

Graphical abstract

Abbreviations: HCC, Hepatocellular carcinoma; H3K4me3, Histone H3 trimethylated at lysine 4; H3K27ac, Histone H3 acetylated at lysine 27; H3K9ac, Histone H3 acetylated at lysine 9; H3K4me2, Histone H3 dimethylated at lysine 4; H3K27me3, Histone H3 trimethylated at lysine 27; H3K9me3, Histone H3 trimethylated at lysine 9; H2AFZ, H2A histone family member Z; H3K4me1, Histone H3 monomethylated at lysine 4; H3K36me3, Histone H3 trimethylated at lysine 36; H3K79me2, Histone H3 dimethylated at lysine 79; H4K20me1, Histone H4 monomethylated at lysine 20; ONCO, Oncogenes; TSG, Tumor suppressor genes; DHLEG, Different highly and lowly expressed genes; TH, The genes are highly expressed in tumor cell line but not in normal cell line; TL, The genes are lowly expressed in tumor cell line but not in normal cell line; NH, The genes are highly expressed in normal cell line but not in tumor cell line; NL, The genes are lowly expressed in normal cell line but not in tumor cell line; NH-TL, The genes are highly expressed in normal cell line and lowly expressed in tumor cell line; NL-TH, The genes are lowly expressed in normal cell line and highly expressed in tumor cell line
Keywords: Histone modification signals, Gene expression, Oncogenes, Tumor suppressor genes, Biomarkers
Abstract
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer death in the world. It has been reported that HCC is closely related to the changes of histone modifications. However, finding histone modification patterns in key genes which related to HCC is still an important task. In our study, the patterns of 11 kinds of histone modifications in the promoter regions for the different types of genes were analyzed by hierarchical screening for hepatocyte (normal) cell line and HepG2 (tumor) cell line. The important histone modifications and their key modification regions in different types of genes were found. The results indicate that these important genes may play a pivotal role in the occurrence of HCC. By analyzing the differences of histone modifications and gene expression levels for these important genes between the two cell lines, we found that the signals of H3K4me3, H3K27ac, H3K9ac, and H3K4me2 in HCC are significantly stronger. The changed regions of important histone modifications in 17 key genes were also identified. For example, the H3K4me3 signals increased 150 times in regions (−1500, −500) bp and (0, 1000) bp of ARHGAP5 in tumor cell line than in normal cell line. Finally, a prognostic risk scoring model was constructed, and the effects of key genes on the prognosis of HCC were verified by the survival analysis. Our results may provide a more precise potential therapeutic targets for identifying key genes and histone modifications in HCC as new biomarkers.
1. Introduction
Hepatocellular carcinoma (HCC) is the most common primary live malignancy [1]. Liver cancer is the sixth most commonly diagnosed cancer and the fourth leading cause of cancer death worldwide in 2018, with about 841,000 new cases and 782,000 deaths annually [2]. For the reason that patients with advanced HCC are unsuitable for curative hepatectomy or hepatic transplantation, even if patients undergo surgical resection, the high recurrence rate is the main cause for the poor 5-years survival rate of HCC patients [3], [4]. The development of HCC is a complex process, and more and more evidence has shown that epigenetic changes are an important reason for the occurrence of HCC [5], [6]. Therefore, understanding the underlying mechanism of epigenetic changes is crucial for finding new effective treatments.
A number of studies about histone modifications of HCC have been reported. The histone modification patterns are dynamically regulated by enzymes that add and remove covalent modifications to histones [7]. Histone deacetylation is closely related to HCC, especially the role of histone deacetylase in the pathogenesis of HCC [8]. It has found that histone deacetylase 3 may be an important factor regulating the proliferation and invasion of HepG2 cell line. HDAC6 suppresses tumors by mediating autophagic cell death in HCC [9]. EZH2 represses gene transcription through histone H3 trimethylated at lysine 27 (H3K27me3). It has also found that EZH2-mediated epigenetic silencing contributes to constitutive activation of Wnt/β-catenin signaling and consequential proliferation of HCC [10]. Moreover, EZH2 and SUV39H2 are closely related to the survival and tumor stage, respectively [11].
The microarray studies of tumor tissue have demonstrated that high H3K27me3 is associated with lower survival and poor prognosis of HCC [12], [13], [14]. Recent studies have identified histone H4 dimethylated at lysine 20 and histone H4 acetylated at lysine 16 as novel biomarkers of microvascular infiltration in HCC [15]. The high signals of H3K4me3 are related to poor prognosis in HCC patients [12]. The aberrant modification of histone 3 phosphorylation has also been found in HCC [16]. The H3K4me3 levels in Oct4, Yap1, and TCF7 promoters are also relevant to the self-renewal of liver the cancer stem cells [17], [18], [19], [20]. These researches provide new directions for the discovery of new biomarkers and potential therapeutic targets in HCC.
The epigenetic modification patterns of HCC are still a mystery throughout the genome, so it is urgent and necessary to analyze the epigenetic modifications in HCC. Although there were many studies about enzymes and the changes of histone modifications, almost all of them were based on experimental levels. At present, there are relatively few studies on the specific patterns of histone modifications and the relationship between histone modifications and gene expression.
With the development of next-generation sequencing technologies, it has been feasible to analyze cancers from the whole genome level [13], [21]. In this study, we used the publicly available data of HepG2 (tumor) cell line and hepatocyte (normal) cell line from ENCODE to identify key genes and the changes of histone modification signals for HCC [22]. The patterns and main features of histone modification signals in the promoter regions were calculated. The important histone modification signals of the key genes were found. We also identified regions where the important histone modification signals have been changed for the key genes and constructed a prognostic risk scoring model related to the expression of key genes, which well validated the impact of these genes on the prognosis of HCC. These results should be important for the development of HCC.
2. Material and methods
2.1. Gene expression data
The gene expression data (RNA-Seq, hg19) of HepG2 and hepatocyte cell lines were downloaded from ENCODE database (https://www.encodeproject.org/) (Supplementary appendix file: Table A1), which are the bam format, then we used the bedtools software to convert data to the bed format [23]. The gene expression levels in both cell lines were calculated according to the reads per kilobase of exon model per million mapped sequence reads (RPKM) [24].
2.2. Data of histone modifications
The ChIP-Seq data (hg19) for 11 histone modifications in HepG2 and hepatocyte were downloaded from ENCODE database. These histone modifications include histone H3 trimethylated at lysine 9 (H3K9me3), histone H3 trimethylated at lysine 4 (H3K4me3), histone H3 trimethylated at lysine 27 (H3K27me3), H2A histone family member Z (H2AFZ), histone H3 monomethylated at lysine 4 (H3K4me1), histone H3 trimethylated at lysine 36 (H3K36me3), histone H3 dimethylated at lysine 4 (H3K4me2), histone H3 acetylated at lysine 27 (H3K27ac), histone H3 dimethylated at lysine 79 (H3K79me2), histone H3 acetylated at lysine 9 (H3K9ac) and histone H4 monomethylated at lysine 20 (H4K20me1) (Supplementary appendix file: Table A1), and these data are also the bam format. The histone modification signals () of k-th histone modification in j-th bin for i-th gene were calculated by following Eq. (1):
| (1) |
where i is i-th gene, j is the j-th bin, k is the k-th histone modification, is the read counts of k-th histone modification that located in j-th bin for i-th gene, is the total read counts of k-th histone modifications, is the length of j-th bin, is used to keep the consistent magnitude with RPKM (In the process of calculating RPKM, the unit of the exon length is kilobase and the counting unit of mapped reads is million.).
