

Abstract
Azadirachta Indica (Neem) extracts have been known for their anti-bacterial and other effects since ancient times. The present work examines the inhibitory activity of Neem extracts on Papain like protease (PLpro) of the novel coronavirus SARS-CoV-2. The activity is analysed by molecular docking study along with molecular dynamics simulation. All the studied Neem compounds showed decent level of inhibitory activity against PLpro of SARS-CoV-2. Among them, desacetylgedunin (DCG) found in Neem seed showed the highest binding affinity towards PLpro. Furthermore, MD-simulation studies supported by standard analysis (e.g. root mean square deviation and fluctuation (RMSD, RMSF), radius of gyration, solvent accessible surface area (SASA)) showed large impact on the structure of PLpro by DCG. We believe that the significant effect of DCG on PLpro may help in therapeutic efforts against SARS-CoV-2.
Keywords: Neem extracts, PLpro, Molecular docking, MD simulation
Graphical abstract
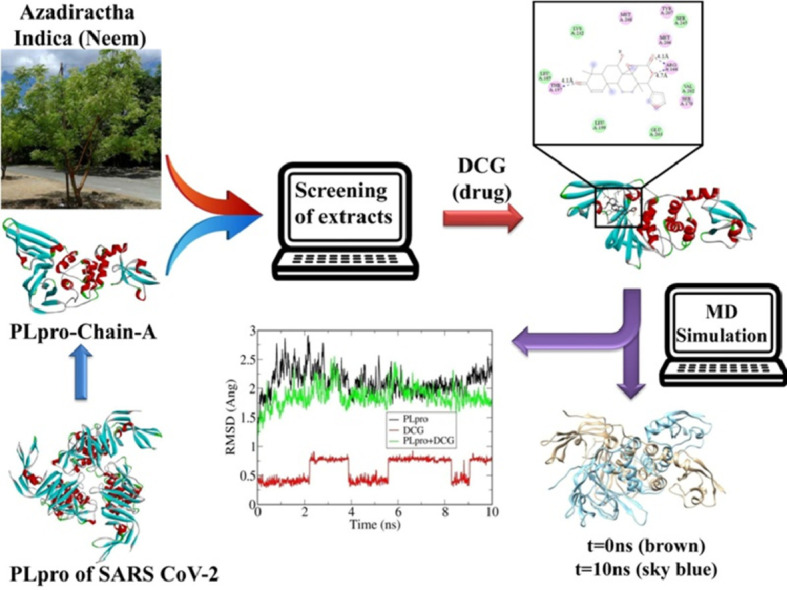
1. Introduction
Recent pandemic caused by the novel coronavirus SARS-CoV-2 (COVID-19 in short) has thrown a big challenge to humanity. COVID-19 belongs to the beta-coronavirus group, very similar to previously reported SARS coronavirus [1]. Supposedly originating from bats and pangolins [2], it spreads very rapidly through human to human community transmission [3]. It is a fairly large virus, with spike glycoproteins on outer surface, a membrane, envelope, and nucleocapsid protein containing a positive sense single strand of RNA, which consists, among others, of a 5′-untranslated region (UTR), a replicase complex (ORF1ab) encoding non-structural proteins (nsps) [4]. It is now known that the virus enters into host cell by the interaction of spike protein to the angiotensin-converting enzyme-2 (ACE-2) receptors in host cell and uses the cellular serine protease TMPRSS2 for S protein priming. It also encodes cysteine protease, a serine protease, besides the main protease (3CLpro) and a papain-like protease (PLpro). These may participate in cleavage of viral polyproteins [5,6], leading to creation of active substances for replication of virus inside the host cell. Therefore, activity of each of these individual entities and binding of the virus to the host cell, and activity of ACE-2 have all been examined in finding potential antidote to COVID-19.
However, despite much effort, there is no potentially active drug or vaccine available till date which can effectively combat SARS-CoV-2. Although several drugs (chloroquine, hydroxy‑chloroquine, remdesivir etc.) are recommended against COVID-19, many adverse effects of these have also been reported [7,8]. These drug molecules target different entities in the virus or its binding with the host cell. It was decided to focus on activity of the papain-like protease PLpro and how to inhibit it in the present work.
