

Summary
Higher-order supramolecular complexes dubbed signalosomes carry out key signaling and effector functions in innate immunity and inflammation. In this review, we present several recently discovered signalosomes that are formed either by stable protein-protein interactions or by dynamic liquid-liquid phase separation. Structural features of these signalosomes are highlighted to elucidate their functions and biological insights.
Introduction
Innate immune signaling is triggered when cell surface or cytosolic pattern recognition receptors (PRRs) recognize exogenous pathogen- and endogenous damage-associated molecular patterns (PAMPs and DAMPs) to initiate immune and inflammatory responses. Upon stimulation, PRRs recruit adaptors and effectors to form supramolecular complexes, or higher-order assemblies, which in turn execute the signal transduction and signal amplification. Collectively known as signalosomes, these complexes are dynamically regulated for their formation and disassembly in different innate immune pathways [1–3], which are critical for shaping the strength and duration of innate immune responses, controlling the intensity of inflammation, as well as for preventing autoimmune diseases.
Elucidating the biophysical principles governing the assembly of higher-order supramolecular structures has uncovered a new paradigm in innate immune signaling pathways [4]. In contrast to a traditional model of signal transduction, which describes the interaction among signaling molecules with simple stoichiometry, higher-order signaling depicts the polymerization of the signalosome components into complexes with variable stoichiometry and conformational heterogeneity. In its early days of discovery, proteins in the death domain (DD) superfamily, which are ubiquitous in innate immune pathways such as tumor necrosis factor receptor (TNFR) and Toll-like receptor pathways, were first found to assemble into helical oligomers or filaments that can be heterogeneous in length and composition [5–7]. Around the same time, multivalent interactions [8] and amyloid structures [9] were also elucidated as effective scaffolds for signalosome formation. In recent years, facilitated by the advance in cryo-electron microscopy (cryo-EM), other DD family domains, from caspase recruitment domain (CARD), pyrin domain (PYD), to death effector domain (DED) [10–19], were found to assemble into helical filaments in inflammasomes and the RIG-I pathway, making higher-order signalosomes a phenomenon in multiple innate immune systems [20,21].
Interestingly, discoveries from structural biology were paralleled by the identification of dynamic higher-order assemblies, which are exemplified as liquid droplets assembled from the process of liquid-liquid phase separation (LLPS) [22–25]. Similar to structured filaments, these droplets are also formed using multivalent interactions from folded domains (SH2 and SH3) or low complexity sequences [22–24]. In fact, interconversion can often occur from LLPS to structured interaction to display a continuum in the dynamics of higher-order complexes [26].
Now, with an increasing number of discoveries in innate immunity and beyond, higher-order signaling is transforming our view of not only cellular communication, but also cellular organization. Mechanistically, many more types of protein domains, including disordered domains, are found to form higher-order signaling scaffolds that can represent stable protein-protein interactions, dynamic liquid-liquid phase separation, or somewhere in between. To present these conceptual advances, we discuss here examples of signalosomes in the pathways of the tumor necrosis factor receptor superfamily (TNFRSF), necrosomes, inflammasomes, and cGAS-STING, which are pivotal components of cellular signaling hub for many physiological processes and inflammatory responses. Some of these complexes are formed from domains that are very different from those observed previously, including single-pass transmembrane domains, suggesting the versatility in the molecular basis of their interactions. Understanding the structures and functions of these higher-order assemblies is critical for revealing the molecular mechanisms that govern innate immune and inflammatory signaling.
Transmembrane helix clustering in TNFRSF-mediated signal transduction
The tumor necrosis factor receptor (TNFR) superfamily (TNFRSF) is essential for activation of immune response, regulation of cellular homeostasis, and induction of morphogenesis [27,28]. TNFRSF members including death receptors are Type I transmembrane proteins. They share an extracellular domain (ECD) composed of multiple cysteine-rich domains (CRDs) that bind ligands such as TNF-related apoptosis-inducing ligand (TRAIL). The ECD is then followed by a transmembrane helix (TMH), and an intracellular domain (ICD) that interacts with downstream adaptor proteins, such as Fas-associated death domain (FADD), TNFR1-associated death domain (TRADD), and TNFR-associated factors (TRAFs) [27,28] (Figure 1a).
Figure 1.
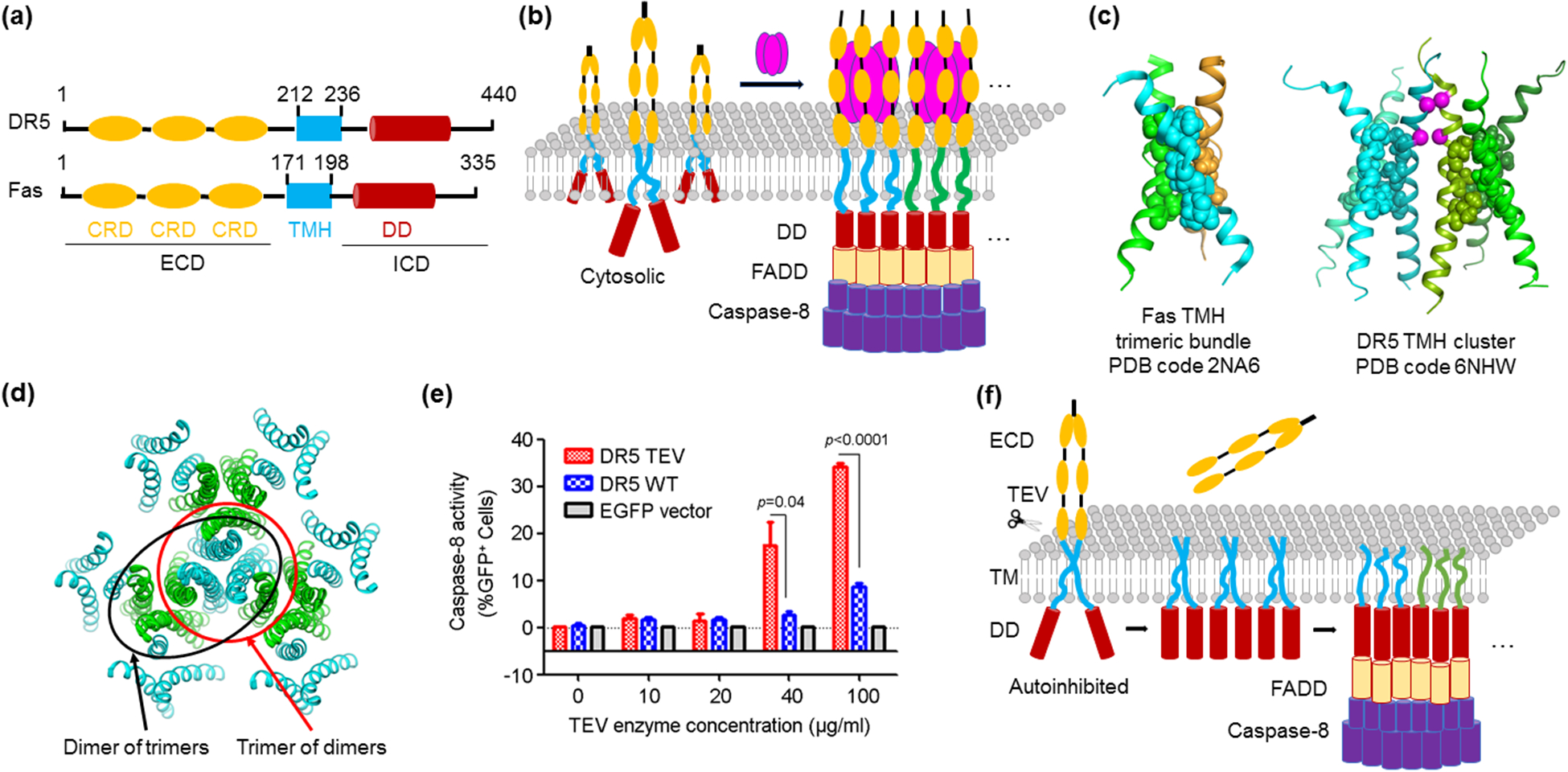
