

Abstract
β-Amino esters are obtained with high levels of enantioselectivity via the conjugate addition of cyclic amines to unactivated α,β-unsaturated esters. A related strategy enables the kinetic resolution of racemic cyclic 2-arylamines, using benzyl acrylate as the resolving agent. Reactions are facilitated by an unprecedented selenourea-thiourea organocatalyst. As elucidated by DFT calculations and 13C kinetic isotope effect studies, the rate-limiting and enantiodetermining step of the reaction is the protonation of a zwitterionic intermediate by the catalyst. This represents a rare case in which a thiourea compound functions as an asymmetric Brønsted acid catalyst.
Graphical Abstract

The prevalence of β-amino acids in nature and the utility of this structural motif in drug discovery have inspired the development of numerous synthetic methods.1 Particularly desirable are approaches that facilitate access to β-amino acid derivatives in a catalytic enantioselective fashion.1 An attractive way in which this can be accomplished is via the well-known conjugate addition of amines to α,β-unsaturated carboxylic acid derivatives.2 Indeed, a range of methods have been reported that facilitate catalytic enantioselective additions of nitrogen-centered nucleophiles to conjugate acceptors (Figure 1).3,4 Examples of such reactions include chiral Lewis-acid-catalyzed conjugate additions of O-alkylhydroxylamine to α,β-unsaturated acylpyrazoles, acylpyrroles, and ketones,5 hydrazoic acid to α,β-unsaturated imides,6 and amines to α,β-unsaturated nitriles,7 α,β-unsaturated imides,8 and maleimides.9 Asymmetric organocatalytic variants include the addition of TMS azide to α,β-unsaturated imides,10 N-silyloxycarbamates,11 and N-heterocycles12 to α,β-unsaturated aldehydes, O-benzylhydroxylamine to α,β-unsaturated acylpyrazoles13 and α,β-unsaturated acids,14 amines to nitroalkenes,15 benzyloxycarbamates to 4,4,4-trifluorocrotonates,16 indolines to α,β-unsaturated ketones,17 and hydroxamic acids to quinone imine ketals.18,19 Intramolecular versions are also known, albeit not with basic amine nucleophiles.20 Organocatalytic enantioselective additions to unactivated α,β-unsaturated esters are rare with any nucleophile,21 likely a consequence of their low electrophilicity.22 There appears to be only a single example of a catalytic enantioselective addition of a challenging basic amine to an α,β-unsaturated ester.23 Specifically, a polymeric Lewis acidic aluminum complex was reported to catalyze the addition of benzylamine to ethylcinnamate, with the corresponding product being obtained in 82% ee.23 Here we report the first examples of catalytic enantioselective conjugate additions of basic, cyclic amines to unactivated α,β-unsaturated esters.
Figure 1.
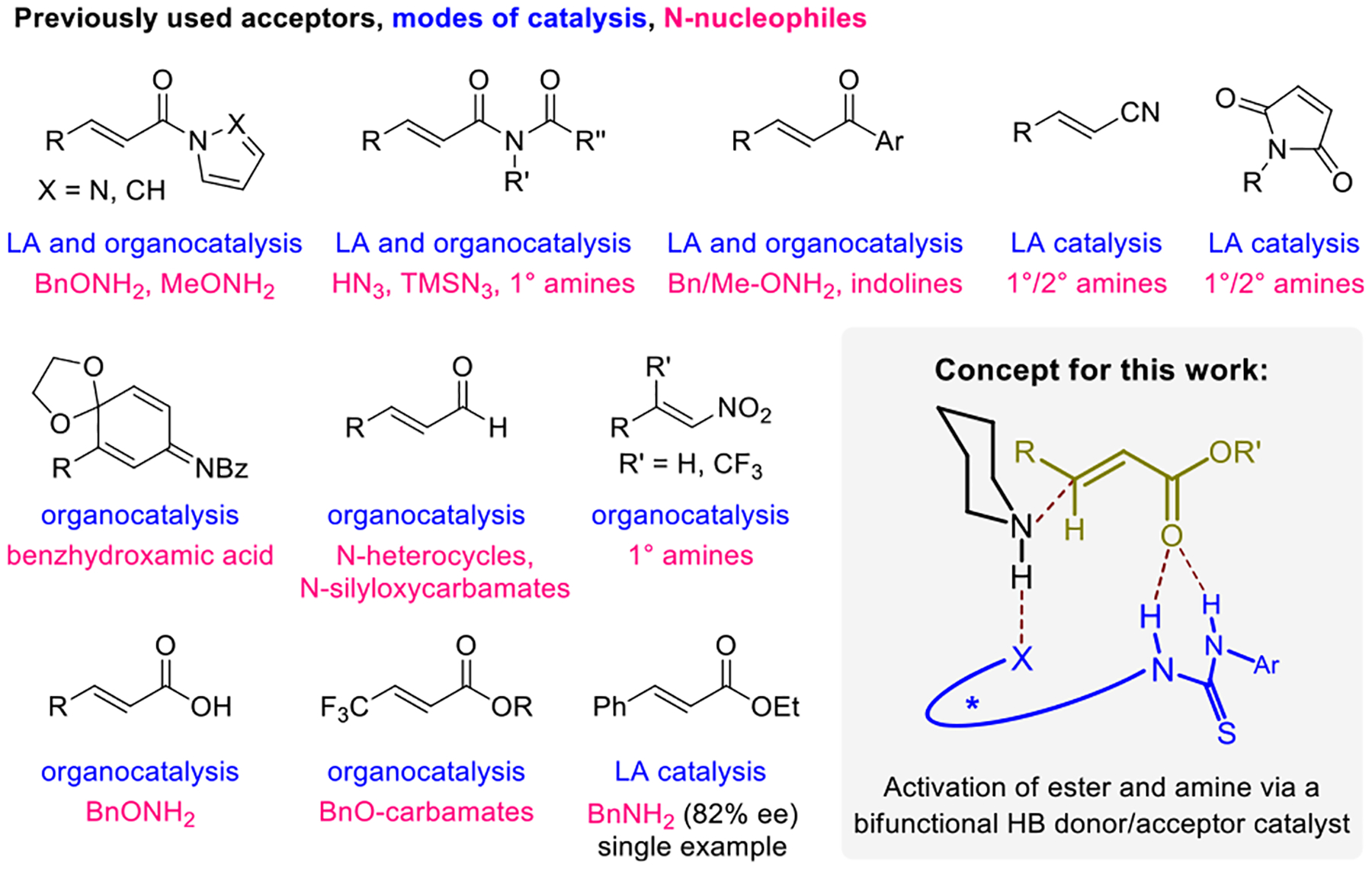
Examples of catalytic enantioselective additions of N-nucleophiles to conjugate acceptors and concept for bifunctional catalysis with α,β-unsaturated ester substrates.
We reasoned that the challenging substrate combination of an unactivated α,β-unsaturated ester and a basic amine nucleophile might be successfully realized through bifunctional organocatalysis (Figure 1).24 Specifically, an organocatalyst featuring an electron-deficient thiourea functionality was envisioned to activate the α,β-unsaturated ester substrate via hydrogen bonding (HB) to the carbonyl oxygen. Such interactions have been observed in X-ray crystal structures25 and are implicated in prior work.21 A Brønsted basic/HB acceptor site on the catalyst could serve to simultaneously activate the amine nucleophile. The absolute stereochemistry of the resulting product would thus be controlled by a network of HB interactions.
