

Abstract
The reaction of carboxylic acid derivatives with amines to form amide bonds has been the most widely used transformation in organic synthesis over the past century. Its utility is driven by the broad availability of the starting materials as well as the kinetic and thermodynamic driving force for amide bond formation. As such, the invention of new reactions between carboxylic acid derivatives and amines that strategically deviate from amide bond formation remains both a challenge and an opportunity for synthetic chemists. This report describes the development of a nickel-catalyzed decarbonylative reaction that couples (hetero)aromatic esters with a broad scope of amines to form (hetero)aryl amine products. The successful realization of this transformation was predicated on strategic design of the cross-coupling partners (phenol esters and silyl amines) to preclude conventional reactivity that forms inert amide by-products.
Graphical Abstract
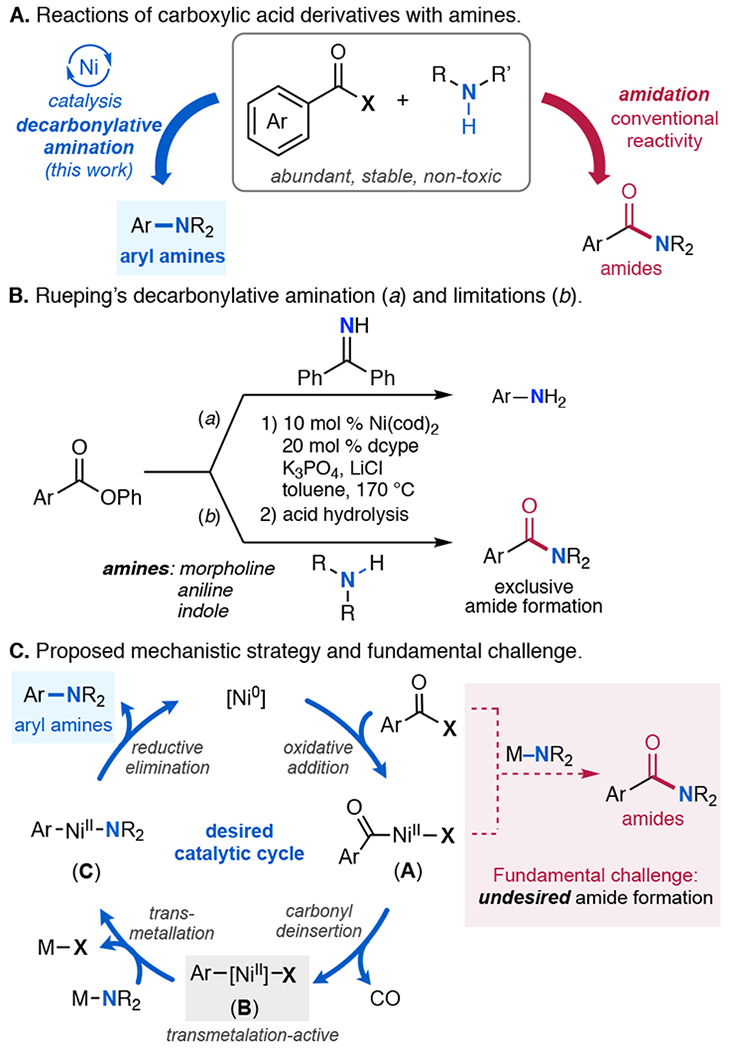
Carboxylic acids are abundant, inexpensive, and stable compounds, and these properties render them attractive building blocks for organic synthesis. As such, metal-catalyzed decarbonylative or decarboxylative reactions that employ aryl carboxylic acid derivatives [ArC(O)X] as coupling partners have gained tremendous attention over the past two decades.1,2 A variety of (hetero)aryl carbon–carbon, carbon–sulfur, carbon–boron, carbon–silicon, carbon–halogen, and other carbon–heteroatom linkages can be formed using this approach.1 However, methods for the decarbonylative or decarboxylative coupling of ArC(O)X (Ar = aryl) with amines to afford aryl C(sp2)–N bonds (Figure 1A) remain limited. While a few such transformations have been reported, they often have a very narrow scope with respect to the carboxylic acid and/or amine coupling partner.3–6 For example, most decarboxylative methods require strong electron withdrawing groups on the arene ring, or weakly nucleophilic N-group to form carbamates.5–6 In decarbonylative reactions, couplings involving non-stabilized 1° or 2° amines remain challenging, largely due to the competing amide formation with these strongly nucleophilic partners (Figure 1A, conventional reactivity).3 This is exemplified by Rueping’s recent report of Ni-catalyzed decarbonylative amination of phenyl esters. As shown in Figure 1B, this transformation is effective for weakly nucleophilic benzophenone imine. In contrast, the use of more nucleophilic amines such as morpholine,3 aniline, and indole results in exclusive formation of the amide product.