2.3. Known cancer genes and human reference genes
We downloaded 235 oncogenes (ONCO) and 222 tumor suppressor genes (TSG) from COSMIC somatic mutation catalog (https://cancer.sanger.ac.uk/cosmic). The RefSeq genes of human genome (hg19) were downloaded from the UCSC database (http://genome.ucsc.edu/cgi-bin/hgTables) (Supplementary appendix file: Table A2). RefSeq genes include name, chrom, strand, txStart, txEnd, cdsStart, cdsEnd, exonCount, exonStarts, exonEnds, score, name2, cdsStartStat, cdsEndStat, and exonFrames. The RefSeq data we downloaded has a total of 68,534 lines of information. The non-coding genes starting with NR in the name column were removed, the coding genes starting with NM (the mature messenger RNA) were retained, leaving 52,162 genes. Then the genes with the same transcription start site were deduplicated, and only one of them was retained, leaving 25,383 genes. The gene names (name2 column) were deduplicated, leaving 19,198 genes. Finally, all genes on the chromosome Y were removed, leaving 19,157 genes to be selected for analysis.
2.4. Partition of promoter regions
We obtained the transcription start site (TSS, txStart) of each gene from RefSeq data (Supplementary appendix file: Table A2). For each gene, the functional region of (−2000, 2000) bp flanking the TSS was divided into 80 bins of 50 bp in size. The histone modification signals in each bin were calculated by Eq. (1), so that we obtained all histone modification signals of 80 bins in the promoter regions.
2.5. Correlation analysis
Since different histone modifications may have cooperation for gene expression, to study the relationship between histone modifications, we computed Spearman’s correlations between histone modification signals () according to Eq. (2). Finally, we got an 11 × 11 correlation coefficients matrix and used the gplots package [25] to plot the heat maps of histone modifications correlations.
| (2) |
where m = 80, n = 19157, Δ=, i is i-th gene, j is the j-th bin, k () is the k-th (-th) histone modification, and were calculated according to Eq. (1), () is the average signals of k-th (-th) histone modification in the j-th bin, () is the rank number of the ().
In addition, the Spearman’s correlations () between the k-th histone modification signals in the j-th bin of promoter regions and gene expression levels (el) were also calculated by Eq. (3), so we got an 11 × 80 data matrix.
| (3) |
where m, n, Δ, i, j, k, are the same as in Eq. (2). is the logarithm of , is the RPKM of i-th gene, is the logarithm of the , () is the rank number of the ().
2.6. Hierarchical screening for genes
The gene expression levels were sorted in the two cell lines, respectively. We selected the top ten percent of genes as the highly expressed genes and the expression value is zero as the lowly expressed genes. Then, we got 1,915 highly expressed genes and 2,922 lowly expressed genes for the normal cell line. For the tumor cell line, we got 1,915 highly expressed genes and 5,352 lowly expressed genes. Subsequently, we selected the differentially expressed genes as the different highly and lowly expressed genes (DHLEG) from the above four gene sets of highly and lowly expressed genes.
2.7. Clinical data and corresponding gene expression data of patients with HCC
Clinical data and corresponding gene expression data of patients with liver hepatocellular carcinoma (LIHC) were downloaded from The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/) [26]. Samples with both clinical data and expression data were selected, while samples without survival information in clinical data were deleted. The selected clinical data are all samples with overall survival time and survival status. These samples data in TCGA were collected from surgical resection of biopsy biospecimens of patients diagnosed with hepatocellular carcinoma (HCC), and had not received prior treatment for their disease (ablation, chemotherapy, or radiotherapy) [26]. Finally, the data of 364 patients with LIHC were used in this study (Supplementary appendix file: Table A3).
2.8. Construction of risk scoring model
To further identify clinically significant prognostic genes, we evaluated the relationship between gene expression levels and overall survival. We performed a multivariate Cox proportional hazard regression analysis for genes [27], the hazard ratio (HR) and 95% confidence interval (CI) were calculated. The genes with P < 0.05 were selected, and a risk scoring model (RS) was constructed by Eq. (4) through weighting multivariable regression coefficients of each gene. Then the risk score was calculated for each sample. Finally, the patients were divided into high-risk group and low-risk group by using the median risk score of all samples as the threshold.
| (4) |
Where i is the i-th gene, m is the number of genes, RS is a prognostic risk score for the HCC patient, is the contribution of i-th gene to prognostic risk scores that were obtained from the regression coefficient of multivariate Cox analysis, represents the expression level of i-th gene.
2.9. Survival analysis
The Kaplan-Meier method was used for survival analysis, the survival rate and median survival of each prognostic risk group were calculated. The survival rates of patients in different risk groups were compared, and the significance of the differences was evaluated using the log-rank test [28]. In order to verify the predictive ability of the risk scoring model, the performance of the RS was also evaluated by the time-dependent receiver operating characteristic (ROC) curve [29].
3. Results
3.1. Histone modification signals of DHLEG
In order to analyze the differences between the highly and lowly expressed genes, we selected the differentially expressed genes as DHLEG [30]. The DHLEG includes the following six situations (Fig. 1A): A. There are 1,051 genes that are highly expressed in tumor cell line but not in normal cell line (TH). B. 2,816 genes are lowly expressed in tumor cell line but not in normal cell line (TL). C. 1,051 genes are highly expressed in normal cell line but not in tumor cell line (NH). D. 386 genes are lowly expressed in normal cell line but not in tumor cell line (NL). E. 81 genes are highly expressed in normal cell line and lowly expressed in tumor cell line (NH-TL). F. Only PEG3 is lowly expressed in normal cell line and highly expressed in tumor cell line (NL-TH).
Fig. 1.

The average histone modification signals of DHLEG. (A) The Venn diagram of DHLEG in two kinds of cell lines. (B) and (C) are the average histone modification signals of the TH (TL) and NH (NL). (D) The average histone modification signals of NH-TL in two kinds of cell lines. Abbreviations: DHLEG: different highly and lowly expressed genes. TH: the genes are highly expressed in tumor cell line but not in normal cell line. NH: the genes are highly expressed in normal cell line but not in tumor cell line. TL: the genes are lowly expressed in tumor cell line but not in normal cell line. NL: the genes are lowly expressed in normal cell line but not in tumor cell line. NH-TL: The genes are highly expressed in normal cell line and lowly expressed in tumor cell line.
By computing the average histone modification signal strength of each bin in the promoter regions for DHLEG, we subsequently got the distributions of each histone modification. It was found that most of the histone modifications are stronger in highly expressed genes than that in lowly expressed genes by comparing the histone modification signals between TH (NH) and TL (NL) (Fig. 1B and C), suggesting that most of the histone modifications play an activating role in the two kinds of cell lines. However, H3K27me3 signals are stronger in lowly expressed genes than that in highly expressed genes for the two kinds of cell lines. These results indicate that the H3K27me3 may play an inhibiting role in the two kinds of cell lines. Moreover, we also observed that the signals of H3K27ac and H3K4me3 are stronger in tumor cell line than that in normal cell line. For the histone modifications of NH-TL (Fig. 1D), it can be seen that the H3K27me3 signals are stronger in tumor cell line than that in normal cell line. Furthermore, the H3K4me3 signals are stronger in tumor cell line, which is consistent with the results of Fig. 1B and C.