Transmembrane helix clustering in TNFRSF-mediated signal transduction. (a) Domain organization of DR5 and Fas, representative DRs which belong to TNFRSF. TNFRSF members share an N-terminal ECD containing CRD repeats, a single-pass TMH, and an ICD that interacts with adaptor proteins and may contain a DD. Abbreviations: ECD - extracellular domain; ICD - intracellular domain; CRD - cysteine-rich domain; TMH - transmembrane helix; DD - death domain. (b) Ligand-induced clustering of DRs on the cell surface. A trimeric ligand induces DR trimerization through the ECD. In turn, the ICD captures the activation signal to induces the higher-order assembly of downstream adaptor proteins such as FADD through DD-DD interactions. Adaptors then relay the signal to effectors such as caspase-8, which also forms higher-order assembly. (c) Oligomers formed by the TMH of DR5 and Fas. While the TMH of Fas forms a stable trimeric bundle, the TMH of DR5 forms more extended architecture that can be interpreted as a trimer of dimers or a dimer of trimers. (d) Higher-order clustering of DR5 TMH. The trimer-of-dimer or dimer-of-trimer TMH architecture enables the integration of numerous DR5 molecules into a network assembly. (e) Activation of an engineered DR5 with a TEV enzyme cleavage site between ECD and TMH by increasing amounts of TEV [35]. (f) Schematic for TMH-mediated higher-order signaling. Removal of ECD by the TEV protease leads to TNFRSF clustering and downstream caspase-8 activation [35], suggesting that TMH higher-order assembly alone is sufficient for signaling.
Earlier light microscopy studies revealed the formation of large cellular puncta by TNFRSF members on the cell surface upon ligand binding [29–31] (Figure 1b). The nature of the molecular interactions that mediate the assembly of these large complexes has been the subject of intensive investigations. First, trimeric interactions of the ECD with trimeric ligands have been revealed from crystal structures of ligand-receptor complexes [32], and surface aggregation by 2-dimensional ligand-receptor complex networks has been implicated [33]. Secondly, for death receptors within TNFRSF, highly oligomeric helical assemblies at the intracellular death domains (DD) with FADD or TRADD are known [7], and for receptors that do not have intracellular DDs, receptor ICD-induced TRAF oligomerization through alternating trimerization and dimerization was proposed [8]. These ECD and ICD interactions have been accepted as responsible for TNFR clustering and activation.
This idea was recently challenged by the discovery that the TMH of TNFRSF members directly participates in higher-order assembly [34,35]. The TMH of Fas forms a tightly packed trimer bundled by intermolecular interactions, which are compromised by cancer-associated mutations at the interface [34] (Figure 1c). More surprisingly, the TMH of DR5 forms an organized dimer-of-trimer or trimer-of-dimer cluster, and both dimer and trimer interactions are required for DR5 activation and signaling [35] (Figure 1c,d). These two structures raised the possibility that TMH alone may be signaling competent. Indeed, when an engineered protease site was introduced at the junction between ECD and TMH of DR5, proteolytic removal of the ECD robustly activated DR5 to an extent similar to ligand-induced stimulation [35] (Figure 1e,f). The same phenomenon was observed for TNFR2 and Ox40, indicating that TMH can be sufficient for receptor activation and that the unliganded ECD may inhibit TMH clustering and activation [35] (Figure 1f). Taken together, these studies suggest that in addition to the ECD and ICD of TNFRSF members, the TMH is a critical driver of receptor higher-order assembly and signal activation.
Novel amyloid structure in the hetero-oligomeric RIPK1-RIPK3 necrosome core complex.
Necroptosis is a regulated form of programmed cell death downstream of death receptor activation, which requires the kinase activities of receptor-interacting protein 1 and 3 (RIPK1 and RIPK3) [36–39]. In the TNFα signaling pathway, RIPK1 is upstream of RIPK3, and acts as a key arbitrator of cell death pathways [37]. TNFα can initiate formation of an RIPK1-based cytosolic complex engaging the adaptor protein FADD and procaspase-8, which can induce apoptotic cell death by activating caspase-8 [40,41]. Alternatively, the RIPK1-based complex can associate with RIPK3, referred to as the RIPK1-RIPK3 necrosome, to trigger necroptosis [37].
The interaction between RIPK1 and RIPK3 is mediated by the RIP homotypic interaction motif (RHIM), a short sequence centered on the IQIG or VQVG tetrapeptide of RIPK1 and RIPK3 respectively [42] (Figure 2a). Biochemical and structural studies showed that the RHIMs of RIPK1 and RIPK3 assemble into filamentous structures that are characteristics of β-amyloids, and cellular studies confirmed that RIPK1 and RIPK3 form Thioflavin T-positive amyloidal clusters in cells during necroptosis [9]. While most known β-amyloids are associated with protein aggregation and neurodegenerative diseases, the RIPK1-RIPK3 amyloid assembly is a functional amyloid in death receptor-induced signal transduction [9].
Figure 2.