Figure 1.
Proposed strategy for the catalytic decarbonylation of carboxylic acid derivatives to aryl amines.
The paucity of methods to convert carboxylic acid derivatives to amines is particularly noteworthy because the products are of high value in medicinal chemistry.7 As such, new, general, and practical approaches to forge (hetero)aryl C(sp2)–N bonds from abundant starting materials have the potential for widespread application.
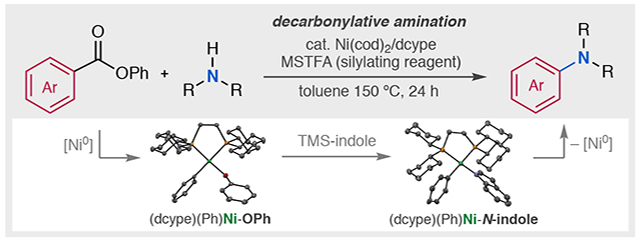
We hypothesized that undesired background amide formation could be mitigated by masking the amine with a main group element (M). Appropriate selection of M would provide an amine nucleophile, M–NR2, that is inert to background acyl transfer but can selectively engage a metal catalyst via transmetalation.8 A catalytic cycle that leverages this approach is shown in Figure 1B. First, ArC(O)X reacts with a low valent metal catalyst via oxidative addition and carbonyl deinsertion to afford an arylmetal intermediate (B).9 Intermediate B then undergoes transmetalation with M–NR2 and subsequent C(sp2)–NR2 coupling to release the targeted aryl amine product. The success of this cycle relies on strategic design of M–NR2, ArC(O)X, and the metal catalyst such that: (i) background acyl transfer between M–NR2 and ArC(O)X is slow; (ii) B undergoes facile transmetalation with M–NR2; (iii) carbonyl deinsertion is fast relative to transmetalation (since reaction between M–NR2 and metal acyl intermediate A would afford the undesired amide by-product; Figure 1B);10 and (iv) other key steps of the catalytic cycle (oxidative addition, C(sp2)–N coupling) are energetically feasible. This report describes the successful realization of this transformation, using a nickel phosphine catalyst to couple aromatic ester electrophiles with in situ-generated silyl amines.
Based on the criteria outlined above, trimethylsilyl (TMS)-substituted amines as the M–NR2 nucleophile are expected to slow background acyl transfer, while facilitating base-free transmetalation8 between TMS–NR2 and B. To identify a suitable ArC(O)X coupling partner, we evaluated the background reaction of TMS–morpholine with three carboxylic acid derivatives: acid chloride 1-Cl, acid fluoride 1-F, and aryl ester 1-OPh. Heating TMS–morpholine with 1-Cl or 1-F at 100 °C for 1 h resulted in undesired acyl transfer to afford amide 3 in high yield (Figure 2A, [M] = TMS). In contrast, the less electrophilic 1-OPh showed <5% amide formation under analogous conditions. Notably, as expected, the use of free morpholine as the nucleophile led to a high yield of the amide product with all three electrophiles (Figure 2A, [M] = H).
Figure 2.
