

Abstract
Aggregation of the islet amyloid polypeptide (IAPP) to form fibrils and oligomers is important in the progression of type 2 diabetes. This paper describes X-ray crystallographic and solution-state NMR studies of peptides derived from residues 11–17 of IAPP that assemble to form tetramers. Incorporation of residues 11–17 of IAPP (RLANFLV) into a macrocyclic β-sheet peptide results in a monomeric peptide that does not self-assemble to form oligomers. Mutation of Arg11 to the uncharged isostere citrulline gives peptide homologues that assemble to form tetramers in both the crystal state and in aqueous solution. The tetramers consist of hydrogen-bonded dimers that sandwich together through hydrophobic interactions. The tetramers share several features with structures reported for IAPP fibrils and demonstrate the importance of hydrogen bonding and hydrophobic interactions in the oligomerization of IAPP-derived peptides.
Graphical Abstract
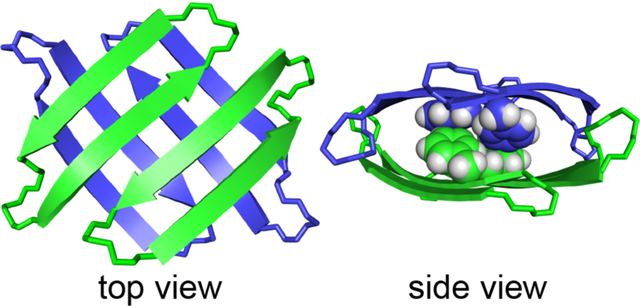
Introduction
Interactions among β-sheets are central to the misfolding and aggregation of peptides and proteins that form toxic oligomers and insoluble fibrils in over 40 amyloid diseases, including type 2 diabetes, Alzheimer’s disease, and Parkinson’s disease.1,2 The fibrils share the common feature of an extended network of hydrogen-bonded β-sheets that are further stabilized through hydrophobic packing.3–11 Less is known about the structures of the smaller, often metastable and polymorphic oligomers, but β-sheets and hydrophobic interactions appear to be important in oligomer formation.12–17
The 37-residue islet amyloid polypeptide (IAPP) aggregates to form fibrils and oligomers that are central to islet β-cell death in the progression of type 2 diabetes.18–23 Tycko and coworkers proposed structural models of IAPP fibrils using constraints from solid-state NMR spectroscopy.4 In these models, a fibril consists of layers of parallel, in-register β-sheets that run perpendicular to the length of the fibril. β-Strands consisting of central residues 8–17 form one of the layers, while β-strands comprising C-terminal residues 28–36 form the other layer. A loop region containing residues 18–27 connects the two β-strands. Using X-ray crystallographic structures derived from fragments of IAPP, Eisenberg and coworkers constructed a similar fibril model in which the side chains are more tightly packed.5,24 Langen and coworkers proposed a model of IAPP fibrils using constraints from EPR spectroscopy in which the β-strands pack more loosely.25 The residues involved in β-sheet formation vary slightly among these different models, but central residues 12–17 and C-terminal residues 31–36 are generally thought to be involved in β-strand formation.
Although the structures of IAPP oligomers are not known at high resolution, they appear to be composed of β-sheets. Through ion mobility-mass spectrometry (IM-MS) studies in conjunction with replica exchange molecular dynamics (REMD), Bowers and coworkers proposed β-hairpin building blocks for human IAPP oligomers.26,27 Hoyer and coworkers obtained an NMR-based structure of IAPP in a β-hairpin conformation bound to an affibody and suggested that this β-hairpin might be involved in IAPP aggregation.23 The β-hairpin comprises residues 12–28; residues 12–18 and residues 22–28 form β-strands, while residues 19–21 form a turn. In a separate study, Zanni and coworkers used 2D IR spectroscopy to show that residues 23–27 have β-sheet structure in the oligomer but form a disordered loop in the fibril.28