3.2. Correlation between histone modifications in highly and lowly expressed genes
To illustrate the cooperative effects of histone modifications for the highly and lowly expressed genes in the two kinds of cell lines, we calculated Spearman’s correlations between histone modifications then plotted heat maps (Fig. 2). In highly expressed genes of tumor cell line, there are two histone modification clusters with strong positive correlation (), including (H3K27me3, H3K9ac, H3K4me3, and H3K27ac, >0.87) and (H4K20me1 and H3K36me3, =0.96), there is just one histone modification cluster between H3K4me1 and (H3K27ac, H3K4me3, and H3K9ac) with strong negative correlation (<−0.76, ) (Fig. 2A). For the lowly expressed genes of tumor cell line, there is only one histone modification cluster (H3K4me3, H3K4me2, H3K4me1, H2AFZ, and H3K9ac) that has strong positive correlation (>0.76, ) (Fig. 2B). Three histone modification clusters have strong positive correlation () in highly expressed genes of normal cell line, including (H3K36me3, H3K79me2, and H4K20me1, >0.86), (H2AFZ and H3K27ac, =0.94) and (H3K4me2, H3K9ac and H3K4me3, >0.86) (Fig. 2C). In lowly expressed genes of normal cell line, there is just one histone modification cluster (H3K79me2, H3K36me3, and H3K9me3) that has strong positive correlation (>0.84, ). However, the Spearman’s correlations between H3K4me2 and (H3K79me2, H3K36me3, and H3K9me3) have strong negative correlation (<−0.58, ) (Fig. 2D). Therefore, heat maps show that different types of genes are modified by different histone modifications clusters.
Fig. 2.

The Spearman correlation coefficient of histone modifications in two kinds of cell lines. (A) The heat map of highly expressed genes in tumor cell line. (B) The heat map of lowly expressed genes in tumor cell line. (C) The heat map of highly expressed genes in normal cell line. (D) The heat map of lowly expressed genes in normal cell line.
3.3. Analysis of the histone modification signals in oncogenes (ONCO) and tumor suppressor genes (TSG) of DHLEG for two kinds of cell lines
In order to further find key genes that are related to HCC in DHLEG, we selected the known cancer genes in DHLEG (Table 1). To illustrate the characteristics of histone modifications in these genes, we calculated the average histone modification signals for tumor and normal cell lines, respectively. For tumor cell line (Fig. 3A), it shows that the average histone modification signals in highly expressed ONCO are generally stronger than that in lowly expressed TSG, especially for H3K4me3, H3K9ac, H3K27ac, and H3K4me2. Whereas for normal cell line (Fig. 3B), it shows that the average histone modification signals in highly expressed TSG are generally stronger than that in lowly expressed ONCO, except for H3K27me3.
Table 1.
Oncogenes and tumor suppressor genes in DHLEG.
| DHLEG | TYPE | NUMBER | GENE SYMBOL |
|---|---|---|---|
| TH | ONCO | 16 | ARHGAP5, CCND1, CHD4, ERBB3, ETV4, HLF, MALT1, MAP2K1, MAPK1, MDM2, MET, MYC, MYD88, PLCG1, WWTR1, XPO1 |
| TSG | 18 | AXIN1, AXIN2, CDKN1B, CHD2, CLTC, EIF3E, FAT1, FH, FUS, MSH2, MSH6, PTPRK, RMI2, RNF43, SMARCB1, SETD2, SDHA, TGFBR2 | |
| TL | ONCO | 15 | ACKR3, ALK, CD79B, CHST11, CTNNA2, ETV1, FEV, FOXR1, GLI1, HOXD13, KCNJ5, MYB, NTRK3, RSPO3, ZNF521 |
| TSG | 4 | CBFA2T3, CDX2, CREB3L1, ROBO2 | |
| NH | ONCO | 14 | ACKR3, AKT1, CCND3, CDK4, DDIT3, FOXA1, FSTL3, HIF1A, IDH2, KDR, SGK1, SH3GL1, STAT3, TFE3 |
| TSG | 13 | BTG1, CDH1, CREB3L1, DNM2, ELF3, ID3, KLF6, PER1, PPP2R1A, SDHAF2, SDHC, SDHD, TNFAIP3 | |
| NL | ONCO | 2 | P2RY8, POU2AF1 |
| TSG | 0 | – | |
| NH-TL | ONCO | 1 | ACKR3 |
| TSG | 1 | CREB3L1 | |
| NL-TH | ONCO | 0 | – |
| TSG | 0 | – |
Abbreviations: DHLEG: different highly and lowly expressed genes. ONCO: oncogenes. TSG: tumor suppressor genes. TH: the genes are highly expressed in tumor cell line but not in normal cell line. TL: the genes are lowly expressed in tumor cell line but not in normal cell line. NH: the genes are highly expressed in normal cell line but not in tumor cell line. NL: the genes are lowly expressed in normal cell line but not in tumor cell line. NH-TL: the genes are highly expressed in normal cell line and lowly expressed in tumor cell line. NL-TH: the genes are lowly expressed in normal cell line and highly expressed in tumor cell line.
Fig. 3.

The histone modification signals and gene expression levels in ONCO and TSG of DHLEG for two kinds of cell lines. (A) The histone modification signals of tumor cell line. (B) The histone modification signals of normal cell line. (C) The distribution of histone modification signals in CREB3L1 that is highly expressed in normal cell line and lowly expressed in tumor cell line. (D) The distribution of histone modification signals in ACKR3 that is highly expressed in normal cell line and lowly expressed in tumor cell line. (E) The heat map of Spearman’s correlations between histone modification signals in the 80 bins of the promoter regions and the gene expression levels for TH-ONCO. (F) The heat map of Spearman’s correlations between histone modification signals in the 80 bins of the promoter regions and the gene expression levels for NH-TSG. Abbreviations: ONCO: oncogenes. TSG: tumor suppressor genes. DHLEG: different highly and lowly expressed genes. TH-ONCO: the highly expressed oncogenes in tumor cell line. NH-TSG: the highly expressed tumor suppressor genes in normal cell line.
For TSG CREB3L1 (Fig. 3C), it shows that the histone modifications of H3K9ac, H3K4me2, and H3K27ac activate the high expression of this gene in the normal cell line, H3K27me3 inhibits the expression of this gene in the tumor cell line. For the gene ACKR3, it is highly expressed in normal cell line and lowly expressed in tumor cell line (NH-TL), and the signals of H3K9ac, H3K4me2, and H3K27ac are stronger in normal cell line than that in tumor cell line (Fig. 3D). It suggests that these histone modifications mainly activated the high expression of this gene in normal cell line. However, H3K27me3 has a higher level of modification in tumor cell line, it inhibits the expression of this gene in tumor cell line.