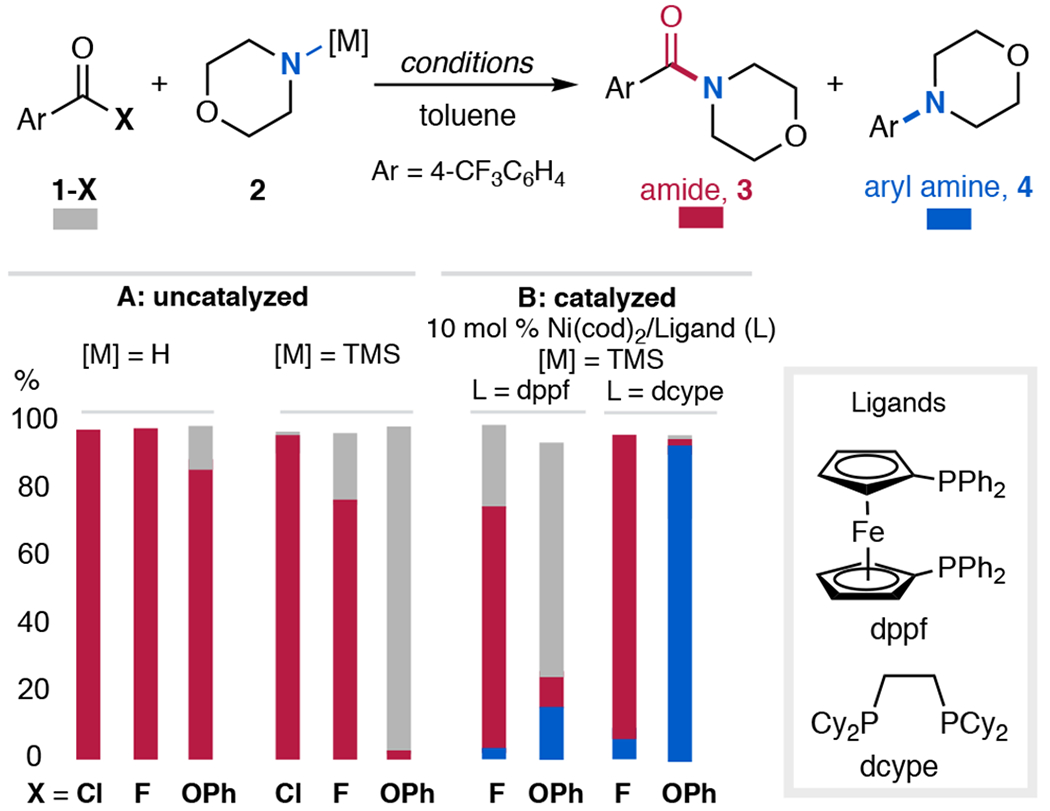
(A) Uncatalyzed reaction of 1-X with morpholine and TMS–morpholine at 100 °C for 1 h. (B) Ni-catalyzed reaction of 1-X with TMS–morpholine at 150 °C for 24 h. Yields determined via 19F NMR spectroscopy.
We next examined the coupling of TMS–morpholine with 1-F and 1-OPh in the presence of Ni-bisphosphine catalysts (Figure 2B). Importantly, Ni phosphine complexes are known to participate in oxidative addition and carbonyl deinsertion with diverse ArC(O)X electrophiles.1 Bisphosphine supporting ligands were chosen based on their ability to effect C(sp2)–N coupling at NiII centers.11 Representative results with dppf and dcype are shown in Figure 2 (see SI for larger ligand screen). Consistent with the fast background reaction, amide formation dominated with 1-Cl and 1-F under catalytic conditions. In contrast, with 1-OPh, decarbonylative coupling to afford aryl amine 4 proceeded in modest to high yield with dppf and dcype, respectively. Under the optimized conditions (10 mol % Ni/dcype in toluene at 150 °C), 1-OPh reacted with TMS–morpholine to afford 4 in 90% yield with >19:1 selectivity for amine 4 versus amide 3.