This concept was evaluated with piperidine and benzyl crotonate as summarized in Table 1. Relatively high substrate concentrations in toluene solvent (2 M in piperidine) were initially employed. Under these conditions, in the absence of any catalyst at room temperature, approximately 50% conversion of piperidine was noted after 24 h (entry 1). Well-known bifunctional catalysts 1a,26 1b,27 1c,28 and 1d29 all modestly accelerated the reaction, albeit with low or no enantioinduction (entries 2–5). Significant rate acceleration was observed with the Nagasawa bisthiourea catalyst 1e.30 Encouragingly, product 2a was obtained with 43% ee (entry 6). Amide-thiourea 1f provided inferior results (entry 7),31 suggesting an important role for the second thiourea functionality and prompting the evaluation of several catalysts containing an electron-rich thiourea moiety in addition to an electron-poor one (entries 8–13).32 Catalysts 1g–l all outperformed 1e, with 1l providing the best results (entry 13). The analogous urea-thiourea 1m was found to be significantly less reactive and provided 2a in lower ee (entry 14). However, the corresponding selenourea-thiourea 1n achieved significantly improved ee while providing further rate acceleration (entry 15). Catalyst 1o containing an additional bromine substituent to further increase the electron-with-drawing character of the aryl substituent proved better still, despite not being fully soluble (entry 16). A reduction in piperidine concentration proved beneficial in regard to product ee with 0.2 M being optimal (entry 18). Under these conditions, there was no detectable background reactivity within 24 h (entry 19). Further evaluation of reaction parameters and additional catalysts resulted in the identification of catalyst 1q, which, at a 10 mol % loading at −10 °C, provided product 2a in 90% yield and 93% ee (entry 27).
Table 1.
Optimization of the Reaction Conditions
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | solvent (M) | time [h] | yield (%) | ee (%) |
| 1 | - | PhMe (2) | 24 | 50 | - |
| 2 | 1a | PhMe (2) | 23 | 86 | 0 |
| 3 | 1b | PhMe (2) | 22 | 94 | 0 |
| 4 | 1c | PhMe (2) | 20 | 87 | 11 |
| 5 | 1d | PhMe (2) | 20 | 90 | −28 |
| 6 | 1e | PhMe (2) | 4 | 93 | 43 |
| 7 | 1f | PhMe (2) | 7 | 91 | 34 |
| 8 | 1g | PhMe (2) | 5 | 93 | 46 |
| 9 | 1h | PhMe (2) | 4 | 93 | 58 |
| 10 | 1i | PhMe (2) | 4 | 92 | 55 |
| 11 | 1j | PhMe (2) | 5 | 95 | 50 |
| 12 | 1k | PhMe (2) | 4 | 94 | 58 |
| 13 | 1l | PhMe (2) | 4 | 92 | 59 |
| 14 | 1m | PhMe (2) | 18 | 92 | 53 |
| 15 | 1n | PhMe (2) | 3 | 93 | 71 |
| 16a | 1o | PhMe (2) | 3 | 93 | 76 |
| 17 | 1o | PhMe (0.5) | 16 | 91 | 84 |
| 18 | 1o | PhMe (0.2) | 20 | 89 | 86 |
| 19 | - | PhMe (0.2) | 24 | trace | - |
| 20 | 1o | TBME (0.2) | 24 | 86 | 85 |
| 21 | 1o | CHCl3 (0.2) | 24 | 92 | 75 |
| 22a | 1o | PhCF3 (0.2) | 24 | 92 | 79 |
| 23 | 1o | PhH (0.2) | 24 | 92 | 84 |
| 24 | 1p | PhMe (0.2) | 22 | 92 | 85 |
| 25 | 1q | PhMe (0.2) | 24 | 92 | 88 |
| 26b | 1q | PhMe (0.2) | 34 | 89 | 88 |
| 27b,c | 1q | PhMe (0.2) | 72 | 90 | 93 |
Reaction mixture was partially heterogeneous.
Reaction was performed with 10 mol % catalyst.
Reaction was performed at −10 °C.
The scope of the reaction is summarized in Scheme 1. A range of cyclic amines participated in the title reaction to provide products 2 with good to excellent levels of enantioselectivity. p-Methoxybenzylamine, a representative primary amine, provided products with slightly reduced ee. Surprisingly, a significant drop in reactivity was noted with benzyl 2-pentenoate, an observation that could be partially rationalized by our computational model (vide infra).33 Acyclic secondary amines such as diethylamine and N-benzylmethylamine provided poor conversions, even in reactions conducted at rt for a period of several days (not shown). α-Branched primary amines such as benzhydrylamine failed to undergo conjugate additions even at a temperature of 40 °C (not shown).
Scheme 1. Substrate Scope.
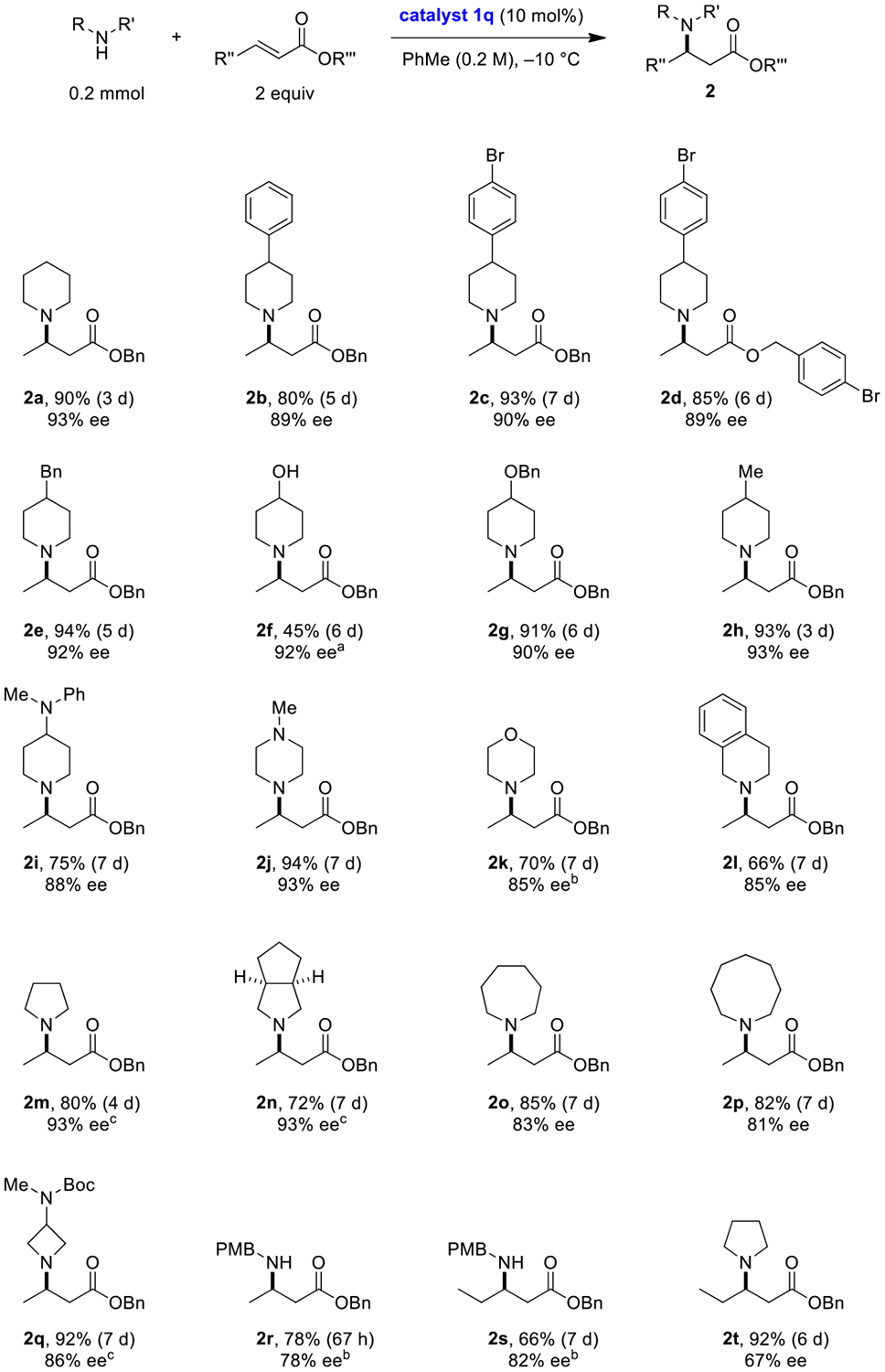
aReaction mixture was partially heterogeneous. bReaction was performed at room temperature. cReaction was performed at a 0.025 M concentration of amine.
