

Abstract
The NLRP3 inflammasome is an innate immune platform that senses various pathogens and sterile insults. NLRP3 stimulation leads to activation of caspase-1, the secretion of pro-inflammatory cytokines and an inflammatory cell death called pyroptosis. Effectors of the NLRP3 inflammasome efficiently drive an immune response, providing protection in physiological settings but also promoting pathology when over-activated. Generation of reactive oxygen species (ROS) and intracellular calcium mobilization can activate the NLRP3 inflammasome. Recent studies suggest that TRPM2 is a calcium-permeable cation channel mediating ROS-dependent NLRP3 activation. Here, we review the role of TRPM2 in NLRP3 inflammasome activation and provide an update on new functional and structural discoveries. Understanding the molecular mechanism of TRPM2 dependent NLRP3 inflammasome activation will shed lights on this complex pathway and help the developing of therapeutic strategies.
The NLRP3 inflammasome
Inflammasomes are supramolecular complexes that activate caspase-1 or other inflammatory caspases. Among inflammasomes, the nucleotide-binding domain (NBD) and leucine-rich repeat (LRR)-containing protein (NLR) family Pyrin domain (PYD)-containing protein 3 (NLRP3) forms the most studied inflammasome, with a central role in the innate immune response. NLRP3 is universally expressed in all immune cells including those in the brain. It is activated by exogenous stimuli, termed pathogen-associated molecular patterns (PAMPs) such as bacterial toxins, and endogenous stimuli, termed damage-associated molecular patterns (DAMPs) which derive from the host [1,2]. NLRP3 activators are chemically diverse, including for example, extracellular ATP [3], bacterial pore forming toxin nigericin [4] and particulate matters such as uric acid crystals, amyloid aggregates and cholesterol crystals [5]. The structurally distinct identities of NLRP3-activating stimuli argue that they might converge to common nodes in NLRP3 activation. Accumulating studies suggest that these common nodes may be represented by three mechanisms: potassium efflux [6], calcium mobilization [7] and the generation and release of mitochondrial, lysosomal or nuclear reactive oxygen species (ROS) [8], although how they lead to the assembly of the NLRP3 inflammasome remains elusive.
Nonetheless, the sensing of these different stimuli triggers a rapid cytosolic inflammatory response by NLRP3, which recruits the adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and pro-caspase-1, culminating in formation of a single perinuclear inflammasome speck [2,9]. Upon inflammasome assembly, pro-caspase-1 is activated by dimerization, which in turn processes pro-inflammatory cytokines pro-interleukin-1 β (pro-IL-1β) and pro-IL-18 as well as the pore-forming protein gasdermin D (GSDMD) [10]. Bioactive IL-1β and IL-18 can be released through the cell membrane GSDMD pores, and pore formation may be followed by cell swelling and the pro-inflammatory, lytic cell death known as pyroptosis. Both cytokine release and lytic cell death contribute to a wide spectrum of acute and chronic inflammatory diseases including obesity [11], diabetes [12], atherosclerosis [13], gout [14,15] and cancer [16].
Ca2+ influx in NLRP3 inflammasome activation
Many studies have identified Ca2+ influx as a common node for NLRP3 inflammasome activation and cytokine release [17,18]. The concentration of Ca2+ is kept low in the cytoplasm (~100 nM), which enables the flux from the extracellular space or intracellular stores. A rapid increase in cytoplasmic Ca2+ leads to the activation of various Ca2+ binding proteins and diverse cellular outcomes [19]. Specifically, artificial abrogation of Ca2+ flux from either the extracellular environment or intracellular pools has been shown to significantly inhibit ASC oligomerization and pro-caspase-1 processing in NLRP3 inflammasome activation [20].