We next explored the scope of this transformation and found that it is general for a variety of electron-deficient and electron-neutral carboxylic acid esters (Figure 3A).12 Substituents such as trifluoromethyl, methyl ester, nitrile, ketone, and phenyl ether (4-9) are well tolerated. Competing cross-coupling is not observed at methyl ester (5) or boronate ester (10) sites. Various N-containing heteroaryl carboxylic acid esters such as pyridine, quinoline, and quinoxaline derivatives are converted to N-heteroaryl amines (12-14) in moderate to excellent yields. S- and O-containing heteroaryl esters such as benzothiophene (15), benzofuran (16), chromone (17), and thiazoles (20) are also converted to the desired amine products. Esters derived from carboxylic acid-containing drugs such as probenecid (18), bexarotene (19) and febuxostat (20) afford good to excellent yields. This transformation is also general with respect to the amine coupling partner (Figure 3B). Utilizing the probenecid ester 18-OPh as the electrophile, various TMS-amines react smoothly to yield 18, 25, 27, and 28. More stable triethylsilyl (TES)- and triisopropylsilyl (TIPPS)-protected amines are effective coupling partners but provide lower yields of 25.
Figure 3.
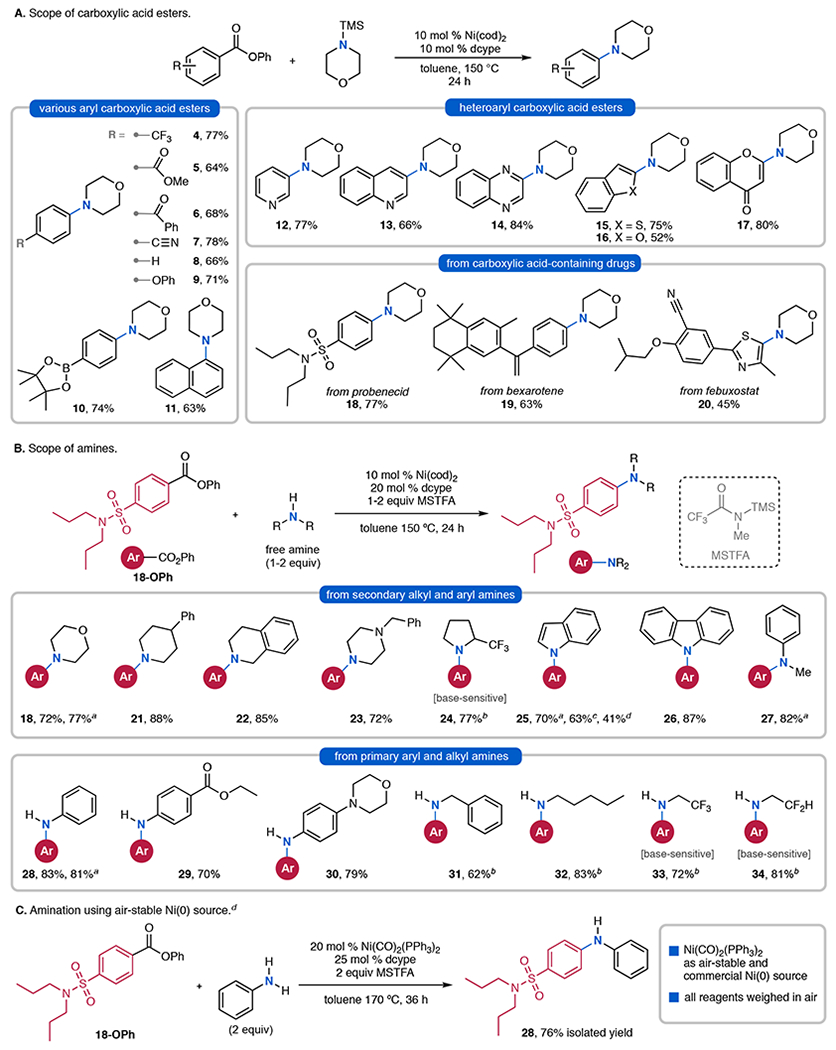
Scope of Ni-catalyzed decarbonylative amination. aUsing TMS–amine. bTMS–amine was generated in situ by premixing the amine with MSTFA. cUsing TES–indole or dTIPPS–indole. dReagents were weighed on benchtop. For additional substrates that were found to be challenging under the optimized conditions, see SI.