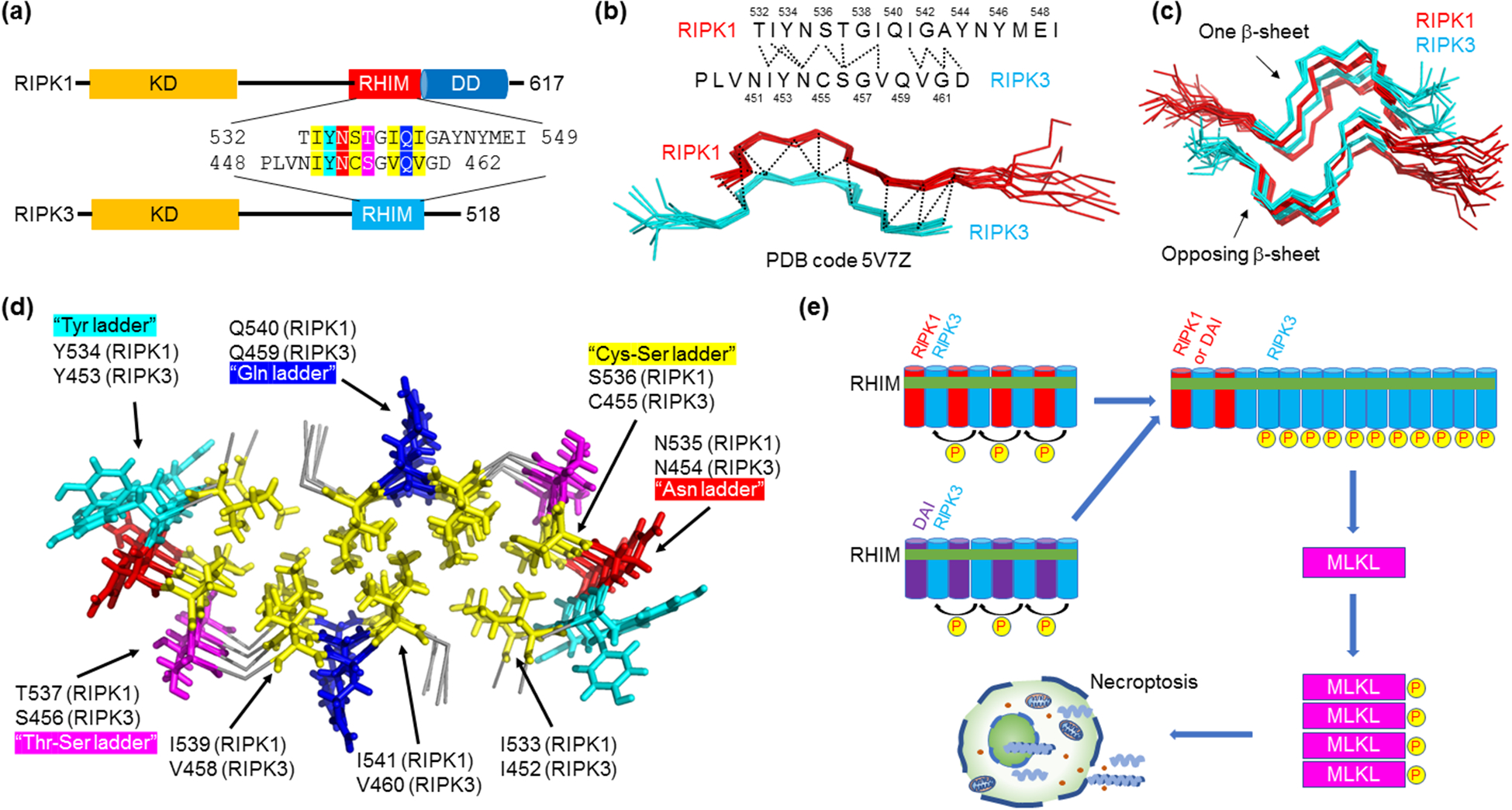
Novel amyloid structure in the hetero-oligomeric RIPK1-RIPK3 necrosome core complex. (a) Domain architecture of RIPK1 and RIPK3. Residue details are shown at the RHIMs, which contain the IQIG and VQVG tetrapeptide sequences for RIPK1 and RIPK3, respectively, at their centers. Abbreviations: KD - kinase domain; RHIM - RIP homotypic interaction motif; DD - death domain. (b) Serpentine fold formed by the RHIMs of RIPK1 and RIPK3. RIPK1 and RIPK3 each contribute a meandering β-strand consisting of short β-segments and turns. Molecular interactions between the two strands observed by NMR are indicated above. (c) Amyloidal assembly of the RIPK1-RIPK3 necrosome. To form a β-sheet, more RIPK1 and RIPK3 strands are integrated alternately and in parallel. Then, two such β-sheets stack in an anti-parallel fashion to complete the complex architecture. (d) A top-down view of the RIPK1-RIPK3 necrosome core with key residues indicated. Yellow: hydrophobic core. Other colors: solvent-exposed interactions. (e) A model for necrosome higher-order signaling. RIPK3 prefers hetero-amyloid assembly with RIPK1 or another RHIM-containing protein ZBP-1. RIPK1 or ZBP-1 therefore acts as a nucleus for RIPK3 homo-polymerization, which results in proximity-induced RIPK3 kinase activation and signal amplification. Active RIPK3 then phosphorylates and activates MLKL, the executioner of necroptosis.
Recent studies using solid-state nuclear magnetic resonance (NMR) revealed the atomic details of the RIPK1-RIPK3 hetero-amyloid core complex [43,44]. The structure is quite complex, with RIPK1 and RIPK3 each forming a meandering β-strand that is broken into four short β-segments separated by turns, which was named the serpentine fold (Figure 2b–d). RIPK1 and RIPK3 strands stack in parallel to form a β-sheet, and two such sheets come together in an antiparallel fashion to complete the fold (Figure 2c). The buried hydrophobic core is oblong-shaped, consisting of not only the two Ile and the two Val residues in the consensus IQIG and VQVG sequences, but also I533 (RIPK1), I452 (RIPK3), and an unusual Cys (RIPK3)-Ser (RIPK1) ladder (Figure 2a,d). Facing outward, numerous solvent-exposed stacking interactions further stabilize the fold, including an Asn ladder and a Gln ladder that are analogous to those known to support other amyloid structures through H-bonding by the side chains, a Tyr ladder, and a Thr (RIPK1)-Ser (RIPK3) ladder. This dense distribution of stacking motifs along the fibril axis may have made these proteins highly amyloidogenic.
RIPK1 and RIPK3 appear to prefer RIPK1-RIPK3 hetero-amyloid assembly instead of RIPK1-RIPK1 or RIPK3-RIPK3 homo-amyloid assembly. Previously, RIPK1 and RIPK3 preferentially formed hetero-amyloid when co-expressed [9]. Energetic calculation based on the structure suggests that the hydrophobic packing between the two β-sheets is more favorable for the hetero-amyloid, providing an explanation for this preference [43]. We speculate that in the necroptotic pathway, the preferred assembly of the hetero-amyloid of RIPK1-RIPK3, or of RIPK3 with another RHIM-containing protein such as ZBP-1, acts as a nucleus to induce the recruitment and polymerization of RIPK3 by the same β-amyloid interaction (Figure 2e). The RIPK3 amyloid results in proximity-facilitated kinase activation to amplify the necroptotic signal [43,44] (Figure 2e). Activated RIPK3 phosphorylates its substrate mixed-lineage kinase domain-like protein (MLKL), induces its aggregation on the cell membrane, and results in MLKL-mediated membrane permeabilization and necroptosis [45]. Therefore, in the RIPK1-RIPK3 necrosome, specific formation of a hetero-amyloidal signalosome serves as the crucial first step to activate the necroptotic pathway.
NEK7 and stress granule in NLRP3 inflammasome assembly and regulation
Inflammasomes are cytosolic multiprotein platforms that operate as a major class of signalosomes in innate immunity [46–48]. Canonical inflammasomes are in general composed of a sensor, which is often a nucleotide-binding domain (NBD) and leucine-rich repeat (LRR)-containing protein (NLR), an adaptor such as ASC or NLRC4, and the effector caspase-1. Inflammasome assembly has previously been shown to possess prion-like property due to the ability of ASC, as well as caspase-1, to polymerize into higher-order, filamentous structures [11,12,18]. Inflammasome assembly leads to proximity-facilitated caspase-1 activation [49,50]. Caspase-1, in turn, proteolytically processes inactive pro-forms of the proinflammatory cytokines interleukin 1β (IL-1β) and IL-18 for their maturation, and cleaves gasdermin D to induce cytokine release and pyroptosis [51–54]. As such, inflammasomes play important roles in protection against infection by activating effective antimicrobial immune responses [46–48]. Tight regulation of inflammasome assembly and signaling is crucial for mounting host defense while preventing overt tissue damage.