3.4. The analysis of the correlations between histone modifications and gene expression levels
In order to study the relationships between the locations of histone modifications and expression levels, we further calculated the Spearman’s correlations between histone modifications in 80 bins of promoter regions and the expression levels of TH-ONCO and NH-TSG, the results are shown in the heat maps. For the TH-ONCO in tumor cell line, there are the obvious negative correlations between gene expression levels and H3K27me3 in the regions (200, 250) bp and (850, 900) bp, while an obvious positive correlation between gene expression levels and H3K27ac in the region (1700, 1750) bp (Fig. 3E). For the NH-TSG in normal cell line, the heat map also shows that there are prominent correlations between gene expression levels and histone modifications, such as H3K9ac in the region (1500, 1550) bp, H3K4me3 in the regions (−1650, −1600) bp and (1800, 1850) bp. In addition, it has the obvious correlations between gene expression levels and H3K27ac in the regions (−1500, −1450), (850, 1000), (1050, 1100), (1150, 1250), (1300, 1450), and (1900, 1950) bp (Fig. 3F), the detailed results are displayed in Table 2. The above results indicate that H3K4me3, H3K9ac, H3K27ac, and H3K27me3 are important histone modifications for gene expression.
Table 2.
The obvious correlation between gene expression levels and histone modification signals.
| GENES | HM | REGION | P-value | |
|---|---|---|---|---|
| TH-ONCO | H3K27me3 | (−1300 bp, −1250 bp) (15-th) | −0.536 | 0.032 |
| (−1150 bp, −1100 bp) (18-th) | −0.518 | 0.040 | ||
| (−700 bp, −650 bp) (27-th) | −0.581 | 0.018 | ||
| (−500 bp, −450 bp) (31-st) | −0.552 | 0.027 | ||
| (200 bp, 250 bp) (45-th) | −0.612 | 0.012 | ||
| (850 bp, 900 bp) (58-th) | −0.630 | 0.009 | ||
| (1100 bp, 1150 bp) (63-rd) | −0.498 | 0.050 | ||
| H3K27ac | (1700 bp, 1750 bp) (75-th) | 0.519 | 0.039 | |
| (1750 bp, 1800 bp) (76-th) | 0.457 | 0.039 | ||
| NH-TSG | H3K9ac | (1500 bp, 1550 bp) (71-st) | 0.669 | 0.012 |
| H3K4me3 | (−1650 bp, −1600 bp) (8-th) | 0.687 | 0.010 | |
| (1800 bp, 1850 bp) (77-th) | 0.615 | 0.025 | ||
| H3K27ac | (−1500 bp, −1450 bp) (11-th) | 0.575 | 0.040 | |
| (850 bp, 900 bp) (58-th) | 0.600 | 0.030 | ||
| (900 bp, 950 bp) (59-th) | 0.598 | 0.031 | ||
| (950 bp, 1000 bp) (60-th) | 0.574 | 0.040 | ||
| (1050 bp, 1100 bp) (62-nd) | 0.580 | 0.040 | ||
| (1150 bp, 1200 bp) (64-th) | 0.617 | 0.025 | ||
| (1200 bp, 1250 bp) (65-th) | 0.721 | 0.005 | ||
| (1300 bp, 1350 bp) (67-th) | 0.639 | 0.019 | ||
| (1350 bp, 1400 bp) (68-th) | 0.727 | 0.005 | ||
| (1400 bp, 1450 bp) (69-th) | 0.756 | 0.003 | ||
| (1900 bp, 1950 bp) (79-th) | 0.712 | 0.006 |
Abbreviations: HM: histone modification.
3.5. Analysis of important histone modifications in key genes related to HCC
To further recognize the differences between histone modification signals in normal and tumor cell lines for the four types of differential genes, i.e. TH-ONCO, TL-TSG, NH-TSG, and NL-ONCO, we calculated the distribution and relative difference of histone modification signals in TH-ONCO for tumor cell line and normal cell line. The signals of H3K4me3 and H3K27ac are very stronger for oncogenes (ERBB3, CCND1, ARHGAP5, PLCG1, HLF, and MYD88) in tumor cell line (Fig. 4). The results are consistent with the histone modification clusters of the TH heat map, it also indicates that the co-modification of the two histones promote the high expression of these ONCO in tumor cell line. Furthermore, by comparing the histone modifications of oncogenes, we also found other important genes, such as MALT1, CHD4, MYC, MDM2, MET, ETV4, and MAP2K1 (Supplementary appendix file: Fig. A1, Table A4).
Fig. 4.

The distribution of histone modification signals in TH-ONCO for two kinds of cell lines. In each figure, the same histone modifications are represented in the same color that the box on the left represents the histone modifications in tumor cell line (T) and the box on the right represents the histone modifications in normal cell line (N). The line represents the relative difference between the same histone modifications. Abbreviations: TH-ONCO: the highly expressed oncogenes in tumor cell line. HMS: the histone modification signals.
Similarly, for the TL-TSG, we found that the signals of H3K9ac, H3K27ac, and H3K4me2 for ROBO2, and H3K9ac, H3K27ac, H2AFZ, and H3K4me2 for CREB3L1 are generally stronger in normal cell line than that in tumor cell line (Supplementary appendix file: Fig. A2, Table A4). For the NH-TSG, the signals of H3K9ac, H3K4me3, H2AFZ, H3K27ac, and H3K4me2 for CREB3L1, and H3K9ac, H3K36me3, H3K79me2, and H3K27ac for CDH1, as well as H3K9ac, H3K4me3, H3K27ac, and H3K4me2 for BTG1 are stronger in normal cell line than that in tumor cell line (Supplementary appendix file: Fig. A3, Table A4).
3.6. Identifying the location of histone modifications in key genes related to HCC
Through our above analysis, we found 17 key genes (13 oncogenes are MYD88, MYC, ARHGAP5, PLCG1, MDM2, MET, ETV4, HLF, ERBB3, MAP2K1, MALT1, CCND1, and CHD4, 4 tumor suppressor genes are CREB3L1, ROBO2, BTG1, and ACKR3) related to important histone modifications regulating gene expression in HCC. Afterward, we got the distributions of histone modifications by calculating the histone modification signals in the promoter regions for each gene. By comparing with normal cell line, we identified the key regions of some important histone modifications in genes that are associated with HCC. The results indicate that the increase of the H3K4me3, H3K27ac, H3K9ac, etc. histone modification signals in oncogenes may be the main reason that leads to the development of HCC. The signals of H3K4me3 in some regions of oncogenes (MYD88, MYC, ARHGAP5, PLCG1, MDM2, MET, HLF, ERBB3, MAP2K1, and MALT1) are significantly stronger.
For key ONCO, the main histone modifications and their regions that have changed in tumor cell line were summarized as the following example (Table 3A).
-
(1)
The H3K4me3 signals are markedly intensified in the regions (−2000, −1300) bp and (−1000, 1000) bp of MYD88, the region (−1000, 1500) bp of PLCG1, and the region (−1000, 2000) bp of ERBB3, etc. Particularly, in the regions (−1500, −500) bp and (0, 1000) bp of ARHGAP5, the H3K4me3 signals increase 150 times in tumor cell line than that in normal cell line. The H3K4me3 signals of these regions in the related genes may result in the HCC.
-
(2)
There are also obvious increases for H3K27ac signals in the region (0, 2000) bp of ERBB3, regions (−1700, −1000) bp and (0, 500) bp of ETV4, regions (−1000, −300) bp, (0, 500) bp and (500, 1500) bp of MDM2, region (0, 2000) bp of HLF. Especially, the signals increase 100 times in the region (0, 1500) bp of ARHGAP5.