While TMS–amines are straightforward to synthesize, their commercial availability is limited. In addition, some derivatives are susceptible to hydrolysis. Thus, the in situ formation of these species from readily available amine starting materials would significantly enhance the practicality of this method. After some optimization, we identified the commercial silyl transfer reagent N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) as effective for the rapid, room temperature conversion of diverse HNR2 to TMS–NR2. Indeed, the direct addition of HNR2 and MSTFA to the standard coupling conditions resulted in effective Ni-catalyzed decarbonylative coupling (Figure 3B). Secondary dialkyl and diaryl N-heterocycles13 such as morpholines (18), piperidines (21, 22), piperazines (23), pyrrolidines (24), indoles (25), and carbazoles (26) underwent coupling in good to excellent yields under these conditions. Furthermore, both primary aryl (28-30), and alkyl amines (31-34) afforded secondary aryl amines products in good yields. In the few cases where background acyl transfer reactivity was observed (31-34), prestirring of HNR2 with MSTFA for 1 h prior to catalysis resulted in selective generation of the desired aryl amine.
Traditional metal-catalyzed couplings between aryl electrophiles and amines require typically stoichiometric quantities of an exogenous inorganic base to promote metalation of the amine coupling partner.14 This is a key limitation of the Buchwald-Hartwig amination of aryl halides, and significant recent effort has focused on identifying milder bases for these transformations.15 In contrast, the current method eliminates the need for an exogenous base for C(sp2)–N coupling. As such, base-sensitive amine substrates16 are well tolerated and deliver aryl amine products (24, 33, 34) in good yields.
We then set out studies focused on eliminating the need for air-sensitive Ni(cod)2 as the nickel source. These investigations revealed that the use of Ni(CO)2(PPh3)2, an air-stable and commercial reagent, as Ni(0) source affords aryl amine 28 in good yield (Figure 3C). All of the catalysts and reagents were weighed on the benchtop without the requirement of an inert glovebox.
Finally, we conducted stoichiometric studies to interrogate the proposed reaction mechanism. The reaction of phenyl ester 8-OPh with Ni(cod)2/dcype in toluene at 80 °C for 3 h resulted in oxidative addition/carbonyl deinsertion to afford (dcype)Ni(Ph)(OPh) (B) in 60% yield (Figure 4A).17 A NiII acyl intermediate (A in Figure 1B) was not detected by 31P NMR spectroscopy during this reaction, suggesting that carbonyl deinsertion is fast under these conditions. Notably, reactions performed at 60 °C or lower did not afford observable conversion of 8-OPh due to slow oxidative addition. The treatment of B with TMS–indole in toluene at room temperature for 1 h resulted in transmetalation to form NiII complex C in quantitative yield (Figure 4B). Complex C is stable and isolable at room temperature. Aryl C(sp2)–N bond-forming reductive elimination was only observed upon heating at 120 °C, which afforded aryl amine product 35 in 65% yield after 16 h. These studies show the feasibility of each proposed step of the proposed catalytic cycle. Furthermore, they demonstrate that in this stoichiometric system, C(sp2)–N bond formation is the most challenging step of the sequence.
Figure 4.
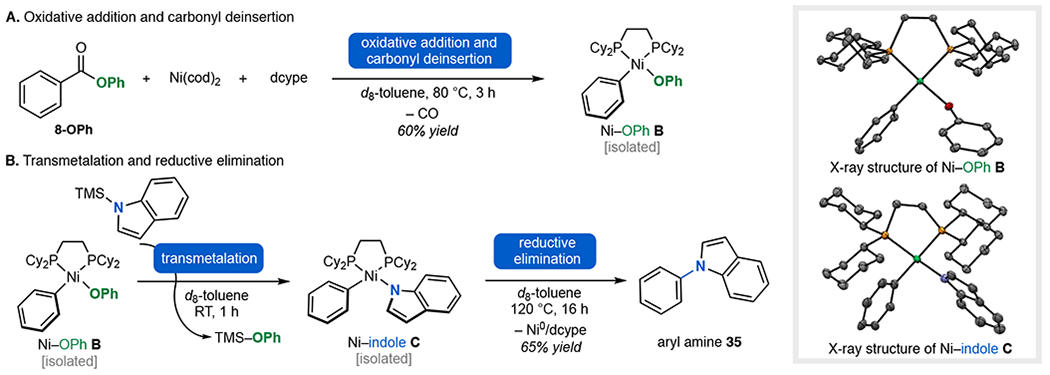
Mechanistic studies: stoichiometric reactions for the fundamental steps in decarbonylative amination. See the SI for details on reaction conditions.