One potential intracellular Ca2+ source is the endoplasmic reticulum (ER), and the inositol trisphosphate receptor (IP3R), a Ca2+ channel mainly localized at the ER, may activate NLRP3 by releasing Ca2+ into the cytoplasm upon stress-related stimuli. Pharmacological inhibition of the IP3R channel has been implicated in preventing NLRP3 inflammasome activation in human and murine cells [21]. Another source of intracellular Ca2+ source is the lysosomal membrane, and destabilization of the membrane by particulate matters can lead to Ca2+ efflux from the organelle into the cytoplasm, enabling NLRP3 inflammasome activation [22]. Growing evidence also indicates that, besides the intracellular stores, Ca2+ influx from the extracellular space can also be involved in NLRP3 activation and cytokine release, making Ca2+ influx into the cytoplasm by different mechanisms a converging point for NLRP3 activation.
The Ca2+-permeable channel TRPM2 as a specific NLRP3 inducer upon oxidative stress
Transient Receptor Potential (TRP) channel family is one of the largest cation channel families, and most TRP channels permeate Ca2+ [23–25]. Based on sequence homology, the TRP family is divided into seven TRP subfamilies, TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPA (ankyrin), TRPP (polycystin), TRPML (mucolipin), and TRPN (NOMPC). All TRP channels share conserved transmembrane (TM) regions containing the voltage-sensor like domain (VSLD, S1-S4), the pore domain (S5-S6), and the TRP domain following S6. However, the cytosolic domains are drastically different among TRP subfamilies, which implicate different cellular functions.
TRPM2 is a member of the TRPM family and possesses a tetrameric architecture containing a large N-terminal TRPM homology region (MHR1–4), the conserved TM region, a rib helix, a pole helix, and a unique NUDT9 homology (NUDT9H) domain, which belongs to the Nudix hydrolase family that hydrolyze ADP-ribose (ADPR) to AMP and ribose-5-P [26,27]. In human TRPM2, it was shown that the NUDT9H domain only binds but does not hydrolyze ADPR [28,29]; this ADPR binding at the cytosolic domain, together with binding of intracellular Ca2+ at the TM at a low resting Ca2+ concentration, can already activate TRPM2 [26]. The resulting TRPM2-mediated Ca2+ permeation leads to higher intracellular Ca2+ concentration, which in turn promotes Ca2+ binding at the TM and positively feeds back to greater TRPM2 opening probability. Physiologically, ADPR production is increased by intracellular ROS levels, through degradation of poly-ADP-ribose in the nucleus or degradation of NAD released from damaged mitochondria in the cytoplasm [30]. As such, TRPM2 is a crucial endogenous sensor of ROS, linking it to Ca2+ influx.
Notably, a direct link has been described between TRPM2 and the NLRP3 inflammasome; knockout or inhibition of TRPM2 abolished ROS-dependent NLRP3 inflammasome activation in macrophages and monocytes [31,32]. On one hand, ROS is considered a signal downstream to nearly all NLRP3-activating stimuli, which modulates NLRP3 inflammasome assembly [8]; on the other hand, TRPM2 links ROS to inflammasome activation in immune cells, such as monocyte, neutrophils, and macrophages, where its expression is abundant [33,34]. As ROS stimulates ADPR production, ADPR is a second messenger that induces Ca2+ influx through TRPM2 and is particularly important for inflammasome activation.
Mechanism of human TRPM2 co-activation by ADPR and Ca2+
Recent years saw the cryo-electron-microscopy (cryo-EM) structure determination of human TRPM2 (hsTRPM2), zebrafish TRPM2 (drTRPM2), and sea anemone TRPM2 (nvTRPM2) [28,35,36]. These structures and biochemical experiments reveal striking differences in the gating mechanisms among the TRPM2 orthologs from different species, and we will focus our discussions here on the hsTRPM2 structures, which were solved in apo state, ADPR-bound state, and ADPR/Ca2+-doubly bound state [28]. The hsTRPM2 structures display an overall architecture of three tiers (Fig. 2). The top/TM tier contains pre-S1, S1-S4 VSLD, and S5-S6 gating domain and the TRP helix. The middle tier consists of the rib helix and the MHR4 domain with mainly stacked α-helices. The bottom/peripheral tier is composed of N-terminal MHR1/2, MHR3, C-terminal NUDT9H domain, and the pole helix. The TRP helix, in particular, bridges the cytosolic domain and the TM domain (Fig. 2).