The present work is to find out potential drugs against COVID-19 from natural sources such as the medicinal plant Neem, (Azadirachta Indica). There have been a lot of investigations on Neem extracts from its leaves, fruits, bark etc. It is known that compounds in Neem extracts have anti-inflammatory, anti-hyperglycaemic, anti-carcinogenic, immune-modulator, anti-mutagenic, anti-oxidant, anti-ulcer and anti-viral effects [9]. Anti-viral effects of Neem extracts include inhibitory action against herpes, [10] smallpox, chickenpox [11,12] and even HIV [13]. Compounds in Neem extract are also active against polio virus, coxackie B group virus, HIV and dengue virus at early step of viral genome replication [12], [13], [14], [15], [16], [17]. In the present work we have selected 19 natural compounds of Neem and evaluated their inhibitory potency against PLpro of SAS-COV-2 through molecular docking and molecular dynamics simulation.
2. Methodology
2.1. Docking of Neem extracts with SARS-CoVs-2 PLpro
The crystal structure of papain like protease (PLpro) of SARS-COV-2 (PDB ID: 6W9C) [18] was obtained from the Protein Data Bank. Only chain-A of PLpro was chosen for docking studies. Structures of 7 drugs in use against COVID-19, and 19 compounds from NEEM extacts, were obtained from the PubChem (National Library of Medicine) and were converted to check the ligand-protein binding interaction using UCSF Chimera [19]. The structure of PLpro was prepared by eliminating hetero-atoms and chain B and C using UCSF Chimera package. Autodock Vina [20] was used to obtain the best docking configuration between PLpro and the drug molecules. Autodock Tools was used to prepare necessary files for docking simulation.
2.2. MD simulation of PLpro-DCG complex
Molecular dynamics (MD) simulation studies were performed using GROMACS (Version 2018.5) [21] with the minimum energy configurations obtained from docking studies using the CHARMM36-mar2019 force-field [22] by TIP3P model [23]. The topology and the parameter files were generated for the ligand desacetylgedunin (DCG) (the compound with highest docking with PLpro) from CHARMM General Force Field server. To maintain periodic boundary conditions a cubical box was generated. The PLpro-DCG complex was at least 1 nm from the edges of the box to maintain at least 2 nm distance between two successive images of the complex. Three Cl- ions were added to neutralize the system. Energy minimization was carried out until the maximum force becomes less than 10 kJmol−1nm−1. The steepest descent algorithm was used followed by conjugate gradient protocol. Then isochoric-isothermal (NVT) ensemble was used for equilibration of the system for 1000 ps at 300 K. 2 fs time step was taken. Isothermal-isobaric or NPT ensemble is then used for equilibration at 300 K for 1000 ps. For the NPT ensemble modified Berendsen thermostat was used. Time step was kept at 2 fs. The electrostatic and van der Waals interaction cut off were kept at 1.0 nm for both NVT and NPT equilibration. Long range interactions were calculated using smooth particle mesh Ewald (PME) method [24]. With the same electrostatic and van der Waals cut off, the equilibrated ensembles were finally subjected to MD simulation for 100 ns. PME method was used to calculate long range electrostatic interactions. A modified Berendsen thermostat and a Parinello-Rahman barostat were used with reference temperature and pressure at 300 K and 1 bar respectively. Snapshots of the trajectory were saved after every nanosecond.
2.3. Analysis of MD simulations
The structural trajectories for PLpro-DCG systems were calculated using trjconv tool. To re-center the protein and other molecules within the cubical box, the same tool was used. gmx energy tool was used to calculate the van der Waals interaction, conformational energy etc. xmgrace tool was used to plot RMSD, RMSF, Radius of gyration and solvent accessible surface area (SASA) plots.
3. Results and discussion
3.1. Molecular docking studies
19 major compounds found in different parts of Neem are chosen to analyze their potential inhibitory capacity against PLpro of corona virus. Table 1 lists the binding affinities of these compounds along with their molecular formula and structure. The highest binding affinity was obtained as −7.3 Kcal/mol for desacetylgedunin (DCG) against PLpro.
Table 1.