The NLRP3 inflammasome is one of the best studied canonical inflammasomes with a domain architecture that includes an N-terminal Pyrin domain (PYD), an NBD-containing domain known as NACHT, and a C-terminal LRR (Figure 3a). While NLRP3 can sense microbial products such as the bacterial toxin nigericin, which is a potassium ionophore, it is also activated by a plethora of endogenous sterile products or damage-associated signals [55]. These include extracellular aggregates, uric acid crystals that cause gout, cholesterol crystals with implications in heart disease, β-amyloids with associations to Alzheimer’s disease, and ATP released from dead cells. For this reason, the NLRP3 inflammasome has been proposed as a general surveillance mechanism of cellular stress and viability [55], likely by sensing cellular potassium efflux, an apparent common feature triggered by all NLRP3 stimuli [56]. Nonetheless, the direct activator of NLRP3 in the inflammasome signaling pathway remains obscure.
Figure 3.
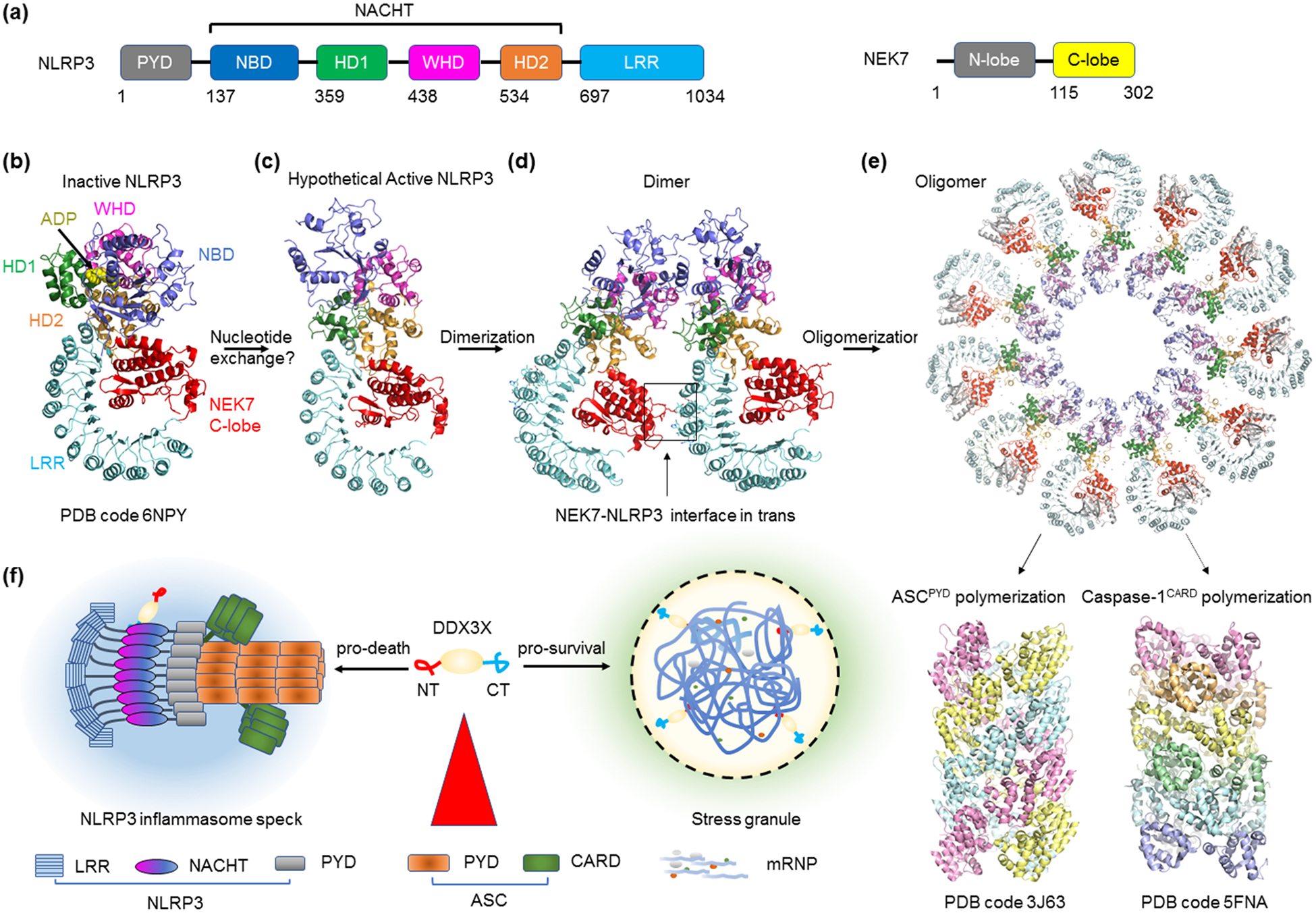
NEK7 and stress granule in NLRP3 inflammasome assembly and regulation. (a) Domain organization of NLRP3 and NEK7. NLRP3 features an N-terminal PYD, a central NACHT domain containing NBD, and a C-terminal LRR domain. NEK7 has two domains, the N-lobe and the C-lobe that directly interacts with NLRP3. Abbreviations: PYD - pyrin domain; NBD - nucleotide-binding domain; LRR - leucine-rich repeat. (b) Cryo-EM structure of the NLRP3-NEK7 complex with ADP bound. The PYD of NLPR3 was truncated for structural determination. With the entire complex shaped like an earring, the C-lobe of NEK7 rests in between the curved LRR and the NACHT domain. Despite nucleotide and NEK7 engagement, NLRP3 is in an autoinhibited conformation. (c) Model of the NLRP3-NEK7 complex in active conformation without ADP. A speculated nucleotide exchange step is required to trigger the activating conformational change. (d) A trans interface revealed by the dimer model of NLRP3-NEK7. The trans interface is formed between NEK7 of an NLRP3-NEK7 complex and NLRP3 of a neighboring complex. (e) Modelled oligomerization of the NLRP3-NEK7 complex into a disk. The 11-blade disk-like architecture was derived from the activated oligomeric NLRC4 inflammasome [62,63]. The disk architecture supports the oligomerization of NLRP3PYD at the central cavity. The NLPR3PYD platform can then seeds ASCPYD filament formation. In addition to PYD, ASC contains a CARD also capable of filament formation. ASCCARD filaments then recruit the effector protein caspase-1 through CARD-CARD interaction, resulting in polymerization and proximity-facilitated activation of caspase-1. (f) NLRP3 regulation by DDX3X. DDX3X interacts with NLRP3 to activate inflammasome signaling and pyroptosis, and sequestration of DDX3X by pro-survival stress granules counters inflammasome activity.