Figure 2.
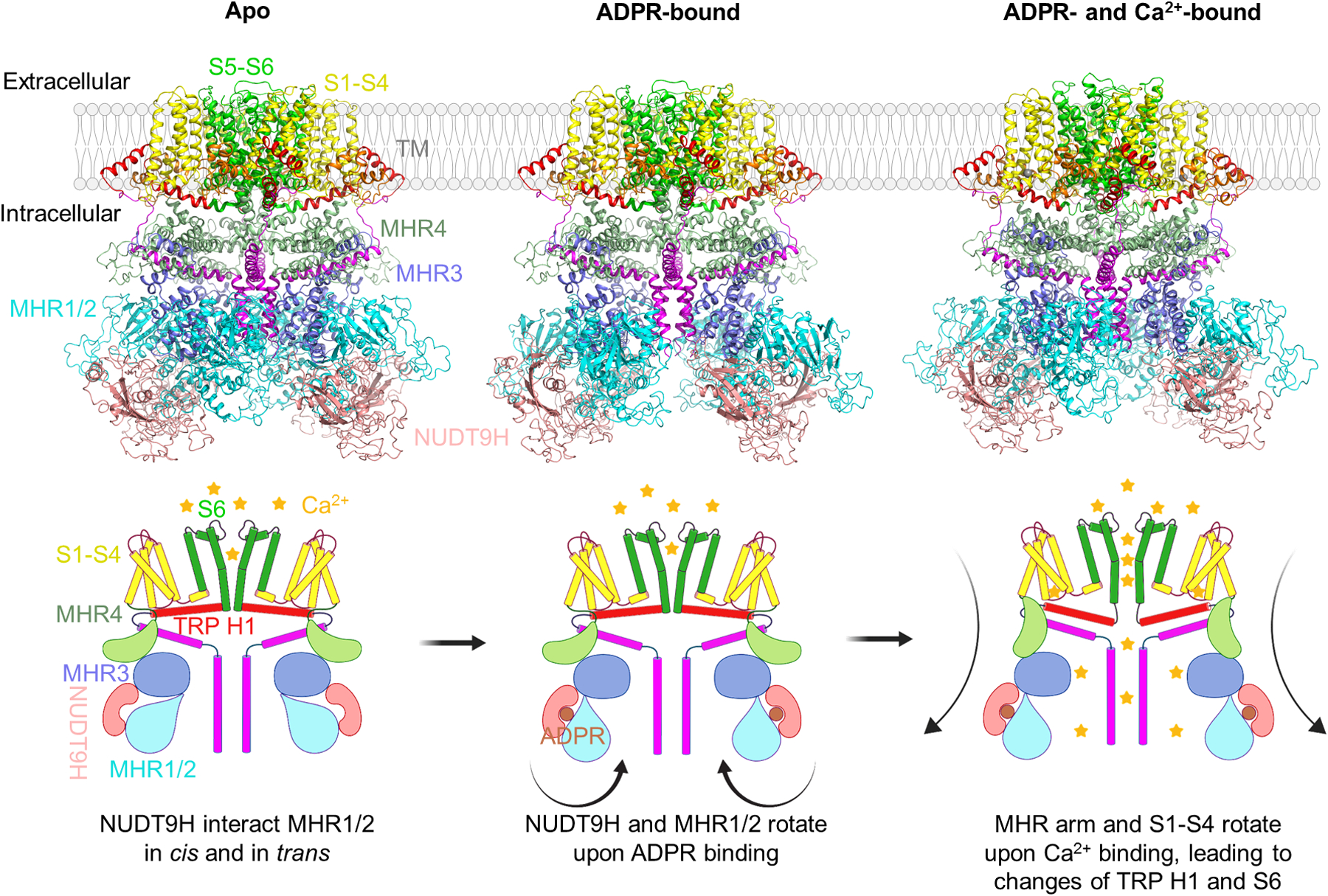
Structures and activation mechanism of human TRPM2.