Our laboratory recently introduced macrocyclic β-sheet peptides as a platform to explore the interactions among β-sheets in amyloidogenic peptides and proteins (Chart 1).28,29 The macrocyclic β-sheet peptides contain a peptide strand and a template strand that are connected by two d-linked ornithine (δOrn) turn units.30,31 The peptide strand contains a heptapeptide sequence derived from an amyloidogenic peptide or protein. The template strand contains four additional a-amino acids and the unnatural amino acid Hao, which mimics a tripeptide β-strand, templates β-sheet formation, and blocks uncontrolled aggregation.32 Our laboratory has used macrocyclic β-sheet peptides containing heptapeptide segments from the β-amyloid peptide Aβ, β2-microglobulin, and asynuclein as inhibitors to control amyloid aggregation and reduce amyloid toxicity.29 Recently, we used NMR spectroscopy to study the homotetramers and heterotetramers formed by macrocyclic β-sheet peptides containing residues 17–23 and 30–36 from Aβ.33,34
Chart 1.
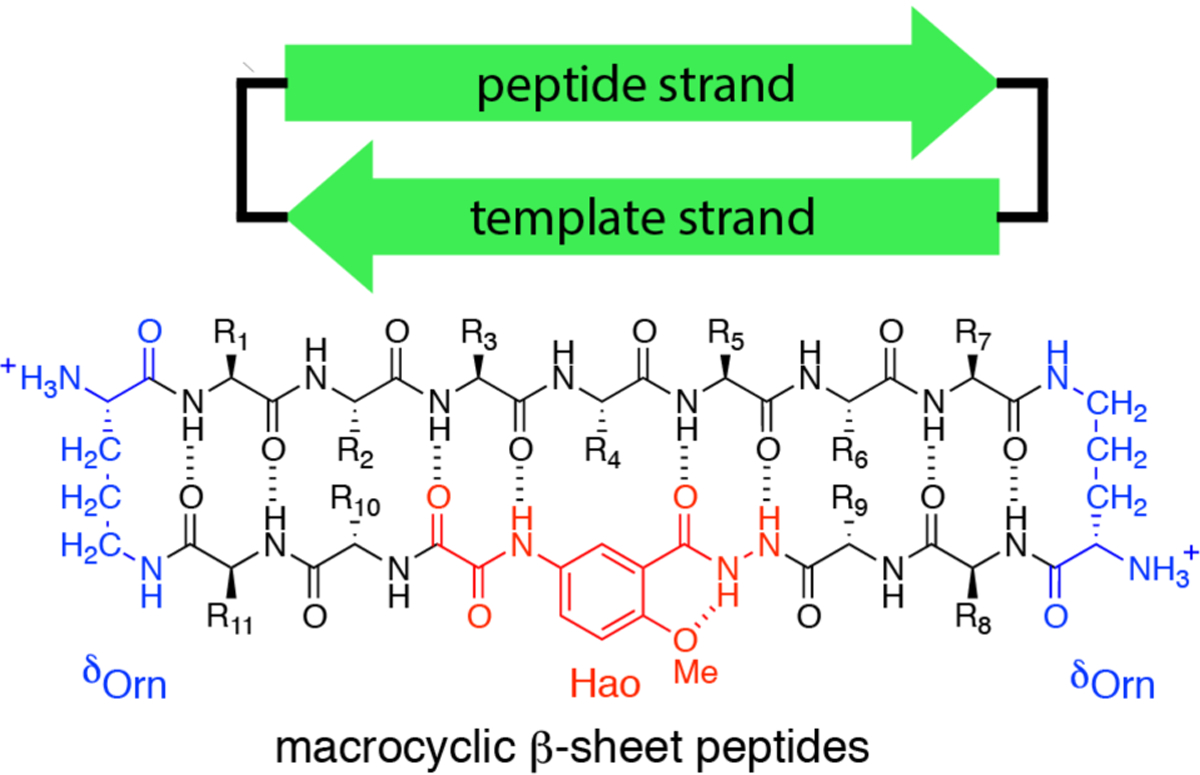
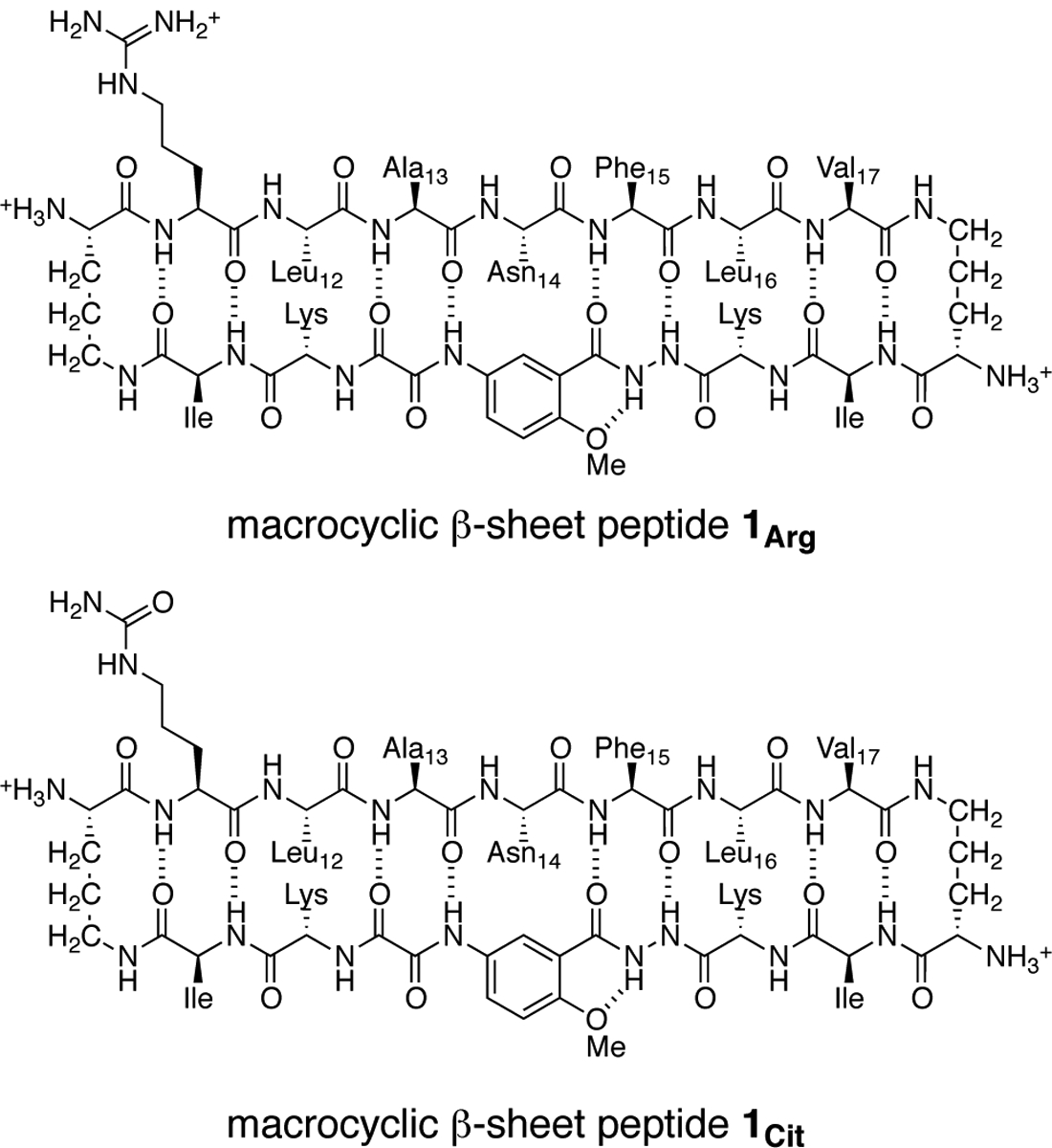
In the current study, we use macrocyclic β-sheet peptides as a platform to explore assembly of the central region of IAPP. We designed peptide 1Arg to contain central residues 11–17 of IAPP (RLANFLV). We chose the IAPP11–17 heptapeptide for its propensity to form a β-strand in fibrils and β-hairpin monomers. We also designed homologue peptide 1Cit, in which we mutated Arg11 to the uncharged isostere of arginine, citrulline (Chart 2). Here we describe NMR spectroscopic studies of peptides 1Arg and 1Cit, report the X-ray crystallographic structure of a tetramer formed by a homologue of peptide 1Cit, and correlate the X-ray crystallographic structure with the solution-phase assembly of the IAPP-derived peptides.
Chart 2.
Results
1. Design and 1H NMR Studies of Peptides 1Arg and 1Cit.
The β-strand formed by IAPP11–17 displays a hydrophobic surface comprising the side chains of Ala13, Phe15, and Val17, as well as Arg11. To reinforce the hydrophobicity of this surface, we incorporated isoleucine residues at positions R8 and R11 of the template strand. We incorporated lysine residues at positions R9 and R10 to increase solubility of the peptide and render the surface displaying the side chains of Leu12, Asn14, and Leu16 hydrophilic.
In aqueous solution, peptide 1Arg is monomeric. The 1H NMR spectrum of peptide 1Arg at 4 mM exhibits a single set of sharp resonances, associated with the monomeric peptide (Fig. 1). The resonances associated with the α-protons are in the 4–5 ppm range, showing little to no downfield shifting relative to the typical values for the corresponding residues in a random coil conformation (Fig. S3).35 At 8 mM, we observe broadening of the resonances but no additional peaks associated with assembly of the peptide (Fig. S1). We hypothesized that the positive charge on Arg11 may prevent peptide 1Arg from oligomerizing. To explore this hypothesis, we replaced Arg11 with the neutrally charged isostere citrulline, reducing the net charge from +5 to +4.
Fig. 1.
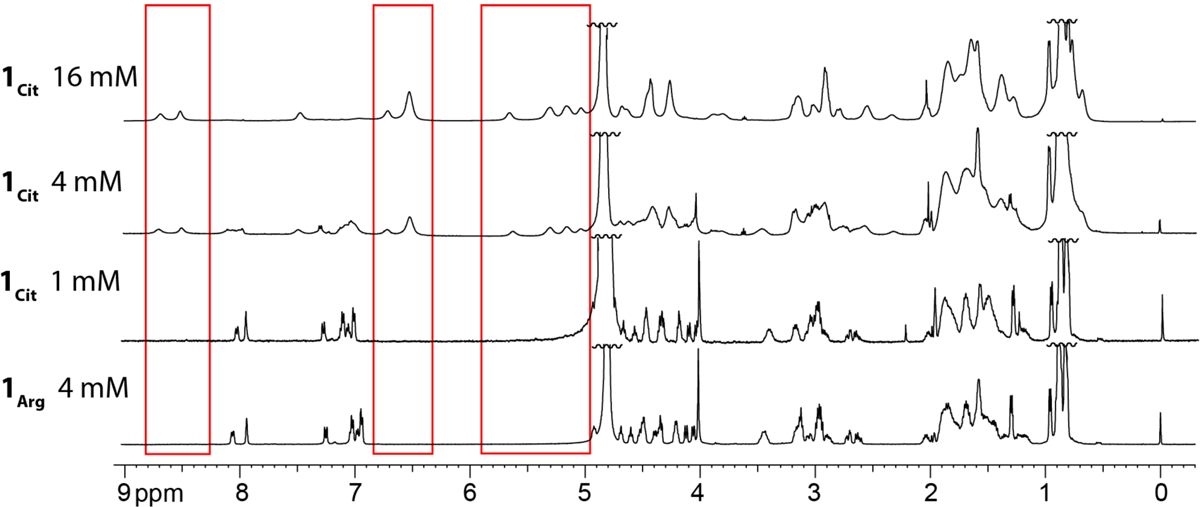
1H NMR spectra of macrocyclic β-sheet peptides 1Arg and 1Cit in 50 mM CD3COOD and 50 mM CD3COONa buffer in D2O at 298 K. The red boxes highlight noteworthy resonances of the tetramer.
Peptide 1Cit is monomeric at low concentrations but forms a tetramer at higher concentrations. At 1 mM, the spectrum is similar to that of peptide 1Arg (Fig. 1 and S2). At 4 mM, peptide 1Cit displays two sets of broad resonances associated with the monomer and the tetramer. At 16 mM, only the resonances associated with the tetramer are visible. The resonances associated with the α-protons of the tetramer are shifted downfield by up to 1 ppm relative to those of random coil values (Fig. S4).35 This downfield shifting reflects the folding and assembly of the peptide into the tetramer. The aromatic resonances associated with the Phe15 side chain are shifted upfield by approximately 0.5 ppm, to 6.5 ppm, suggesting that the side chains of Phe15 pack to form a hydrophobic core within the tetramer.
