

Summary
Robust synthetic biology applications rely heavily on the design and assembly of DNA parts with specific functionalities based on engineering principles. However, the assembly standards adopted by different communities vary considerably, thus limiting the interoperability of parts, vectors and methods. We hereby introduce the SEVA 3.1 platform consisting of the SEVA 3.1 vectors and the Golden Gate‐based ‘SevaBrick Assembly’. This platform enables the convergence of standard processes between the SEVA platform, the BioBricks and the Type IIs‐mediated DNA assemblies to reduce complexity and optimize compatibility between parts and methods. It features a wide library of cloning vectors along with a core set of standard SevaBrick primers that allow multipart assembly and exchange of short functional genetic elements (promoters, RBSs) with minimal cloning and design effort. As proof of concept, we constructed, among others, multiple sfGFP expression vectors under the control of eight RBSs, eight promoters and four origins of replication as well as an inducible four‐gene operon expressing the biosynthetic genes for the black pigment proviolacein. To demonstrate the interoperability of the SEVA 3.1 vectors, all constructs were characterized in both Pseudomonas putida and Escherichia coli. In summary, the SEVA 3.1 platform optimizes compatibility and modularity of inserts and backbones with a cost‐ and time‐friendly DNA assembly method, substantially expanding the toolbox for successful synthetic biology applications in Gram‐negative bacteria.
We hereby introduce the SEVA 3.1 platform consisting of the SEVA 3.1 vectors and the Golden Gate‐based ‘SevaBrick assembly’. This platform enables the convergence of standard processes between the SEVA platform, the BioBricks and Type IIs‐mediated DNA assemblies to reduce complexity and optimize compatibility between parts and methods.
![]()
Introduction
Synthetic biology is a fast‐growing field that incorporates biological and engineering principles to extend or modify the capabilities of organisms or biological systems towards new applications (Andrianantoandro et al., 2006). To increase the speed and predictability of any synthetic biology application, it is critical for it to be designed in accordance with the principles of standardization, decoupling, abstraction and modularity (Andrianantoandro et al., 2006; Way et al., 2014). Often, these applications require the combination of multiple genetic elements such as promoters, ribosomal binding sites (RBSs) or coding sequences (CDSs) into new and complex DNA‐encoded molecular devices (Ellis et al., 2011). To this end, DNA parts need to be modular, standardized and based on rational design in order to minimize redundancy and unpredictability (Kelly et al., 2009). Evolution has driven biological systems to be inherently dynamic and even though complex, having a very well‐balanced behaviour (Purnick and Weiss, 2009). Disturbing this balance using poorly designed, incompatible or uncharacterized DNA parts could disrupt the whole biological system with unpredictable and often detrimental effects (Gardner et al., 2000). Although de novo DNA synthesis could become a solution to the problem, as parts can be directly ordered and synthesized in a matter of days, double‐stranded DNA synthesis is still relatively expensive and at times impossible when it comes to complex DNA structures (Ma et al., 2012; Hughes and Ellington, 2017; Perkel et al., 2019). Consequently, synthetic biology still relies on robust DNA assembly methods to avoid de novo DNA synthesis of long and complex parts and to facilitate high‐throughput exchange of DNA parts at low cost and within a reasonable short time. Moreover, the design of the DNA parts to be assembled requires adherence to standard rules independently of each specific part, thereby allowing interchangeability between laboratories and automation of construction. In addition, given that a wide range of hosts are used in synthetic biology applications, with new organisms being used as chassis, a holistic approach towards the interoperability of parts and vectors has become imperative (Adams, 2016).
Currently, DNA parts can be assembled using several in vitro and in vivo methods. Popular protocols to this end include, inter alia: (i) homology‐based in vitro methods [Overlap Extension PCR (Higuchi et al., 1988), Gibson assembly (Gibson et al., 2009), USER (Bitinaite et al., 2007)], (ii) Golden Gate‐based (Engler et al., 2008) methods [MoClo (Weber et al., 2011), GoldenBraid (Sarrion‐Perdigones et al., 2011; Sarrion‐Perdigones et al., 2013), Mobius (Andreou and Nakayama, 2018), Loop (Pollak et al., 2019)] and (iii) in vivo methods [Yeast DNA assembly (Gibson et al., 2008; Chandran and Shapland, 2017), Bacillus subtillis DNA assembly (Tsuge et al., 2003; Itaya et al., 2018), recombineering in E. coli (Sharan et al., 2009)]. Even though these methods can result in highly functional constructs, they require specific restrictions and/or design per part. Regarding homology‐based and in vivo methods, each part needs to be prepared in advance with individualized primers every time the specific part has to be assembled in a new construct. Consequently, this process could hamper the concept of standardization and interoperability. On the other hand, mainstream Type IIS (Golden Gate‐based) assembly methods require the use of at least two restriction enzymes whose restriction sites must be mutated. Additionally, parts need to be cycled between different vectors during the assembly process in order to result in complex constructs. Such diverse requirements often limit the throughput of constructs and the collaboration in time and space among synthetic biology communities.
In this context, the BioBricks™ platform (Shetty et al., 2011) was one of the initial attempts to standardize the assembly process of interchangeable DNA parts using standardized flanks which enable a simple and universal restriction/ligation. By using BioBricks, it became possible to store and share DNA parts which could be easily assembled by the synthetic biology community. As a result of the popularity of the iGEM competition, the Registry of Standard Biological Parts has become one of largest public DNA libraries with more than 20.000 parts (http://parts.igem.org/Main_Page). However, despite the high simplicity of the standardized assembly, the proposed 3A assembly (Shetty et al., 2011) is a laborious and time‐consuming process, while backbone availability is limited and poorly characterized.
In addition to standardizing DNA parts, vectors also require corresponding standardization and robustness. The key elements of an engineered vector that make it important for biotechnological purposes are the origin of replication (Ori), the antibiotic resistance (AR) and the cloning module. Various vector collections have been developed in recent years, with unique features for specific applications (e.g. high gene expression, reporter genes). The Standard European Vector Architecture (SEVA) (Silva‐Rocha et al., 2013; Martínez‐García et al., 2015, 2020) is a well‐characterized and curated public platform of vectors for use in Gram‐negative bacteria. This platform contains a large number of vectors, combining seventeen Ori and eight AR genes, which follow the same format and nomenclature. However, the proposed assembly method of SEVA vectors is based on traditional restriction/ligation in which each part has to be individually amplified with specific primers, digested and purified for any particular application (Martínez‐García et al., 2015). Therefore, although SEVA vectors are widely preferred for their reliability and variety, their performance as assembly vectors for multiple DNA constructs is not optimal.
To combine the unique features of SEVA vectors and BioBricks, eliminating their drawbacks, we developed the SEVA 3.1 platform. The concept of the SEVA 3.1 vectors arose as an attempt to bridge (i) the backbone (Ori and AR) flexibility of the SEVA vectors, (ii) the part abundance and interchangeability of BioBricks and (iii) the practicality of the Type IIS restriction enzymes. The SEVA 3.1 vectors consist of any SEVA backbone (Ori, AR, terminators) merged with the standard BioBrick cloning site (prefix, suffix) resulting in the new SEVA 3.1 cloning site called MCS 2.0 (Modular Cloning Site). The end‐product named pSEVAb carrying SevaBricks is a vector with maximized host flexibility, compatible with the BioBrick assembly protocol (RFC10). The SEVA standard was the ideal candidate due to its modular design, lack of BsaI restriction sites, well‐curated modules, multi‐origin compatibility, SBOL (Galdzicki et al., 2014) compatibility and widespread scientific acceptance (Kuepper et al., 2015; Calero et al., 2016; Kim et al., 2019). In addition, we present the SevaBrick Assembly which emulates the 3A Assembly by further simplifying and upgrading the assembly process. This Golden Gate‐based assembly method consists of standardized protocols and primers that enable simple and straightforward one‐step assembly of single or multiple BioBricks into the SEVA 3.1 backbones. A critical characteristic of the method is the use of a sole restriction enzyme (BsaI) for the whole process. To our knowledge, the proposed method is the first Golden Gate‐based method which allows direct construction of vectors with different standardized elements such as promoters, RBSs, CDSs, ARs and origins of replication using one restriction enzyme and a single ligation step.