In the apo state (left), the channel is in a closed conformation with S6 helix (green) forming the lower gate and NUDT9H (pink) interacting with both MHR1/2 (cyan) and MHR3 (marine) domains in cis and the MHR1/2 domain from a neighboring subunit in trans. Upon ADPR binding (middle), rotation of MHR1/2 and disengagement of the trans interaction prime the channel for opening. In the open conformation (right) doubly bound to ADPR and Ca2+, the Ca2+ site directly engages S2 and S3 helices (yellow) and TRP H1 (red), leading to a tilt at TRP H1 and partial melting at the S6-TRP junction to trigger S6 rotation and channel opening. Arrows indicate conformational transitions.
In the apo state of hsTRPM2, the C-terminal NUDT9H domain forms extensive interactions with N-terminal MHR domains both within the subunit (in cis) and between the subunits (in trans) (Fig. 2). Through these interfaces, NUDT9H may restrict inter-subunit movement and stabilize MHR arms (MHR1/2, MHR3, and MHR4) in the absence of ADPR binding. In the ADPR-bound state, large conformational rearrangements occur at the bottom tier of the channel: the NUDT9H and MHR1/2 domains rotate by about 27°. The rotation disrupts the trans interactions between NUDT9H and MHR1/2 to prime the channel for opening. In the ADPR/Ca2+-doubly bound state, the Ca2+ ion directly engages S2, S3, and TRP H1 helices, leading to a tilt at TRP H1 and partial melting at the S6-TRP junction to trigger the S6 rotation for channel opening. The Ca2+-induced conformational change at TRP H1 is not possible without the signal of ADPR binding, because the tightly coupled MHR domains would not be able to tilt with TRP H1 in the presence of the trans interactions between NUDT9H and MHR1/2 before ADPR binding. We propose that the TRP domain acts as an allosteric center to integrate stimulations from the TM and cytosolic regions for gating.
Positive feedback and lack of inactivation
In contrast to hsTRPM2, the cryo-EM map of full-length nvTRPM2 does not show density corresponding to the NUDT9H domain, which indicates that the NUDT9H is flexibly linked to the main body of nvTRPM2. Moreover, it was demonstrated that the NUDT9H domain of nvTRPM2 hydrolyzes ADPR, whereas the NUDT9H domain of hsTRPM2 only binds to ADPR but has no enzymatic activity [28,29]. These evidence suggests that nvTRPM2 and hsTRPM2 have fundamentally different gating mechanisms. Indeed, a recent study shows dramatic difference in the kinetics of APDR/Ca2+-induced currents between the two channels [37]. The ADPR/Ca2+-induced current of hsTRPM2 accumulates slowly and shows very little inactivation for several minutes, whereas nvTRPM2 reaches maximal current immediately followed by rapid inactivation. The different kinetics of TRPM2 channels in different species are likely suited for their different biological functions. nvTRPM2 does not response to ROS and may function as a chemosensor, in which inactivation is often observed. For hsTRPM2, its Ca2+-dependent positive feedback mechanism may ensure continuous cytosolic Ca2+ mobilization for inflammasome activation and cell death induction.
Concluding remarks
Human TRPM2 has been implicated in inflammatory diseases, neurological disorders, and cancers, although it is yet to be discovered whether the TRPM2/NLRP3 axis is responsible for these serious diseases. Nevertheless, TRPM2 is an attractive therapeutic target for human disease modulation. Tremendous progress has been made recently on the mechanism of TRPM2 function. However, there are still many missing pieces on the structure, biology and regulation of the TRPM2 channel. Further studies are needed to help us better understand this sophisticated cation channel and substantially speed up the development of TRPM2-targeting therapies.