Comparison of 1H NMR spectra of peptide 1Cit at 1 mM and 16 mM shows that folding accompanies oligomerization. The magnetic anisotropy of the diastereotopic δ-protons of the δOrn turn units reflects β-sheet folding in macrocyclic β-sheet peptides.30,31 In a well-folded macrocyclic β-sheet peptide, each diastereotopic pro-S δ-proton resonance is about 0.6 ppm downfield of the corresponding pro-R δ-proton resonance. The monomer subunits of the tetramer formed by peptide 1Cit are well folded, with the difference in chemical shifts of the diastereotopic pro-S and pro-R δ-protons being 0.66 and 0.70 ppm. In contrast, monomeric peptide 1Cit is partially folded, with the difference in chemical shifts of the diastereotopic pro-S and pro-R δ-protons being 0.19 and 0.29 ppm.
2. X-ray Crystallographic Structure of a Tetramer Derived from IAPP11–17.
To further characterize the structure of the tetramer formed by peptide 1Cit, we turned to X-ray crystallography. To facilitate X-ray crystallographic phase determination, we designed and synthesized homologue peptide 2Cit (Chart 3). Peptide 2Cit contains the leucine isostere, (2-bromoallyl)glycine, in place of one of the isoleucines in the template strand to allow X-ray crystallographic phase determination by means of single-wavelength anomalous diffraction phasing.13 Peptide 2Cit afforded crystals suitable for X-ray diffraction in conditions containing sodium citrate buffer, isopropanol, and poly(ethylene glycol) (PEG) 4,000.
Chart 3.
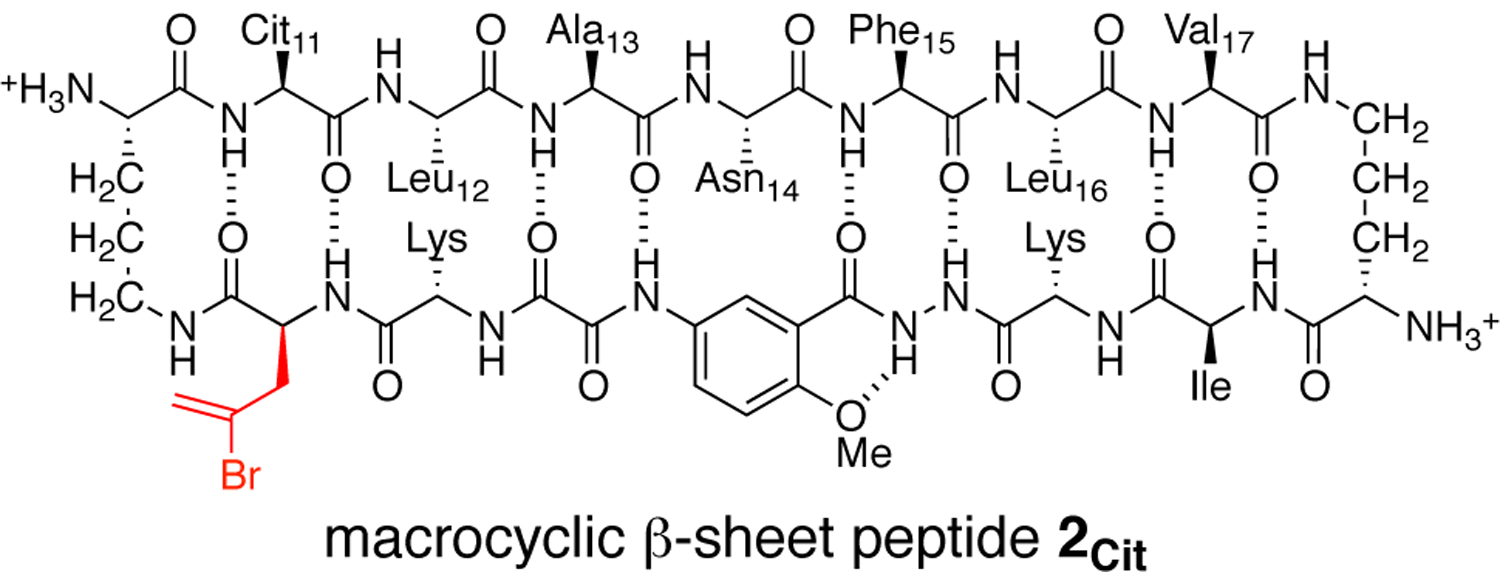
The asymmetric unit contains four folded macrocyclic β-sheets, which pack to form a tetramer. The heptapeptide β-strand of each monomer subunit is hydrogen bonded to the template strand through a network of eight hydrogen bonds (Fig. 2A). The monomers in the asymmetric unit are similar in conformation with little variation in the side chains. One notable difference is in the Phe15 residues, which exhibit two rotamers (Fig. 2B).
Fig. 2.
X-ray crystallographic structure of peptide 2Cit (PDB 5UHR). (A) Representative monomer subunit. (B) Overlay of the four monomers in the asymmetric unit.
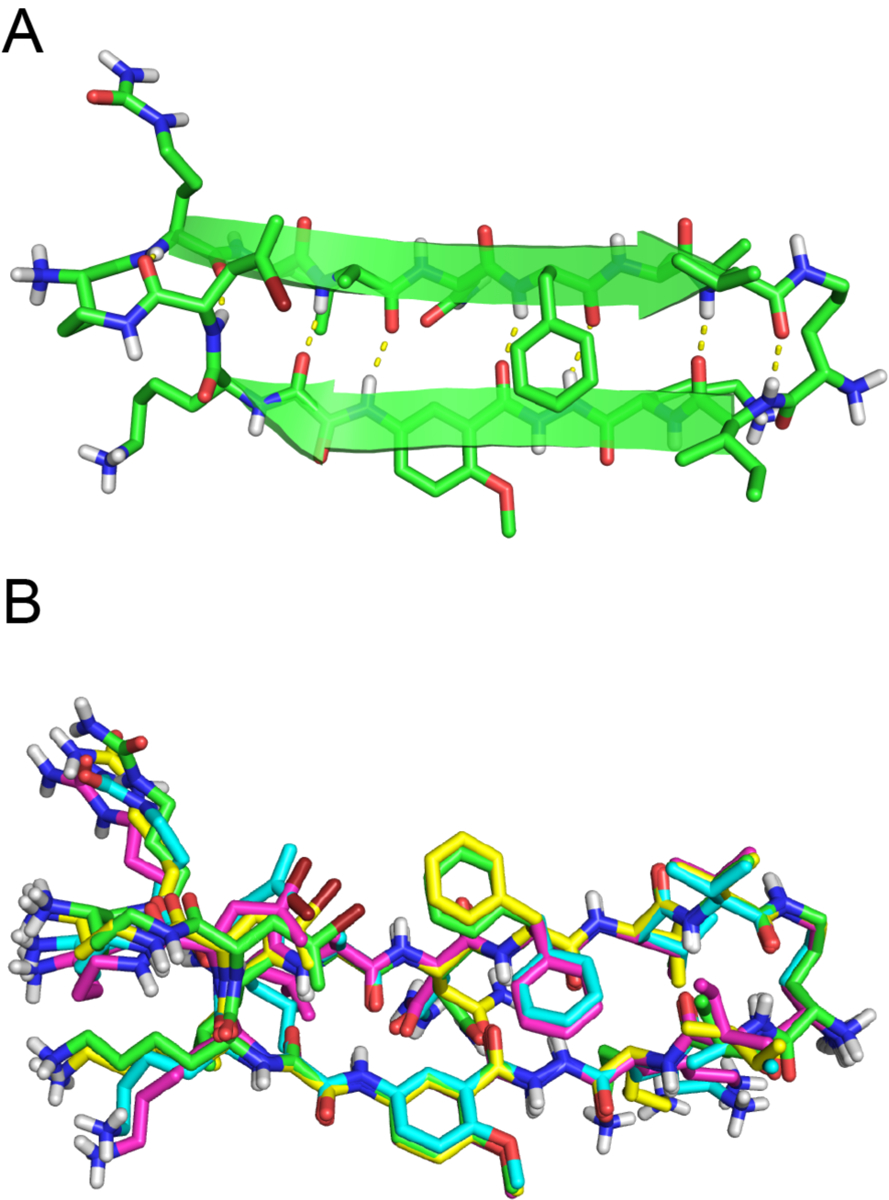
The monomer subunits pair through edge-to-edge hydrogen bonding to form antiparallel β-sheet dimers (Fig. 3). The two heptapeptide β-strands in the dimer are shifted by two residues towards the C-termini. Six intermolecular hydrogen bonds form between the two monomers. The dimer presents two surfaces. One surface is hydrophobic and displays the side chains of Cit11, Ala13, Phe15, and Val17 of the peptide strand, as well as the Ile and (2-bromoallyl)glycine of the template strand (Fig. 3A). The two Phe15 side chains on the hydrophobic surface adopt different rotamers. The other surface is hydrophilic and displays the Leu12, Asn14, and Leu16 side chains of the peptide strand, as well as the two Lys residues of the template strand (Fig. 3B).