We recently reported a simple one-step method to access cyclic 2-arylamines in racemic form34 and wondered whether such substrates could be resolved via a conjugate addition strategy. With few exceptions,35 small-molecule-based catalytic strategies for the kinetic resolution of basic amine substrates typically rely on acylation and are limited to primary amines.36 There are few solutions to the kinetic resolution of cyclic amines,37 and no general strategies exist to resolve cyclic 2-arylamines. Following an extensive screen of readily available conjugate acceptors, using 2-phenylpiperidine as the model substrate,38 commercial benzyl acrylate was identified as a suitable resolving agent. As summarized in Scheme 2, catalyst 1q facilitated the kinetic resolution of a number of 2-arylpiperidines and related substrates with good to excellent selectivities.39 In situ Boc protection of the unreacted starting material was performed to facilitate product isolation and s-factor analysis.
Scheme 2.

Kinetic Resolution of Cyclic 2-Arylamines
Our next efforts were directed toward understanding the mechanism and origin of enantioselectivity of this novel bifunctional selenourea-thiourea organocatalyzed reaction. The proposed catalytic cycle for the addition of secondary amines to α,β-unsaturated esters involves: [1] initial activation of the ester through H-bonding to the selenourea-thiourea catalyst; [2] conjugate addition of the amine to the activated complex, in the key C–N bond-forming (stereogenic-center-forming) step; and either [3a] direct C-protonation of the zwitterionic enolate intermediate (2azwit) to form the N-protonated β-amino ester (2aprot), which is subsequently deprotonated to deliver product 2a and regenerate catalyst 1q (red pathway, Figure 2), or [3b] the zwitterionic enolate intermediate (2azwit) undergoes an intramolecular proton transfer to form 2aenol followed by tautomerization to give 2a and regenerate 1q (blue pathway, Figure 2).
Figure 2.
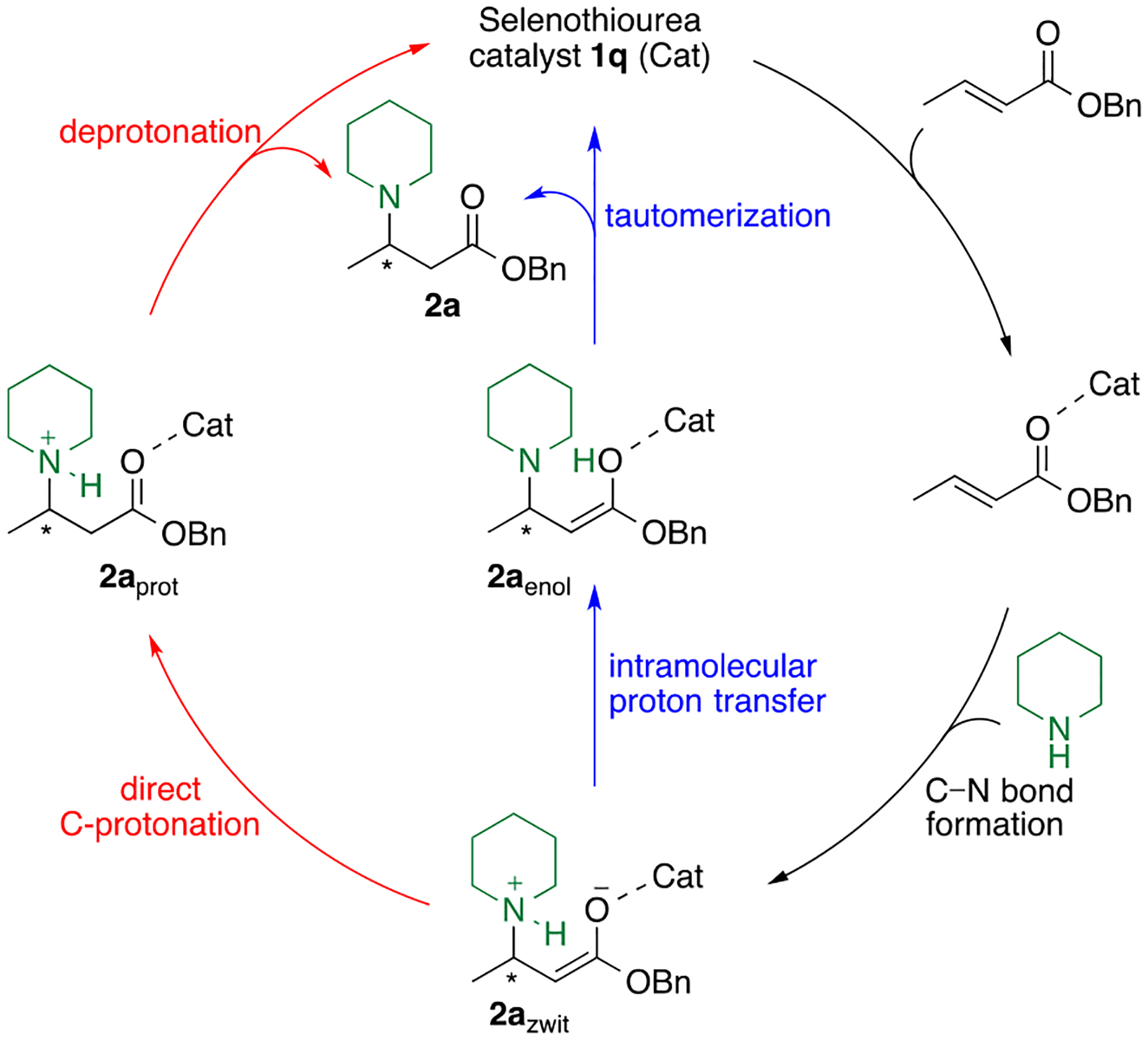
Proposed catalytic cycle for the addition of amines to α,β-unsaturated esters catalyzed by a selenourea-thiourea catalyst. The stereogenic center is formed during C–N bond formation. Subsequent proton transfers may follow an intramolecular proton transfer path (shown in blue) or direct C-protonation (shown in red).
In order to understand the origin of enantioselectivity, we decided to model the stereogenic-center-forming step in the reaction of piperidine and benzyl crotonate catalyzed by 1q using density functional theory (DFT) calculations. Transition structures (TSs) for the C–N bond-forming step leading to formation of the R- and S-enantiomers of the β-amino ester (2azwit) were computed using the B3LYP method40 with a split 6–31G* (C,H,O,N,F)/6–31+G** (S, Se) basis set as implemented in Gaussian ‘09.41 Single-point energy calculations were performed using B3LYP-D3(BJ)42/6–311++G** with a PCM solvent model43 for toluene. Relative energies presented herein are the extrapolated Gibbs free energy obtained by adding the free energy correction to the high-level single-point energy computed for each structure. The free energies were corrected using Grimme’s quasi rigid rotor-harmonic oscillator (qRRHO) approach, which raises vibrational frequencies that are below 100 cm−1.44 This approach is routinely utilized to evaluate reactivity and selectivity in similar catalytic systems.45
Following a thorough conformational search conducted via systematic variation of catalyst and reactant geometries,46 we identified the lowest energy C–N bond-forming transition structures leading to both enantiomers of 2azwit—R-TSC–N and S-TSC–N (Figure 3). Both TSs involve β-attack of piperidine on the s-cis conformation of benzyl crotonate, which is more favored than the corresponding s-trans conformation.47 In both of these TSs, the thiourea NHs activate the ester carbonyl by dual H-bonding (1.84 Å/1.97 Å R-TSC–N and 1.81 Å/1.90 Å S-TSC–N), while the selenourea directs the attack of piperidine via an H-bonding interaction between the selenium and the amine proton of piperidine (2.54 Å R-TSC–N and 2.56 Å S-TSC–N). This catalyst conformation is the most favored since it is stabilized by an intramolecular H-bonding interaction between the two arms of the catalyst, i.e., between the thiourea sulfur and the NH of the selenourea moiety. A careful analysis of R-TSC–N and S-TSC–N reveals that the key C–N bond-forming distances (1.87 Å R-TSC–N and 1.83 Å S-TSC–N) and all H-bonding interactions primarily responsible for transition state stabilization (vide supra) are almost identical for both TSs. Slightly stronger dual H-bonding interactions in S-TSC–N make it lower in energy than R-TSC–N, resulting in a predicted ee of 55% (S) at −10 °C. This is inconsistent with the experimental ee of 93% (R).
Figure 3.