In this study, the efficiency and flexibility of the newly developed SEVA 3.1 platform was showcased through the construction and characterization of several genetic constructs with varying degrees of complexity. Furthermore, in order to prove the broad host nature of the methodology, all constructed expression vectors were tested in both E. coli DHα and the considerably promising as microbial chassis P. putida KT2440 (Poblete‐Castro et al., 2012; Calero and Nikel, 2019), with successful observation of reporter molecules.
Results and discussion
Design and construction of the SEVA 3.1 platform
The herein described SEVA 3.1 platform converges existing standards of vectors, DNA parts and assemblies into one universal standard which can be used for efficient downstream synthetic biology applications. The required elements of the SEVA 3.1 platform are the SevaBrick Assembly (Fig. 1B) and the SEVA 3.1 vectors (Fig. 1A). The SevaBrick Assembly is a method where all the parts and backbones to be assembled are PCR amplified from any SEVA 3.1 or BioBrick vector, using a core set of standard long primers. All SevaBrick primers (Table 1) anneal on standard sequences of the SEVA 3.1 or BioBrick vectors, introducing BsaI recognition sites for directional multipart assembly via Golden Gate. The unique characteristic of the process is an in‐house biphasic PCR protocol, same for all primer sets, which enables these long primers to successfully amplify any part or backbone of the SEVA 3.1 and BioBricks platforms (see Experimental procedures). A SEVA 3.1 vector is constructed through the assembly of three modules, the SEVA 3.1 cloning site module (MCS 2.0), the AR module and the Ori module. The construction of the entire vector library is based on PCR amplification of all modules (MCS 2.0, AR, Ori) using three sets of standard primers with subsequent SevaBrick Assembly (Fig. 2). The MCS 2.0 module is amplified from any BioBrick, preferably a reporter transcription unit (TU), using the Ep/Pp insert primers. The remaining two sets are used to amplify the AR (AR‐F/AR‐R) and Ori (Ori‐F/Ori‐R) modules from any SEVA vector, while adding extra DNA sequences which after the assembly with the BioBrick amplicon reconstruct the BioBrick prefix and suffix. Additionally, the Ori‐R and AR‐F primers carry 23 nt and 27 nt of the upstream and downstream BioBrick terminators respectively (Fig. 2). These additional DNA sequences are essential elements of the new MCS 2.0 as they function as annealing sequences for the SevaBrick Assembly primers. All three primer sets anneal on standard sequences of the template vectors and are therefore compatible with all available SEVA and BioBrick plasmids, allowing any possible combination of all modules. In terms of design, the new vectors have to be fully compatible with the BioBricks assembly protocol and due to this prerequisite, the SEVA AR module had to be modified during PCR amplification in order to remove the SpeI restriction site within it. For the construction of any possible combination, the method requires only a selected number of the template SEVA plasmids carrying all modules (AR, Ori) and one BioBrick. Therefore, we amplified all modules (Fig. S1) required for this study and constructed a wide set of SEVA 3.1 vectors (Table S1) which we make available through the SEVA database (http://seva.cnb.csic.es).
Fig. 1.
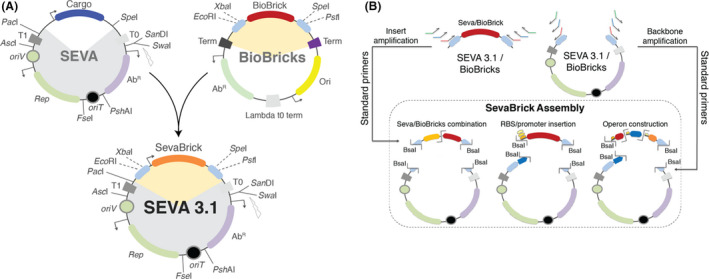
The SEVA 3.1 platform.
A. The SEVA 3.1 vectors are generated by merging: (i) the backbone of the SEVA vectors (grey shade), containing the antibiotic resistance module (purple), the origin of replication module (green), origin of transfer (black) and two transcriptional terminators (dark and light grey) and (ii) the cloning site of the BioBrick vectors (orange shade) comprised by: the BioBrick prefix and suffix (light blue) and a BioBrick (red).
B. Graphical overview of the SevaBrick Assembly. Inserts and backbones are amplified using a core set of standard primers which introduce compatible Bsal sites, allowing the construction of expression vectors with varying degrees of complexity via Golden Gate.
Table 1.
SevaBrick Assembly standard primers.
| Purpose | Insert primers a | Backbone primers b |
|---|---|---|
| Basic assembly | ||
| Convergence | Brk‐F/Brk‐R c | Ev/Pv |
| Single part | Ep/Pp | Ev/Pv |
| Double part | Ep/Sp, Xp/Pp | Ev/Pv |
| Extended assembly | ||
| Single‐gene constitutive TU | OP1‐F/Pp | Evpromoter/Pv |
| Double‐gene constitutive TU | OP1‐F/OP1‐R, OP2‐F/Pp | Evpromoter/Pv |
| Triple‐gene constitutive TU | OP1‐F/OP1‐R, OP2‐F/OP2‐R, OP3‐F/Pp | Evpromoter/Pv |
| Quadruple‐gene constitutive TU | OP1‐F/OP1‐R, OP2‐F/OP2‐R, OP3‐F/OP3‐R, OP4‐F/Pp | Evpromoter/Pv |
| n‐gene inducible TU d | Ep/Sp, OPn‐F/OP(n–1)‐R + Pp | Ev/Pv |
Templates for insert primers: Seva/BioBricks for Basic Assembly, Seva/BioBrick CDSs for Extended assembly, converged inducible expression systems for inducible TUs of Extended Assembly.
Templates for backbone primers: SEVA 3.1 and BioBrick vectors.
Individualized primers, based on standard rules, for part convergence.
Ep/Sp for the amplification of the inducible expression system. n = number of genes in TU.
Fig. 2.
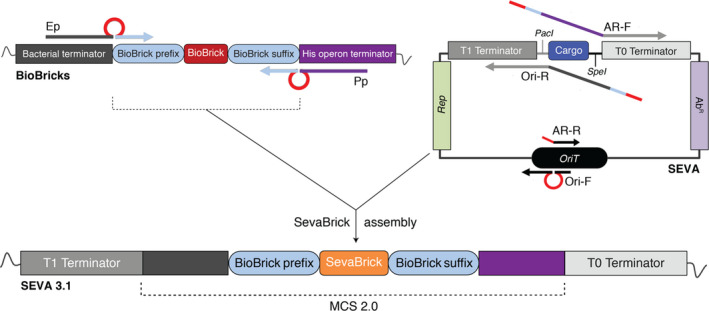
Construction of SEVA 3.1 vectors. The Ep/Pp insert primers anneal on the standard BioBrick prefix and suffix, respectively, amplifying any BioBrick. The AR‐F/AR‐R and Ori‐F/Ori‐R primers sets anneal on the SEVA transcriptional terminators (T0 and T1, respectively) and on the origin of transfer (OriT), amplifying any AR and Ori module, respectively, when pSEVA is used as template. The amplified parts contain two BsaI recognition sites (red loop or line) required for SevaBrick Assembly. The AR‐F and the Ori‐R primers carry overhang sequences that reconstruct the BioBrick prefix and suffix after SevaBrick Assembly. In addition, they carry nucleotides of the upstream and downstream BioBrick terminators which are essential sequences of the new MCS 2.0.
The nomenclature of the SEVA 3.1 vectors complies with SEVA’s (Silva‐Rocha et al., 2013). However, as the configuration of the SEVA 3.1 vectors is not identical with SEVA’s (SpeI restriction site has been moved to the BioBrick suffix), the vector’s name has changed from pSEVA to pSEVAb. Thus, all SEVA 3.1 plasmids are named pSEVAb, while the nomenclature of the standard modules was maintained. For instance, vector pSEVAb23 carries the AR kanamycin [2] and the Ori pBBR1 [3].