Our understanding of the NLRP3 inflammasome was greatly enhanced by the identification using genetic methods of the mitotic Ser/Thr kinase NEK7 (Figure 3a) as a required ligand for NLRP3 activation [57–59]. Recent reconstitution of the NLRP3 (ΔPYD)-NEK7 complex using recombinant proteins and the cryo-EM structure determination revealed an earring-shaped, ADP-bound NLRP3 with the C-terminal lobe of NEK7 nestling against the curved LRR and one side of the NACHT domain [60] (Figure 3b). Structure-based mutagenesis confirmed the importance of the NLRP3-NEK7 interface for NLRP3 signaling in mouse bone marrow-derived macrophages [60]. Based on comparison with the previously reported autoinhibited conformation of NLRC4 [61], another NLR, the structure is in an inactive conformation. Therefore, NEK7 binding does not activate NLRP3, but is required for NLRP3 activation, most likely by acting as a licensing factor in the pathway [60].
To elucidate why and how NEK7 may support NLRP3 inflammasome assembly, the previously reported active conformation of NLRC4 in its oligomeric form [62,63] was used to generate an oligomeric model of an active NLRP3-NEK7 complex [60] (Figure 3c–e). Surprisingly, this modeling exercise brings NEK7 from one NLRP3-NEK7 complex to the apposition of NLRP3 with an adjacent NLRP3-NEK7 complex in the oligomer (Figure 3d). Structure-based mutagenesis of this NLRP3-NEK7 interface in trans confirmed its role in NLRP3 activation, suggesting that NEK7 may be important for stimulus-dependent NLRP3 activation by bridging the NLRP3-NEK7 heterodimer to form an oligomer (Figure 3e).
Our data suggests that NLRP3 inflammasome assembly requires at least two steps although details of the process await further studies (Figure 3b–e). The first step may involve conformational activation of NLRP3 by nucleotide exchange from ADP to ATP, with or without NEK7 binding. In the second step, the NLRP3-NEK7 heterodimer in the active conformation oligomerizes to form the disk-like assembly [60], which should present the PYDs of NLRP3 as an oligomeric platform to nucleate filament formation of the PYD of the adaptor ASC [12] (Figure 3e). ASC also has a caspase recruitment domain (CARD), which in turn recruits and caspase-1 by CARD-mediated polymerization [10,11] (Figure 3e).
The NLRP3 inflammasome is regulated by a plethora of other factors either by direct interaction or post-translational modifications such as phosphorylation and ubiquitination [64]. A recent study also suggests a crosstalk between the NLRP3 inflammasome and stress granules, which are cytoplasmic organelles that form upon various stressors [65,66]. The stress granule protein DDX3X, a DEAD box protein, was identified as an NLRP3 interactor and drives NLRP3 inflammasome activation (Figure 3f). Induction of stress granules sequesters DDX3X, and thus specifically inhibits NLRP3 inflammasome activation, ASC speck formation, and pyroptosis. This finding indicates a competition between two contrasting cell-fate decisions upon stress sensing, the pro-survival stress granules and the pro-pyroptotic inflammasome activation.
Higher-order assemblies in the cGAS-STING pathway
Presence of foreign DNA in intracellular compartments signals pathogen invasion. Cyclic GMP-AMP (cGAMP) synthase (cGAS) is a cytosolic DNA sensor that belongs to the nucleotidyltransferase family [67,68]. Catalytic activation by cytosolic DNA enables cGAS to generate the second messenger 2′3′-cyclic GMP-AMP (cGAMP) from ATP and GTP. Human and mouse cGAS both have a disordered N-terminal domain highly enriched in positively charged residues and a C-terminal catalytic domain (CD) (Figure 4a). Crystallographic studies of cGAS catalytic domain with or without dsDNA revealed dramatic DNA-induced conformational changes, which cause rearrangement of the active site to turn on cGAS [20,69]. Initial analysis implicated the monomeric cGAS-dsDNA interaction as the cause for allosteric activation, but subsequent analysis showed two independent dsDNA-interacting sites on each cGAS molecule in the context of a cGAS-dsDNA dimer [20,69] (Figure 4b), or even three independent dsDNA-interacting sites on each cGAS molecule in the context of a crystal lattice of the cGAS-dsDNA trimer [70] (Figure 4c). It was further observed that cGAS senses long DNA or U-turn DNA by forming protein-DNA ladders [71].
Figure 4.
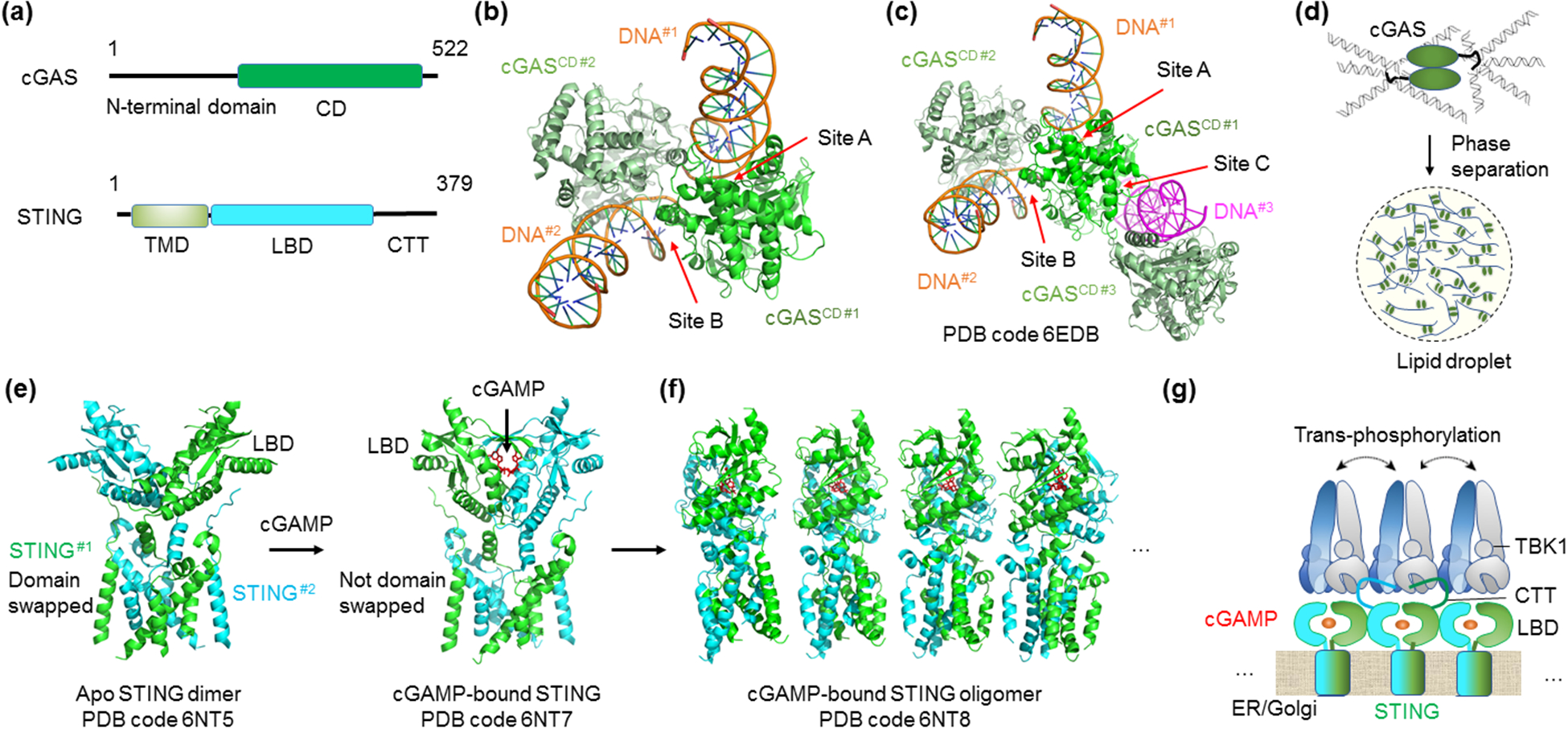
Supramolecular assembly in the cGAS-STING pathway. (a) Domain organization of cGAS and STING. cGAS has a disordered, positively charged N-terminal domain and a C-terminal CD. STING is an ER membrane protein with an N-terminal TMD, a central cytoplasmic LBD, followed by a CTT that interacts with TBK1. Abbreviations: CD - catalytic domain; TMD - transmembrane domain; LBD - ligand-binding domain; CTT - C-terminal tail. (b) Two DNA-binding sites shown by the crystal lattice interaction of a cGASCD-dsDNA complex. (c) An additional DNA-binding site (site C) observed in the crystal lattice of a cGASCD-dsDNA complex, making a total of three DNA-contacting sites on cGAS. (d) A model for DNA-induced cGAS condensation and phase separation through multivalent interactions. (e) c-GAMP-induced STING activation. Comparison between apo and cGAMP-bound STING dimer structures shows a dramatic conformational change at the LBD of STING upon ligand engagement. (f) Higher-order assembly of STING shown with a tetramer-of-dimer structure model. The side of a STING dimer serves as an oligomerizing surface for more STING dimers to add on. The side-to-side packing allows the formation of an extended STING network. (g) Schematic for cGAMP-mediated STING phosphorylation by TBK1. cGAMP-induced higher-order assembly of STING allows trans-phosphorylation of STING CTT by TBK1 bound to an adjacent STING.