Lowest energy transition structures for C–N bond formation leading to (R)- and (S)-enantiomers of 2azwit.
Since the extensive transition state search for C–N bond formation structures results in an incorrect prediction of enantioselectivity, we contemplated that the rate- and enantiodetermining step occurs after the stereogenic-center-forming step. To test this hypothesis, we measured 13C kinetic isotope effects (KIEs) for benzyl crotonate using Singleton’s 13C NMR methodology for starting materials at natural abundance.48 Two independent reactions of benzyl crotonate and piperidine were taken as 75 ± 2% and 79 ± 2% conversion with respect to the ester. Unreacted benzyl crotonate was reisolated, and the 13C isotopic composition was compared to samples of benzyl crotonate not subjected to reaction conditions. From the changes in relative isotopic composition and the fractional conversion, 13C KIEs were determined in a standard way.38
The key results are the unity KIE observed at the β-carbon and the normal KIE of ~1.5% on the α-carbon (Figure 4). If C–N bond formation is the first irreversible step in the catalytic cycle for benzyl crotonate, a normal KIE on the β-carbon is expected; however, the observed unity KIE is qualitatively consistent with reversible C–N bond formation. Second, an observed normal KIE of ~1.5% on the α-carbon suggests that α-protonation is likely the first irreversible step in the catalytic cycle. The results qualitatively validate our hypothesis that the C–N bond-forming step is reversible and that the rate- and enantiodetermining step occurs after this stereogenic-center-forming step—a finding that has important consequences for our future efforts in expanding the scope of this reaction.
Figure 4.

Experimental 13C KIEs for benzyl crotonate (numbers in parentheses represent the standard deviation in the last digit as determined from six independent measurements).
Experimental 13C KIEs suggest that the origin of enantioselectivity is best understood by analysis of the enantiomeric transition states for the α-carbon protonation step. In the absence of a more acidic proton on the catalyst or an external Brønsted acid in the system, we considered that one of the thiourea NHs is most likely involved in the α-protonation event (Figure 5). Based on this assumption, two possibilities emerge that are qualitatively consistent with a normal 13C KIE on the α-carbon-catalyst-mediated tautomerization (Figure 5, bottom panel) or catalyst-mediated direct C-protonation (Figure 5, top panel).49
Figure 5.
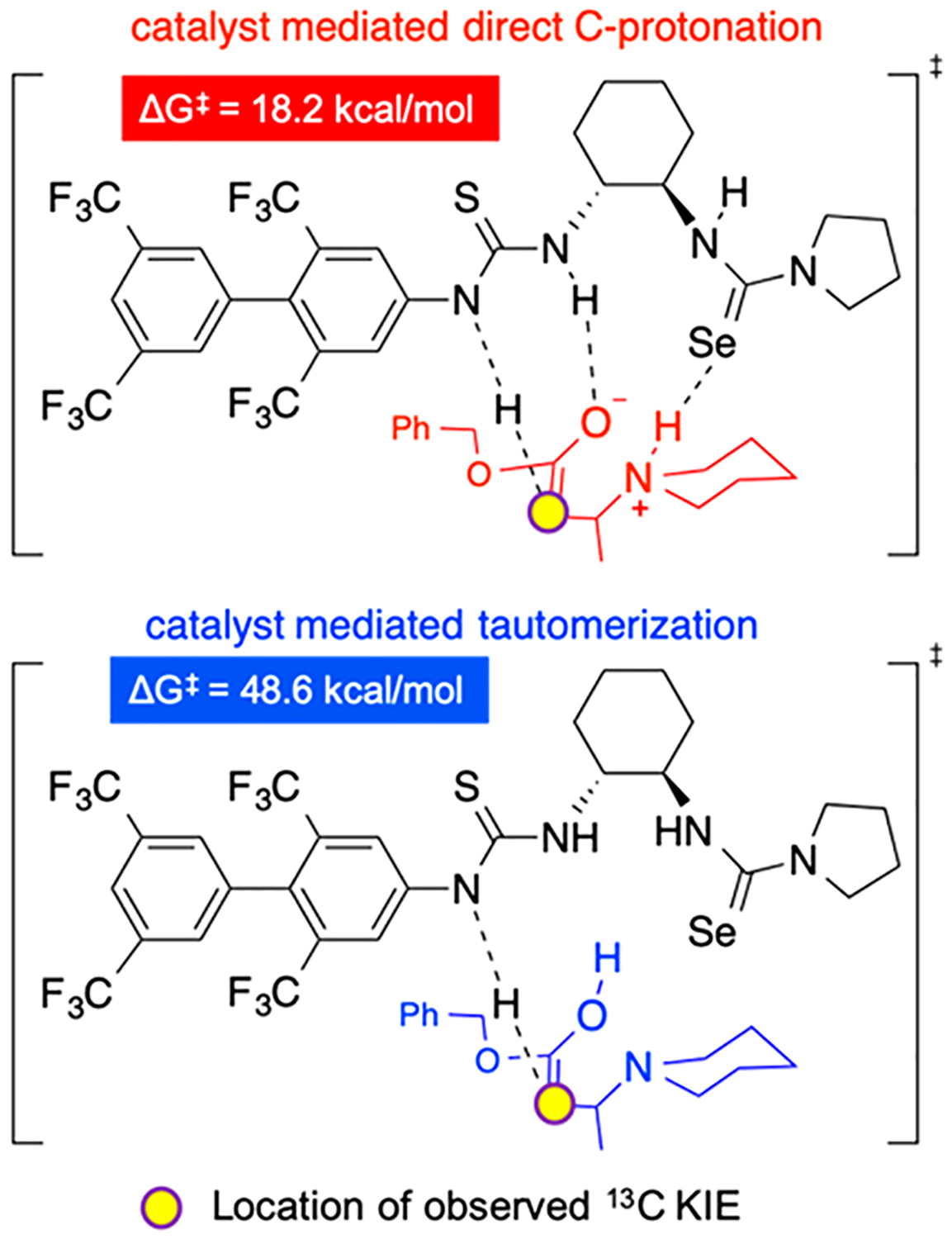
Possible transition states consistent with experimental KIEs.
Accordingly, we investigated both pathways for α-protonation using DFT calculations (vide supra). While intramolecular proton transfer from N to O of 2azwit to 2aenol is facile (not shown, ΔG‡ = 18.1 kcal/mol for proton transfer from R-2azwit), the ensuing catalyst-mediated tautomerization of 2aenol to 2a is prohibitively high in energy (Figure 5, ΔG‡ = 48.6 kcal/mol).45 On the other hand, the catalyst-mediated C-protonation is energetically accessible (Figure 5, ΔG‡ = 18.2 kcal/mol). Inaccessibility of the intramolecular proton transfer/tautomerization pathway led us to turn our explorations to the catalyst-mediated C-protonation as the rate- and enantioselectivity-determining step. Involvement of the thiourea catalyst directly as a Brønsted acid in the mechanism has only been reported in two instances in the literature,50 thus representing a new mode of catalysis, which should be considered in the development of future systems.51
In the lowest energy transition structure for direct protonation of R-2azwit (Figure 6, R-TSC-prot), the enolate adopts a geometry with strong intramolecular H-bonding interactions between the protonated piperidine and the enolate oxygen (1.81 Å). One of the thiourea NHs is loosely bound to the enolate oxygen (2.35 Å), while the other (more acidic) thiourea NH is transferred to the α-carbon of the ester. Selenium is engaged in a weak nonconventional CH⋯Se interaction with one of the α-CHs of the piperidine moiety (2.76 Å). A significantly altered arrangement is observed for the lowest energy transition structure for direct protonation of S-2azwit (Figure 6, S-TSC-prot). The enolate oxygen is bound via a strong H-bonding interaction with one of the thiourea NHs (1.94 Å) during the protonation event. Protonated piperidine NH is engaged in an H-bonding interaction with selenium (2.49 Å) (unlike R-TSC-prot, where the same NH is engaged in a stronger intramolecular H-bond with the enolate oxygen). Another key difference between the two TSs is the extent of proton transfer from the thiourea NH to the α-carbon of the enolate (1.35 and 1.26 Å for the breaking thiourea N–H bond in R-TSC-prot and S-TSC-prot, respectively). These differences in stabilizing interactions at the enantiomeric transition states for catalyst-mediated direct α-C-protonation result in R-TSC-prot being 1.9 kcal/mol lower in free energy than S-TSC-prot; this corresponds to a predicted 95% ee (R) at −10 °C, which is in excellent agreement with the 93% ee (R) observed experimentally. Additional support for direct α-C-protonation as the rate- and enantioselectivity-determining step was obtained by modeling the two transition structures by replacing the selenium atom with sulfur. With this sulfur analogue of catalyst 1q, we predicted a 59% ee which is in good agreement with the experimental ee of 62%.38 Finally, we also probed the effect of varying the β-substituent of the ester from methyl to ethyl by modeling the R-TSC-prot and S-TSC-prot for the β-ethyl-substituted benzyl ester. These analogous transition structures gave a drop in the predicted ee to 80%, which is a slight overestimation (0.7 kcal/mol) of the experimental ee of 47%. However, these calculations qualitatively predict the drop in ee experimentally observed with the bulkier β-ethyl substituent.38
Figure 6.