Docking scores of Neem extracts with chain A of PLpro.
| Compounds | Pubchem CID | MW (g/mol) | MF | Structure | Docking Score (Kcal/mol) | |
|---|---|---|---|---|---|---|
| Neem Leav-es | Quercetin (flavonoid) | 5,280,343 | 302.23 | C15H10O7 | 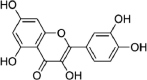 |
−6.1 |
| Nimbosterol (β- sitosterol) | 222,284 | 414.7 | C29H50O | 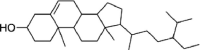 |
−6.8 | |
| Nimbolide | 12,313,376 | 466.5 | C27H30O7 | 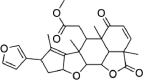 |
−7.1 | |
| Neem Flow-ers | Palmitic acid | 985 | 256.42 | C16H32O2 | −4.1 | |
| Oleic acid | 445,639 | 282.5 | C18H34O2 | 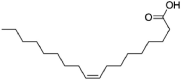 |
−5.4 | |
| Linoleic acid | 5,280,450 | 280.4 | C18H32O2 | 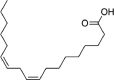 |
−4.6 | |
| Neem Barks | Margocin | 21,632,833 | 298.4 | C20H26O2 | 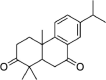 |
−6.8 |
| Nimbidiol | 11,334,829 | 274.35 | C17H22O3 | 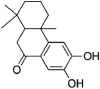 |
−6.2 | |
| Nimbione | 189,404 | 286.4 | C18H22O3 | 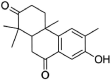 |
−6.6 | |
| Neem Seeds | Azadiradione | 12,308,714 | 450.6 | C28H34O5 | 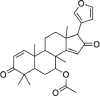 |
−6.4 |
| Fraxinellone | 124,039 | 232.27 | C14H16O3 | 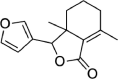 |
−5.6 | |
| Salannin | 6,437,066 | 596.7 | C34H44O9 | 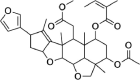 |
−5.7 | |
| Salannol | 157,144 | 556.7 | C32H44O8 | 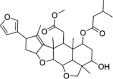 |
−5.8 | |
| Vepinin | 185,552 | 452.6 | C28H36O5 | 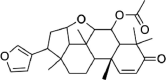 |
−6.6 | |
| Azadirone | 10,906,239 | 436.6 | C28H36O4 | 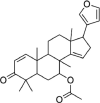 |
−6.4 | |
| Gedunin | 12,004,512 | 482.6 | C28H34O7 | 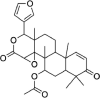 |
−6.4 | |
| Azadirachtin H | 16,722,121 | 662.7 | C33H42O14 | 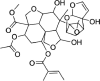 |
−6.5 | |
| Nimbin | 108,058 | 540.6 | C30H36O9 | 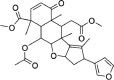 |
−6.6 | |
| Desacetylgedunin | 3,034,112 | 440.5 | C26H32O6 | 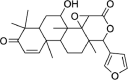 |
−7.3 | |
Table 2 shows the binding affinities of some well known potentially active drugs against PLpro. It is clear from the Table 1 and 2 that desacetylgedunin showed higher binding affinity than all the known and active drugs used.
Table 2.
Docking scores of some potential drugs of SARS-COV-2 with PLpro-Chain-A.
| Some potentially active drug for repurposing | Pubchem CID | MW (g/mol) | MF | Docking Score (Kcal/mol) |
|---|---|---|---|---|
| Chloroquine | 2719 | 319.9 | C18H26ClN3 | −5.3 |
| Hydroxychloroquine | 3652 | 335.9 | C18H26ClN3O | −5.5 |
| Remdesivir | 121,304,016 | 602.6 | C27H35N6O8P | −6.3 |
| Ribavirin (tribavirin) | 37,542 | 244.2 | C8H12N4O5 | −5.6 |
| Favipiravir (Avigan) | 492,405 | 157.1 | C5H4FN3O2 | −5.4 |
| Ritonavir | 392,622 | 720.9 | C37H48N6O5S2 | −5.7 |
| Nitazoxanide | 41,684 | 307.28 | C12H9N3O5S | −6.2 |
Furthermore to predict the toxicity level of the studied compounds, we have performed standard absorption-distribution-metabolism-excretion (ADME) toxicity analysis by pkCSM online server [25].