Fig. 3.
X-ray crystallographic structure of the dimer formed by peptide 2Cit. (A) View of the hydrophobic surface with side chains shown as spheres. (B) View of the hydrophilic surface with side chains shown as spheres.
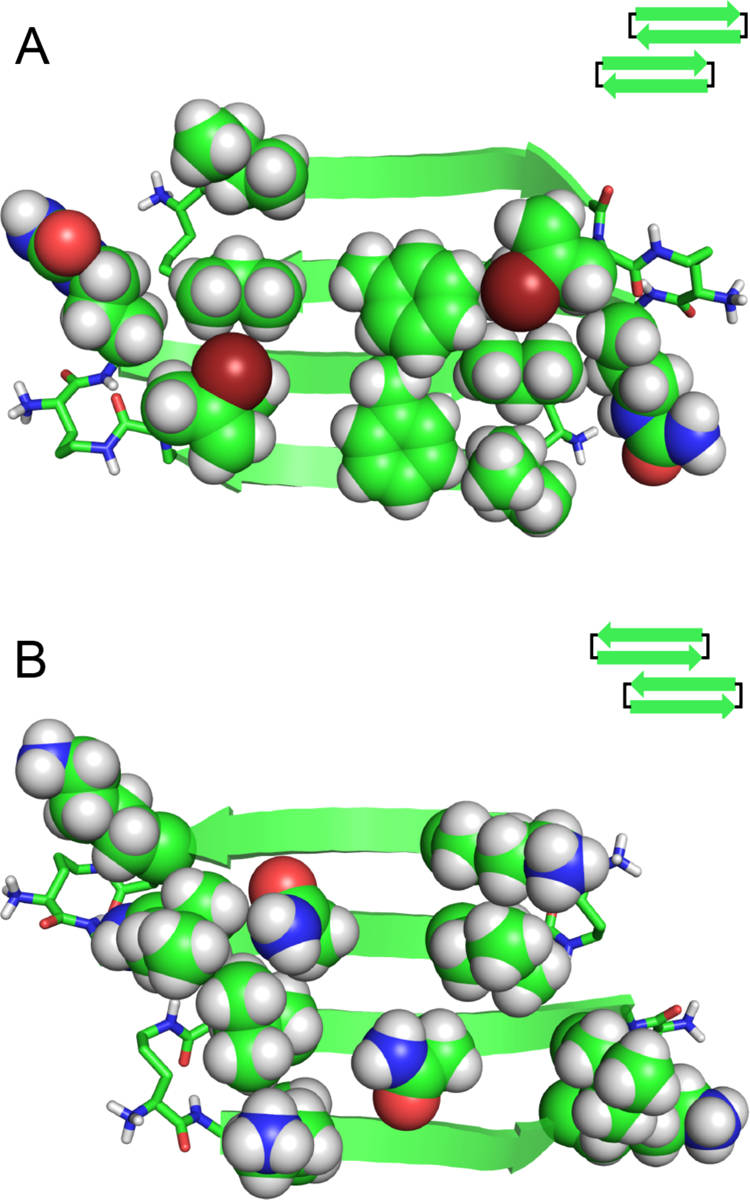
Two hydrogen-bonded dimers sandwich together on their hydrophobic surfaces to form a tetramer (Fig. 4). In the tetramer, the β-sheets of the dimers are nearly orthogonal. The side chains of Ala13, Phe15, Val17, Ile, and (2-bromoallyl)glycine create a hydrophobic core within the sandwich, with the four phenyl groups packing at the center of the sandwich. The two rotamers of the Phe15 side chains allow the phenyl groups to pack together tightly through hydrophobic interactions and aromatic stacking at the center of the hydrophobic core. The side chains of Ala13, Val17, Ile, and (2-bromoallyl)glycine surround the four Phe15 side chains, packing through hydrophobic interaction to stabilize the tetramer. The hydrophilic side chain of Cit11 extends out of the hydrophobic core. The hydrophilic surfaces of the dimer subunits that comprise the tetramer are exposed to solvent.
Fig 4.
X-ray crystallographic structure of the tetramer of peptide 2Cit. (A) Top view. (B) Side view. Sphere representations of Phe15 show packing of the aromatic side chains in the hydrophobic core.
3. Solution-State Studies of a Tetramer Derived from IAPP11–17.
Diffusion-ordered spectroscopy (DOSY) provides a useful tool for assessing the relative oligomerization state of macrocyclic β-sheets peptides.33,34,36,37 Oligomers diffuse more slowly than the monomers, with dimers having a diffusion coefficient of about 0.75–0.79 times that of the monomer and tetramers having a diffusion coefficient of about 0.58–0.63 times that of the monomer.33,37,38 At 298 K, the diffusion coefficient of peptide 1Arg is 19.6 × 10−7 cm2/s in deuterioacetate buffered D2O (Table 1). At 1 mM, peptide 1Cit is monomeric, with a diffusion coefficient of 20.0 × 10−7 cm2/s, which is the same as peptide 1Arg within the limits of experimental error. At 2 mM, peptide 1Cit is largely monomeric, with a diffusion coefficient of 18.9 × 10−7 cm2/s.
Table 1.
Diffusion coefficients (D) of peptides 1Arg and 1Cit in 50 mM CD3COOD and 50 mM CD3COONa buffer in D2O at 298 K
| MWmonomer (Da) |
MWtetramer (Da) |
Conc. (mM) |
D (× 10−7 cm2/s) |
Oligomer state | |
|---|---|---|---|---|---|
| 1Arg | 1759 | 7036 | 4 | 19.6 ± 0.6 | monomer |
| 1cit | 1760 | 7040 | 1 | 20.0 ± 2.0 | monomer |
| 1cit | 1760 | 7040 | 2 | 18.9 ± 1.2 | monomer |
| 1cit | 1760 | 7040 | 4 | 17.4 ± 1.2 | mixture |
| 1cit | 1760 | 7040 | 8 | 14.2 ± 0.3 | mixture |
| 1cit | 1760 | 7040 | 16 | 12.4 ± 0.3 | tetramer |
As the concentration is increased and the resonances from the oligomer grow in, the diffusion coefficient of peptide 1Cit decreases. At 4 mM and 8 mM, discrete diffusion coefficients cannot be measured for the resonances associated with the monomer and the oligomer, because exchange occurs at a rate similar to the 75-ms time scale of the experiment. Instead, an averaged diffusion coefficient for the mixture is observed. At 4 mM, the diffusion coefficient is 17.4 × 10−7 cm2/s, and at 8 mM the diffusion coefficient is 14.2 × 10−7 cm2/s. At 16 mM, the oligomer predominates vastly, and only resonances for the oligomer are observed. At 16 mM, the diffusion coefficient is 12.4 × 10−7 cm2/s. This value is about 0.6 times that of the monomer of peptide 1Cit, and is consistent with a tetramer.
NOESY NMR spectroscopy establishes that the structure of the tetramer formed by peptide 1Cit in aqueous solution is similar to the tetramer observed for peptide 2Cit in the crystal. We observe NOEs from the folding of the monomers and formation of the dimers. Two strong crosspeaks in the NOESY spectrum reflect folding of macrocyclic β-sheet peptide 1Cit, one between the α-protons of Leu and Lys, and one between the α-proton of Asn14 and the proton at the 6-position of the unnatural amino acid Hao (Fig. 5). NOEs between the α-proton and pro-S δ-proton of the δOrn turn units further reflect folding of the macrocycle. A strong crosspeak between the α-protons of Ala13 and Val17 reflect shifted antiparallel dimer formation. An additional NOE between the methoxy protons of Hao and the Ile side chain reflect packing of the dimers in a sandwich-like fashion (Fig. 6). The interlayer NOE between the Hao-methoxy and Ile side chain is characteristic of sandwich-like tetramers of Hao-containing macrocyclic β-sheet peptides.33 These NOEs associated with folding, dimerization, and packing reflect the congruence of the X-ray crystallographic and solution-state tetramers.
Fig 5.
