013e3182a1aac7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edwards JCW, Szechinski J, Emery P, Shaw T. Efficacy of B-cell–targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 63.Hauser SL, Arnold DL, Fox RJ, Sarkar N, Smith CH. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 64.Reske D, Haupt WF. Use of rituximab in multiple sclerosis: current progress and future perspectives. Expert Rev Clin Immunol. 2008;4:573–582. doi: 10.1586/1744666X.4.5.573. [DOI] [PubMed] [Google Scholar]
- 65.Salvi M, Vannucchi G, Campi I, Beck-Peccoz P. Rituximab in the treatment of thyroid eye disease: science fiction? Orbit. 2009;28:251–255. [PubMed] [Google Scholar]
- 66.Cree BA, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64:1270–1272. doi: 10.1212/01.WNL.0000159399.81861.D5. [DOI] [PubMed] [Google Scholar]
- 67.Bedi GS, Brown AD, Delgado SR, Usmani N, Lam BL, Sheremata WA. Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult Scler J. 2011;17:1225–1230. doi: 10.1177/1352458511404586. [DOI] [PubMed] [Google Scholar]
- 68.Collongues N, Brassat D, Maillart E, Labauge P, Ouallet J, Carra-Dalliere C, et al. Efficacy of rituximab in refractory neuromyelitis optica. Mult Scler J. 2016;22:955–959. doi: 10.1177/1352458515602337. [DOI] [PubMed] [Google Scholar]
- 69.Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70:1110–1117. doi: 10.1001/jamaneurol.2013.3071. [DOI] [PubMed] [Google Scholar]
- 70.Memon AB, Javed A, Caon C, Srivastawa S, Bao F, Bernitsas E, et al. Long-term safety of rituximab induced peripheral B-cell depletion in autoimmune neurological diseases. PLoS One. 2018;13:e0190425. doi: 10.1371/journal.pone.0190425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol. 2017;264:2003–2009. doi: 10.1007/s00415-017-8590-0. [DOI] [PubMed] [Google Scholar]
- 72.Ciron J, Audoin B, Bourre B, Brassat D, Durand-Dubief F, Laplaud D, et al. Recommendations for the use of Rituximab in neuromyelitis optica spectrum disorders. Rev Neurol. 2018;174:255–264. doi: 10.1016/j.neurol.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 73.Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a Review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009;10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 74.Clifford DB. Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol. 2011;68:1156–1164. doi: 10.1001/archneurol.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsutsumi Y. Hepatitis B virus reactivation with a rituximab-containing regimen. World J Hepatol. 2015;7:2344. doi: 10.4254/wjh.v7.i21.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sharman JP, Farber CM, Mahadevan D, Schreeder MT, Brooks HD, Kolibaba KS, et al. Ublituximab (TG-1101), a novel glycoengineered anti-CD20 antibody, in combination with ibrutinib is safe and highly active in patients with relapsed and/or refractory chronic lymphocytic leukaemia: results of a phase 2 trial. Br J Haematol. 2017;176:412–420. doi: 10.1111/bjh.14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Konno Y, Kobayashi Y, Takahashi K, Takahashi E, Sakae S, Wakitani M, et al. Fucose content of monoclonal antibodies can be controlled by culture medium osmolality for high antibody-dependent cellular cytotoxicity. Cytotechnology. 2012;64:249–265. doi: 10.1007/s10616-011-9377-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Agius MA, Klodowska-Duda G, Maciejowski M, Potemkowski A, Li J, Patra K, et al. Safety and tolerability of inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: Results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Mult Scler. 2017;25:235–245. doi: 10.1177/1352458517740641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tedder TF. CD19: a promising B cell target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:572–577. doi: 10.1038/nrrheum.2009.184. [DOI] [PubMed] [Google Scholar]
- 80.Schuh E, Berer K, Mulazzani M, Feil K, Meinl I, Lahm H, et al. Features of human CD3+ CD20+ T cells. J Immunol. 2016;197:1111–1117. doi: 10.4049/jimmunol.1600089. [DOI] [PubMed] [Google Scholar]
- 81.Palanichamy A, Jahn S, Nickles D, Derstine M, Abounasr A, Hauser SL, et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol. 2014;193:580–586. doi: 10.4049/jimmunol.1400118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hammer O. CD19 as an attractive target for antibody-based therapy. mAbs. 2012;4:571–577. doi: 10.4161/mabs.21338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schiopu E, Chatterjee S, Hsu V, Flor A, Cimbora D, Patra K, et al. Safety and tolerability of an anti-CD19 monoclonal antibody, MEDI-551, in subjects with systemic sclerosis: a phase I, randomized, placebo-controlled, escalating single-dose study. Arthritis Res Ther. 2016;18:131. doi: 10.1186/s13075-016-1021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394:1352–1363. doi: 10.1016/S0140-6736(19)31817-3. [DOI] [PubMed] [Google Scholar]