Lowest energy transition structures for catalyst-mediated direct C-protonation of (R)- and (S)-enantiomers of 2azwit.
As a key step in the quantitative interpretation of our experimental KIEs, we predict 13C KIEs from the scaled vibrational frequencies using the program ISOEFF98 for both R-TSC–N and R-TSC-prot, then applying a Wigner tunneling correction to all predicted KIEs.52–54 The large 13C KIE of 1.035 predicted for the β-carbon in R-TSC–N (Figure 7, red numbers) quantitatively eliminates C–N bond formation as the rate-determining step. On the other hand, the predicted KIEs for R-TSC-prot (Figure 7, green numbers) are in excellent agreement with experimental KIEs for all carbons, lending further support for rate-determining catalyst-mediated direct α-C-protonation.
Figure 7.

Comparison of experimental and predicted KIEs for the C–N bond-forming transition structure R-TSC–N (shown in red) and the direct C-protonation transition structure R-TSC-prot (shown in green).
Finally, the computed reaction coordinate diagram summarizes the relevant energies from our theoretical investigation (Figure 8). Energies of all TSs and intermediates are computed relative to the free energy of separated catalyst and starting materials (Figure 8, catalyst + piperidine + benzyl crotonate). To the left of the starting materials is the reaction pathway leading to (S)-2a (minor product), and to the right is the corresponding pathway leading to (R)-2a (major product). As discussed earlier, the C–N bond-forming step is reversible, and the enantioselectivity is determined at the rate-determining direct C-protonation step.55
Figure 8.
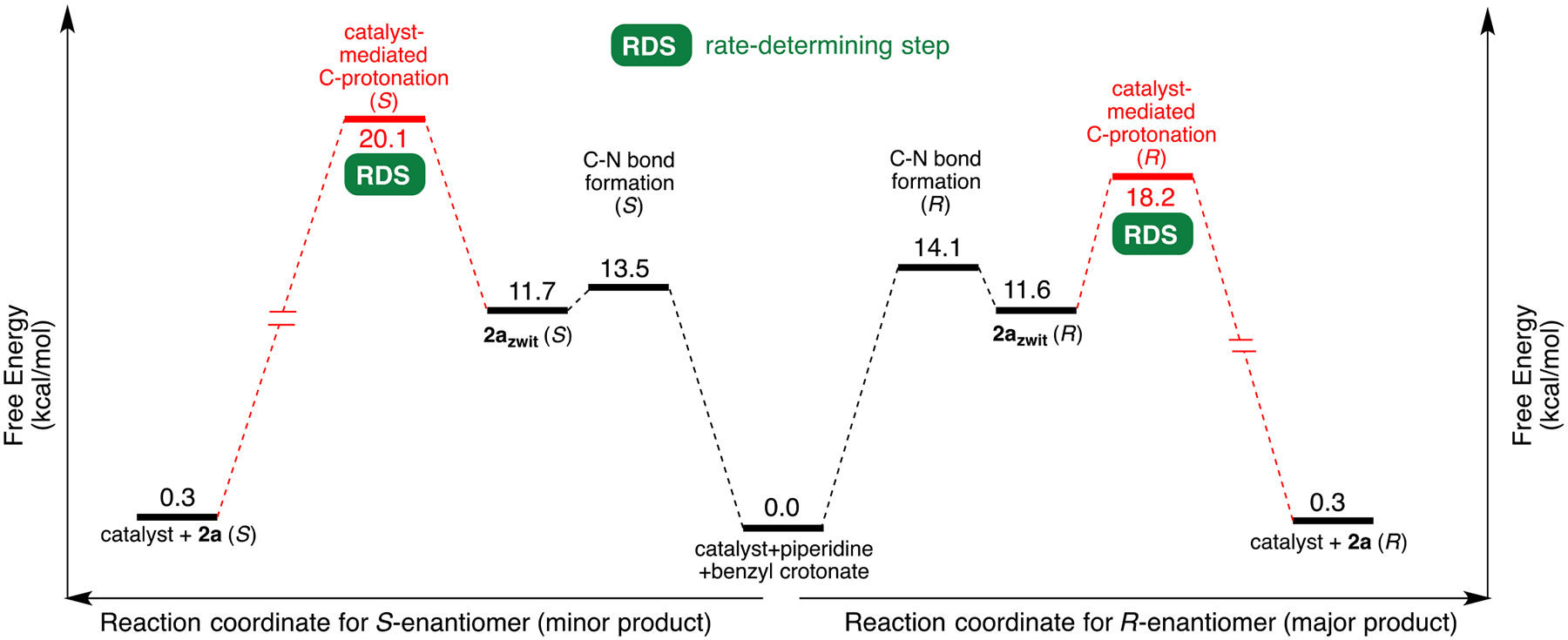
Computed reaction coordinate diagram (qRRHO) depicting the proposed pathway leading to (R)- and (S)-enantiomers of 2a.
In summary, we have achieved highly enantioselective conjugate additions of cyclic amines to unactivated α,β-unsaturated esters. This strategy is applicable to the kinetic resolution of cyclic 2-arylamines. A novel bifunctional selenourea-thiourea was identified in the course of this study. Experimental and predicted KIEs, free energy estimates, and enantioselectivity predictions all lend strong support to a reaction mechanism that proceeds via reversible C–N bond formation to form a β-amino enolate. This is followed by rate- and enantioselectivity-determining protonation by one of the thiourea NHs, which functions as a Brønsted acid. The transition structure, in which a thiourea compound functions as an asymmetric Brønsted acid, should serve as a guide for further development of this new mode of catalysis by chiral thiourea organocatalysts.
Supplementary Material
ACKNOWLEDGMENTS
This material is based upon work supported by the National Science Foundation under CHE-1806747 and CHE-1856613 (grants to D.S.). M.J.V. and J.S.H. acknowledge support from NIGMS - R01 GM126283 and the XSEDE Science Gateways Program, which is supported by the National Science Foundation grant number ACI-1548562. We further acknowledge the National Science Foundation (grant# 1828064 to K.A.A.) and the University of Florida for funding the purchase of the X-ray equipment.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b12457.
Experimental procedures and characterization data, kinetic isotope effect studies, details of the theoretical studies along with coordinates, and energies of all calculated structures (PDF)
X-ray crystal structure of catalyst 1q (CIF)
X-ray crystal structure of product 2d (CIF)
The authors declare no competing financial interest.
Contributor Information
Yingfu Lin, Center for Heterocyclic Compounds, Department of Chemistry, University of Florida, Gainesville, Florida 32611, United States; Department of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, United States.
William J. Hirschi, Department of Chemistry, Binghamton University, Binghamton, New York 13902, United States
Anuj Kunadia, Department of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, United States.
Anirudra Paul, Center for Heterocyclic Compounds, Department of Chemistry, University of Florida, Gainesville, Florida 32611, United States; Department of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, United States.
Ion Ghiviriga, Center for NMR Spectroscopy, Department of Chemistry, University of Florida, Gainesville, Florida 32611, United States.