-
(3)
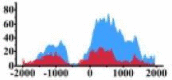
H3K9ac signals in the region (0, 1500) bp of ARHGAP5, regions (−500, −200) bp and (0, 1500) bp of PLCG1, regions (300, 800) bp and (1000, 2000) bp of ERBB3 and region (0, 2000) bp of HLF also increase.
-
(4)
H3K4me2 signals in the regions (−1500, −500) bp and (1000, 2000) bp of ARHGAP5 are also stronger. It can be seen that these histone modifications in these genes are at least 50 times greater in tumor cell line than that in normal cell line. This indicates that the increase of H3K4me2 in these genes may significantly affect the occurrence of HCC.
Table 3.
Genes and important distribution regions of histone modification signals that related to HCC. (A) is the oncogenes, (B) is the tumor suppressor genes.
| (A) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GENE (ONCO) | H3K4me3 |
H3K9ac |
H3K27ac |
H3K4me2 |
H3K79me2 |
||||||||||
| DISTRIBUTION | DHMR (bp) | Δ | DISTRIBUTION | DHMR (bp) | Δ | DISTRIBUTION | DHMR (bp) | Δ | DISTRIBUTION | DHMR (bp) | Δ | DISTRIBUTION | DHMR (bp) | Δ | |
| MYD88 | 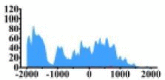 |
(−2000, −1300) (−1000, 1000) |
↑ | 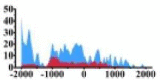 |
(−2000, −1500) (−1000, 0) |
↑ | |||||||||
| MYC | 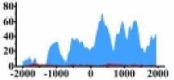 |
(−1300, −700) (−500, 2000) |
↑ |  |
(0, 1500) | ↑ | 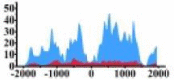 |
(−1800, −800) (−800, −100) (200, 1500) |
↑ | ||||||
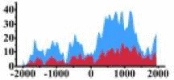
| ARHGAP5 | 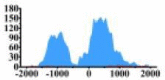 |
(−1500, −500) (0, 1000) |
↑ |  |
(0, 1500) | ↑ | 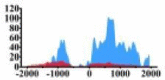 |
(−1000, −500) (0, 1500) |
↑ | 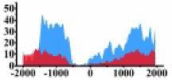 |
(−1500, −500) (1000, 2000) |
↑ |  |
(500, 2000) | ↑ |
| PLCG1 | 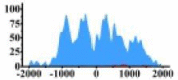 |
(−1000, 1500) | ↑ | 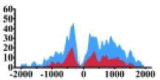 |
(−500, −200) (0, 1500) |
↑ | 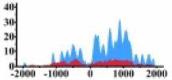 |
(0, 1200) | ↑ | 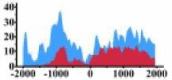 |
(−1500, −500) | ↑ | |||
| MDM2 | 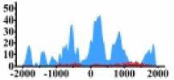 |
(−1000, −300) (0, 500) (500, 1000) |
↑ | 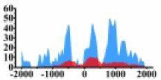 |
(−1000, −300) (0, 500) (500, 1500) |
↑ | |||||||||
| MET | 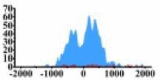 |
(−800, 800) | ↑ | 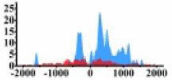 |
(−500, 0) (0, 1200) |
↑ | |||||||||
| ETV4 | 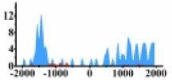 |
(−1700, −1200) (1000, 2000) |
↑ | 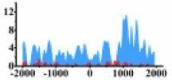 |
(1000, 2000) | ↑ | 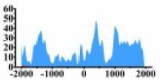 |
(−1700, −1000) (0, 500) (1000, 2000) |
↑ | ||||||
| HLF |  |
(0, 2000) | ↑ | 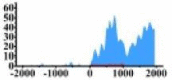 |
(0, 2000) | ↑ | 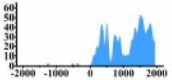 |
(0, 2000) | ↑ |  |
(0, 1000) (1200, 2000) |
↑ | |||
| ERBB3 | 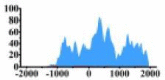 |
(−1000, 2000) | ↑ | 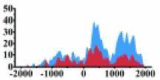 |
(300, 800) (1000, 2000) |
↑ | 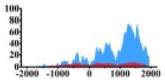 |
(0, 2000) | ↑ | ||||||
| MAP2K1 | 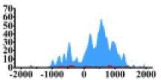 |
(0, 1500) | ↑ | 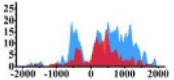 |
(−700, −300) (800, 1500) |
↑ | 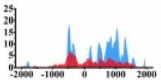 |
(500, 1500) | ↑ |  |
(700, 2000) | ↑ | |||
| MALT1 | 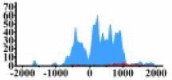 |
(−800, 1000) | ↑ | 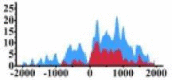 |
(200, 1500) | ↑ | 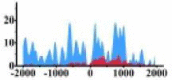 |
(−2000, 0) (0, 500) (700, 1100) |
↑ |  |
(500, 2000) | ↑ | |||
| CCND1 | 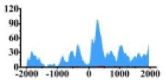 |
(−2000, −1500) (−1000, −2000) |
↑ | 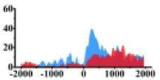 |
(0, 500) | ↑ | 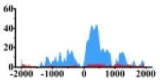 |
(0, 800) | ↑ | ||||||
| CHD4 |  |
(−2000, −1500) | ↑ | 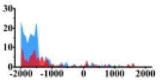 |
(−2000, −1400) | ↑ |  |
(−2000, −1400) | ↑ | ||||||
| (B) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GENE (TSG) | H3K4me3 |
H3K9ac |
H3K27ac |
H3K4me2 |
H3K27me3 | ||||||||||
| DISTRIBUTION | DHMR (bp) | Δ | DISTRIBUTION | DHMR (bp) | Δ | DISTRIBUTION | DHMR (bp) | Δ | DISTRIBUTION | DHMR (bp) | Δ | DISTRIBUTION | DHMR (bp) | Δ | |
| CREB3L1 | 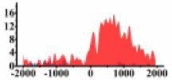 |
(0, 1500) | ↓ | 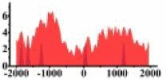 |
(−1500, −300) (300, 2000) |
↓ | 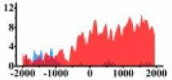 |
(−500, 2000) | ↓ | 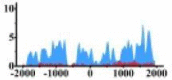 |
(−1800, −500) (−300, 2000) |
↑ | |||
| ROBO2 | 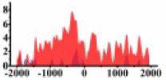 |
(−1500, 500) | ↓ | 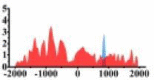 |
(−1500, 500) | ↓ | 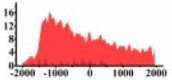 |
(−1500, 2000) | ↓ | 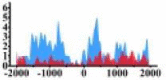 |
(−1500, −500) (0, 500) |
↑ | |||
| BTG1 | 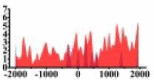 |
(−2000, −500) (700, 2000) |
↓ | 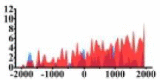 |
(1000, 2000) | ↓ | 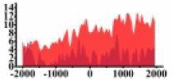 |
(−1000, 2000) | ↓ | ||||||
| ACKR3 | 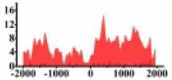 |
(−2000, −800) (0, 2000) |
↓ | 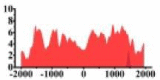 |
(−2000, 1300) | ↓ | 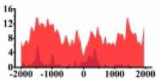 |
(−1500, 500) (500, 2000) |
↓ | 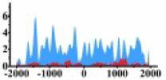 |
(−1500, 2000) | ↑ | |||
In the distribution maps of histone modification signals, the blue represents the distribution of histone modification signals in tumor cell line and the red represents the distribution of histone modification signals in normal cell line. The Δ represents the change of histone modification signals in tumor cell line relative to normal cell line, the ↑ represents increasing of histone modification signals, the ↓ represents weakening of histone modification signals. Abbreviations: HM: histone modification. DHMR: differentially histone modification regions in tumor cell line.