- 85.Stüve O, Warnke C, Deason K, Stangel M, Kieseier BC, Hartung H-P, et al. CD19 as a molecular target in CNS autoimmunity. Acta Neuropathol. 2014;128:177–190. doi: 10.1007/s00401-014-1313-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25:1256–1264. doi: 10.1038/nbt1344. [DOI] [PubMed] [Google Scholar]
- 87.Hengstman GJD, Wesseling P, Frenken CWGM, Jongen PJH. Neuromyelitis optica with clinical and histopathological involvement of the brain. Mult Scler J. 2007;13:679–682. doi: 10.1177/1352458506070145. [DOI] [PubMed] [Google Scholar]
- 88.Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang H, Bennett JL, Verkman AS. Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann Neurol. 2011;70:943–954. doi: 10.1002/ana.22551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jore MM, Johnson S, Sheppard D, Barber NM, Li YI, Nunn MA, et al. Structural basis for therapeutic inhibition of complement C5. Nat Struct Mol Biol. 2016;23:378–386. doi: 10.1038/nsmb.3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, et al. Eculizumab in aquaporin-4–positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381:614–625. doi: 10.1056/NEJMoa1900866. [DOI] [PubMed] [Google Scholar]
- 94.Tanaka T, Narazaki M, Kishimoto T. Therapeutic targeting of the interleukin-6 receptor. Annu Rev Pharmacol Toxicol. 2012;52:199–219. doi: 10.1146/annurev-pharmtox-010611-134715. [DOI] [PubMed] [Google Scholar]
- 95.Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A. 2011;108:3701–3706. doi: 10.1073/pnas.1017385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Uzawa A, Mori M, Sawai S, Masuda S, Muto M, Uchida T, et al. Cerebrospinal fluid interleukin-6 and glial fibrillary acidic protein levels are increased during initial neuromyelitis optica attacks. Clin Chim Acta. 2013;421:181–183. doi: 10.1016/j.cca.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 97.Wang H, Wang K, Zhong X, Dai Y, Qiu W, Wu A, et al. Notable increased cerebrospinal fluid levels of soluble interleukin-6 receptors in neuromyelitis optica. Neuroimmunomodulation. 2012;19:304–308. doi: 10.1159/000339302. [DOI] [PubMed] [Google Scholar]
- 98.Kieseier BC, Stüve O, Dehmel T, Goebels N, Leussink VI, Mausberg AK, et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol. 2013;70:390. doi: 10.1001/jamaneurol.2013.668. [DOI] [PubMed] [Google Scholar]
- 99.Komai T, Shoda H, Yamaguchi K, Sakurai K, Shibuya M, Kubo K, et al. Neuromyelitis optica spectrum disorder complicated with Sjogren syndrome successfully treated with tocilizumab: A case report. Mod Rheumatol. 2016;26:294–296. doi: 10.3109/14397595.2013.861333. [DOI] [PubMed] [Google Scholar]
- 100.Araki M. Blockade of IL-6 signaling in neuromyelitis optica. Neurochem Int. 2018;2018:104315. doi: 10.1016/j.neuint.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 101.Ringelstein M, Ayzenberg I, Harmel J, Lauenstein A-S, Lensch E, Stögbauer F, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. 2015;72:756. doi: 10.1001/jamaneurol.2015.0533. [DOI] [PubMed] [Google Scholar]
- 102.Ayzenberg I, Kleiter I, Schröder A, Hellwig K, Chan A, Yamamura T, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol. 2013;70:394–397. doi: 10.1001/jamaneurol.2013.1246. [DOI] [PubMed] [Google Scholar]
- 103.Qian P, Lancia S, Alvarez E, Klawiter EC, Cross AH, Naismith RT. Association of neuromyelitis optica with severe and intractable pain. Arch Neurol. 2012;69:1482–1487. doi: 10.1001/archneurol.2012.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ohtori S, Miyagi M, Eguchi Y, Inoue G, Orita S, Ochiai N, et al. Efficacy of epidural administration of anti-interleukin-6 receptor antibody onto spinal nerve for treatment of sciatica. Eur Spine J. 2012;21:2079–2084. doi: 10.1007/s00586-012-2183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Beauchemin P, Carruthers R. MS arising during Tocilizumab therapy for rheumatoid arthritis. Mult Scler J. 2016;22:254–256. doi: 10.1177/1352458515623862. [DOI] [PubMed] [Google Scholar]
- 106.Gabay C, McInnes IB, Kavanaugh A, Tuckwell K, Klearman M, Pulley J, et al. Comparison of lipid and lipid-associated cardiovascular risk marker changes after treatment with tocilizumab or adalimumab in patients with rheumatoid arthritis. Ann Rheum Dis. 2016;75:1806–1812. doi: 10.1136/annrheumdis-2015-207872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gout T, Östör AJK, Nisar MK. Lower gastrointestinal perforation in rheumatoid arthritis patients treated with conventional DMARDs or tocilizumab: a systematic literature review. Clin Rheumatol. 2011;30:1471–1474. doi: 10.1007/s10067-011-1827-x. [DOI] [PubMed] [Google Scholar]