Figure 1.
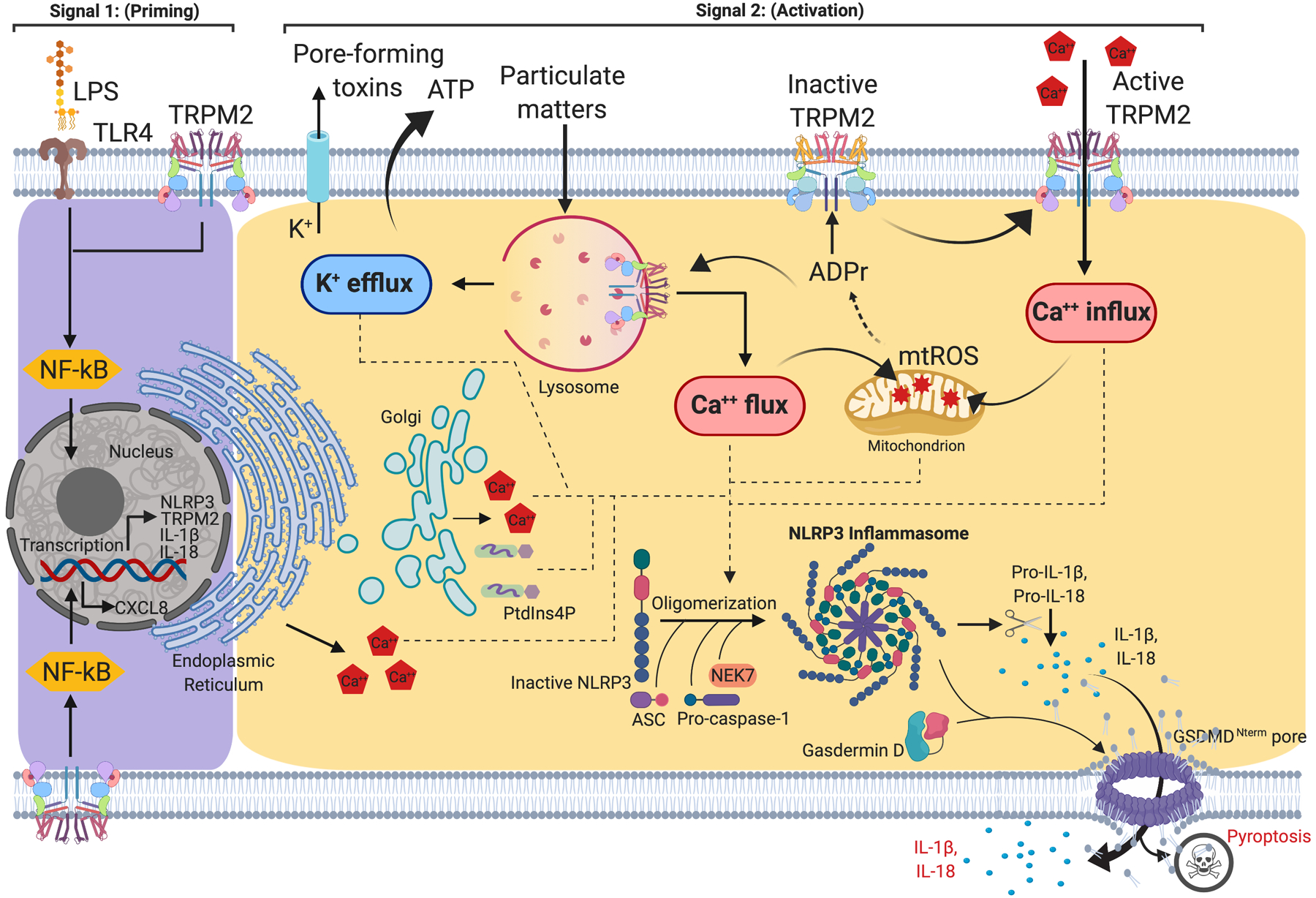
NLRP3 inflammasome priming and activation.