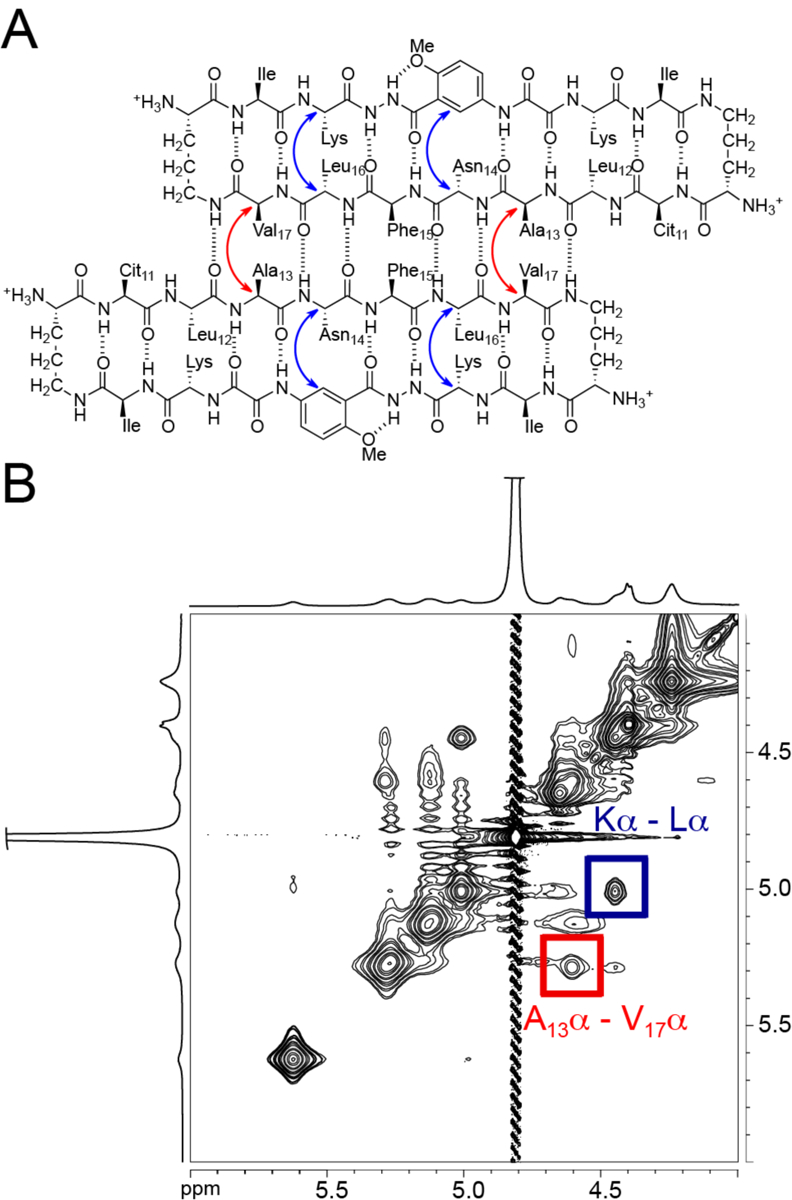
NOEs associated with the dimer subunit of peptide 1Cit. (A) Shifted, antiparallel hydrogen-bonded dimer. Blue arrows indicate intramolecular NOEs and red arrows indicate intermolecular NOE observed in the NOESY spectrum. (B) Expansion of the NOESY spectrum showing selected intramolecular (blue) and intermolecular (red) NOE crosspeaks.
Fig 6.
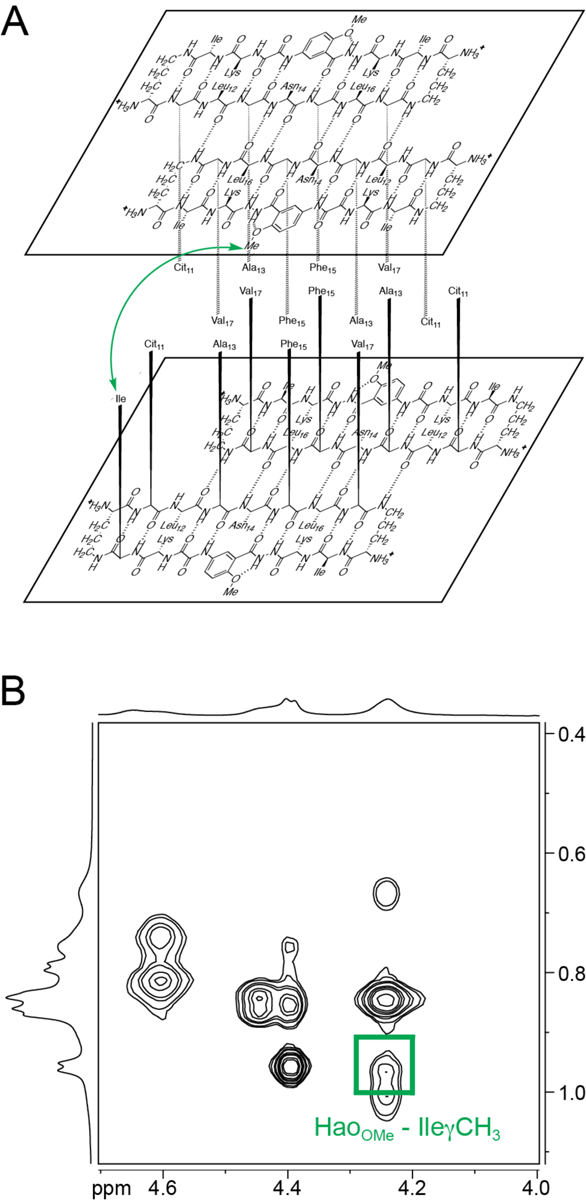
NOE associated with the tetramer of peptide 1Cit. (A) Green arrow indicates interlayer NOE observed in the NOESY spectrum. (B) NOE crosspeak between the Ile side chain and the methoxy protons of the unnatural amino acid Hao.
Discussion
To understand why peptide 1Cit forms tetramers but peptide 1Arg does not, we used the X-ray crystallographic structure of peptide 2Cit to generate molecular models of tetramers of peptides 1Cit and 1Arg. We modeled the tetramer formed by peptide 1Cit by mutating the (2-bromoallyl)glycine residue to isoleucine and minimizing the resulting structure in MacroModel with the MMFFs force field with GB/SA water solvation (Fig. S5). We modeled a tetramer of peptide 1Arg in a similar fashion, mutating both the (2-bromoallyl)glycine and the citrulline residues. Both molecular models overlay well with the X-ray crystallographic structure of the tetramer of peptide 2Cit (RMSD ~1 Å). Inspection of the dimer subunits suggests an explanation of the differing stabilities of the 1Cit and 1Arg tetramers (Fig. 7). In the dimer subunit of peptide 1Arg, the cationic guanidinium group of each arginine is near the ammonium group of one of the δOrn turn units, while in the dimer subunit of peptide 1Cit, the neutral urea group of citrulline is near the ammonium group. Thus, it appears that charge-charge repulsion destabilizes the dimer and hence the tetramer of peptide 1Arg.
Fig 7.