In conclusion, we developed a Ni-catalyzed decarbonylative conversion of esters to aryl amines. The generality, selectivity, and base-free nature of this transformation render it complementary to existing Pd/Ni-catalyzed methods for the construction of (hetero)aryl amines. Current limitations, including low reactivity of electron rich and sterically hindered aryl esters (see SI), and the requirement of high temperatures and catalyst loading, will be addressed by future mechanistic and catalysis development studies.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge J. W. Kampf for X-ray crystallographic analyses of compounds B and C. We acknowledge financial support from NIH NIGMS (GM073836), the Danish National Research Foundation (Carbon Dioxide Activation Center; CADIAC), and the Spanish Ministry of Science (PhD grant to M.B.; BES-2016-076349).
Footnotes
Supporting Information
Supporting Information is available free of charge on the ACS Publications website.
Experimental details, characterization data, and NMR spectra of compounds (PDF)
X-ray crystal structure files (CIF)
The authors declare no competing financial interest.
REFERENCES
- 1.For recent reviews, see:; (a) Ogiwara Y; Sakai N Acyl fluorides in late transition-metal catalysis. Angew. Chem. Int. Ed 2019, DOI: 10.1002/ange.201902805 [DOI] [PubMed] [Google Scholar]; (b) Blanchard N; Bizet V Acid fluorides in transition-metal catalysis: a good balance between stability and reactivity. Angew. Chem. Int. Ed 2019, 58, 2. [DOI] [PubMed] [Google Scholar]; (c) Guo L; Rueping M Decarbonylative cross-couplings: nickel catalyzed functional group interconversion strategies for the construction of complex organic molecules. Acc. Chem. Res 2018, 51, 1185. [DOI] [PubMed] [Google Scholar]; (d) Meng G; Szostak M N-Acyl-glutarimides: privyleged scaffolds in amide N–C bond cross-coupling. Eur. J. Org. Chem 2018, 2352 [Google Scholar]; (e) Takise R; Muto K; Yamaguchi J Cross-coupling of aromatic esters and amides. Chem. Soc. Rev 2017, 46, 5864. [DOI] [PubMed] [Google Scholar]; (f) Rodriguez N; Gooβen LJ Decarboxylative coupling reactions: a modern strategy for C–C bond formation. Chem. Soc. Rev 2011, 40, 5030. [DOI] [PubMed] [Google Scholar]
- 2.For seminal report on decarboxylative biaryl synthesis:; Gooβen LJ; Deng G; Levy LM Synthesis of biaryls via catalytic decarboxylative coupling. Science 2006, 313, 662. [DOI] [PubMed] [Google Scholar]
- 3.(a) Yue H; Guo L; Liao H-H; Cai Y; Zhu C; Rueping M Catalytic ester and amide to amine interconversion: nickel-catalyzed decarbonylative amination of esters and amides by C–O and C–C bond activation. Angew. Chem. Int. Ed 2017, 56, 4282. [DOI] [PubMed] [Google Scholar]; (b) Lee S-C; Guo L; Yue H; Liao H-H; Rueping M Nickel-catalyzed decarbonylative silylation, borylation, and amination of arylamides via a deaminative reaction pathway. Synlett 2017, 28, 2594. [Google Scholar]
- 4.For intramolecular decarbonylation of aryl amides, see:; (a) Liu X; Yue H; Jia J; Guo L; Rueping M Synthesis of amidines from amides using nickel-catalyzed decarbonylative amination through CO extrusion intramolecular recombination fragment coupling. Chem. Eur. J 2017, 23, 11771. [DOI] [PubMed] [Google Scholar]; (b) Morioka T; Nakatani S; Sakamoto Y; Kodama T; Ogoshi S; Chatani N; Tobisu M Nickel-catalyzed decarbonylation of N-acylated N-heteroarenes. Chem. Sci 2019, 10, 6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For catalytic decarboxylative formation of aryl C(sp2)–N bonds, see:; (a) Zhang Y; Patel S; Mainolfi N Copper-catalyzed