Signal 1 (priming, left) is mediated by the engagement of the Toll-like receptor (TLR) signaling pathway, which detects structurally conserved molecules derived from microbes and leads to NF-κB-mediated up-regulation of intracellular level of NLRP3, pro-IL-1β and pro-IL-18. Signal 2 (activation; right) is provided by a wide range of pathogens-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), such as pore forming toxins, particulate matters and ATP, that converge to K+ efflux, Ca2+ influx, lysosomal disruption and mitochondrial reactive oxygen species (mtROS) production. The assembly of NLRP3 inflammasome requires NEK7 and culminates in the activation of caspase-1, which in turn converts pro-IL-1β and pro-IL-18 into their mature forms. Caspase-1 also cleaves gasdermin D (GSDMD), whose active N-terminal fragment inserts and forms pores in the plasma membrane, inducing pyroptosis as well as releasing IL-1β and IL-18 to the extracellular milieu.
Highlights.
ROS is both a trigger and an effector of NLRP3 inflammasome activation
Ca2+ influx is upstream of NLRP3 inflammasome activation
ADP-ribose (ADPR) activates TRPM2 under ROS in the NLRP3 pathway
ADPR and Ca2+ co-activate TRPM2
There exist drastic differences in TRPM2’s structure and function during evolution
References
References and recommended reading Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
• • of outstanding interest
- 1.Martinon F, Agostini L, Meylan E, Tschopp J: Identification of Bacterial Muramyl Dipeptide as Activator of the NALP3/Cryopyrin Inflammasome. Current Biology 2004, 14:1929–1934. [DOI] [PubMed] [Google Scholar]
- 2.•.Swanson KV, Deng M, Ting JPY: The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol 2019, doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review summarizes recent research progress on the regulation of NLRP3 activation and discusses potential strategies to target the NLRP3 inflammasome.
- 3.Amores-Iniesta J, Barberà-Cremades M, Martínez CM, Pons JA, Revilla-Nuin B, Martínez-Alarcón L, Di Virgilio F, Parrilla P, Baroja-Mazo A, Pelegrín P: Extracellular ATP Activates the NLRP3 Inflammasome and Is an Early Danger Signal of Skin Allograft Rejection. Cell Rep 2017, 21:3414–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greaney AJ, Leppla SH, Moayeri M: Bacterial Exotoxins and the Inflammasome. Front. Immunol 2015, 6:1013–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He Y, Hara H, Núñez G: Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends in Biochemical Sciences 2016, 41:1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G: K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38:1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.•.Lee G-S, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ: The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that intracellular Ca2+ plays important roles in NLRP3 inflammasome activation.
- 8.Zhou R, Yazdi AS, Menu P, Tschopp J: A role for mitochondria in NLRP3 inflammasome activation. Nature 2010, 469:221–225. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, Wang H, Kouadir M, Song H, Shi F: Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis 2019, 10:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieberman J, Wu H, Kagan JC: Gasdermin D activity in inflammation and host defense. Sci Immunol 2019, 4:eaav1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Febbraio MA: Role of interleukins in obesity: implications for metabolic disease. Trends in Endocrinology & Metabolism 2014, 25:312–319. [DOI] [PubMed] [Google Scholar]
- 12.Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD: The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med 2011, 17:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grebe A, Hoss F, Latz E: NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res 2018, 122:1722–1740. [DOI] [PubMed] [Google Scholar]
- 14.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440:237–241. [DOI] [PubMed] [Google Scholar]
- 15.Alberts BM, Bruce C, Basnayake K, Ghezzi P, Davies KA, Mullen LM: Secretion of IL-1β From Monocytes in Gout Is Redox Independent. Front. Immunol 2019, 10:503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karki R, Kanneganti T-D: Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 2019, 19:197–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.••.Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, Schubert K, Schöneberg T, Schaefer M, Krügel U, et al. : Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun 2012, 3:417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that increased extracellular Ca2+ leads to increased intracellular Ca2+ concentration, and activates the NLRP3 inflammasome.