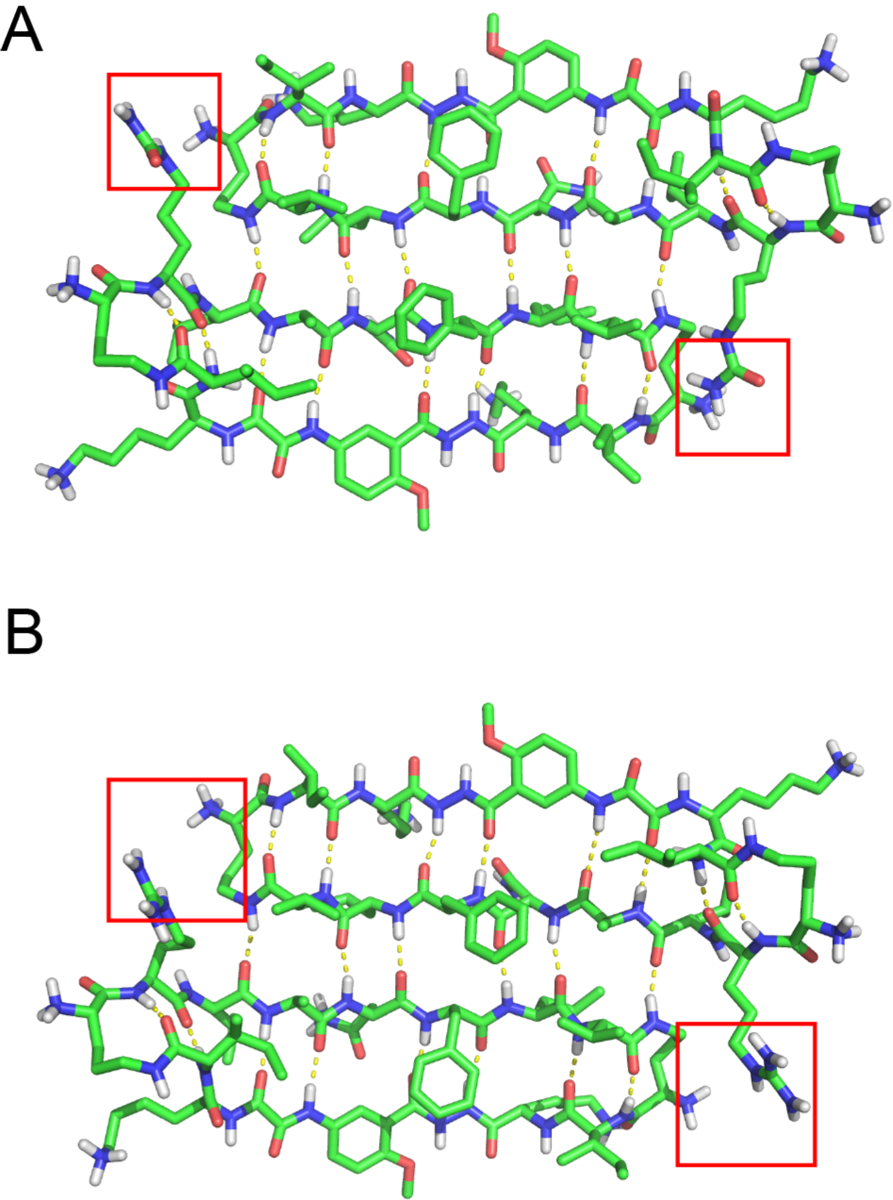
Crystallographically based molecular models of the dimer subunits of the tetramers of peptide 1Cit (A) and 1Arg (B). The red boxes highlight interactions between the ammonium groups of the δOrn turn units and the Arg11 or Cit11 side chains.
The X-ray crystallographic structure and molecular model of the tetramers formed by peptides 2Cit and 1Cit share several features with structures reported for IAPP fibrils. Like the tetramers, the full atomic models of IAPP described by Tycko and coworkers consist of layered β-sheets in which hydrophobic interactions stabilize the layered structure. In contrast to the tetramers of peptides 2Cit and 1Cit, the fibrils consist of parallel β-sheets formed by IAPP rather than antiparallel β-sheets. In the two different fibril models proposed by Tycko and coworkers, the Phe15 side chains are on either the interior or the exterior of the layers. The fragment-based model of IAPP fibrils proposed by Eisenberg and coworkers also consists of layered parallel β-sheets, with the side chains of Phe15 on the exterior of the layers. Recently, Eisenberg and coworkers reported that IAPP15–25 forms fibrils consisting of loosely laminated antiparallel β-sheets.39 Hydrophobic interactions of Phe15 appear to be more important in the NMR structure of the IAPP β-hairpin bound to an affibody reported by Hoyer et al., in which the Phe15 and Phe23 pack tightly against one of the phenylalanine residues in the affibody to form a central core.23 This mode of assembly is similar to the packing of Phe15 in the tetramers formed by peptides 1Cit and 2Cit.
The tetramers formed by peptides 1Cit and 2Cit share common features with assemblies formed by other Hao-containing macrocyclic β-sheet peptides that our laboratory has studied (Chart 4). Several Hao-containing macrocyclic β-sheet peptides containing heptapeptide sequences from various amyloidogenic peptides and proteins form hydrogen-bonded assemblies laminated through hydrophobic interactions in the crystal state.29,40 We have also identified two Hao-containing macrocyclic β-sheet peptides containing heptapeptide sequences from the β-amyloid peptide Aβ, which assemble to form tetramers in solution.33,34 The flat, hydrophobic Hao group appears to facilitate the formation of these layered structures. These structures differ from those that we have observed for N-methylated macrocyclic β-sheet peptides, which we have also studied (Chart 4). Most of the N-methylated macrocyclic β-sheet peptides containing sequences from various amyloidogenic peptides and proteins that we have observed in the crystal state form compact higher-order oligomers, such as hexamers, octamers, nonamers, and dodecamers.15,16,41,42 The N-methylated macrocyclic β-sheet peptides are generally more twisted than the Hao-containing macrocyclic β-sheet peptides. This twisting appears to facilitate assembly through interactions between curved hydrophobic surfaces, in addition to hydrogen bonding. The flat sandwich-like structures formed by the Hao-containing macrocyclic β-sheets resemble the laminated structures of amyloid fibrils, while the compact globular structures formed by the N-methylated macrocyclic β-sheets might offer a glimpse into the structures of amyloid oligomers.
Chart 4.
Conclusion
The 1H NMR, X-ray crystallographic, and molecular modeling studies of peptides 1Arg, 1Cit, and 2Cit described above demonstrate the importance of hydrogen bonding and hydrophobic interactions in the oligomerization of IAPP-derived peptides. Peptide 1Arg remains monomeric in aqueous solution, whereas peptides 1Cit and 2Cit assemble to form tetramers in aqueous solution and in the crystal state. The differences between peptides 1Arg and 1Cit suggest that charge-charge interactions can modulate oligomer formation.
Our laboratory has previously used NMR spectroscopy and X-ray crystallography as complimentary techniques to investigate the assembly of different macrocyclic β-sheet peptides derived from Aβ.37,43 In these studies, the macrocyclic β-sheet peptides assembled to form tetramers both in solution and in the crystal, but the structure of the tetramer formed in solution differed from the structure formed in the crystal. The findings of the current paper are significant because they demonstrate that macrocyclic β-sheet peptides can assemble to form the same structure in solution and in the crystal state. Furthermore, the tetramer described here might serve as a structural model for understanding important contacts within oligomers and fibrils formed by full-length IAPP.
Experimental Section
Synthesis of Macrocyclic β-Sheet Peptides.
Synthesis and purification of the macrocyclic β-sheet peptides were performed as previously described.33 The peptides were purified by reverse-phase HPLC and the pure fractions were lyophilized to give 22 mg of peptide 1Arg, 37 mg of peptide 1Cit, and 19 mg of 2Cit. Based on resin loading, the yields are 9%, 14%, and 7%, respectively.
Macrocyclic β-Sheet peptide 1Arg:
1H NMR, (500 MHz, 4 mM in D2O, 298K) δ 8.06 (d, J = 8.6 Hz, 1H), 7.94 (s, 1H), 7.25 (d, J = 9.1 Hz, 1H), 6.92–7.05 (m, 5H), 4.92 (t, J = 7.2 Hz, 1H), 4.69 (t, J = 6.7 Hz, 1H), 4.60 (t, J = 7.1 Hz, 1H), 4.46–4.55 (m, 3H), 4.32–4.42 (m, 3H), 4.21 (m, 2H), 4.12 (d, J = 9.3 Hz, 1H), 4.06 (d, J = 8.7 Hz, 1H), 4.02 (s, 3H), 3.38–3.50 (m, 2H), 3.09–3.20 (m, 4H), 3.05 (dd, J = 14.2, 5.5 Hz, 1H), 2.92–3.02 (m, 4H), 2.88 (dd, J = 14.0, 6.6 Hz, 1H), 2.71 (dd, J = 15.3, 5.4 Hz, 1H), 2.62 (dd, J = 15.4, 8.8 Hz, 1H), 1.11–2.10 (m, 37H), 1.31 (d, J = 6.8 Hz, 3H), 0.96 (d, J = 6.4 Hz, 3H), 0.79–0.92 (m, 27H); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C83H139N24O18 1760.0699; Found 1760.0710.
Macrocyclic β-Sheet peptide 1Cit:
1H NMR, (500 MHz, 1 mM in D2O, 298K) δ 7.99 (d, J = 8.9 Hz, 1H), 7.91 (s, 1H), 7.25 (d, J = 9.2 Hz, 1H), 6.96–7.12 (m, 5H), 4.65 (t, J = 6.0 Hz, 1H), 4.56 (t, J = 7.3 Hz, 1H), 4.42–4.50 (m, 3H), 4.28–4.38 (m, 3H), 4.17 (t, J = 6.2 Hz, 2H), 4.09 (d, J = 8.3 Hz, 1H), 4.03 (d, J = 8.3 Hz, 1H), 4.00 (s, 3H), 3.33–3.44 (m, 2H), 3.12–3.21 (m, 2H), 2.88–3.10 (m, 8H), 2.71 (dd, J = 14.9, 5.5 Hz, 1H), 2.63 (dd, J = 15.7, 8.5 Hz, 1H), 1.35–2.07 (m, 35H), 1.28 (d, J = 7.1 Hz, 3H), 1.11–1.25 (m, 2H), 0.96 (d, J = 6.5 Hz, 3H), 0.78–0.93 (m, 27H); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C83H137N23O19Na 1783.0359; Found 1783.0337.