Khalil A. Abboud, Center for X-ray Crystallography, Department of Chemistry, University of Florida, Gainesville, Florida 32611, United States
Rachael W. Karugu, Department of Chemistry, Binghamton University, Binghamton, New York 13902, United States
Mathew J. Vetticatt, Department of Chemistry, Binghamton University, Binghamton, New York 13902, United States;.
Jennifer S. Hirschi, Department of Chemistry, Binghamton University, Binghamton, New York 13902, United States;
Daniel Seidel, Center for Heterocyclic Compounds, Department of Chemistry, University of Florida, Gainesville, Florida 32611, United States; Department of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, United States;.
REFERENCES
- (1).Selected reviews on β-amino acids:; (a) Cardillo G; Tomasini C Asymmetric synthesis of β-amino acids and α-substituted β-amino acids. Chem. Soc. Rev 1996, 25, 117–128. [Google Scholar]; (b) Liu M; Sibi MP Recent advances in the stereoselective synthesis of β-amino acids. Tetrahedron 2002, 58, 7991–8035. [Google Scholar]; (c) Seebach D; Beck AK; Capone S; Deniau G; Grošelj U; Zass E Enantioselective Preparation of β2-Amino Acid Derivatives for β-Peptide Synthesis. Synthesis 2009, 2009, 1–32. [Google Scholar]; (d) Weiner B; Szymanski W; Janssen DB; Minnaard AJ; Feringa BL Recent advances in the catalytic asymmetric synthesis of beta-amino acids. Chem. Soc. Rev 2010, 39, 1656–1691. [DOI] [PubMed] [Google Scholar]
- (2).Rulev AY Aza-Michael reaction: achievements and prospects. Russ. Chem. Rev 2011, 80, 197. [Google Scholar]
- (3).Selected reviews on asymmetric conjugate additions with N-nucleophiles:; (a) Xu L-W; Xia C-G A Catalytic Enantioselective Aza-Michael Reaction: Novel Protocols for Asymmetric Synthesis of β-Amino Carbonyl Compounds. Eur. J. Org. Chem 2005, 2005, 633–639. [Google Scholar]; (b) Vicario JL; Badía D; Carrillo L; Etxebarria J; Reyes E; Ruiz N The asymmetric aza-Michael reaction. A review. Org. Prep. Proced. Int 2005, 37, 513–538. [Google Scholar]; (c) Enders D; Wang C; Liebich JX Organocatalytic Asymmetric Aza-Michael Additions. Chem. - Eur. J 2009, 15, 11058–11076. [DOI] [PubMed] [Google Scholar]; (d) Krishna PR; Sreeshailam A; Srinivas R Recent advances and applications in asymmetric aza-Michael addition chemistry. Tetrahedron 2009, 65, 9657–9672. [Google Scholar]; (e) Wang J; Li P; Choy PY; Chan ASC; Kwong FY Advances and Applications in Organocatalytic Asymmetric aza-Michael Addition. ChemCatChem 2012, 4, 917–925. [Google Scholar]; (f) Vinogradov MG; Turova OV; Zlotin SG Recent advances in the asymmetric synthesis of pharmacology-relevant nitrogen heterocycles via stereoselective aza-Michael reactions. Org. Biomol. Chem 2019, 17, 3670–3708. [DOI] [PubMed] [Google Scholar]
- (4).Mailyan AK; Eickhoff JA; Minakova AS; Gu Z; Lu P; Zakarian A Cutting-Edge and Time-Honored Strategies for Stereoselective Construction of C-N Bonds in Total Synthesis. Chem. Rev 2016, 116, 4441–4557. [DOI] [PubMed] [Google Scholar]
- (5).(a) Sibi MP; Shay JJ; Liu M; Jasperse CP Chiral Lewis Acid Catalysis in Conjugate Additions of O-Benzylhydroxylamine to Unsaturated Amides. Enantioselective Synthesis of β-Amino Acid Precursors. J. Am. Chem. Soc 1998, 120, 6615–6616. [Google Scholar]; (b) Sugihara H; Daikai K; Jin XL; Furuno H; Inanaga J Catalytic conversion of conjugated enones into optically active α-keto aziridines using chiral rare earth metal complexes. Tetrahedron Lett. 2002, 43, 2735–2739. [Google Scholar]; (c) Jin XL; Sugihara H; Daikai K; Tateishi H; Jin YZ; Furuno H; Inanaga J Chiral rare earth metal complex-catalyzed conjugate addition of O-alkylhydroxylamines. An efficient synthetic entry into optically active 2-acyl aziridines. Tetrahedron 2002, 58, 8321–8329. [Google Scholar]; (d) Yamagiwa N; Matsunaga S; Shibasaki M Heterobimetallic Catalysis in Asymmetric 1,4-Addition of O-Alkylhydroxylamine to Enones. J. Am. Chem. Soc 2003, 125, 16178–16179. [DOI] [PubMed] [Google Scholar]; (e) Yamagiwa N; Qin H; Matsunaga S; Shibasaki M Lewis Acid-Lewis Acid Heterobimetallic Cooperative Catalysis: Mechanistic Studies and Application in Enantioselective Aza-Michael Reaction. J. Am. Chem. Soc 2005, 127, 13419–13427. [DOI] [PubMed] [Google Scholar]
- (6).Myers JK; Jacobsen EN Asymmetric Synthesis of β-Amino Acid Derivatives via Catalytic Conjugate Addition of Hydrazoic Acid to Unsaturated Imides. J. Am. Chem. Soc 1999, 121, 8959–8960. [Google Scholar]
- (7).Fadini L; Togni A Ni(II) Complexes containing chiral tridentate phosphines as new catalysts for the hydroamination of activated olefins. Chem. Commun 2003, 30–31. [DOI] [PubMed] [Google Scholar]
- (8).Hamashima Y; Somei H; Shimura Y; Tamura T; Sodeoka M Amine-Salt-Controlled, Catalytic Asymmetric Conjugate Addition of Various Amines and Asymmetric Protonation. Org. Lett 2004, 6, 1861–1864. [DOI] [PubMed] [Google Scholar]
- (9).Uno BE; Dicken RD; Redfern LR; Stern CM; Krzywicki GG; Scheidt KA Calcium(II)-catalyzed enantioselective conjugate additions of amines. Chem. Sci 2018, 9, 1634–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Horstmann TE; Guerin DJ; Miller SJ Asymmetric Conjugate Addition of Azide to α,β-Unsaturated Carbonyl Compounds Catalyzed by Simple Peptides. Angew. Chem., Int. Ed 2000, 39, 3635–3638. [DOI] [PubMed] [Google Scholar]; (b) Guerin DJ; Miller SJ Asymmetric Azidation-Cycloaddition with Open-Chain Peptide-Based Catalysts. A Sequential Enantioselective Route to Triazoles. J. Am. Chem. Soc 2002, 124, 2134–2136. [DOI] [PubMed] [Google Scholar]
- (11).Chen YK; Yoshida M; MacMillan DWC Enantioselective Organocatalytic Amine Conjugate Addition. J. Am. Chem. Soc 2006, 128, 9328–9329. [DOI] [PubMed] [Google Scholar]
- (12).Dinér P; Nielsen M; Marigo M; Jørgensen KA Enantioselective Organocatalytic Conjugate Addition of N Heterocycles to α,β-Unsaturated Aldehydes. Angew. Chem., Int. Ed 2007, 46, 1983–1987. [DOI] [PubMed] [Google Scholar]
- (13).Sibi MP; Itoh K Organocatalysis in Conjugate Amine Additions. Synthesis of b-Amino Acid Derivatives. J. Am. Chem. Soc 2007, 129, 8064–8065. [DOI] [PubMed] [Google Scholar]
- (14).(a) Hayama N; Kuramoto R; Földes T; Nishibayashi K; Kobayashi Y; Pápai I; Takemoto Y Mechanistic Insight into Asymmetric Hetero-Michael Addition of α,β-Unsaturated Carboxylic Acids Catalyzed by Multifunctional Thioureas. J. Am. Chem. Soc 2018, 140, 12216–12225. [DOI] [PubMed] [Google Scholar]; (b) Michigami K; Murakami H; Nakamura T; Hayama N; Takemoto Y Catalytic asymmetric aza-Michael addition of fumaric monoacids with multifunctional thiourea/boronic acids. Org. Biomol. Chem 2019, 17, 2331–2335. [DOI] [PubMed] [Google Scholar]