Convergence of new DNA parts
A critical step of the SEVA 3.1 platform is the convergence of new DNA parts. This is the only step of the platform where individualized primers have to be designed, using standard rules and flank sequences (Table S2). In terms of structure, all new parts have to comply with the RFC10 protocol of BioBricks, which means being cloned into a BioBrick repository vector and flanked with the appropriate prefix and suffix sequences (http://parts.igem.org/Help:Prefix‐Suffix). Although any SEVA 3.1 vector could serve as repository vector, we chose the BioBrick vector pSB1C3, as it is the main repository vector of the iGEM registry (http://parts.igem.org/Help:2019_DNA_Distribution). During convergence, the repository vector is amplified using the standard backbone linearization primers, Ev/Pv, while the part under construction is amplified with primers which add either the CDS or non‐CDS BioBrick prefix and suffix along with BsaI recognition sites (Fig. 3). The generated sticky ends are compatible with the sticky ends of the linearized vector making the parts of the assembly compatible. Therefore, the only requirement for a part to be SEVA 3.1 compatible is to be free of BsaI recognition sites. BsaI recognition sites can be mutated during the convergence process through a multipart domestication assembly of the new part. Domestication primers are designed to implement the mutation through a loop on the one of the two primers and concurrently add compatible BsaI restriction sites between amplified parts. This method is an iteration of the method proposed in GoldenBraid (Sarrion‐Perdigones et al., 2011). However, instead of adding the mutation via sticky‐end manipulation on both primers, the change is performed through one primer (Fig. S2). In the case of DNA synthesis, the part can be suitably designed to be pre‐domesticated and ready for convergence by carrying the required flanks and sticky ends prior to ordering. After the preparation and final assembly of all parts, the final product is a scarless and domesticated BioBrick. As proof of concept, we have converged, inter alia (Table S3), the (i) E. coli rhamnose‐induced expression system RhaRS/PrhaB (Egan and Schleif, 1993), (ii) the sfGFP CDS from the BBa_K515105 BioBrick and (iii) the XylS/Pm expression system from the SEVA collection (Marques et al., 1999). All parts were stored in the pSB1C3 repository vector as most iGEM parts and further used in downstream constructs. In particular, in the case of the XylS/Pm expression system, which carries a BsaI recognition site on its XylS regulator, the domestication process was carried out in parallel with convergence. All primers used for the convergence process are listed in Table S4.
Fig. 3.
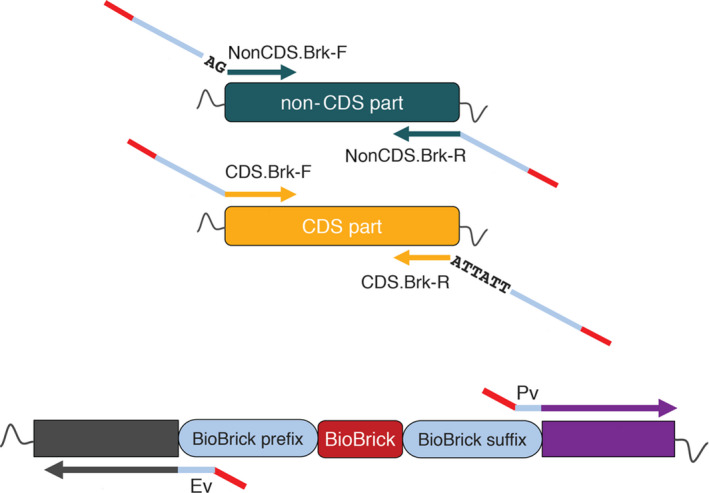
Convergence of DNA parts into SEVA 3.1 platform. A non‐CDS part is converged using the non‐CDS.Brk‐F and the non‐CDS.Brk‐R primers. The non‐CDS.Brk‐F carries the non‐CDS BioBrick prefix (blue) while the non‐CDS.Brk‐R carries the BioBrick suffix (blue). A CDS part is converged using the CDS.BrK‐F and CDS.Brk‐R primers. The CDS.BrK‐F carries the CDS BioBrick prefix while the CDS.Brk‐R carries the BioBrick suffix and a double stop codon (TAATAA). All primers carry a BsaI recognition site (red) at the 5’‐ or 3’‐end which enables assembly with the amplified with Ev/Pv primers repository vector (pSB1C3). After the assembly the BioBrick suffix and prefix are reconstructed.
Basic SevaBrick Assembly for Seva/BioBricks
The SevaBrick Assembly for BioBricks is an alternative method to the 3A Assembly proposed for BioBrick construction. Since all converged parts of the SEVA 3.1 platform are stored into pSB1C3, we sought to engineer a method that could assemble Seva/BioBricks together while removing the complexity of the 3A assembly. The construction of repository parts, as described in the convergence process, can be considered as the simplest single part SevaBrick Assembly application, equivalent to constructing basic BioBricks. To construct composite parts, in addition to the backbone and insert amplification primers (Ev/Pv and Ep/Pp, respectively), two additional standard primers (Sp and Xp) were designed to allow double part assembly. Each of these primers anneals on the standard cloning sequence of SEVA 3.1 (MCS 2.0) and BioBrick compatible vectors (Fig. 4A). The unique amplicon overhangs were designed so that after the final assembly, the plasmid contains a regenerated prefix and suffix and a newly generated BioBrick scar between the assembled parts as it occurs after the 3A assembly. The fidelity of the method was tested by assembling two TUs of the reporter proteins amilCP, mRFP and counting the blue‐purple (only amilCP), light red (mRFP), dark red (amilCP, mRFP) and white (vector self‐assembly) colonies (with dark red expected to be produced by the correct composite part). Initially, the amilCP and mRFP TUs were amplified from two in‐house vectors (the construction of both vectors is described below) using the Ep/Sp and Xp/Pp primers, respectively, and later cloned into the pSEVAb23 vector linearized with the Ev/Pv primers. Both TUs consist of the BBa_BJ23100 promoter, the BBa_B0034 RBS and the corresponding reporter. The assembly procedure was repeated three times, and the ligation efficiency was calculated based on the dark red colonies to 90% (Fig. 4B). Thus, SevaBrick Assembly enables the construction of double TU cassettes using three standard primer sets and one restriction enzyme (BsaI), whereas 3A assembly requires at least four restriction enzymes (EcoRI, XbaI, SpeI, PstI) and the individual digestion of each part.
Fig. 4.
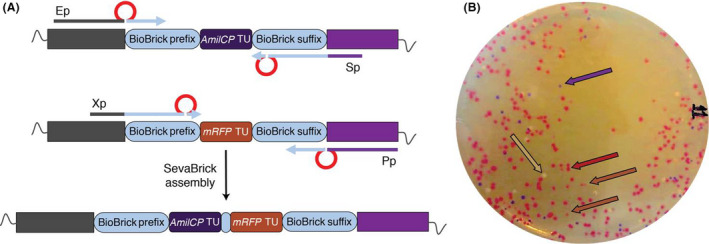
Illustration of Basic SevaBrick Assembly.
A. Design of the Basic SevaBrick Assembly primers. Each primer anneals on the standard MCS 2.0 sequence and they all carry Bsal recognition sites (red) which allow directional cloning of two parts after digestion with Bsal. Primer sets i) Ep/Sp: amplification of first part, ii) Xp/Pp: amplification of second part.
B. Representative picture showing the efficiency of the Basic SevaBrick Assembly to ligate two parts (amilCP TU, mRFP TU). White colonies: vector self‐assembly, blue‐purple colonies: amilCP TU, light red: mRFP TU, dark red: amilCP TU + mRFP TU.