For TSG (Table 3B), we found that the signal strength of H3K9ac, H3K4me2, and H3K27ac are weaker in tumor cell line than normal cell line, such as H3K9ac in the region (0, 1500) bp of CREB3L1. In contrast, the signal strength of H3K27me3 increases in tumor cell line relative to normal cell line. It indicates that the increase of H3K27me3 and the decrease of H3K9ac, H3K4me2, and H3K27ac in tumor cell line inhibit the expression of TSG.
3.7. Functional enrichment analysis of ONCO (TSG) for DHLEG
To further analyze the action of these important genes in the development of HCC, we performed GO and KEGG pathway enrichment analysis using Metascape [31] (http://metascape.org/) (Fig. 5). The Pathways in cancer (hsa05200) is rich in many significant genes (CCND1, MAP2K1, MAPK1, MDM2, MET, MYC, PLCG1, CDH1, etc.). These analyses also reveal that these key genes are related to transcriptional misregulation in cancer (hsa05202), the negative regulation of cell differentiation (GO: 0045596), mesenchyme development (GO: 0060485), the regulation of DNA binding transcription factor activity (GO: 0051090), cell surface receptor signaling pathway involved in cell–cell signaling (GO: 1905114), and the maintenance of DNA repeat elements (GO: 0043570) (Fig. 5A). GO enrichment cluster analysis shows that these genes are indeed related to cancer, such as the cluster in PI3K-Akt signaling pathway, Pathways in cancer, MAPK family signaling cascades, epithelial cell proliferation and differentiation, the positive regulation of cell death, etc. (Fig. 5B). We also found some links between these genes (Fig. 5C). All these enrichment results indicate that these important genes do have an impact on the occurrence of cancer.
Fig. 5.

Functional enrichment analysis of key genes. (A) Heat maps showing enrichment of key genes. Length of bars represent based on the best-scoring term within each cluster, the color key from shallow to deep indicates high to low P values, respectively. (B) Enrichment network of these genes. Each term is indicated by a circular node. The number of these genes falling into that term is represented by the circle size and the nodes of the same color belonged to the same cluster. (C) Circos plots of these key genes. The related genes are linked by lines.
3.8. Survival analysis of key genes
To further validate the prognostic value of the key genes we found for HCC, we performed survival analysis using the clinical data and corresponding expression data of 364 LIHC patients from the TCGA database (including the HCC-related genes CDK4 and AKT1 that have been found [32], [33], [34], [35]. First, the effect of high and low expression of these genes on patient survival was verified. It was found that the survival rate in high expression group is lower than that in low expression group for ONCO (MYD88, ARHGAP5, PLCG1, ETV4, ERBB3, MAP2K1, CHD4, CDK4, AKT1) of DHLEG, and the survival rate in low expression group is lower than that in high expression group for TSG (ROBO2, BTG1) of DHLEG, but the difference is not significant (Supplementary appendix file: Fig. A4). It may be because the expression of a single gene is not sufficient to significantly change the survival of the patient.
Therefore, we further performed multiple Cox regression analyses (log-rank test, ) on the expression of these genes (Fig. 6A), finally constructed a prognostic model related to the expression of six genes, as shown below:
Fig. 6.

Survival analysis of key genes. (A) Forest plot with multiple Cox analysis results. The horizontal line corresponds to the 95% confidence interval and the vertical line indicates a C-index of 1. (B) Kaplan-Meier curve for high-risk and low-risk groups. (C) The ROC curve corresponding to the risk scoring model.
Among them, ARHGAP5, ETV4, ACKR3, and CDK4 are protection factors, MAP2K1 and BTG1 are risk factors. The risk scores of all patients were calculated by using the risk scoring model. According to the median risk score of 1.3093, the patients were divided into high-risk and low-risk groups. Kaplan-Meier survival analysis shows that patients in the high-risk group has significantly lower survival rates than patients in the low-risk group (P = 0.0001) (Fig. 6B). The ROC curve was used to evaluate the risk scoring model (AUC = 0.74), which proves that the risk scoring model has a good predictive ability for patient survival (Fig. 6C).
In addition, we performed the survival analysis of patients in different groups at high-risk and low-risk based on age, gender, tumor stage, T, N, and M stages, combined with clinical pathological factors. It was found that the high-risk and low-risk groups divided by the risk scoring model in different age groups (age54, age54), male, different pathological stages (I/II, III/IV), different T stages (T1/T2, T3/T4), M0, N0 stages, the survival rate of patients in the high-risk group is significantly lower than those in the low-risk group (Fig. 7). The results show that the predictive ability of our risk scoring model has nothing to do with other pathological factors, is a better independent predictor, and can well predict the survival rate of patients.
Fig. 7.

Kaplan–Meier curve to assess the independent prognostic performance of risk scoring models in different categories of patients.
4. Discussion
In this study, we aimed at revealing the key genes that are related to important histone modifications as well as the vital regions of these histone modifications that resulted in HCC. We analyzed the histone modification patterns of DHLEG and found that the H3K4me3, H3K27ac, and H3K9ac are activating modifications, while H3K27me3 is an inhibitory modification. By analyzing the ONCO and TSG of DHLEG, we found that some key genes may be important for the occurrence of HCC, that including MYC, CCND1, CHD4, MAP2K1, MDM2, MET, AKT1, CDK4, ARHGAP5, ETV4, ERBB3, HLF, MALT1, MYD88, and PLCG1. Besides, in further researches of the distribution patterns of histone modifications within these ONCO and TSG, it was observed that H3K4me3, H3K27ac, H3K9ac, and H3K27me3 play important roles for oncogenes expression. Finally, the key genes were analyzed for survival, and a prognostic risk scoring model related to the expression of six genes was established, which can better predict the survival rate of patients.
In fact, some of the genes (MYC, CCND1, CHD4, MDM2, and MET) were found in our research that have been reported in previous studies. For example, MYC is a transcription factor, regulates many programs which are related to the occurrence of cancer [36], [37], [38], [39]. MYC and CCND1 are also canonical Wnt targets, often occur overexpression and genomic amplification in HCC [10], [40], [41], [42], [43], [44], [45]. Studies have also reported CHD4 is a good target for the eradication of HCC [46]. MDM2 has been shown to function in HCC [47]. MET frequently undergoes copy number variation and has been identified as biomarkers in HCC [48], [49], [50], [51], [52]. These researches not only have proved that the key genes obtained in our study are associated with HCC, but also have demonstrated that our results are credible.