- 18.••.Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T: Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proceedings of the National Academy of Sciences 2012, 109:11282–11287. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights the role of Ca2+ mobilization in NLRP3 inflammasome activation.
- 19.Bagur R, Hajnóczky G: Intracellular Ca 2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66:780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Meszaros G, He W-T, Xu Y, de Fatima Magliarelli H, Mailly L, Mihlan M, Liu Y, Puig Gámez M, Goginashvili A, et al. : Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med 2017, 214:2671–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong T, Yang Y, Jin T, Jiang W, Zhou R: Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends Immunol. 2018, 39:393–406. [DOI] [PubMed] [Google Scholar]
- 22.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E: Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008, 9:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han H, Yi F: New insights into TRP channels. Channels 2013, 8:13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nilius B, Owsianik G: The transient receptor potential family of ion channels. Genome Biol. 2011, 12:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venkatachalam K, Montell C: TRP channels. Annu. Rev. Biochem 2007, 76:387–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, et al. : ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411:595–599. [DOI] [PubMed] [Google Scholar]
- 27.Shen BW, Perraud AL, Scharenberg A, Stoddard BL: The crystal structure and mutational analysis of human NUDT9. J. Mol. Biol 2003, 332:385–398. [DOI] [PubMed] [Google Scholar]
- 28.••.Wang L, Fu T-M, Zhou Y, Xia S, Greka A, Wu H: Structures and gating mechanism of human TRPM2. Science 2018, 362:eaav4809–35. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors report the cryo-EM structures of human TRPM2 in apo, primed and open states, propose a model for TRPM2 gating, and uncover mechanistic differences in TRPM2 from different species.
- 29.Iordanov I, Mihályi C, Tóth B, Csanády L: The proposed channel-enzyme transient receptor potential melastatin 2 does not possess ADP ribose hydrolase activity. Elife 2016, 5:25059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knowles H, Li Y, Perraud AL: The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol. Res 2013, 55:241–248. [DOI] [PubMed] [Google Scholar]
- 31.••.Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, Qiao L: TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun 2013, 4:252–24. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors show TRPM2 as a key factor for NLRP3 inflammasome activation and a potential target for NLRP3 inflammasome-associated diseases.
- 32.Tseng HHL, Vong CT, Kwan YW, Lee SM-Y, Hoi MPM: TRPM2 regulates TXNIP-mediated NLRP3 inflammasome activation via interaction with p47 phox under high glucose in human monocytic cells. Sci Rep 2016, 6:47–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di A, Gao X-P, Qian F, Kawamura T, Han J, Hecquet C, Ye RD, Vogel SM, Malik AB: The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol 2011, 13:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, et al. : TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med 2008, 14:738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.•.Zhang Z, Tóth B, Szollosi A, Chen J, Csanády L: Structure of a TRPM2 channel in complex with Ca2+ explains unique gating regulation. Elife 2018, 7:213. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the structure of sea anemone TRPM2, in which the NUDT9H domain is disordered.
- 36.•.Huang Y, Winkler PA, Sun W, x000FC L W, Du J: Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 2018, doi: 10.1038/s41586-018-0558-4. [DOI] [PubMed] [Google Scholar]; The authors report the cryo-EM structures of zebrafish TRPM2 in apo and open states, and identify a new ADPR binding site at MHR1/2 domain.
- 37.•.Kühn FJP, Watt JM, Potter BVL, Lückhoff A: Different substrate specificities of the two ADPR binding sites in TRPM2 channels of Nematostella vectensis and the role of IDPR. Sci Rep 2019, 9:4985. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes dramatic differences between human and sea anemone TRPM2, with an example in which two antagonists of human TRPM2 are agonists for sea anemone TRPM2.