Introduction of short functional genetic elements via PCR amplification
The ‘Registry of Standard Biological Parts’ contains a plethora of genes (metabolic, reporters, regulators etc.) which are stored as CDSs starting at the 5'‐end by a start codon (ATG) and ending at the 3'‐end with a double stop codon (TAATAA). They are stored as basic parts which encode for proteins, without additional functional elements such as promoters or RBSs. This set‐up allows the user to combine BioBricks with the functional genetic elements of choice (RBSs, promoters) in order to achieve the desired transcription–translation levels. Specifically, in synthetic biology and metabolic engineering approaches, the right combination of RBSs and promoters has a significant impact on the required performance. The process of constructing and selecting optimal promoter‐RBS‐CDS combinations can be very complicated and often requires high‐throughput strategies in which random combinations need to be constructed and screened. To simplify and standardize the process of introducing short functional DNA sequences into any BioBrick, we engineered two primers that introduce to the parts such functional sequences within a single‐step PCR amplification. These two long standard primers are (i) the CDS amplification primer (OP1‐FRBS) and (ii) the promoter‐backbone amplification primer (Evpromoter). In the first case, an RBS is introduced to the amplified CDS BioBrick, while in the second case, a short constitutive promoter is introduced to any SEVA 3.1 or BioBrick backbone (Fig. 5A). As shown in Figure 5A, the OP1‐FRBS primer consists of one standard and one modular sequence region (yellow) in which any RBS of choice is introduced. The primer anneals on the standard MCS 2.0 sequence and carries a loop with the RBS, flanked by a BsaI recognition site followed by standard sticky ends. The Evpromoter primer anneals on the standard non‐CDS BioBrick prefix sequence while carrying the promoter sequence at its 5’‐end along with the required sticky ends and a BsaI recognition site.
Fig. 5.
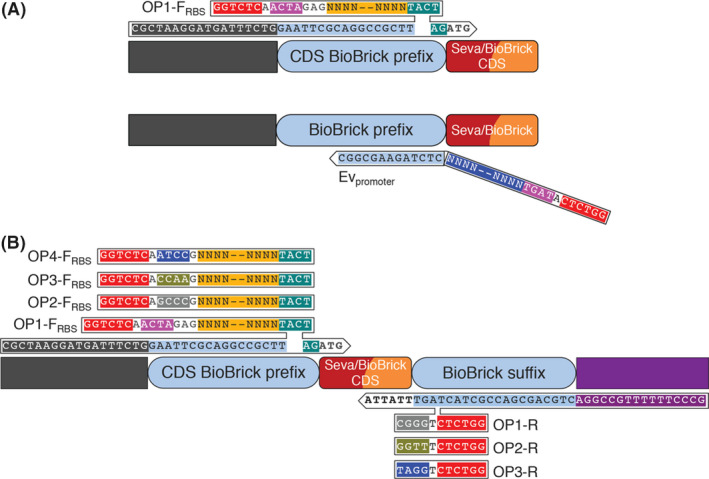
Structure of the OP1‐FRBS and Evpromoter primers.
A. The OP1‐FRBS primer consists of one standard and one modular sequence region; Standard sequence region: annealing sequence [black, light blue, green (AG), start codon (ATG)], Bsal recognition site (red), sticky ends (pink), RBS scar [green (TACTAG)]. Modular sequence region: RBS sequence (yellow). The Evpromoter primer also consists of one standard and one modular sequence region; Standard sequence region: annealing sequence (light blue), Bsal recognition site (red), compatible with the OP1‐FRBS sticky ends (pink). Modular sequence region: promoter sequence (dark blue).
B. Graphical representation of four OP primer sets: all the forward and reverse primers share the same annealing sequence, which is represented by the OP1‐FRBS and OP1‐R primers, respectively. The OP‐FRBS primers use the ATG of the stored CDSs for annealing to the 3’‐end while the OP‐R utilizes the standard double stop codon (TAATAA) used in all BioBrick CDSs. Unique sticky ends are introduced to each primer enabling the directional assembly of each amplified CDS. The sticky ends of the OP1‐FRBS and Pp (used to amplify the last gene in the operon) are compatible with the sticky ends of the backbone linearization primers, Evpromoter/Pv.
Introduction of RBSs and short promoters
Using the standard primers described above, we amplified several chromoproteins and fluorescent proteins registered as CDSs in the iGEM registry and constructed functional TUs in the pSEVAb23 vector. All reporter proteins were amplified using the OP1‐FBBa_B0034 primer, for introducing the RBS BBa_B0034 and the reverse insert primer Pp. The backbone of choice, pSEVA23b, was linearized using the forward Pv primer and the reverse EvBBa_J23100 primer which introduces the BBa_J23100 promoter allowing directional cloning of any RBS‐CDS module. Following the PCRs, all amplified CDSs were cloned into vector pSEVAb23 BBa_J23100 through a one‐step SevaBrick Assembly resulting in the TU structure shown in Figure 6A. In this way, we constructed expression vectors for the reporter proteins mCherry (BBa_J06504), gfasPurple (BBa_K1033919), aeBlue (BBa_K1864401), mOrange (BBa_E2050), amil‐GFP (BBa_K592010), amilCP (BBa_K592009), mRFP (BBa_E1010) and amiLime (BBa_K1033916) which were later transformed in P. putida. All reporter proteins were successfully expressed and maturated in P. putida giving colour in < 24 h in rich LB medium (Fig. 6D). Consequently, the SevaBrick Assembly allowed the construction of eight expression vectors for both E. coli and P. putida using only two sets of standard primers and a single ligation step. In addition, although we did not conduct additional experiments to further evaluate these reporter proteins, Liljeruhm et al. (2018) and Shaner et al. (2004), demonstrated their potential as quantitative reporters of gene expression and promoter strength in E. coli. It was shown that most of them have a comparable maturation time with the widely used fluorescent protein, mRFP.
Fig. 6.
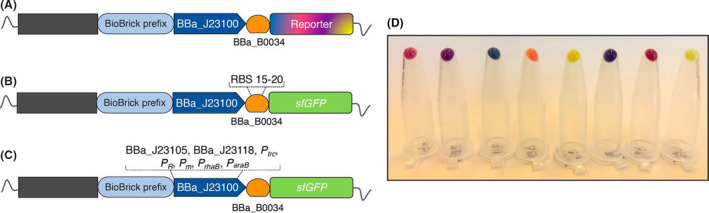
Graphical representation of constructed TUs using SevaBrick Assembly and the standard primers for introduction of short functional genetic elements.
A. Each reporter CDS, amplified from the iGEM registry, was assembled with the constitutive promoter BBa_J23100 and the RBS BBa_B0034.
B. sfGFP was assembled with BBa_J23105 promoter and either the BBa_B0034 RBS or six in silico predicted RBSs (RBS 15–20).
C. sfGFP was assembled with either five short constitutive promoters or three inducible expression systems and the RBS BBa_B0034.
D. P. putida transformed with eight different reporter expression vectors. From right to left: mCherry, gfasPurple, aeBlue, mOrange, amilGFP, amilCP, mRFP and amajLime.
Modular exchange and screening of functional genetic elements
Next, to demonstrate the ability of the OP1‐FRBS and Evpromoter primers to easily exchange genetic functional elements, we constructed several plasmids expressing the sfGFP under the control of various RBSs or promoters. At first, we predicted RBS sequences suitable for the translation of the sfGFP protein in P. putida by using the freely available tool ‘Salis RBS calculator’ (Salis, 2011). Six RBSs with different predicted strengths, from high to low (RBS.15 > RBS.20), were selected and variants of the OP1‐FRBS primer were synthesized. The standard sequence region of the OP1‐FRBS primer was maintained and only the modular RBS sequence region was exchanged by each individual predicted RBS. Using as template a pSB1C3 plasmid carrying the sfGFP as CDS BioBrick, we amplified the sfGFP with all OP1‐FRBS.15–20 primers separately, resulting in multiple RBS‐sfGFP amplicons. In parallel, backbone pSEVAb23 was linearized with EvBBa_J23105 and Pv primers. In a single SevaBrick Assembly step, we constructed sfGFP expression vectors under the control of the BBa_J23105 promoter and six in silico predicted RBSs. RBS BBa_B0034 was used as an internal reference for the normalization of the fluorescence levels. In vivo fluorescence assays in P. putida showed correlation between the predicted RBSs strength and the fluorescence levels. The predicted as strongest RBS.15 provided the highest fluorescence levels at 8 h, similar to the internal standard RBS BBa_B0034, while the predicted as the weakest RBS.20 was considerably the least strong (Fig. 7D,G). Moreover, to highlight the interoperability of the SEVA 3.1 vectors, all constructs were subsequently tested in E. coli (Fig. 7A) where, unlike P. putida, RBS BBa_B0034 exhibited ≈ 2.5‐folds higher fluorescence levels than RBS.15. Last, we sought to randomize the RBS sequence and construct a pooled library of sfGFP expression vectors. To do this, primer OP1‐FRBS.15 was synthesized with two random nucleotides within the RBS sequence (‐TTATAAGGNNG‐) resulting in a new CDS amplification primer (OP1‐FRBS.NN). Following the PCR, the random RBS‐sfGFP amplicon was assembled with pSEVAb23BBa_J23105 and transformed into E. coli (Fig. 7G). Ten obviously high fluorescent colonies were selected and sent for sequencing. Out of the ten sequenced colonies, we were able to recover five different RBS sequences (Fig. S6) highlighting the feature of the SevaBrick Assembly primers to construct random RBS libraries within a PCR step.