- (15).(a) Uraguchi D; Nakashima D; Ooi T Chiral Arylamino-phosphonium Barfates as a New Class of Charged Brønsted Acid for the Enantioselective Activation of Nonionic Lewis Bases. J. Am. Chem. Soc 2009, 131, 7242–7243. [DOI] [PubMed] [Google Scholar]; (b) Wang L; Chen J; Huang Y Highly Enantioselective Aza-Michael Reaction between Alkyl Amines and β-Trifluoromethyl β-Aryl Nitroolefins. Angew. Chem., Int. Ed 2015, 54, 15414–15418. [DOI] [PubMed] [Google Scholar]
- (16).Weiß M; Borchert S; Rémond E; Jugé S; Gröger H Asymmetric addition of a nitrogen nucleophile to an enoate in the presence of a chiral phase-transfer catalyst: A novel approach toward enantiomerically enriched protected β-amino acids. Heteroat. Chem 2012, 23, 202–209. [Google Scholar]
- (17).Ghosh AK; Zhou B Bifunctional cinchona alkaloid-squaramide-catalyzed highly enantioselective aza-Michael addition of indolines to α,β-unsaturated ketones. Tetrahedron Lett. 2013, 54, 3500–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hashimoto T; Gálvez AO; Maruoka K Boronic Acid-Catalyzed, Highly Enantioselective Aza-Michael Additions of Hydroxamic Acid to Quinone Imine Ketals. J. Am. Chem. Soc 2015, 137, 16016–16019. [DOI] [PubMed] [Google Scholar]
- (19).For an enantioselective radical-based approach to β-amination, see:; Zhou Z; Li Y; Han B; Gong L; Meggers E Enantioselective catalytic β-amination through proton-coupled electron transfer followed by stereocontrolled radical-radical coupling. Chem. Sci 2017, 8, 5757–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Sánchez-Roselló M; Aceña JL; Simón-Fuentes A; del Pozo C A general overview of the organocatalytic intramolecular aza-Michael reaction. Chem. Soc. Rev 2014, 43, 7430–7453. [DOI] [PubMed] [Google Scholar]; (b) Roy TK; Parhi B; Ghorai P Cinchonamine Squaramide Catalyzed Asymmetric aza-Michael Reaction: Dihydroisoquinolines and Tetrahydropyridines. Angew. Chem., Int. Ed 2018, 57, 9397–9401. [DOI] [PubMed] [Google Scholar]; (c) Metsänen TT; Lexa KW; Santiago CB; Chung CK; Xu Y; Liu Z; Humphrey GR; Ruck RT; Sherer EC; Sigman MS Combining traditional 2D and modern physical organic-derived descriptors to predict enhanced enantioselectivity for the key aza-Michael conjugate addition in the synthesis of Prevymis (letermovir). Chem. Sci 2018, 9, 6922–6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).; Yang J; Farley AJM; Dixon DJ Enantioselective bifunctional iminophosphorane catalyzed sulfa-Michael addition of alkyl thiols to unactivated β-substituted-α,β-unsaturated esters. and references cited therein. Chem. Sci 2017, 8, 606–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Allgäuer DS; Jangra H; Asahara H; Li Z; Chen Q; Zipse H; Ofial AR; Mayr H Quantification and Theoretical Analysis of the Electrophilicities of Michael Acceptors. J. Am. Chem. Soc 2017, 139, 13318–13329. [DOI] [PubMed] [Google Scholar]
- (23).Sundararajan G; Prabagaran N A New Polymer-Anchored Chiral Catalyst for Asymmetric Michael Addition Reactions. Org. Lett 2001, 3, 389–392. [DOI] [PubMed] [Google Scholar]
- (24).Selected reviews:; (a) Takemoto Y Recognition and activation by ureas and thioureas: stereoselective reactions using ureas and thioureas as hydrogen-bonding donors. Org. Biomol. Chem 2005, 3, 4299–4306. [DOI] [PubMed] [Google Scholar]; (b) Doyle AG; Jacobsen EN Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev 2007, 107, 5713–5743. [DOI] [PubMed] [Google Scholar]; (c) Connon SJ Asymmetric catalysis with bifunctional cinchona alkaloid-based urea and thiourea organocatalysts. Chem. Commun 2008, 2499–2510. [DOI] [PubMed] [Google Scholar]; (d) Siau W-Y; Wang J Asymmetric organocatalytic reactions by bifunctional amine-thioureas. Catal. Sci. Technol 2011, 1, 1298–1310. [Google Scholar]; (e) Serdyuk OV; Heckel CM; Tsogoeva SB Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem 2013, 11, 7051–7071. [DOI] [PubMed] [Google Scholar]; (f) Fang X; Wang C-J Recent advances in asymmetric organocatalysis mediated by bifunctional amine-thioureas bearing multiple hydrogen-bonding donors. Chem. Commun 2015, 51, 1185–1197. [DOI] [PubMed] [Google Scholar]
- (25).Examples:; (a) Busschaert N; Bradberry SJ; Wenzel M; Haynes CJE; Hiscock JR; Kirby IL; Karagiannidis LE; Moore SJ; Wells NJ; Herniman J; Langley GJ; Horton PN; Light ME; Marques I; Costa PJ; Felix V; Frey JG; Gale PA Towards predictable transmembrane transport: QSAR analysis of anion binding and transport. Chem. Sci 2013, 4, 3036–3045. [Google Scholar]; (b) Vallavoju N; Selvakumar S; Pemberton BC; Jockusch S; Sibi MP; Sivaguru J Organophotocatalysis: Insights into the Mechanistic Aspects of Thiourea-Mediated Intermolecular [2 + 2] Photocycloadditions. Angew. Chem., Int. Ed 2016, 55, 5446–5451. [DOI] [PubMed] [Google Scholar]
- (26).Okino T; Hoashi Y; Takemoto Y Enantioselective Michael reaction of malonates to nitroolefins catalyzed by bifunctional organocatalysts. J. Am. Chem. Soc 2003, 125, 12672–12673. [DOI] [PubMed] [Google Scholar]
- (27).Vakulya B; Varga S; Csámpai A; Sooós, T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts. Org. Lett 2005, 7, 1967–1969. [DOI] [PubMed] [Google Scholar]
- (28).Herrera RP; Sgarzani V; Bernardi L; Ricci A Catalytic enantioselective Friedel-Crafts alkylation of indoles with nitroalkenes by using a simple thiourea organocatalyst. Angew. Chem., Int. Ed 2005, 44, 6576–6579. [DOI] [PubMed] [Google Scholar]
- (29).Reisman SE; Doyle AG; Jacobsen EN Enantioselective Thiourea-Catalyzed Additions to Oxocarbenium Ions. J. Am. Chem. Soc 2008, 130, 7198–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sohtome Y; Tanatani A; Hashimoto Y; Nagasawa K Development of bis-thiourea-type organocatalyst for asymmetric Baylis-Hillman reaction. Tetrahedron Lett. 2004, 45, 5589–5592. [Google Scholar]
- (31).Klauber EG; De CK; Shah TK; Seidel D Merging Nucleophilic and Hydrogen Bonding Catalysis: An Anion Binding Approach to the Kinetic Resolution of Propargylic Amines. J. Am. Chem. Soc 2010, 132, 13624–13626. [DOI] [PubMed] [Google Scholar]
- (32).Zhao C; Chen SB; Seidel D Direct Formation of Oxocarbenium Ions under Weakly Acidic Conditions: Catalytic Enantioselective Oxa-Pictet-Spengler Reactions. J. Am. Chem. Soc 2016, 138, 9053–9056. [DOI] [PubMed] [Google Scholar]
- (33).Other α,β-unsaturated ester substrates also provided unfavorable results. For instance, addition of piperidine to benzyl cinnamate proved sluggish and provided the corresponding product in only 51% ee (reaction conducted at rt). α,β-Unsaturated esters with α-substituents such as benzyl tiglate were found to be unreactive.