- 108.Iwasa T, Nakamura K, Ogino H, Itaba S, Akiho H, Okamoto R, et al. Multiple ulcers in the small and large intestines occurred during tocilizumab therapy for rheumatoid arthritis. Endoscopy. 2011;43:70–72. doi: 10.1055/s-0030-1255931. [DOI] [PubMed] [Google Scholar]
- 109.Paul F, Murphy O, Pardo S, Levy M. Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin Investig Drugs. 2018;27:265–271. doi: 10.1080/13543784.2018.1443077. [DOI] [PubMed] [Google Scholar]
- 110.Kaplon H, Reichert JM. Antibodies to watch in 2018. mAbs. 2018;10:183–203. doi: 10.1080/19420862.2018.1415671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381:2114–2124. doi: 10.1056/NEJMoa1901747. [DOI] [PubMed] [Google Scholar]
- 112.Wang Y, Fei D, Vanderlaan M, Song A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis. 2004;7:335–345. doi: 10.1007/s10456-004-8272-2. [DOI] [PubMed] [Google Scholar]
- 113.Shimizu F, Sano Y, Takahashi T, Haruki H, Saito K, Koga M, et al. Sera from neuromyelitis optica patients disrupt the blood–brain barrier. J Neurol Neurosurg Psychiatry. 2012;83:288–297. doi: 10.1136/jnnp-2011-300434. [DOI] [PubMed] [Google Scholar]
- 114.Vincent T, Saikali P, Cayrol R, Roth AD, Bar-Or A, Prat A, et al. Functional consequences of neuromyelitis optica-igg astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J Immunol. 2008;181:5730–5737. doi: 10.4049/jimmunol.181.8.5730. [DOI] [PubMed] [Google Scholar]
- 115.Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A. 2009;106:1977–1982. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, et al. Neuromyelitis optica: Pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–643. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- 118.Phuan PW, Zhang H, Asavapanumas N, Leviten M, Rosenthal A, Tradtrantip L, et al. C1q-targeted monoclonal antibody prevents complement-dependent cytotoxicity and neuropathology in in vitro and mouse models of neuromyelitis optica. Acta Neuropathol. 2013;125:829–840. doi: 10.1007/s00401-013-1128-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bourre B, Marignier R, Zephir H, Papeix C, Brassat D, Castelnovo G, et al. Neuromyelitis optica and pregnancy. 2012;78:875–879. doi: 10.1212/WNL.0b013e31824c466f. [DOI] [PubMed] [Google Scholar]
- 120.Kim W, Kim S-H, Nakashima I, Takai Y, Fujihara K, Leite MI, et al. Influence of pregnancy on neuromyelitis optica spectrum disorder. Neurology. 2012;78:1264–1267. doi: 10.1212/WNL.0b013e318250d812. [DOI] [PubMed] [Google Scholar]
- 121.Kimby E, Sverrisdottir A, Elinder G. Safety of rituximab therapy during the first trimester of pregnancy: a case history. Eur J Haematol. 2004;72:292–295. doi: 10.1111/j.1600-0609.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 122.Ojeda-Uribe M, Afif N, Dahan E, Sparsa L, Haby C, Sibilia J, et al. Exposure to abatacept or rituximab in the first trimester of pregnancy in three women with autoimmune diseases. Clin Rheumatol. 2013;32:695–700. doi: 10.1007/s10067-012-2156-4. [DOI] [PubMed] [Google Scholar]
- 123.Ponte P, Lopes MJP. Apparent safe use of single dose rituximab for recalcitrant atopic dermatitis in the first trimester of a twin pregnancy. J Am Acad Dermatol. 2010;63:355–356. doi: 10.1016/j.jaad.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 124.Ringelstein M, Harmel J, Distelmaier F, Ingwersen J, Menge T, Hellwig K, et al. Neuromyelitis optica and pregnancy during therapeutic B cell depletion: infant exposure to anti-AQP4 antibody and prevention of rebound relapses with low-dose rituximab postpartum. Mult Scler J. 2013;19:1544–1547. doi: 10.1177/1352458513498125. [DOI] [PubMed] [Google Scholar]
- 125.Simister N. Placental transport of immunoglobulin G. Vaccine. 2003;21:3365–3369. doi: 10.1016/s0264-410x(03)00334-7. [DOI] [PubMed] [Google Scholar]
- 126.Das G, Damotte V, Gelfand JM, Bevan C, Cree BAC, Do L, et al. Rituximab before and during pregnancy: A systematic review, and a case series in MS and NMOSD. Neurol Neuroimmunol. Neuroinflamm. 2018;5:e453. doi: 10.1212/NXI.0000000000000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shi K, Wang Z, Liu Y, Gong Y, Fu Y, Li S, et al. CFHR1-modified neural stem cells ameliorated brain injury in a mouse model of neuromyelitis optica spectrum disorders. J Immunol. 2016;197:3471–3480. doi: 10.4049/jimmunol.1600135. [DOI] [PubMed] [Google Scholar]
- 128.Wang Z, Guo W, Liu Y, Gong Y, Ding X, Shi K, et al. Low expression of complement inhibitory protein CD59 contributes to humoral autoimmunity against astrocytes. Brain Behav Immun. 2017;65:173–182. doi: 10.1016/j.bbi.2017.04.023. [DOI] [PubMed] [Google Scholar]