Fig. 7.
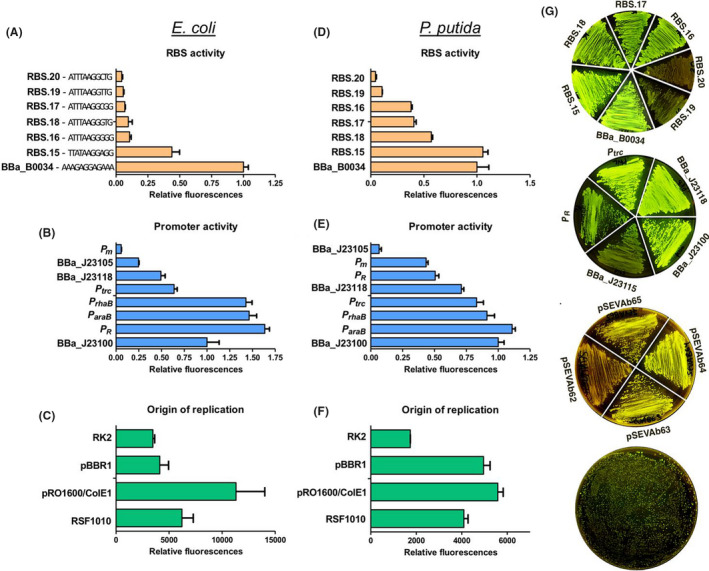
Part characterization in E. coli and P. putida. (A, D) A selected range of predicted RBS, (B, E) constitutive and inducible promoters, (C, F) origins of replication. The BBa_B0034 and BBa_J23100 used for normalization of promoter and RBS levels respectively. For the RBS screening, sfGFP was placed under the control of the BBa_J23105 promoter. For the promoter screening sfGFP was placed under the control of the BBa_B0034 RBS. For the origins of replication screening the sfGFP was placed under the control of the BBa_J23105 promoter and the predicted RBS.20. (G) Visual appearance of sfGFP production on LB plates. From top to bottom RBSs (P. putida), constitutive promoters (P. putida), origins of replication (P. putida) and random RBSs (E. coli).
A similar strategy was followed for the characterization of constitutive and inducible promoters. In this case, the vector pSEVAb23 was linearized using variants of the Evpromoter primer in which the modular promoter sequence region was exchanged by three constitutive promoters of the iGEM Anderson collection as well as the well‐characterized constitutive promoters Ptrc (without the lac operator) and PR (without the cI repressor, BBa_R0051). The sfGFP was amplified with OP1‐FBBa_B0034/Pp and later cloned into pSEVAb23promoter. Moreover, three inducible expression systems [Xyls/Pm, RhaS/PrhaB, AraC/ParaB (Guzman et al., 1995)] were cloned upstream the RBS‐sfGFP module. All three inducible expression systems, previously converged to pSB1C3 repository vector, were amplified using the Ep/Sp primers and cloned directly with the RBS‐sfGFP amplicon into a pSEVAb23, pre‐linearized with Ev/Pv primers. All expression vectors were then characterized in both E. coli and P. putida (Fig. 7B,E,G). As shown in Figure 7B,E, all Anderson promoters showed similar expression patterns with the previously reported levels starting from the BBa_J23100 as the strongest to BBa_J23105 as the weakest (Kelly et al., 2009). The highest expression levels were achieved by the PR promoter for E. coli and the AraC/ParaB expression system for P. putida. However, in our hands and in contrast to previously published work in P. putida, the Xyls/Pm expression system showed less than the half expression level of the AraC/ParaB (Calero et al., 2016).
Finally, the applicability of the Evpromoter primer to easily amplify and introduce short promoters to any SEVA 3.1 backbone was showcased by assembling the sfGFP into four different expression vectors with either the RK2, pBBR1, pRO1600/ColE1 or RFS1010 origins of replication (Silva‐Rocha et al, 2013). Vectors pSEVAb62, pSEVAb63, pSEVAb64 and pSEVAb65 were linearized using EvBBa_J23105/Pv and assembled with the sfGFP harbouring the RBS.20. All vectors were transformed in P. putida and E. coli, and the sfGFP fluorescence levels were measured. As expected, vector pSEVAb62 (low copy number) showed the least sfGFP fluorescence, while the highest fluorescence levels were achieved from pSEVAb64 for both E. coli and P. putida (Fig. 7C,F,G). The observed fluorescence levels in E. coli are similar to those presented by Jahn and colleagues (2016).
Therefore, the modular design of the OP1‐FRBS and Evpromoter primers allows the user to easily introduce and exchange RBS or promoter sequences, respectively, within one standard PCR step and with reasonably minimal design.
Extended SevaBrick Assembly for operon construction
The basic version of the SevaBrick Assembly, as explained above, is a quick and easy method of assembling parts, however its main limitation is the small number of fragments that can be simultaneously assembled; two parts at a time. To further extend the capabilities of SevaBrick Assembly to construct more complex genetic designs, such as multigene operons, we propose another set of standard primers based on the structure of the OP1‐FRBS primer (Fig. 5B). Using the sequence of the OP1‐FRBS primer as the basic template for the forward primer, we designed four primer sets (OP1 to OP4) that allow amplification of any BioBrick CDS while introducing RBSs and compatible sticky ends for sequential assembly of up to four CDSs. To maximize assembly efficiency, the sticky ends for this process were selected from a high‐fidelity library proposed by New England Biolabs (NEB) (Potapov et al., 2018). These unique sticky ends allow directional cloning of each particular amplicon into a final operon through SevaBrick Assembly. For instance, the OP2‐FRBS primer introduces sticky ends compatible with the sticky ends of the OP1‐R primer, while the OP2‐R introduces sticky ends compatible with the OP3‐FRBS. In this way, the position of each particular gene can be easily determined using the corresponding OP primer set (OP1/position 1, OP2/position 2 etc). The last gene in the operon has to be amplified with the OP‐Fwrn (being n the number of the genes in the operon) and the basic assembly primer Pp. The sequence between the RBS and the ATG was kept constant (TACTAG) for standardization purposes since this sequence has a significant impact on the translation initiation. Based on this specific design, constitutive promoters can be introduced to the operon via PCR amplification of the backbone, as it is described above. In the case of inducible operons, the inducible expression system (stored in repository vector) and the backbone of interest have to be amplified using the primers Ep/Sp and the Ev/Pv respectively. The Evpromoter and Sp primers carry identical sticky ends allowing the construction of either constitutive or inducible operon by using the same RBS‐CDS amplicons.