In addition, we also found some important genes for HCC that hardly reported in previous studies, such as ARHGAP5, ETV4, ERBB3, MAP2K1, HLF, MALT1, MYD88, and PLCG1. Although there is no evidence that ERBB3 is carcinogenic, it has found that ERBB2 is associated with this gene, which has genomic amplification in breast cancer [53]. Moreover, the major histone co-modification patterns of the genes were also been found in both cell lines. Some important histone modifications (H3K4me3, H3K27ac, H3K9ac, and H3K4me2) and their modification regions in each gene were identified. Our results will help study the pathogenesis of HCC and discover new biomarkers as well as using epigenetic modifications as novel target drugs.
5. Conclusion
In this study, we analyzed the changes of histone modifications in two kinds of cell lines and the effects of histone modifications on the levels of gene expression from the perspective of bioinformatics. The important histone modifications and 17 key genes were identified, the changes of regions for histone modifications in these key genes were located, the survival analysis was used to further verify the impact of key genes on prognosis. These results may help identify histone modifications as biomarkers and find more potential therapeutic targets for HCC.
Author contributions
Yu-Xian Liu designed the study, collected data, analyzed data, and wrote the article. Qian-Zhong Li conceived the idea and was involved in the study, discussion, writing, and revision of the whole article. Yan-Ni Cao collected part of the data, discussed part of the results and revised the whole article. Lu-Qiang Zhang discussed part of the results and revised the article. They all approved final version of the manuscript.
Financial support
This work was supported by the National Natural Science Foundation of China [grant nos. 31870838 and 61861035].
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant nos. 31870838 and 61861035].
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.09.013.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Llovet J.M., Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48(4):1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Bosch F.X., Ribes J., Díaz M., Cléries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127(5):S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 5.Naoshi N., Ajay G. Genetic and epigenetic signatures in human hepatocellular carcinoma: a systematic review. Curr Genom. 2011;12(2):130–137. doi: 10.2174/138920211795564359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao W, Kondo Y, Shen L, Shimizu Y, Sano T, Yamao K. Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas. Carcinogenesis. 2008;29(10):1901–1910. doi: 10.1093/carcin/bgn170. [DOI] [PubMed] [Google Scholar]
- 7.Sharma S., Kelly T.K., Jones P.A. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H., Shang Y.-P., Chen H.-Y., Li J. Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatol Res. 2017;47(2):149–159. doi: 10.1111/hepr.12757. [DOI] [PubMed] [Google Scholar]
- 9.Jung K.H., Noh J.H., Kim J.K., Eun J.W., Bae H.J., Chang Y.G., Kim M.G., Park W.S., Lee J.Y., Lee S.-Y., Chu I.-S., Nam S.W. Histone deacetylase 6 functions as a tumor suppressor by activating c-Jun NH2-terminal kinase-mediated beclin 1-dependent autophagic cell death in liver cancer. Hepatology. 2012;56(2):644–657. doi: 10.1002/hep.25699. [DOI] [PubMed] [Google Scholar]
- 10.Cheng A.S., Lau S.S., Chen Y., Kondo Y., Li M.S., Feng H. EZH2-mediated concordant repression of Wnt antagonists promotes β-catenin-dependent hepatocarcinogenesis. Cancer Res. 2011;71(11):4028–4039. doi: 10.1158/0008-5472.CAN-10-3342. [DOI] [PubMed] [Google Scholar]
- 11.Hung S.Y., Lin H.H., Yeh K.T., Chang J.G. Histone-modifying genes as biomarkers in hepatocellular carcinoma. Int J Clin Exp Pathol. 2014;7(5):2496–2507. [PMC free article] [PubMed] [Google Scholar]
- 12.He C., Xu J., Zhang J., Xie D., Ye H., Xiao Z., Cai M., Xu K., Zeng Y., Li H., Wang J. High expression of trimethylated histone H3 lysine 4 is associated with poor prognosis in hepatocellular carcinoma. Hum Pathol. 2012;43(9):1425–1435. doi: 10.1016/j.humpath.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Dhanasekaran R., Bandoh S., Roberts L.R. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Res. 2016;5:879. doi: 10.12688/f1000research10.12688/f1000research.6946.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai M.-Y., Hou J.-H., Rao H.-L., Luo R.-Z., Li M., Pei X.-Q., Lin M.C., Guan X.-Y., Kung H.-F., Zeng Y.-X., Xie D. High expression of H3K27me3 in human hepatocellular carcinomas correlates closely with vascular invasion and predicts worse prognosis in patients. Mol Med. 2011;17(1-2):12–20. doi: 10.2119/molmed.2010.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poté N., Alexandrov T., Le Faouder J., Laouirem S., Léger T., Mebarki M., Belghiti J., Camadro J.-M., Bedossa P., Paradis V. Imaging mass spectrometry reveals modified forms of histone H4 as new biomarkers of microvascular invasion in hepatocellular carcinomas: hepatology. Hepatology. 2013;58(3):983–994. doi: 10.1002/hep.26433. [DOI] [PubMed] [Google Scholar]
- 16.Sistayanarain A., Tsuneyama K., Zheng H., Takahashi H., Nomoto K., Cheng C. Expression of Aurora-B kinase and phosphorylated histone H3 in hepatocellular carcinoma. Anticancer Res. 2006;26(5A):3585–3593. [PubMed] [Google Scholar]
- 17.Zhu P., Fan Z. Cancer stem cells and tumorigenesis. Biophys Rep. 2018;4(4):178–188. doi: 10.1007/s41048-018-0062-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu P., Wang Y., Huang G., Ye B., Liu B., Wu J., Du Y., He L., Fan Z. lnc-β-Catm elicits EZH2-dependent β-catenin stabilization and sustains liver CSC self-renewal. Nat Struct Mol Biol. 2016;23(7):631–639. doi: 10.1038/nsmb.3235. [DOI] [PubMed] [Google Scholar]
- 19.Zhu P., Wang Y., Wu J., Huang G., Liu B., Ye B. LncBRM initiates YAP1 signalling activation to drive self-renewal of liver cancer stem cells. Nat Commun. 2016;7(1):13608. doi: 10.1038/ncomms13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y., He L., Du Y., Zhu P., Huang G., Luo J., Yan X., Ye B., Li C., Xia P., Zhang G., Tian Y., Chen R., Fan Z. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell. 2015;16(4):413–425. doi: 10.1016/j.stem.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Furuta M., Ueno M., Fujimoto A., Hayami S., Yasukawa S., Kojima F., Arihiro K., Kawakami Y., Wardell C.P., Shiraishi Y., Tanaka H., Nakano K., Maejima K., Sasaki-Oku A., Tokunaga N., Boroevich K.A., Abe T., Aikata H., Ohdan H., Gotoh K., Kubo M., Tsunoda T., Miyano S., Chayama K., Yamaue H., Nakagawa H. Whole genome sequencing discriminates hepatocellular carcinoma with intrahepatic metastasis from multi-centric tumors. J Hepatol. 2017;66(2):363–373. doi: 10.1016/j.jhep.2016.09.021. [DOI] [PubMed] [Google Scholar]