Prediction of ADME- Evaluation of pharmacokinetic properties of the studied compounds suggests that they are effectively absorbed by the gastro-intestinal part with low blood-brain-barrier (BBB) permeability value and these drugs do not affect CYP2D6 substrate, CYP2C9 inhibitor and CYP2D6 inhibitor cytochromes. Skin permeability for these compounds is found to be between −2.314 to −3.599 units. The toxicity table is shown in Table S1.
Prediction of toxicity- All compounds show negative Ames toxicity and none of them have shown hERGI inhibition activity. The LD50 values of the studied compounds are between 1.417 and 3.71 mol/kg. The chronic oral rat toxicity (LOAEL) values lie between 0.391 and 3.259 (log mg/kg_bw/day). Most of the Neem extracts do not show hepatotoxicity. Few of the compounds show skin sensitization. T. pyriformis and minnow toxicity values are tabulated in Table 3 .
Table 3.
Toxicity prediction of compounds of Neem extracts along with some potential drugs.
| Compound | AMES toxicity | Max. tolerated dose (human) | hERG I inhibitor | hERG II inhibitor | Oral Rat Acute Toxicity (LD50) (mol/kg) | Oral Rat Chronic Toxicity (LOAEL) (log mg/kg_bw/day) | Hepato-toxicity | Skin Sensitisation | T.Pyriformis toxicity (log ug/L) | Minnow toxicity (log mM) |
|---|---|---|---|---|---|---|---|---|---|---|
| Quercetin (flavonoid) | No | 0.499 | No | No | 2.471 | 2.612 | No | No | 0.288 | 3.721 |
| Nimbosterol (β- sitosterol) | No | −0.621 | No | Yes | 2.552 | 0.855 | No | No | 0.83 | −1.802 |
| Nimbolide | No | −0.476 | No | No | 2.374 | 1.554 | No | No | 0.328 | 0.318 |
| Palmitic acid | No | −0.708 | No | No | 1.44 | 3.181 | No | Yes | 0.84 | −1.083 |
| Oleic acid | No | −0.81 | No | No | 1.417 | 3.259 | No | Yes | 0.676 | 1.438 |
| Linoleic acid | No | −0.827 | No | No | 1.429 | 3.187 | Yes | Yes | 0.701 | −1.31 |
| Margocin | No | −0.144 | No | No | 1.935 | 1.972 | No | No | 2.686 | −0.058 |
| Nimbidiol | No | −0.434 | No | No | 2.4 | 2.174 | No | No | 1.36 | 0.805 |
| Nimbione | No | −0.514 | No | No | 2.279 | 1.618 | No | No | 1.722 | 0.939 |
| Azadiradione | No | −0.224 | No | Yes | 2.677 | 0.391 | No | No | 0.364 | −0.253 |
| Fraxinellone | No | 0.557 | No | No | 2.57 | 1.913 | No | No | 0.821 | 0.9 |
| Salannin | No | −0.378 | No | No | 2.825 | 1.066 | Yes | No | 0.286 | 0.656 |
| Salannol | No | −0.482 | No | No | 2.817 | 1.377 | No | No | 0.297 | 0.203 |
| Vepinin | No | −0.132 | No | No | 2.77 | 0.092 | No | No | 0.317 | 0.026 |
| Azadirone | No | −0.295 | No | No | 2.472 | 1.174 | No | No | 0.444 | −0.658 |
| Gedunin | No | −0.736 | No | No | 2.998 | 0.195 | No | No | 0.291 | 0.456 |
| Azadirachtin H | No | −1.12 | No | No | 3.71 | 2.858 | No | No | 0.285 | 9.192 |
| Nimbin | No | −0.371 | No | No | 2.48 | 1.57 | No | No | 0.295 | 1.269 |
| Desacetylgedunin | No | −0.671 | No | No | 2.907 | 1.448 | Yes | No | 0.307 | 0.121 |
| Chloroquine | Yes | −0.167 | No | Yes | 2.85 | 1.026 | Yes | No | 1.558 | 0.747 |
| Hydroxychloroquine | Yes | −0.091 | No | Yes | 2.656 | 1.407 | Yes | No | 1.061 | 1.325 |
| Remdesivir | No | 0.15 | No | Yes | 2.043 | 1.639 | Yes | No | 0.285 | 0.291 |
| Ribavirin | No | 1.011 | No | No | 1.988 | 3.096 | No | No | 0.285 | 4.626 |
| Favipiravir | No | 1.291 | No | No | 1.941 | 2.023 | No | No | 0.099 | 3.407 |
| Ritonavir | No | 0.096 | No | Yes | 2.703 | 2.231 | Yes | No | 0.285 | 1.787 |
| Nitazoxanide | Yes | 0.761 | No | No | 2.399 | 0.945 | Yes | No | 0.409 | 1.163 |
From the above analysis it is clear that the studied compounds of Neem extract showed low toxicity level compared to standard anti-COVID-19 drugs. From the binding affinity and toxicity level analysis we have selected desacetylgedunin (DCG) for further analysis. Fig. 1 represents docked cavities of Neem compounds against PLpro with high binding affinity.