Recent studies showed that binding of dsDNA to full-length cGAS induced liquid-liquid phase separation (LLPS) or formation of cGAS-DNA condensates, which were observed as cytoplasmic foci [70,72] Figure 4d). Several important elements are involved in eliciting the LLPS. First, the disordered and positively charged N-terminal domain, which not only interacts with DNA but also stabilizes dimerization of DNA-free full-length cGAS, promotes LLPS [70,72] (Figure 4a). Second, all three DNA-interacting sites in the catalytic domain promote LLPS and structure-based mutants at these sites reduced propensity for phase condensate formation [70]. Third, long DNA was more efficient than short DNA in promoting LLPS. Mechanistically, the multiple sites of interactions increase the valency of the cGAS-DNA interaction to enhance LLPS. Within the condensates, the enhanced local concentrations of proteins and DNA likely in turn promote formation of the proper cGAS-DNA higher-order complexes to stimulate catalytic production of cGAMP.
Further downstream, the second messenger cGAMP binds to and activates the adaptor protein stimulator of interferon genes (STING) that is an endoplasmic reticulum (ER) membrane protein containing four helices as the transmembrane domain (TMD) followed by a cytoplasmic cyclic dinucleotide-binding domain or ligand-binding domain (LBD), and a signaling domain or C-terminal tail (CTT) that recruits the kinase TBK1 [73] (Figure 4a). Previous crystallographic studies of the STING LBD revealed a symmetrical dimer, and each dimer interacts with one molecule of cGAMP or another cyclic dinucleotide [20,69]. However, despite conformational and affinity analyses of the STING-cGAMP interaction in the context of the STING LBD [20,69,74], the exact nature of STING activation remained elusive.
The cryo-EM structures of full-length STING provided a plausible model of cGAMP-induced STING activation [75]. It revealed a dramatic structural rearrangement of the LBD relative to the TMD upon cGAMP binding. In the inactive dimeric apo STING, the LBD and the TMD exist in an integrated, domain-swapped dimeric assembly with crossover at the junction between these two domains (Figure 4e). Upon cGAMP binding, at least two conformational changes occur. First, a helix of the LBD is pushed by the ligand to make the crossover impossible, leading to rotation of the LBD by 180° relative to the TMD (Figure 4e). Second, a surface loop changes conformation, allowing the juxtaposition of adjacent STING dimers through side-by-side packing to form tetramers and higher-order oligomers [75] (Figure 4f).
Requirement for higher-order assembly of STING in inducing the interferon response is further explained by the crystal and cryo-EM structures of STING in complex with a TBK1 dimer [76,77]. TBK1 is supposed to phosphorylate Ser366 in the STING CTT, and phosphorylated STING in turn recruits IRF3 for phosphorylation by TBK1 [78]. The STING-TBK1 complex structures showed the binding of a short peptide in the STING CTT (residues Asp369-Asp377) into a groove on TBK1 formed by the kinase domain of one TBK1 subunit and the scaffold and dimerization domain of the other TBK1 subunit. In this structure, Ser366, which is only 3 residues away from the peptide, cannot access either kinase domain in the TBK1 dimer. Therefore, cGAMP-induced oligomerization of STING is needed to allow trans-phosphorylation of STING CTT by an adjacent STING-bound TBK1 (Figure 4g).
Conclusion and future remarks
Higher-order assemblies have shifted the paradigm for signal transduction and presented an elegant explanation for many important signaling behaviors and mechanisms. The polymerization of receptors, adaptors, and effectors into supramolecular complexes allows signal transduction, signal amplification, and proximity-facilitated enzyme activation. The requirement for proteins to form cooperative assemblies for effector activation, whether of stable interactions or of dynamic phase-separated organelles, is consistent with digital, threshold-like signaling response. It is templating to speculate that innate immunity may especially benefit from such threshold behavior because the decision to respond or not to respond to a danger signal is greatly simplified, and once such decision is made, cells will respond fully. In a classical study on TNFα-induced NF-κB signaling, cell activation under different TNFα doses is a digital process at the single-cell level with fewer cells responding at lower doses and more cells responding at higher doses [79]. In all cases, the higher-order assemblies drive high local protein concentrations, thereby enabling proximity-facilitated enzyme activation and spatial regulation of signaling. With the signalosome examples in innate immunity and inflammation presented here, we have only seen the tip of the iceberg of higher-order signaling, which is awaiting more discoveries and may represent a general principle in cell biology.
Acknowledgements
This work was supported by the US National Institutes of Health (HD087988, Al124491 and AI045937 to H.W.) and China Scholarship Council (CSC 201806125051 to M.S.). We apologize to authors whose work could not be cited due to space limitation.