To validate the functionality of the OP primers and SevaBrick Assembly to construct multigene operons, we assembled the protoviolaceinic acid biosynthesis operon by introducing the RBS of choice to each particular gene. Protoviolaceinic is an intermediate of the violacein biosynthesis pathway, originated from the soil bacterium Chromobacterium violaceum (August et al., 2000). Violacein and its precursors have notable antimicrobial activity, while the genes involved in its biosynthesis are arranged in an operon consisting of vioA, vioB, vioC, vioD and vioE. L‐tryptophan is converted to protoviolaceinic acid via four steps catalysed by the VioA, VioB, VioD, VioE. Protoviolaceinic acid is then converted to either violacein via VioC or is being oxidized to proviolacein, a black pigment. Therefore, we reconstructed the pathway towards proviolacein and assessed the efficiency of the extended SevaBrick Assembly by counting the black versus the white colonies. Initially, plasmid BBa_K598020 was used as template to amplify and converge each particular gene into the repository vector pSB1C3, following the convergence strategy described above and the primers listed in Table S4. As all CDSs were stored in the repository vectors, a second PCR step was performed to introduce the RBS BBa_B0034 using the OP primers. The order of the CDSs into the operon was determined as vioA‐vioB‐vioD‐vioE by using the primer sets OP1‐FBBa_B0034/OP1‐R, OP2‐FBBa_B0034/OP2‐R, OP3‐FBBa_B0034/OP3‐R and OP4‐FBBa_B0034/Pp respectively. Finally, all RBS‐CDS amplicons were assembled into a linear pSEVAb23BBa_J23105 vector resulting in the operon structure shown in Fig. 8A. The assembly was performed three times and the efficiency was calculated by measuring the ratio of black to white colonies. Appearing black colonies (Fig. 8B) were to 60–65% of the total colonies, indicating the high efficiency of the SevaBrick Assembly to assembly up to five parts in a single ligation step. Several black colonies were selected for plasmid sequencing, which resulted in the correct operon sequence.
Fig. 8.
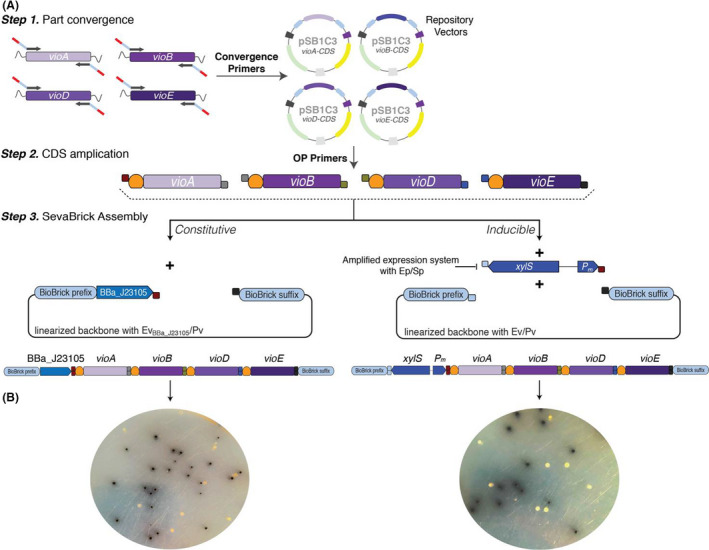
Overview of the vioABDE operon construction via SevaBrick Assembly.
A. Step 1: Convergence of each CDS to the repository vector pSB1C3. Each CDS was amplified with unique convergence primers, following the strategy described in the part convergence paragraph, and later cloned into pSB1C3. Step 2: PCR amplification of each particular CDS using the OP primers (OP1‐OP4) to introduce the RBS BBa_B0034 and unique sticky ends for directional cloning of all CDS in the order of vioA‐vioB‐vioD‐vioE. Step 3: All RBS‐CDS amplicons were assembled with either (i) a linear pSEVAb23BBa_J23105 vector resulting in the constitutive vioABDE operon or (ii) a pre‐linearized with Ev/Pv pSEVAb23 vector and the pre‐amplified with Ep/Sp XylS/Pm expression system, resulting in the inducible vioABDE operon.
B. Cells transformed with the constitutive vioABDE operon (left) and the inducible XylS/Pm‐vioABDE operon (right) form black colonies due to the production of proviolacein.
A notable characteristic of the SevaBrick Assembly is the high modularity and reusability of the amplicons to be assembled. To highlight this feature and additionally evaluate the efficiency of the method to assemble six parts at once, we constructed an inducible version of the protoviolocein operon. To do this, the [vioABDE + RBS] amplicons were assembled with the XylS/Pm inducible expression system into vector pSEVAb23. This time, vector pSEVAb23 was pre‐linearized with the standard Ev/Pv primers, while the XylS/Pm cassette was amplified with Ep/Sp. Both parts were already prepared and utilized in previous applications. All parts were assembled via SevaBrick Assembly and the transformed E. coli cells were plated on LB agar with the XylS/Pm inducer (3‐methylbenzoate). Based on the ratio of the black to white colonies, as shown in Figure 8B, the efficiency of the 6‐part assembly calculated to 53%. The overall procedure of the operon construction is summarized in Figure 8A. Due to the modular design of the SevaBrick Assembly primers which allows the use of multiple sets of ligation sites and the numerous high‐fidelity combinations provided by NEB, the number of the OPRBS primer sets could be further increased. In addition, although in this work we used the standard BBa_B0034 RBS for all four genes, the OP primer design allows the use of different or even random RBSs as previously described.
Conclusions
The SEVA 3.1 platform was developed to provide a modular and versatile Golden Gate‐based assembly method in combination with the part abundance of BioBricks and the flexibility of SEVA vectors. To this end, the SEVA 3.1 platform offers a newly engineered vector standard consisting of the SEVA backbone (AR, Ori, terminators) and the standard BioBrick cloning site. Any SEVA 3.1 vector is compatible with any available BioBrick part either via the BioBrick 3A Assembly or the proposed SevaBrick Assembly. The SevaBrick Assembly comprises a set of standard PCR primers which anneal to any SEVA 3.1/BioBrick compatible vector and enables Golden Gate assembly of single or multiple parts with minimal cloning effort and design. This was underscored by the fact that only fourteen primer sets had to be individually designed per part (convergence primers) for all 52 plasmids constructed in this study, while all assemblies performed using exclusively one restriction enzyme, BsaI. The capabilities of the SEVA 3.1 platform, were demonstrated by constructing several genetic constructs of various complexities. Using the standard primer sets and protocols (same for all applications) of the SevaBrick Assembly, we successfully cloned and expressed single and multigene TUs both in P. putida and E. coli. Additionally, to highlight the modularity of the SevaBrick primers, we cloned the sfGFP under the control of multiple genetic functional elements such as promoters, RBSs and origins of replication. Thus, the newly engineered SEVA 3.1 vectors, along with its proposed assembly, extend and simplify the applicability of the SEVA vectors and BioBricks, while maintaining their unique features and standards.
Experimental procedures
Bacterial strains and growth conditions
E. coli and P. putida were routinely grown in lysogeny broth (LB) medium with kanamycin (50 μg ml−1), chloramphenicol (35 μg ml−1) or gentamycin (10 μg ml−1) as needed at 37 °C and 30°C respectively. Solid media additionally contained 1.5% (w/v) agar. 1 mM of 3‐methylbenzoate, 3 mM rhamnose and 10 mM arabinose were used as inducers of the Pm, PrhaB and ParaB promoters respectively. M9 minimal medium (6 g l−1 Na2HPO4, 3 g l l−1 KH2PO4, 1.4 g l−1 (NH4)2SO4, 0.5 g l−1 NaCl, 0.2 g l l−1 MgSO4, 2.5 ml l−1 trace elements solution) (Nikel and de Lorenzo, 2014) supplemented either with 2% of glycerol (E. coli) or 2% glucose (P. putida) was used for the fluorescence assays.
Transformation
E. coli DH5α chemical competent cells were prepared and transformed as described by Green and Rogers (2013). For transforming P. putida, 10 ml overnight LB culture was washed three times with 1 ml of 300 mM sucrose (filter‐sterilized) and resuspended in 400 µl of 300 mM sucrose. Later, 100 ng plasmid was electroporated into 100 µl cell suspension aliquots with a voltage of 2.5 kV, 25 μF capacitance and 200 Ω resistance.