Fig. 1.

Docked structures of some Neem compounds with PLpro having high binding affinity.
From Table 1 it is clear that DCG has the highest docking score so the binding sites and the 2D-contour plot of DCG against PLpro are shown in Fig. 2 .
Fig. 2.

Docked structure of DCG with PLpro.
Fig. 2 illustrates the docked structure of DCG with PLpro along with the neighbouring residues interacting with it at the left panel and the type of interactions of the protease and target drug at the right panel. In the stable conformation of the docked structure, there are three H-bonding interactions. Along with that, dominant electrostatic interaction and van der Waal interactions are also present. This was the starting point of molecular dynamics simulation to investigate the interaction between PLpro and DCG in greater detail.
Evolution of root mean squared deviations (RMSD) of PLpro alone and with DCG is shown in Fig. 3 . After 40 ns, the RMSD fluctuation of PLpro-DCG becomes much more prominent indicating huge conformational and structural changes of the docked structure. We note that the fluctuation is maximized at 100 ns which is reflected from the structural changes as discussed in Fig. 6. Post MD-simulation docking analysis with PLpro, captured for each 10 ns was also performed to understand the binding affinity of DCG against PLpro during MD trajectory which is shown in Table 4 .
Fig. 3.

Root mean square deviation (RMSD) plot of PLpro, DCG and PLpro+DCG.
Fig. 6.

Structural changes during MD-simulation of undocked (brown) and docked (sky blue) PLpro.
Table 4.
Docking score analysis during MD-simulation between DCG and PLpro after each 10 ns period.
| Time (ns) | Docking score (Kcal/mol) |
|---|---|
| 10 | −7.2 |
| 20 | −6.7 |
| 30 | −6.9 |
| 40 | −7.2 |
| 50 | −6.8 |
| 60 | −7.2 |
| 70 | −6.7 |
| 80 | −6.9 |
| 90 | −6.8 |
| 100 | −6.4 |
During MD-simulation the binding affinity ranges from −7.2 to −6.4 Kcal/mol. This clearly shows the prominent effect of the drug within the PLpro cavity.
Fig. 4 represents the root mean square fluctuations (RMSF) of PLpro before and after docking with DCG during MD simulation. It is clear that the fluctuation of the docked structure is more compared to the undocked structure, similar to the RMSD plot (Fig. 3). In the undocked PLpro, fluctuations arise more in residues such as ARG82, LYS190, GLN194, HIS255. In the docked PLpro, more fluctuations are seen in residues e.g. ARG3 to THR54, LYS94, LYS126, PHE127, ASN128, LYS228, TYR268, GLN269, CYS270, GLY271, TYR305. It is clear that there is higher fluctuation in the docked vis-a-vis undocked PLpro. In other words, the docked PLpro structure is less stable compared to the undocked structure, as observed from the RMSD plot.
Fig. 4.

Root mean square fluctuation (RMSF) of PLpro in docked and undocked state.