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Li S, Hong Z, Wang Z, Li F, Mei J, Huang L, Lou X, Zhao S, Song L, Chen W, et al. : The Cyclopeptide Astin C Specifically Inhibits the Innate Immune CDN Sensor STING. Cell Rep 2018, 25:3405–3421 e3407. [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Nour M, Carneiro LAM, Downey J, Tsalikis J, Outlioua A, Prescott D, Da Costa LS, Hovingh ES, Farahvash A, Gaudet RG, et al. : The heme-regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science 2019, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi M, Zhang Y, Liu L, Zhang T, Han F, Cleveland J, Wang F, McKeehan WL, Li Y, Zhang D: MAP1S Protein Regulates the Phagocytosis of Bacteria and Toll-like Receptor (TLR) Signaling. J Biol Chem 2016, 291:1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.•.Wu H: Higher-order assemblies in a new paradigm of signal transduction. Cell 2013, 153:287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This is the first time that the new form of signal transduction in innate immunity by higher-order oligomerization was proposed, implicating molecular mechanisms for proximity-driven enzyme activation, threshold behavior, signal amplification, and temporal and spatial control of signal transduction.
- 5.Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H: Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 2007, 128:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin SC, Lo YC, Wu H: Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465:885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, Yang JK, Kabaleeswaran V, Rice AJ, Cruz AC, Park AY, Yin Q, Damko E, Jang SB, Raunser S, et al. : The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat Struct Mol Biol 2010, 17:1324–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin Q, Lin SC, Lamothe B, Lu M, Lo YC, Hura G, Zheng L, Rich R, Campos AD, Myszka DG, et al. : E2 interaction and dimerization in the crystal structure of TRAF6 Nat Struct Mol Biol 2009, 16:658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et al. : The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Fu TM, Lu A, Witt K, Ruan J, Shen C, Wu H: Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc Natl Acad Sci U S A 2018, 115:10845–10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu A, Li Y, Schmidt FI, Yin Q, Chen S, Fu TM, Tong AB, Ploegh HL, Mao Y, Wu H: Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat Struct Mol Biol 2016, 23:416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, Egelman EH: Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156:1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen C, Lu A, Xie WJ, Ruan J, Negro R, Egelman EH, Fu TM, Wu H: Molecular mechanism for NLRP6 inflammasome assembly and activation. Proc Natl Acad Sci U S A 2019, 116:2052–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.David L, Li Y, Ma J, Garner E, Zhang X, Wu H: Assembly mechanism of the CARMA1-BCL10-MALT1-TRAF6 signalosome. Proc Natl Acad Sci U S A 2018, 115:1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu TM, Li Y, Lu A, Li Z, Vajjhala PR, Cruz AC, Srivastava DB, DiMaio F, Penczek PA, Siegel RM, et al. : Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex. Mol Cell 2016, 64:236–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu A, Yang L, Yin Q, Ruan J, Yu X, Egelman E, Wu H: Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discovery 2015, 1:15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiao Q, Yang C, Zheng C, Fontan L, David L, Yu X, Bracken C, Rosen M, Melnick A, Egelman EH, et al. : Structural Architecture of the CARMA1/Bcl10/MALT1 Signalosome: Nucleation-Induced Filamentous Assembly. Mol Cell 2013, 51:766–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, Chen ZJ: Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014, 156:1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu B, Peisley A, Tetrault D, Li Z, Egelman EH, Magor KE, Walz T, Penczek PA, Hur S: Molecular Imprinting as a Signal-Activation Mechanism of the Viral RNA Sensor RIG-I. Mol Cell 2014, 55:511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin Q, Fu TM, Li J, Wu H: Structural biology of innate immunity. Annu Rev Immunol 2015, 33:393–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.•.Kagan JC, Magupalli VG, Wu H: SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol 2014, 14:821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]; *In this article, the authors proposed the concept of supramolecular organizing centres (SMOCs) as location-specific higher-order complexes in innate immunity.
- 22.Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, et al. : Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483:336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, et al. : Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149:753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J, King DS, Taunton J, Rosen MK, Vale RD: Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 2016, 352:595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alberti S, Gladfelter A, Mittag T: Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176:419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu H, Fuxreiter M: The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell 2016, 165:1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vanamee ES, Faustman DL: Structural principles of tumor necrosis factor superfamily signaling. Sci Signal 2018, 11. [DOI] [PubMed] [Google Scholar]
- 28.Wu H, Hymowitz SG: Structure and function of tumor necrosis factor (TNF) at the cell surface In Handbook of cell signaling, edn 2nd edition Edited by Bradshaw RA, Dennis EA: Academic Press; 2009:265–275. vol 1.] [Google Scholar]
- 29.Algeciras-Schimnich A, Shen L, Barnhart BC, Murmann AE, Burkhardt JK, Peter ME: Molecular ordering of the initial signaling events of CD95. Mol Cell Biol 2002, 22:207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM: FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81:505–512. [DOI] [PubMed] [Google Scholar]
- 31.Siegel RM, Muppidi JR, Sarker M, Lobito A, Jen M, Martin D, Straus SE, Lenardo MJ: SPOTS: signaling protein oligomeric transduction structures are early mediators of death receptor-induced apoptosis at the plasma membrane. J Cell Biol 2004, 167:735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banner DW, D’Arcy A, Janes W, Gentz R, Schoenfeld HJ, Broger C, Loetscher H, Lesslauer W: Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell 1993, 73:431–445. [DOI] [PubMed] [Google Scholar]
- 33.Mukai Y, Nakamura T, Yoshikawa M, Yoshioka Y, Tsunoda S, Nakagawa S, Yamagata Y, Tsutsumi Y: Solution of the structure of the TNF-TNFR2 complex. Sci Signal 2010, 3:ra83. [DOI] [PubMed] [Google Scholar]
- 34.•.Fu Q, Fu TM, Cruz AC, Sengupta P, Thomas SK, Wang S, Siegel RM, Wu H, Chou JJ: Structural Basis and Functional Role of Intramembrane Trimerization of the Fas/CD95 Death Receptor. Mol Cell 2016, 61:602–613. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Using NMR spectrocopy, the authors determined the transmembrane helix structure of Fas, which revealed a proline-containing motif-mediated trimer assembly essential for Fas signaling.
- 35.••.Pan L, Fu TM, Zhao W, Zhao L, Chen W, Qiu C, Liu W, Liu Z, Piai A, Fu Q, et al. : Higher-Order Clustering of the Transmembrane Anchor of DR5 Drives Signaling. Cell 2019, 176:1477–1489 e1414. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Using NMR spectrocopy, the authors determined the transmembrane helix structure of DR5, which revealed a higher-order clustering by trimerization and dimerization. They further showed that in the absence of ligand binding, the extracellular domain is autoinhibitory to transmembrane helix clustering, and removal of the extracellular domain activates DR5 signaling.