DNA manipulation
Plasmids used in this study are listed in Table S5. General cloning procedures, such as endonuclease restriction digest, ligation and PCR, were performed with enzymes and buffers from New England Biolabs® (NEB; Ipswich, MA, USA) or ThermoScientifc™ (Waltham, MA, USA) according to the respective protocols. Q‐5 hot start® polymerase was used for PCR if the resulting fragment was further used, otherwise for colony PCR, Phire® was the polymerase of choice. PCR purification was performed with the Macherey‐Nagel purification kit. All primers were synthesized by IDT. Primer sequences are provided in the Table S4.
Biphasic PCR protocol with standard SevaBrick Assembly primers
For the preparation of the parts to be assembled and due to the complexity and length of the engineered primers, the standard PCR protocol had to be modified to a biphasic one: 98 °C, 2 min; Phase 1: (98 °C, 20 s; 50 °C, 20 s; 72 °C, part dependent) × 10; Phase 2: (98 °C, 20 s; 76 °C, 20 s; 72 °C, part dependent) × 25; 72 °C, 2 min. The final product is obtained using an initial amplification at low annealing temperature (50 °C) for 10 cycles and a subsequent amplification at an annealing temperature of 76 °C (higher than the standard annealing temperature range) for another 25 cycles. Each primer consists of a short 3’‐end annealing sequence, a modular sequence in a non‐annealing loop and optionally another standardized 5’‐anchor sequence. The short 3’‐end sequence requires a low annealing temperature (Phase 1) to be correctly hybridized with the template, while the anchor sequence, which optimizes the annealing fidelity elevates the Tm. High annealing temperature leads to negative results, while the low temperature cycling provides PCR amplified DNA but at a low yield. This low DNA concentration is due to the structural complexity of the long primers at this temperature. Running Phase 1 for 10 cycles provides enough PCR product which carries the additional sequences to be used as the template for Phase 2. At Phase 2, 76 °C is used for 25 additional cycles providing enough DNA to be used downstream in the process. 25 µl reactions followed by agarose gel purification are recommended for optimal results.
SevaBrick Assembly
All DNA assemblies were performed via Golden Gate in 8 µl reaction comprised of 2 µl of the standard SevaBrick Assembly mix [12 µl BsaI, 10 µl T4 ligase (NEB), 18 µl T4 ligase buffer (NEB), 1 µl DpnI (NEB), 1 µl of 20 mg ml−1 BSA (NEB)] and 1 µl of entry parts and backbone with final concentration of 1nM. An Excel file was programmed to automatically calculate DNA amounts and dilutions (Appendix S1). The SevaBrick Assembly protocol follows the thermocycling condition 37 °C, 20 min; (16 °C, 4 min; 37 °C, 3 min) × 30; 50 °C, 10 min; 80 °C, 10 min. 8 μl of the assembly reaction mix was transformed into 50 μl of chemically competent E. coli DH5α cells using heat shock transformation. After recovery in 500 μl of SOC medium, cells were plated onto LB antibiotic plates and grown at 37 °C overnight.
sfGFP fluorescence and growth measurements
All the genetic functional elements such as RBS, promoter and origin of replication parts were characterized in P. putida and E. coli. Single colonies were picked in triplicate and grown overnight at 30 °C (P. putida) or 37 °C (E. coli) in 10 ml LB with antibiotics. Cell density was measured with IMPLEN OD600 photometer at 600 nm and cells were diluted into 200 μl of M9 medium with antibiotics, and inducers where needed, in a 96‐well Greiner plate to a starting OD600 of 0.1. The plate was incubated at 30 °C or 37 °C for 12 h in a Synergy plate reader (Biotek). OD600 and sfGFP measurements were recorded every 10 min. Fluorescence was determined with the following settings: Ex. 467 nm, Em. 508 nm and the levels were corrected with the fluorescence signal of a blank sample. Relative sfGFP production was quantified after 8 h for P. putida or 5 h for E. coli from an average of triplicate data.
Funding information
Funding was provided by European Union’s Horizon2020 Research and Innovation Programme under grant agreement Nos. 635536 (EmPowerPutida) and 730976 (IBISBA) to V.A.P.M.d.S.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Table S1. List of available pSEVAb vectors.
Table S2. Standard flanks of the Convergence, OP1‐FRBS and Evpromoter primers.
Table S3. List of converged parts (BioBricks).
Table S4. List of oligonucleotides used in this study.
Table S5. Plasmids used and constructed in this study.
Fig. S1. PCR amplified SEVA modules.
Fig. S2. Graphical representation of XylS/Pm domestication primers.
Fig. S3. Graphical representation of SevaBrick primers attached on MCS 2.0
Fig. S4. DNA sequencing summary of predicted RBSs + promoter BBa_J23105 + sfGFP variants from Figure 7.
Fig. S6. DNA sequencing summary of random RBSs + sfGFP variants from the last plate of Figure 7G.
Fig. S7. DNA sequencing summary of constitutive promoters + RBS BBa_0034 + sfGFP variants from Fig. 7.
Appendix S1. Calculation of DNA parts concentrations for SevaBrick assembly.
Acknowledgements
We thank Dr. Esteban Martínez‐García (CNB‐CSIC, Madrid, Spain) and Dr. Tomas Aparicio (CNB‐CSIC, Madrid, Spain) for sharing research materials and for fruitful discussions. We also thank the Wageningen iGEM teams 2017, 2018 for providing us the iGEM distribution kit plates. Funding was provided by European Union’s Horizon2020 Research and Innovation Programme under grant agreement Nos. 635536 (EmPowerPutida) and 730976 (IBISBA) to V.A.P.M.d.S.
Microbial Biotechnology (2020) 13(6), 1793–1806
References
- Adams, B.L. (2016) The next generation of synthetic biology chassis: moving synthetic biology from the laboratory to the field. ACS Synth Biol 5: 1328–1330. [DOI] [PubMed] [Google Scholar]
- Andreou, A.I. , and Nakayama, N. (2018) Mobius Assembly: A versatile Golden‐Gate framework towards universal DNA assembly. PLoS One 13: e0189892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianantoandro, E. , Basu, S. , Karig, D.K. , and Weiss, R. (2006) Synthetic biology: New engineering rules for an emerging discipline. Mol Syst Biol 2: 2006.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- August, P. R. , Grossman, T. H. , Minor, C. , Draper, M. P. , MacNeil, I. A. , Pemberton, J. M. , et al (2000) Sequence analysis and functional characterization of the violacein biosynthetic pathway from Chromobacterium violaceum . J Mol Microbiol Biotechnol 2: 513–519. [PubMed] [Google Scholar]
- Bitinaite, J. , Rubino, M. , Varma, K. , Schildkraut, I. , Vaisvila, R. , and Vaiskunaite, R. (2007) USER friendly DNA engineering and cloning method by uracil excision. Nucleic Acids Res 35: 1992–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calero, P. , and Nikel, P.I. (2019) Chasing bacterial chassis for metabolic engineering: A perspective review from classical to non‐traditional microorganisms. Microb Biotechnol 12: 98–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calero, P. , Jensen, S.I. , and Nielsen, A.T. (2016) Broad‐host‐range ProUSER vectors enable fast characterization of inducible promoters and optimization of p‐coumaric acid production in Pseudomonas putida KT2440. ACS Synth Biol 15: 741–753. [DOI] [PubMed] [Google Scholar]
- Chandran, S. , and Shapland, E. (2017) Efficient assembly of DNA using yeast homologous recombination (YHR). Methods Mol Biol 1472: 187–192. [DOI] [PubMed] [Google Scholar]
- Egan, S.M. , and Schleif, R.F. (1993) A regulatory cascade in the induction of rhaBAD. J Mol Biol 234: 87–98. [DOI] [PubMed] [Google Scholar]
- Ellis, T. , Adie, T. , and Baldwin, G.S. (2011) DNA assembly for synthetic biology: From parts to pathways and beyond. Integr Biol 3: 109–118. [DOI] [PubMed] [Google Scholar]