Macrocyclic β-Sheet peptide 2Cit:
1H NMR, (500 MHz, 1 mM in D2O, 298K) δ 7.96 (d, J = 2.7 Hz, 1H), 7.90 (dd, J = 8.9, 2.6 Hz, 1H), 7.24 (d, J = 9.2 Hz, 1H), 7.02–7.16 (m, 5H), 5.77 (s, 1H), 5.57 (s, 1H), 4.69 (t, J = 6.5 Hz, 1H), 4.62–4.66 (m, J = 2H), 4.32–4.50 (m, 5H), 4.22–4.30 (m, 2H), 4.15 (t, J = 5.8 Hz, 1H), 4.10 (t, J = 5.7 Hz, 1H), 4.01 (d, J = 8.2 Hz, 1H), 3.98 (s, 3H), 3.25–3.37 (m, 1H), 3.20–3.26 (m, 1H), 3.13–3.20 (m, 1H), 3.00–3.10 (m, 3H), 2.99 (d, J = 7.4 Hz, 3H), 2.83–2.97 (m, 4H), 2.68 (qd, J = 6.1, 16 Hz 1H), 1.40–2.05 (m, 34H), 1.24 (d, J = 6.8 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H), 0.80–0.91 (m, 21H). HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C82H132N23O19Br 1822.9331; Found 1822.9333.
NMR Spectroscopy of the Macrocyclic β-Sheet Peptides
Sample Preparation.
NMR spectroscopy of the macrocyclic β-sheet peptides was performed in D2O (D, 99.96%; Cambridge Isotope Laboratories, Inc.) with 50 mM CD3COOD (D, 99.5%; Cambridge Isotope Laboratories, Inc.) and 50 mM CD3COONa (D, 99%; Cambridge Isotope Laboratories, Inc.). The solutions were prepared by dissolving a weighed portion of the peptide in the appropriate volume of solvent. The molecular weights of the peptides were calculated as the TFA salts with all amino groups assumed to be protonated (1Arg, M.W. 2330.24 mg/mL, 1Cit, M.W. 2331.24 mg/mL, and 2Cit, M.W. 2394.10 mg/mL). Each solution contained 4,4-dimethyl-4-silapentane-1-ammonium trifluoroacetate (DSA) as an internal standard for referencing chemical shifts.44 The solutions were allowed to stand for 24 h to allow complete hydrogen to deuterium exchange of the amide NH protons.
1H NMR, TOCSY, ROESY, and NOESY Data Collection.
NMR spectra were recorded on a Bruker 500 MHz spectrometer with a TCI cryoprobe. TOCSY spectra were recorded with 2048 points in the f2 dimension and 512 increments in the f1 dimension with a 150-ms spin-lock mixing time. TOCSY spectra were recorded with 2048 points in the f2 dimension and 512 increments in the f1 dimension with either a 100- or 150-ms spin-lock mixing time. NOESY spectra were recorded with 2048 points in the f2 dimension and 512 increments in the f1 dimension with a 150-ms mixing time.
1H NMR, TOCSY, and NOESY Data Processing.
NMR spectra were processed with BrukerXwinNMR software. Automatic baseline correction was applied in both dimensions after phasing the spectra. TOCSY spectra were Fourier transformed to a final matrix size of 4096 × 1024 real points using a Qsine weighting function and forward linear prediction. NOESY and ROESY spectra were Fourier transformed to a final matrix size of 4096 × 2048 real points using a Qsine weighting functionand forward linear prediction.
Diffusion-Ordered Spectroscopy (DOSY) Experiments.
DOSY experiments were performed on a Bruker 500 MHz spectrometer equipped with a TCI cryoprobe, with a diffusion delay (Δ) of 75-ms and a diffusion gradient length (δ) of 2.5-ms. Sixteen sets of FIDs were recorded with the gradient strength incremented from 5%–95% using a linear ramp. The combined FIDs were Fourier transformed in Bruker’s TopSpin™ software to give a pseudo-2D spectrum. After phasing and performing baseline correction, each pseudo-2D spectrum was processed with logarithmic scaling on the Y-axis. The Y-axis was calibrated to the diffusion coefficient of the residual HOD peak in D2O (1.9 × 10−9 m2/s at 298 K).45 The diffusion coefficients of the peptides were read and converted from logarithmic values to linear values.
Crystallization of Peptide 2Cit
The procedures in this section follow closely to those our laboratory has previously published.15 Initial crystallization conditions for peptide 2Cit were determined using the hanging-drop vapor-diffusion method. Crystallization conditions were screened using three crystallization kits in a 96-well plate format (Hampton Index, PEG/Ion, and Crystal Screen). Three 150 nL hanging drops that differed in the ratio of peptide to well solution were made per condition in each 96-well plate for a total of 864 experiments. Hanging drops were made by combining an appropriate volume of peptide 2Cit (10 mg/mL in deionized water) with an appropriate volume of well solution to create three 150-nL hanging drops with 1:1, 1:2, and 2:1 peptide:well solution. The hanging drops were made using a TTP LabTech Mosquito nanodisperse instrument. Crystals of peptide 2Cit grew in ~48 h in a solution of 0.1 M sodium citrate at pH 5.0, 20% (v/v) isopropanol, and 18% PEG 4000.
Crystallization conditions for peptide 2Cit were optimized using a 4×6 matrix Hampton VDX 24-well plate. The sodium citrate pH was varied in each row in increments of 0.5 pH units (4.0, 4.5, 5.0, and 5.5) and the isopropanol concentration in each column in increments of 2% (26%, 24%, 22%, 20%, 18%, 16%). The first well in the 4×6 matrix was prepared by combined 100 μL of 1 M sodium citrate at pH 4.0, 260 μL of isopropanol, 360 μL of 50% (w/v) PEG 4000, and 280 μL of deionized water. The other wells were prepared in analogous fashion, by combining 100 μL of sodium citrate of varying pH, 360 μL of 50% (w/v) PEG 4000, isopropanol in varying amounts, and deionized water for a total volume of 1 mL in each well.
Three hanging-drops were prepared per borosilicate glass slide by combining a solution of peptide 2Cit (10 mg/mL in deionized water) and the well solution in the following amounts: 1 μL:1 μL, 2 μL:1 μL, and 1 μL:2 μL. Slides were inverted and pressed firmly against the silicone grease surrounding each well. Crystals of peptide 2Cit suitable for X-ray diffraction grew in ~3 days. Crystallization conditions were further optimized using smaller variations in sodium citrate pH (in increments of 0.2 pH units) and isopropanol concentrations (in increments of 1%). Crystals were harvested with a nylon loop attached to a copper or steel pin and flash frozen in liquid nitrogen prior to data collection. The optimized crystallization conditions for peptide 2Cit are summarized in Table S1.
X-ray Crystallographic Data Collection, Data Processing, and Structure Determination for Peptide 2Cit.
Diffraction data for peptide 2Cit were collected at the Stanford Synchrotron Radiation Lightsource (SSRL) with a synchrotron source at 0.92-Å wavelength. Diffraction data were scaled and merged using XDS.46 Coordinates for the anomalous signal were determined by HySS in the Phenix software suite 1.10.1.47 The electron density map was generated in Phaser using the coordinates of the bromine anomalous signal. Molecular manipulation of the model was performed with Coot. Coordinates were refined with phenix.refine.
Molecular Modeling of the Macrocyclic β-Sheet Peptides
Molecular models of the tetramers of peptides 1Arg and 1Cit were generated from the X-ray crystallographic structure of the homologous peptide 2Cit (PDB: 5UHR). We modeled the tetramer formed by peptides 1Cit by mutating the (2-bromoallyl)glycine residue to isoleucine and the side chain torsion angles were adjusted to match the resulting NOE. We modeled a tetramer of peptide 1Arg in a similar fashion, mutating both the (2-bromoallyl)glycine and the citrulline residues. The coordinates were exported from PyMOL and the file was imported into MacroModel with the Maestro user interface. Atom types and bond orders were edited as needed to correct errors in bond type and charge. Minimization was performed with the MMFFs force field and GB/SA water solvation.