- 36.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G: Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010, 11:700–714. [DOI] [PubMed] [Google Scholar]
- 37.Moquin D, Chan FK: The molecular regulation of programmed necrotic cell injury. Trends Biochem Sci 2010, 35:434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK: Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137:1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shan B, Pan H, Najafov A, Yuan J: Necroptosis in development and diseases. Genes Dev 2018, 32:327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Du F, Wang X: TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133:693–703. [DOI] [PubMed] [Google Scholar]
- 41.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, Macfarlane M, Cain K, et al. : The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol Cell 2011, 43:432–448. [DOI] [PubMed] [Google Scholar]
- 42.Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM: Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem 2002, 277:9505–9511. [DOI] [PubMed] [Google Scholar]
- 43.••.Mompean M, Li W, Li J, Laage S, Siemer AB, Bozkurt G, Wu H, McDermott AE: The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173:1244–1253 e1210. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Using solid-state NMR, the authors determined the high-resolution structure of the RIPK1-RIPK3 amyloid core, and provided a potential explanation for the specificity of hetero-amyloid formation in the RIPK1-RIPK3 necrosome.
- 44.Mompean M, Bozkurt G, Wu H: Mimicry by a viral RHIM. EMBO Rep 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinlich R, Oberst A, Beere HM, Green DR: Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 2017, 18:127–136. [DOI] [PubMed] [Google Scholar]
- 46.Broz P, Dixit VM: Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016, 16:407–420. [DOI] [PubMed] [Google Scholar]
- 47.Shen C, Sharif H, Xia S, Wu H: Structural and mechanistic elucidation of inflammasome signaling by cryo-EM. Curr Opin Struct Biol 2019, 58:18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evavold CL, Kagan JC: Inflammasomes: Threat-Assessment Organelles of the Innate Immune System. Immunity 2019, 51:609–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Castro-Jorge LA, de Carvalho RVH, Klein TM, Hiroki CH, Lopes AH, Guimaraes RM, Fumagalli MJ, Floriano VG, Agostinho MR, Slhessarenko RD, et al. : The NLRP3 inflammasome is involved with the pathogenesis of Mayaro virus. PLoS Pathog 2019, 15:e1007934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamkanfi M, Dixit VM: Mechanisms and functions of inflammasomes. Cell 2014, 157:1013–1022. [DOI] [PubMed] [Google Scholar]
- 51.Xia S, Hollingsworth LRt, Wu H: Mechanism and Regulation of Gasdermin-Mediated Cell Death. Cold Spring Harb Perspect Biol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F: Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526:660–665. [DOI] [PubMed] [Google Scholar]
- 53.Khanova E, Wu R, Wang W, Yan R, Chen Y, French SW, Llorente C, Pan SQ, Yang Q, Li Y, et al. : Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology 2018, 67:1737–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lieberman J, Wu H, Kagan JC: Gasdermin D activity in inflammation and host defense. Sci Immunol 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaidt MM, Hornung V: The NLRP3 Inflammasome Renders Cell Death Pro-inflammatory. J Mol Biol 2018, 430:133–141. [DOI] [PubMed] [Google Scholar]
- 56.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G: K(+) Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38:1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, Hornung V: A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem 2016, 291:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He Y, Zeng MY, Yang D, Motro B, Nunez G: NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, et al. : NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 2016, 17:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.••.Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, Hauenstein AV, Wu Z, Nunez G, Mao Y, et al. : Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570:338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The authors determined the cryo-EM structure of the complex between ADP-bound, inactive NLRP3 and NEK7. Modeling of an active conformation of the NLRP3-NEK7 complex suggested that NEK7 bridges NLRP3 molecules in an NLRP3-NEK7 oligomer, providing an explanation for the requirement of NEK7 in NLRP3 inflammasome signaling.
- 61.Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, Wang R, Zhang Y, Martinon F, Miao D, et al. : Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 2013, 341:172–175. [DOI] [PubMed] [Google Scholar]
- 62.Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, Li Y, David L, Lu A, Wang WL, et al. : Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 2015, 350:404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, Shao F, Wang HW, Sui SF, Chai J: Structural and biochemical basis for induced self-propagation of NLRC4. Science 2015, 350:399–404. [DOI] [PubMed] [Google Scholar]
- 64.Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E: Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 2018, 17:688. [DOI] [PubMed] [Google Scholar]
- 65.••.Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, Guy CS, Briard B, Place DE, Bhattacharya A, et al. : DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 2019, 573:590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The authors showed that the stress granule protein DDX3X is required for NLRP3 inflammasome activation. Therefore, competivie recruitment of DDX3X to the NLRP3 inflammasome or to stress granuels acts as a central decision maker in the formation of pro-survival stress granules or the pro-death NLRP3 inflammasome.
- 66.Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP: Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun L, Wu J, Du F, Chen X, Chen ZJ: Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339:786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burdette DL, Vance RE: STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol 2013, 14:19–26. [DOI] [PubMed] [Google Scholar]
- 69.Kato K, Omura H, Ishitani R, Nureki O: Cyclic GMP-AMP as an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Annu Rev Biochem 2017, 86:541–566. [DOI] [PubMed] [Google Scholar]
- 70.••.Xie W, Lama L, Adura C, Tomita D, Glickman JF, Tuschl T, Patel DJ: Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc Natl Acad Sci U S A 2019, 116:11946–11955. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The authors identify the N-terminal disordered domain and an additional cGAS-DNA interface (site-C) in the catalytic domain as important for liquid-liquid phase separation of the cGAS-DNA complex and for cGAS activation.
- 71.Andreeva L, Hiller B, Kostrewa D, Lassig C, de Oliveira Mann CC, Jan Drexler D, Maiser A, Gaidt M, Leonhardt H, Hornung V, et al. : cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 2017, 549:394–398. [DOI] [PubMed] [Google Scholar]
- 72.••.Du M, Chen ZJ: DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018, 361:704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The authors reported that DNA binding to cGAS induces the formation of liquid-like droplets within which cGAS is activated. The DNA-induced phase transition of cGAS further promotes cGAMP production and innate immune signaling.
- 73.Shang G, Zhu D, Li N, Zhang J, Zhu C, Lu D, Liu C, Yu Q, Zhao Y, Xu S, et al. : Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat Struct Mol Biol 2012, 19:725–727. [DOI] [PubMed] [Google Scholar]
- 74.Zhang X, Shi H, Wu J, Sun L, Chen C, Chen ZJ: Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell 2013, 51:226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.••.Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X: Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019, 567:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The authors presented the cryo-EM structures of STING and revealed formation of STING tetramers and higher-order oligomers upon cGAMP binding. Ligand-induced conformational changes are required for STING activation.
- 76.••.Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ: Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567:394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The authors determined the cryo-EM structure of the STING-TBK1 complex, which showed the binding of STING C-terminal tail to both subunits of the TBK1 dimer. This binding, however, cannot support phosphorylation of a residue in the STING C-terminal tail by TBK1 because of its far distance to the kinase active site. Therefore, STING-TBK1 oligomerization is needed to achieve STING phosphorylation by TBK1 in trans.
- 77.••.Zhao B, Du F, Xu P, Shu C, Sankaran B, Bell SL, Liu M, Lei Y, Gao X, Fu X, et al. : A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 2019, 569:718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The authors showed that a conserved PLPLRT/SD motif within the C-terminal tail of STING mediates the recruitment of TBK1 by interacting with the dimer interface of TBK1. Oligomerization of cGAMP-bound STING-TBK1 complex is an essential step to promote STING phosphorylation by TBK1 in trans to activate STING-mediated signalling.
- 78.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, et al. : Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347:aaa2630. [DOI] [PubMed] [Google Scholar]
- 79.Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, Covert MW: Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature 2010, 466:267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]