Fig. 5a illustrates how the radius of gyration of PLpro changes with time before and after docking with DCG. Radius of gyration is a measure of compactness of a system. From Fig. 5a, it is clear that the docked structure is less compact compared to the undocked one. From the beginning of the simulation process it is concluded from Fig. 5a that the docked structure has lesser compactness compared to undocked one and at the end of simutation higher fluctuation indicates loss of compactness. This is in agreement with previous observations. Fig. 5b indicates the change in solvent accessible surface area (SASA) with time of the two structures. Here also, the values fluctuate more in case of docked structure. SASA values of undocked PLpro seem to remain a little less than that of the docked structure, indicating less compactness of the latter.
Fig. 5.

Radius of Gyration of undocked and docked PLpro (a) and solvent accessible surface area (SASA) plot for undocked and docked PLpro (b).
Conformational energy, number of H-bonds and average H-bond distance of undocked and docked PLpro are presented in Table 5 . Greater conformational energy in the composite system suggests greater flexibility of it in comparison to PLpro alone. The number of H- bonds increases in the docked structure vis-a-vis the undocked one. The average H-bond distance becomes shorter in docked structure after MD.
Table 5.
Conformational energy and number of H-bonds along with average distances in undocked and docked PLpro.
| System | Conformational Energy (kJ/mol) | Number of H-bond | Average distance (Å) of H-bond |
|---|---|---|---|
| PLpro | 18,034.873 | 248 | 2.047 |
| PLpro+DCG | 18,511.076 | 258 | 2.038 |
Fig. 6 indicates structural changes after each 10 ns period in undocked (brown) and docked (sky blue) PLpro during MD simulation. It is observed that DCG creates a noticeable structural deformation in chain A of PLpro to destabilize its structure. Huge impact of DCG is seen after 40 ns of docked structure as compared to undocked one during the simulation as shown in the Fig. 6. The structures of PLpro and PLpro-DCG complex are seen to alter after each 10 nanosecond. After 40 ns of MD simulation, conformational change of docked structure is noticeable in concordance with Fig. 3. From the above analysis, we can conclude that while docking takes place in the first few nanoseconds, destabilization of the docked structure takes place from 40 ns onwards.
4. Conclusion
The inhibitory effect of several molecules extracted from Neem (A. Indica) on PLpro protein of the SARS-CoV-2 (COVID-19) has been studied in the present work. All the 19 molecules studied showed potential docking ability with PLpro, along with toxicity and other values, while being nontoxic themselves, in comparison with 7 drugs currently being tested against COVID-19. Among them, desacetylgedunin (DCG) gives the highest docking score. Molecular dynamics study reveals that there is a large impact on PLpro structure by DCG. Bonding between the two is facilitated by H-bonds, electrostatic and van der Waals interactions. Deformation of PLpro structure upon docking with DCG is confirmed from RMSD, RMSF, SASA plots, as well as from direct comparison between resulting structures of MD-simulation of PLpro-DCG vs PLpro after every nanosecond. Based on the results, it can be said that DCG causes a huge impact on PLpro upon binding with the latter. This calls for further investigation on the system, as well as with other molecules of Neem extract on other aspects of COVID-19.
Data availability
Data is available upon request to the corresponding author.
CRediT authorship contribution statement
Nabajyoti Baildya: Conceptualization, Data curation, Formal analysis, Methodology, Validation, Visualization, Writing - original draft. Abdul Ashik Khan: Data curation, Investigation, Formal analysis. Narendra Nath Ghosh: Conceptualization, Methodology, Project administration, Software, Supervision, Writing - original draft. Tanmoy Dutta: Data curation, Investigation, Writing - original draft, Visualization. Asoke P. Chattopadhyay: Project administration, Software, Supervision, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors wish to acknowledge infrastural support from Department of Chemistry, University of Kalyani, Kalyani, Nadia, India.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2020.129390.