decarboxylative C–N coupling for N-arylation. Chem. Sci 2012, 3, 3196 [Google Scholar]; (b) Sheng W-J; Ye Q; Yu W-B; Liu R-R; Xu M; Gao J-R; Jia Y-X CuSO4-mediated decarboxylative C–N cross-coupling of aromatic carboxylic acids with amides and anilines. Tetrahedron Lett. 2015, 56, 599 [Google Scholar]; (c) Dai Q; Li P; Ma N; Hu C Palladium-catalyzed decarboxylative synthesis of arylamines. Org. Lett 2016, 18, 5560. [DOI] [PubMed] [Google Scholar]; (d) Ghorbani-Choghamarani A ; Taherinia Z High catalytic activity of peptide nanofibers decorated with Ni and Cu nanoparticles for the synthesis of 5-substituted 1H-tetrazoles and N-arylation of amines. Aus. J. Chem 2017, 70, 1127 [Google Scholar]; (e) Pichette Drapeau M; Bahri J; Lichte D; Gooβen LJ Decarboxylative ipso amination of activated benzoic acids. Angew. Chem. Int. Ed 2019, 58, 892. [DOI] [PubMed] [Google Scholar]
- 6.For decarboxylation to alkyl C(sp3)–N bonds, see:; Trapani G; Reho A; Latrofa A Trimethylamine-borane as useful reagent in the N-acylation or N-alkylation of amines by carboxylic acids. Synthesis 1983, 12, 1013 [Google Scholar]; (b) Zhao W; Wurz RP; Peters JC; Fu GC Photoinduced, copper-catalyzed decarboxylative C–N coupling to generate protected amines: an alternative to Curtius rearrangement. J. Am. Chem. Soc 2017, 139, 12153. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liang Y; Zhang X; MacMillan DWC Decarboxylative sp3 C–N coupling via dual copper and photoredox catalysis. Nature 2018, 559, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mao R; Frey A; Balon J; Hu X Decarboxylative C(sp3)–N cross-coupling via synergetic photoredox and copper catalysis. Nat. Catal 2018, 1, 120 [Google Scholar]; (e) Li P; Ma N; Wang Z; Dai Q; Hu C Base-mediated intramolecular decarboxylative synthesis of alkylamines from alkanoyloxycarbamates. J. Org. Chem 2018, 83, 8233. [DOI] [PubMed] [Google Scholar]
- 7.For selected recent reviews, see:; (a) Forero-Cortés PA; Haydl AM The 25th anniversary of the Buchwald–Hartwig amination: development, applications, and outlook. Org. Process Res. Dev 2019, 23, 1478 [Google Scholar]; (b) Ruiz-Castillo P; Buchwald SL Applications of palladium-catalyzed C–N cross-coupling reactions. Chem. Rev 2016, 116, 12564. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) West MJ; Fyfe JWB; Vantourout JC; Watson AJB Mechanistic development and recent applications of the Chan-Lam amination. Chem Rev. 2019, 119, 12491. [DOI] [PubMed] [Google Scholar]
- 8.(a) Malapit CA; Bour JR; Brigham CE; Sanford MS Base-free nickel-catalysed decarbonylative Suzuki-Miyaura coupling of acid fluorides. Nature 2018, 563, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Malapit CA; Bour JR; Laursen SR; Sanford MS Mechanism and scope of nickel-catalyzed decarbonylative borylation of carboxylic acid fluorides. J. Am. Chem. Soc 2019, 141, 17322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Initial oxidative addition could occur into either the C(carbonyl)–O or C(carbonyl)–C(aryl) bond of the ester:; (a) Ben Halima T; Zhang W; Yalaoui I; Hong X; Yang Y-F; Houk KN; Newman SG Palladium-catalyzed Suzuki-Miyaura coupling of aryl esters. J. Am. Chem. Soc 2017, 139, 1311–1318 [DOI] [PubMed] [Google Scholar]; (b) Chatupheeraphat A; Liao H-H; Srimontree W; Guo L; Minenkov Y; Poater A; Cavallo L; Rueping M Ligand-controlled chemoselective C(acyl)–O bond vs. C(aryl)–C bond activation of aromatic eters in nickel catalyzed C(sp2)–C(sp3) cross-couplings. J. Am. Chem. Soc 2018, 140, 3724–3735. [DOI] [PubMed] [Google Scholar]