- (34).(a) Chen W; Ma L; Paul A; Seidel D Direct α-C-H bond functionalization of unprotected cyclic amines. Nat. Chem 2018, 10, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paul A; Seidel D α-Functionalization of Cyclic Secondary Amines: Lewis Acid Promoted Addition of Organometallics to Transient Imines. J. Am. Chem. Soc 2019, 141, 8778–8782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Das S; Majumdar N; De CK; Kundu DS; Döhring A; Garczynski A; List B Asymmetric Catalysis of the Carbonyl-Amine Condensation: Kinetic Resolution of Primary Amines. J. Am. Chem. Soc 2017, 139, 1357–1359. [DOI] [PubMed] [Google Scholar]
- (36).Selected examples:; (a) Arai S; Bellemin-Laponnaz S; Fu GC Kinetic resolution of amines by a nonenzymatic acylation catalyst. Angew. Chem., Int. Ed 2001, 40, 234–236. [DOI] [PubMed] [Google Scholar]; (b) Arnold K; Davies B; Herault D; Whiting A Asymmetric direct amide synthesis by kinetic amine resolution: a chiral bifunctional aminoboronic acid catalyzed reaction between a racemic amine and an achiral carboxylic acid. Angew. Chem., Int. Ed 2008, 47, 2673–2676. [DOI] [PubMed] [Google Scholar]; (c) Mittal N; Lippert KM; De CK; Klauber EG; Emge TJ; Schreiner PR; Seidel D A Dual-Catalysis Anion-Binding Approach to the Kinetic Resolution of Amines: Insights into the Mechanism via a Combined Experimental and Computational Study. J. Am. Chem. Soc 2015, 137, 5748–5758. [DOI] [PubMed] [Google Scholar]
- (37).(a) Binanzer M; Hsieh SY; Bode JW Catalytic Kinetic Resolution of Cyclic Secondary Amines. J. Am. Chem. Soc 2011, 133, 19698–19701. [DOI] [PubMed] [Google Scholar]; (b) Allen SE; Hsieh S-Y; Gutierrez O; Bode JW; Kozlowski MC Concerted Amidation of Activated Esters: Reaction Path and Origins of Selectivity in the Kinetic Resolution of Cyclic Amines via N-Heterocyclic Carbenes and Hydroxamic Acid Cocatalyzed Acyl Transfer. J. Am. Chem. Soc 2014, 136, 11783–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wanner B; Kreituss I; Gutierrez O; Kozlowski MC; Bode JW Catalytic Kinetic Resolution of Disubstituted Piperidines by Enantioselective Acylation: Synthetic Utility and Mechanistic Insights. J. Am. Chem. Soc 2015, 137, 11491–11497. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lu R; Cao L; Guan H; Liu L Iron-Catalyzed Aerobic Dehydrogenative Kinetic Resolution of Cyclic Secondary Amines. J. Am. Chem. Soc 2019, 141, 6318–6324. [DOI] [PubMed] [Google Scholar]
- (38). See the Supporting Information for details.
- (39).2-Arylamines with different ring sizes were resolved with lower efficiency. For instance, 2-phenylpyrrolidine and 2-phenylazepane were resolved with s-factors of 9.4 and 5.7, respectively.
- (40).Becke AD Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- (41).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas ö.; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]; 3D geometries in the manuscript were generated using CYLview, 1.0b; Legault, C. Y., Universiteó de Sherbrooke, 2009. (http://www.cylview.org). [Google Scholar]
- (42).(a) Grimme S; Ehrlich S; Goerigk L Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]; (b) Krishnan R; Binkley JS; Seeger R; Pople JA Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys 1980, 72, 650–654. [Google Scholar]
- (43).Tomasi J; Mennucci B; Cammi R Quantum Mechanical Continuum Solvation Models. Chem. Rev 2005, 105, 2999–3093. [DOI] [PubMed] [Google Scholar]
- (44).Grimme S Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem. - Eur. J 2012, 18, 9955–9964. [DOI] [PubMed] [Google Scholar]
- (45).Selected recent examples:; (a) Klausen RS; Kennedy CR; Hyde AM; Jacobsen EN Chiral Thioureas Promote Enantioselective Pictet-Spengler Cyclization by Stabilizing Every Intermediate and Transition State in the Carboxylic Acid-Catalyzed Reaction. J. Am. Chem. Soc 2017, 139, 12299–12309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Izzo JA; Myshchuk Y; Hirschi JS; Vetticatt MJ Transition state analysis of an enantioselective Michael addition by a bifunctional thiourea organocatalyst. Org. Biomol. Chem 2019, 17, 3934–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Izzo JA; Poulsen PH; Intrator JA; Jørgensen KA; Vetticatt MJ Isotope Effects Reveal an Alternative Mechanism for “Iminium-Ion” Catalysis. J. Am. Chem. Soc 2018, 140, 8396–8400. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Roytman VA; Karugu RW; Hong Y; Hirschi JS; Vetticatt MJ 13C Kinetic Isotope Effects as a Quantitative Probe To Distinguish between Enol and Enamine Mechanisms in Aminocatalysis. Chem. - Eur. J 2018, 24, 8098–8102. [DOI] [PubMed] [Google Scholar]; (e) Jarvis CL; Hirschi JS; Vetticatt MJ; Seidel D Catalytic Enantioselective Synthesis of Lactams through Formal [4 + 2] Cycloaddition of Imines with Homophthalic Anhydride. Angew. Chem., Int. Ed 2017, 56, 2670–2674. [DOI] [PubMed] [Google Scholar]
- (46). See the Supporting Information for full computational details.
- (47). The lowest energy transition structures with s-trans configuration of the ester were 3.4 (S) and 3.5 (R) kcal/mol higher in energy than the corresponding s-cis transition structures. See SI for coordinates.
- (48).Singleton DA; Thomas AA High-Precision Simultaneous Determination of Multiple Small Kinetic Isotope Effects at Natural Abundance. J. Am. Chem. Soc 1995, 117, 9357–9358. [Google Scholar]
- (49).We also modeled the protonation of the enolate by a protonated piperidine. However, the increased molecularity resulted in a significantly higher free energy barrier.
- (50).(a) Madarasz A; Dosa Z; Varga S; Soos T; Csampai A; Papai I Thiourea Derivatives as Brønsted Acid Organocatalysts. ACS Catal. 2016, 6, 4379–4387. [Google Scholar]; (b) Bradshaw GA; Colgan AC; Allen NP; Pongener I; Boland MB; Ortin YE; McGarrigle M Stereoselective organocatalyzed glycosylations - thiouracil, thioureas and monothiophthalimide act as Brønsted acid catalysts at low loadings. Chem. Sci 2019, 10, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51). A comparison can be drawn to the mechanism of Takemoto’s O-benzylhydroxylamine addition to α,β-unsaturated acids, reactions that are catalyzed by a thiourea catalyst containing both arylboronic acid and tertiary amine functionalities (see ref 14). In these reactions, protonation was also found to be involved in the enantiodetermining step. However, in contrast to the present study where C–N bond formation and protonation represent distinct steps, C–N bond formation is coupled to protonation which occurs in a concerted asynchronous fashion and involves an external carboxylic acid acting as a proton shuttle. Consistent with the different natures of the two catalytic processes, acidic additives proved detrimental in our case. For instance, addition of catalytic amounts of benzoic acid retarded reaction rates and led to lower enantioselectivities.
- (52).ISOEFF98. A program for studies of isotope effects using Hessian modifications. Anisimov V; Paneth P J. Math. Chem 1999, 26, 75–86. [Google Scholar]
- (53).The frequencies were scaled by 0.9614.; Scott AP; Radom L Harmonic Vibrational Frequencies: An Evaluation of Hartree-Fock, Møller-Plesset, Quadratic Configuration Interaction, Density Functional Theory, and Semiempirical Scale Factors. J. Phys. Chem 1996, 100, 16502–16513. [Google Scholar]
- (54).Bell RP The Tunnel Effect in Chemistry; Chapman & Hall: London, 1980. [Google Scholar]
- (55). The final deprotonation of 2aprot (the intermediate resulting from the catalyst mediated direct C-protonation) to regenerate the catalyst and deliver product (R)-2a is expected to be facile and is not included in the energy diagram shown in Figure 8.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.