Appendix. Supplementary materials
References
- 1.Xu X., Chen P., Wang J., Feng J., Zhou H., Li X., Zhong W., Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Science China Life Sciences. 2020;63(3):457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng J. SARS-CoV-2: an emerging coronavirus that causes a global threat. Int. J. Biol. Sci. 2020;16(10):1678. doi: 10.7150/ijbs.45053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.C.S.G. of the International The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology. 2020;5(4):536. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belouzard S., Millet J.K., Licitra B.N., Whittaker G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012;4(6):1011–1033. doi: 10.3390/v4061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Báez-Santos Y.M., John S.E.S., Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharif-Yakan A., Kanj S.S. Emergence of MERS-CoV in the Middle East: origins, transmission, treatment, and perspectives. PLoS Pathog. 2014;10(12) doi: 10.1371/journal.ppat.1004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baildya N., Ghosh N.N., Chattopadhyay A.P. Inhibitory activity of hydroxychloroquine on COVID-19 main protease: an insight from MD-simulation studies. J Mol Struct. 2020 doi: 10.1016/j.molstruc.2020.128595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elfiky A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci. 2020 doi: 10.1016/j.lfs.2020.117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subapriya R., Nagini S. Medicinal properties of neem leaves: a review. Current Medicinal Chemistry-Anti-Cancer Agents. 2005;5(2):149–156. doi: 10.2174/1568011053174828. [DOI] [PubMed] [Google Scholar]
- 10.Tiwari V., Darmani N.A., Yue B.Y., Shukla D. In vitro antiviral activity of neem (Azardirachta indica L.) bark extract against herpes simplex virus type‐1 infection. Phytotherapy Research. 2010;24(8):1132–1140. doi: 10.1002/ptr.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar V.S., Navaratnam V. Neem (Azadirachta indica): prehistory to contemporary medicinal uses to humankind. Asian Pac J Trop Biomed. 2013;3(7):505–514. doi: 10.1016/S2221-1691(13)60105-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Council N.R. The Minerva Group, Inc.; 2002. Neem: a tree for solving global problems. [Google Scholar]
- 13.Udeinya I., Mbah A., Chijioke C., Shu E. An antimalarial extract from neem leaves is antiretroviral. Trans. R. Soc. Trop. Med. Hyg. 2004;98(7):435–437. doi: 10.1016/j.trstmh.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 14.Jalil A., Ashfaq U.A., Shahzadi S., Javed M.R., Rasul I., Rehman S.-u., Shah M., Masoud M.S. Screening and design of anti-diabetic compounds sourced from the leaves of neem (Azadirachta indica) Bioinformation. 2013;9(20):1031. doi: 10.6026/97320630091031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.A.K. Srivastava, R. Maurya, Antihyperglycemic activity of compounds isolated from Indian medicinal plants, (2010). [PubMed]
- 16.Koley K., Lal J. Pharmacological effects of Azadirachta indica (neem) leaf extract on the ECG and blood pressure of rat. Indian J. Physiol. Pharmacol. 1994;38 223-223. [PubMed] [Google Scholar]
- 17.Khan M., Qasim M., Ashfaq U.A., Idrees S., Shah M. Computer aided screening of Accacia nilotica phytochemicals against HCV NS3/4a. Bioinformation. 2013;9(14):710. doi: 10.6026/97320630009710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osipiuk J., Jedrzejczak R., Tesar C., Endres M., Stols L., Babnigg G., Kim Y., Michalska K., Joachimiak A. The crystal structure of papain-like protease of SARS CoV-2. RSCB PDB. 2020 [Google Scholar]
- 19.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 20.Trott O., Olson A.J., Vina AutoDock. improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berendsen H.J., van der Spoel D., van Drunen R., GROMACS a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91(1–3):43–56. [Google Scholar]
- 22.Lee S., Tran A., Allsopp M., Lim J.B., Hénin J.r.m., Klauda J.B. CHARMM36 united atom chain model for lipids and surfactants. The Journal of Physical Chemistry B. 2014;118(2):547–556. doi: 10.1021/jp410344g. [DOI] [PubMed] [Google Scholar]
- 23.Boonstra S., Onck P.R., van der Giessen E. CHARMM TIP3P water model suppresses peptide folding by solvating the unfolded state. The journal of physical chemistry B. 2016;120(15):3692–3698. doi: 10.1021/acs.jpcb.6b01316. [DOI] [PubMed] [Google Scholar]
- 24.Abraham M.J., Gready J.E. Optimization of parameters for molecular dynamics simulation using smooth particle‐mesh Ewald in GROMACS 4.5. Journal of Computational Chemistry. 2011;32(9):2031–2040. doi: 10.1002/jcc.21773. [DOI] [PubMed] [Google Scholar]
- 25.Pires D.E., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58(9):4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is available upon request to the corresponding author.