- 10.For transition-metal-catalyzed amidation of esters and amides, see:; (a) Baker EL; Yamano MM; Zhou Y; Anthony SM; Garg NK A two-step approach to achieve secondary amide transamidation enabled by nickel catalysis. Nat. Commun 2016, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Malapit CA; Caldwell DR; Sassu N; Milbin S; Howell AR Pd-catalyzed acyl C–O bond activation for selective ring-opening of α-methylene-β-lactones with amines. Org. Lett 2017, 19, 1966. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ben Halima T; Masson-Makdissi J; Newman SG Nickel-catalyzed amide bond formation from methyl esters. Angew. Chem. Int. Ed 2018, 57, 12925. [DOI] [PubMed] [Google Scholar]
- 11.Lavoie CM; Stradiotto M Bisphosphines: a prominent ancillary ligand class for application in nickel-catalyzed C–N cross-coupling. ACS Catal. 2018, 8, 7228. [Google Scholar]
- 12.The more electron rich substrate phenyl 4-methoxybenzoate showed low reactivity. Under the standard reaction conditions, only starting material was detected. We hypothesize that this may be a result of slow oxidative addition (see SI).
- 13.Vitaku E; Smith DT; Njardarson JT Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- 14.(a) Sunesson Y; Lime E; Nilsson Lill SO; Meadows RE; Norrby P-O Role of the base in Buchwald-Hartwig amination. J. Org. Chem 2014, 79, 11961. [DOI] [PubMed] [Google Scholar]; (b) Shekhar S; Hartwig JF Effects of bases and halides on the amination of chloroarenes catalyzed by Pd(PtBu3)2. Organometallics 2007, 26, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Brusoe AT; Hartwig JF Palladium-catalyzed arylation of fluoroalkylamines. J. Am. Chem. Soc 2015, 137, 8460. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dennis JM; White NA; Liu RY; Buchwald SL Breaking the base barrier: an electron-deficient palladium catalyst enables the use of a common soluble base in C–N coupling. J. Am. Chem. Soc 2018, 140, 4721. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dennis JM; White NA; Liu RY; Buchwald SL Pd-Catalyzed C–N coupling reactions facilitated by organic bases: mechanistic investigation leads to enhanced reactivity in the arylation of weakly binding amines. ACS Catal. 2019, 9, 3822. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Beutner GL; Coombs JR; Green RA; Inankur B; Lin D; Qiu J; Roberts F; Simmons EM; Wisniewski SR Palladium-catalyzed amidation and amination of (hetero)aryl chlorides under homogeneous conditions enabled by a soluble DBU/NaTFA dual-base system. Org. Process Res. Dev 2019, 23, 1529 [Google Scholar]; (e) Kawamata Y; Vantourout JC; Hickey DP; Bai P; Chen L; Hou Q; Qiao W; Barman K; Edwards MA; Garrido-Castro AF; deGruyter JN; Nakamura H; Knouse K; Qin C; Clay KJ; Bao D; Li C; Starr JT; Garcia-Irizarry C; Sach N; White HS; Neurock M; Minteer SD; Baran PS Electrochemically driven, Ni-catalyzed aryl amination: scope, mechanism, and applications. J. Am. Chem. Soc 2019, 141, 6392. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu RY; Dennis JM; Buchwald SL The quest for the ideal base: rational design of a nickel precatalyst enables mild, homogeneous C–N cross-coupling. J. Am. Chem. Soc 2010, 142, 4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.A recent report by Hartwig identified these β-fluoroamines as base sensitive. See ref. 15a.
- 17.Complex B does not undergo C(sp2)–O bond-forming reductive elimination even upon heating at 150 °C for 24 h. This is consistent with the observation that diaryl ether side products are not detected during catalysis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.