- 22.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 25.Warnes G.R., Bolker B., Bonebakker L., Gentleman R., Huber W., Liaw A. gplots: various R programming tools for plotting data. R Package Version. 2009;2:1. [Google Scholar]
- 26.Ally A., Balasundaram M., Carlsen R. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169(7):1327–1341.e23. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.COX D.R. Partial likelihood. Biometrika. 1975;62(2):269–276. [Google Scholar]
- 28.Therneau T.M., Lumley T. Package 'survival'. Survival analysis Published on CRAN. 2014;2:3. [Google Scholar]
- 29.Heagerty P J, Lumley T, Pepe M S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56(2):337–344. doi: 10.1111/j.0006-341x.2000.00337.x. [DOI] [PubMed] [Google Scholar]
- 30.Bardou P., Mariette J., Escudié F., Djemiel C., Klopp C. jvenn: an interactive Venn diagram viewer. BMC Bioinf. 2014;15(1):293. doi: 10.1186/1471-2105-15-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colak D., Chishti M.A., Al-Bakheet A.-B., Al-Qahtani A., Shoukri M.M., Goyns M.H., Ozand P.T., Quackenbush J., Park B.H., Kaya N. Integrative and comparative genomics analysis of early hepatocellular carcinoma differentiated from liver regeneration in young and old. Mol Cancer. 2010;9(1):146. doi: 10.1186/1476-4598-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Q., Li J., Xing J., Li W., Li H., Ke X., Zhang J., Ren T., Shang Y., Yang H., Jiang J., Chen Z. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J Hepatol. 2014;61(4):859–866. doi: 10.1016/j.jhep.2014.04.035. [DOI] [PubMed] [Google Scholar]
- 34.Joo M, Kang YK, Kim MR, Lee HK, Jang JJ. Cyclin D1 overexpression in hepatocellular carcinoma. Liver Int. 2001;21(2):89–95. doi: 10.1034/j.1600-0676.2001.021002089.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang G.-L., Salisbury E., Shi X., Timchenko L., Medrano E.E., Timchenko N.A. HDAC1 promotes liver proliferation in young mice via interactions with C/EBPβ. J Biol Chem. 2008;283(38):26179–26187. doi: 10.1074/jbc.M803545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moulos P., Sarris M.E., Talianidis I. Mechanism of gene-specificity of oncogenic regulators. Cell Cycle. 2016;15(17):2227–2228. doi: 10.1080/15384101.2016.1191254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. CSH Perspect Med. 2014;4(6):a014241. doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shachaf C.M., Kopelman A.M., Arvanitis C., Karlsson A., Beer S., Mandl S. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431(7012):1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 39.Stine Z.E., Walton Z.E., Altman B.J., Hsieh A.L., Dang C.V. MYC, metabolism, and cancer. Cancer Discov. 2015;5(10):1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang K., Lim H.Y., Shi S., Lee J., Deng S., Xie T., Zhu Z., Wang Y., Pocalyko D., Yang W.J., Rejto P.A., Mao M., Park C.-K., Xu J. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology. 2013;58(2):706–717. doi: 10.1002/hep.26402. [DOI] [PubMed] [Google Scholar]
- 41.Villanueva A., Newell P., Chiang D., Friedman S., Llovet J. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27(1):055–076. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 42.Guichard C., Amaddeo G., Imbeaud S., Ladeiro Y., Pelletier L., Maad I.B., Calderaro J., Bioulac-Sage P., Letexier M., Degos F., Clément B., Balabaud C., Chevet E., Laurent A., Couchy G., Letouzé E., Calvo F., Zucman-Rossi J. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zucmanrossi J., Benhamouche S., Godard C., Boyault S., Grimber G., Balabaud C. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene. 2007;26:774–780. doi: 10.1038/sj.onc.1209824. [DOI] [PubMed] [Google Scholar]
- 44.Hoshida Y., Toffanin S., Lachenmayer A., Villanueva A., Minguez B., Llovet J. Molecular classification and novel targets in hepatocellular carcinoma: recent advancements. Semin Liver Dis. 2010;30(01):035–051. doi: 10.1055/s-0030-1247131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lachenmayer A., Alsinet C., Savic R., Cabellos L., Toffanin S., Hoshida Y., Villanueva A., Minguez B., Newell P., Tsai H.-W., Barretina J., Thung S., Ward S.C., Bruix J., Mazzaferro V., Schwartz M., Friedman S.L., Llovet J.M. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin Cancer Res. 2012;18(18):4997–5007. doi: 10.1158/1078-0432.CCR-11-2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nio K., Yamashita T., Okada H., Kondo M., Hayashi T., Hara Y., Nomura Y., Zeng S.S., Yoshida M., Hayashi T., Sunagozaka H., Oishi N., Honda M., Kaneko S. Defeating EpCAM+ liver cancer stem cells by targeting chromatin remodeling enzyme CHD4 in human hepatocellular carcinoma. J Hepatol. 2015;63(5):1164–1172. doi: 10.1016/j.jhep.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 47.Iakova P., Timchenko L., Timchenko N.A. Intracellular signaling and hepatocellular carcinoma. Semin Cancer Biol. 2011;21(1):28–34. doi: 10.1016/j.semcancer.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts L.R., Gores G.J. Hepatocellular carcinoma: molecular pathways and new therapeutic targets. Semin Liver Dis. 2005;25(2):212–225. doi: 10.1055/s-2005-871200. [DOI] [PubMed] [Google Scholar]
- 49.Allaire M., Nault J.-C. Molecular targets for HCC and future treatments. J Hepatol. 2017;66(1):234–235. doi: 10.1016/j.jhep.2016.07.034. [DOI] [PubMed] [Google Scholar]
- 50.Santoro A., Rimassa L., Borbath I., Daniele B., Salvagni S., Van Laethem J.L., Van Vlierberghe H., Trojan J., Kolligs F.T., Weiss A., Miles S., Gasbarrini A., Lencioni M., Cicalese L., Sherman M., Gridelli C., Buggisch P., Gerken G., Schmid R.M., Boni C., Personeni N., Hassoun Z., Abbadessa G., Schwartz B., Von Roemeling R., Lamar M.E., Chen Y., Porta C. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14(1):55–63. doi: 10.1016/S1470-2045(12)70490-4. [DOI] [PubMed] [Google Scholar]
- 51.Jia D., Wei L., Guo W., Zha R., Bao M., Chen Z., Zhao Y., Ge C., Zhao F., Chen T., Yao M., Li J., Wang H., Gu J., He X. Genome-wide copy number analyses identified novel cancer genes in hepatocellular carcinoma. Hepatology. 2011;54(4):1227–1236. doi: 10.1002/hep.24495. [DOI] [PubMed] [Google Scholar]
- 52.Woo H.G., Park E.S., Lee J.-S., Lee Y.-H., Ishikawa T., Kim Y.J., Thorgeirsson S.S. Identification of potential driver genes in human liver carcinoma by genomewide screening. Cancer Res. 2009;69(9):4059–4066. doi: 10.1158/0008-5472.CAN-09-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marty M., Cognetti F., Maraninchi D., Snyder R., Mauriac L., Tubiana-Hulin M. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2–positive metastatic breast cancer administered as first-line treatment: the M77001 study group. J Clin Oncol. 2005;23(19):4265–4274. doi: 10.1200/JCO.2005.04.173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.