Supplementary Material
Acknowledgement
We thank the National Science Foundation (NSF CHE-1507840) for funding and the Stanford Synchrotron Radiation Lightsource (SSRL) for synchrotron data collection. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b01116.
Supplementary figures and table.
Procedures for NMR spectroscopy and molecular modeling of macrocyclic β-sheet peptides.
HPLC and MS characterization data for peptides 1Arg, 1Cit, 2Cit; and NMR spectroscopic data for peptides.
Crystallographic coordinates of peptide 2Cit in CIF and PDB format.
Crystallographic coordinates of peptide 2Cit were deposited into the Protein Data Bank (PDB) with code 5UHR.
The authors declare no competing financial interest.
References
- (1).Chiti F; Dobson CM Annu. Rev. Biochem 2006, 75, 333–366. [DOI] [PubMed] [Google Scholar]
- (2).Knowles TPJ; Vendruscolo M; Dobson CM Nat. Rev. Mol. Cell. Bio 2014, 15, 384–396. [DOI] [PubMed] [Google Scholar]
- (3).Nelson R; Sawaya MR; Balbirnie M; Madsen AO; Riekel C; Grothe R; Eisenberg D Nature 2005, 435, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Luca S; Yau WM; Leapman R; Tycko R Biochemistry 2007, 46, 13505–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wiltzius JJW; Sievers SA; Sawaya MR; Cascio D; Popov D; Riekel C; Eisenberg D Protein Sci 2008, 17, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lu JX; Qiang W; Yau WM; Schwieters CD; Meredith SC; Tycko R Cell 2013, 154, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Xiao YL; Ma BY; McElheny D; Parthasarathy S; Long F; Hoshi M; Nussinov R; Ishii Y Nat. Struct. Mol. Biol 2015, 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lendel C; Bjerring M; Dubnovitsky A; Kelly RT; Filippov A; Antzutkin ON; Nielsen NC; Hard T Angew. Chem. Int. Ed 2014, 53, 12756–12760. [DOI] [PubMed] [Google Scholar]
- (9).Xiao YL; Ma BY; McElheny D; Parthasarathy S; Long F; Hoshi M; Nussinov R; Ishii Y Nat. Struct. Mol. Biol 2015, 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Walti MA; Ravotti F; Arai H; Glabe CG; Wall JS; Bockmann A; Guntert P; Meier BH; Riek R Proc. Natl. Acad. Sci. U.S.A 2016, 113, E4976–E4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Colvin MT; Silvers R; Ni QZ; Can TV; Sergeyev I; Rosay M; Donovan KJ; Michael B; Wall J; Linse S; Griffin RG J. Am. Chem. Soc 2016, 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sandberg A; Luheshi LM; Sollvander S; de Barros TP; Macao B; Knowles TPJ; Biverstal H; Lendel C; Ekholm-Petterson F; Dubnovitsky A; Lannfelt L; Dobson CM; Hard T Proc. Natl. Acad. Sci. U.S.A 2010, 107, 15595–15600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Laganowsky A; Liu C; Sawaya MR; Whitelegge JP; Park J; Zhao ML; Pensalfini A; Soriaga AB; Landau M; Teng PK; Cascio D; Glabe C; Eisenberg D Science 2012, 335, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mirecka EA; Shaykhalishahi H; Gauhar A; Akgul S; Lecher J; Willbold D; Stoldt M; Hoyer W Angew. Chem. Int. Ed 2014, 53, 4227–4230. [DOI] [PubMed] [Google Scholar]
- (15).Spencer RK; Kreutzer AG; Salveson PJ; Li H; Nowick JS J. Am. Chem. Soc 2015, 137, 6304–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Salveson PJ; Spencer RK; Nowick JS J. Am. Chem. Soc 2016, 138, 4458–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kreutzer AG; Harnza IL; Spencer RK; Nowick JS J. Am. Chem. Soc 2016, 138, 4634––4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Janson J; Ashley RH; Harrison D; McIntyre S; Butler PC Diabetes 1999, 48, 491–498. [DOI] [PubMed] [Google Scholar]
- (19).Meier JJ; Kayed R; Lin CY; Gurlo T; Haataja L; Jayasinghe S; Langen R; Glabe CG; Butler PC Am. J. Physiol. Endocrinol. Metab 2006, 291, E1317–E1324. [DOI] [PubMed] [Google Scholar]
- (20).Konarkowska B; Aitken JF; Kistler J; Zhang SP; Cooper GJS Febs Journal 2006, 273, 3614–3624. [DOI] [PubMed] [Google Scholar]
- (21).Westermark P; Andersson A; Westermark GT Physiol. Rev 2011, 91, 795–826. [DOI] [PubMed] [Google Scholar]
- (22).Bram Y; Frydman-Marom A; Yanai I; Gilead S; Shaltiel-Karyo R; Amdursky N; Gazit E Sci. Rep 2014, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mirecka EA; Feuerstein S; Gremer L; Schroder GF; Stoldt M; Willbold D; Hoyer W Sci. Rep 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Soriaga AB; Sangwan S; Macdonald R; Sawaya MR; Eisenberg DJ Phys. Chem. B 2016, 120, 5810–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bedrood S; Li YY; Isas JM; Hegde BG; Baxa U; Haworth IS; Langen RJ Biol. Chem 2012, 287, 5235–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dupuis NF; Wu C; Shea JE; Bowers MT J. Am. Chem. Soc 2009, 131, 18283–18292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dupuis NF; Wu C; Shea JE; Bowers MT J. Am. Chem. Soc 2011, 133, 7240–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Buchanan LE; Dunkelberger EB; Tran HQ; Cheng PN; Chiu CC; Cao P; Raleigh DP; de Pablo JJ; Nowick JS; Zanni MT Proc. Natl. Acad. Sci. U.S.A 2013, 110, 19285–19290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cheng PN; Liu C; Zhao ML; Eisenberg D; Nowick JS Nat. Chem 2012, 4, 927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nowick JS; Lam KS; Khasanova TV; Kemnitzer WE; Maitra S; Mee HT; Liu RW J. Am. Chem. Soc 2002, 124, 4972–4973. [DOI] [PubMed] [Google Scholar]
- (31).Nowick JS; Brower JO J. Am. Chem. Soc 2003, 125, 876–877. [DOI] [PubMed] [Google Scholar]
- (32).Nowick JS; Chung DM; Maitra K; Maitra S; Stigers KD; Sun YJ Am. Chem. Soc 2000, 122, 7654–7661. [Google Scholar]
- (33).Truex NL; Wang YL; Nowick JS J. Am. Chem. Soc 2016, 138, 13882–13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Truex NL; Nowick JS J. Am. Chem. Soc 2016, 138, 13891–13900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wishart DS; Sykes BD Methods Enzymol. 1994, 239, 363–392. [DOI] [PubMed] [Google Scholar]
- (36).Khakshoor O; Demeler B; Nowick JS J. Am. Chem. Soc 2007, 129, 5558–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pham JD; Demeler B; Nowick JS J. Am. Chem. Soc 2014, 136, 5432–5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Polson AJ Phys. Colloid Chem 1950, 54, 649–652. [Google Scholar]
- (39).Krotee P; Rodriguez JA; Sawaya MR; Cascio D; Reyes FE; Shi D; Hattne J; Nannenga BL; Oskarsson ME; Philipp S; Griner S; Jiang L; Glabe CG; Westermark GT; Gonen T; Eisenberg DS Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Liu C; Zhao ML; Jiang L; Cheng PN; Park J; Sawaya MR; Pensalfini A; Gou DW; Berk AJ; Glabe CG; Nowick J; Eisenberg D Proc. Natl. Acad. Sci. U.S.A 2012, 109, 20913–20918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Spencer RK; Li H; Nowick JS J. Am. Chem. Soc 2014, 136, 5595–5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Yoo S; Kreutzer AG; Truex NL; Nowick JS Chem. Sci 2016, 7, 6946–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Pham JD; Chim N; Goulding CW; Nowick JS J. Am. Chem. Soc 2013, 135, 12460–12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Nowick JS; Khakshoor O; Hashemzadeh M; Brower JO Org Lett. 2003, 5, 3511–3513. [DOI] [PubMed] [Google Scholar]
- (45).Longsworth LG J. Phys. Chem 1960, 64, 1914–1917. [Google Scholar]
- (46).Kabsch W Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.