- Engler, C. , Kandzia, R. , and Marillonnet, S. (2008) A one pot, one step, precision cloning method with high throughput capability. PLoS One 3: e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdzicki, M. , Clancy, K.P. , Oberortner, E. , Pocock, M. , Quinn, J.Y. , Rodriguez, C.A , et al (2014) The Synthetic Biology Open Language (SBOL) provides a community standard for communicating designs in synthetic biology. Nat Biotechnol 32: 545–550. [DOI] [PubMed] [Google Scholar]
- Gardner, T.S. , Cantor, C.R. , and Collins, J.J. (2000) Construction of a genetic toggle switch in Escherichia coli . Nature 403: 339–342. [DOI] [PubMed] [Google Scholar]
- Gibson, D.G. , Gwynedd A.B., Axelrod, K.C. , Zaveri, J. , Algire, M.A. , Moodie, M. , et al (2008) One‐step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome. Proc Natl Acad Sci USA 105: 20404–20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, D.G. , Young, L. , Chuang, R.Y. , Venter, J.C. , Hutchison, C.A. , et al (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. [DOI] [PubMed] [Google Scholar]
- Green, R. , and Rogers, E.J. (2013) Transformation of chemically competent E. coli . Methods Enzymol 529: 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman, L.M. , Belin, D. , Carson, M.J. , and Beckwith, J. (1995) Tight regulation, modulation, and high‐level expression by vectors containing the arabinose P(BAD) promoter. J Bacteriol 177: 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi, R. , Krummel, B. , and Saiki, R.K. (1988) A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res 16: 7351–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, R.A. , and Ellington, A.D. (2017) Synthetic DNA synthesis and assembly: putting the synthetic in synthetic biology. Cold Spring Harb Perspect Biol 9: a023812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itaya, M. , Sato, M. , Hasegawa, M. , Kono, N. , Tomita, M. , and Kaneko, S. (2018) Far rapid synthesis of giant DNA in the Bacillus subtilis genome by a conjugation transfer system. Sci Rep 8: 8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn, M. , Vorpahl, C. , Hübschmann, T. , Harms, H. and Müller, S. . (2016) Copy number variability of expression plasmids determined by cell sorting and Droplet Digital PCR. Microb Cell Fact 15: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, J.R. , Rubin, A.J. , Davis, J.H. , Ajo‐Franklin, C.M. , Cumbers, J. , Czar, M.J. , et al (2009) Measuring the activity of BioBrick promoters using an in vivo reference standard. J Biol Eng 3: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.K. , Yoon, P.K. , Kim, S.J. , Woo, S.G. , Rha, E. , Lee, H. , et al (2020) CRISPR interference‐mediated gene regulation in Pseudomonas putida KT2440. Microb Biotechnol 1: 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuepper, J. , Dickler, J. , Biggel, M. , Behnken, S. , Jäger, G. , Wierckx, N. , and Blank, L.M. (2015) Metabolic engineering of Pseudomonas putida KT2440 to produce anthranilate from glucose. Front Microbiol 6: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljeruhm, J. , Funk, S.K. , Tietscher, S. , Edlund, A.D. , Jamal, S. , Wistrand‐Yuen, P. , et al (2018) Engineering a palette of eukaryotic chromoproteins for bacterial synthetic biology. J Biol Eng 12: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, S. , Tang, N. , and Tian, J. (2012) DNA synthesis, assembly and applications in synthetic biology. Curr Opin Chem Biol 16: 260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques, S. , Manzanera, M. , Gonzalez‐Perez, M.‐M. , Gallegos, M.‐T. , and Ramos, J.L. (1999) The XylS‐dependent Pm promoter is transcribed in vivo by RNA polymerase with sigma32 or sigma38 depending on the growth phase. Mol Microbiol 31: 1105–1113. [DOI] [PubMed] [Google Scholar]
- Martínez‐García, E. , Aparicio, T. , Goñi‐Moreno, A. , Fraile, S. , and de Lorenzo, V. (2015) SEVA 2.0: an update of the Standard European Vector Architecture for de‐/re‐construction of bacterial functionalities. Nucleic Acids Res 43: D1183–D1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐García, E. , Goñi‐Moreno, A. , Bartley, B. , McLaughlin, J. , Sánchez‐Sampedro, L. , Pascual del Pozo, H. , et al. (2020) SEVA 3.0: an update of the standard European vector architecture for enabling portability of genetic constructs among diverse bacterial hosts. Nucleic Acids Res 48: D1164–D1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel, P.I. , and de Lorenzo, V. (2014) Robustness of Pseudomonas putida KT2440 as a host for ethanol biosynthesis. N Biotechnol 31: 562–571. [DOI] [PubMed] [Google Scholar]
- Perkel, J.M. (2019) The race for enzymatic DNA synthesis heats up. Nature 566: 565. [DOI] [PubMed] [Google Scholar]
- Poblete‐Castro, I. , Becker, J. , Dohnt, K. , dos Santos, V. M. , and Wittmann, C. (2012) Industrial biotechnology of Pseudomonas putida and related species. Appl Microbiol Biotechnol 93: 2279–2290. [DOI] [PubMed] [Google Scholar]
- Pollak, B. , Cerda, A. , Delmans, M. , Álamos, S. , Moyano, T. , West, A. (2019) Loop assembly: a simple and open system for recursive fabrication of DNA circuits. New Phytol 222: 628–640. [DOI] [PubMed] [Google Scholar]
- Potapov, V. , Ong, J. L. , Kucera, R. B. , Langhorst, B. W. , Bilotti, K. , Pryor, J. M. et al. (2018) Comprehensive profiling of four base overhang ligation fidelity by T4 DNA ligase and application to DNA assembly. ACS Synth Biol 11: 2665–2674. [DOI] [PubMed] [Google Scholar]
- Purnick, P.E.M. , and Weiss, R. (2009) The second wave of synthetic biology: from modules to systems. Nat Rev Mol Cell Biol 10: 410–422. [DOI] [PubMed] [Google Scholar]
- Salis, H.M. (2011) The ribosome binding site calculator. Methods Enzymol 498: 19–42. [DOI] [PubMed] [Google Scholar]
- Sarrion‐Perdigones, A. , Falconi, E. E. , Zandalinas, S. I. , Juárez, P. , Fernández‐del‐Carmen, A. , Granell, A. , and Orzaez, D. (2011) GoldenBraid: an iterative cloning system for standardized assembly of reusable genetic modules. PLoS One 6: e21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrion‐Perdigones, A. , Vazquez‐Vilar, M. , Palací, J. , Castelijns, B. , Forment, J. , Ziarsolo, P. , Blanca, J. et al (2013) GoldenBraid 2.0: a comprehensive DNA assembly framework for plant synthetic biology. Plant Physiol 162, 1618–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner, N.C. , Campbell, R.E. , Steinbach, P.A. , Giepmans, B.N. , Palmer, A.E. , and Tsien, R.Y . (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22: 1567–1572. [DOI] [PubMed] [Google Scholar]
- Sharan, S.K. , Thomason, L.C. , Kuznetsov, S.G. , and Court, D.L. (2009) Recombineering: a homologous recombination‐based method of genetic engineering. Nat Protoc 4: 206–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty, R. , Lizarazo, M. , Rettberg, R. , and Knight, T.F. (2011) Assembly of BioBrick standard biological parts using three antibiotic assembly. Methods Enzymol. 498: 311–326. [DOI] [PubMed] [Google Scholar]
- Silva‐Rocha, R. , Martínez‐García, E. , Calles, B. , Chavarría, M. , Arce‐Rodríguez, A. , de Las Heras, A. (2013) The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res 41: D666–D675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuge, T. , Matsui, K. , and Itaya, M. (2003) One step assembly of multiple DNA fragments with a designed order and orientation in Bacillus subtilis plasmid. Nucleic Acids Res 31: e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way, J.C. , Collins, J.J. , Keasling, J.D. , and Silver, P.A. (2014) Integrating biological redesign: where synthetic biology came from and where it needs to go. Cell 157: 151–161. [DOI] [PubMed] [Google Scholar]
- Weber, E. , Engler, C. , Gruetzner, R. , Werner, S. , and Marillonnet, S. (2011) A modular cloning system for standardized assembly of multigene constructs. PLoS One 6: e16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of available pSEVAb vectors.
Table S2. Standard flanks of the Convergence, OP1‐FRBS and Evpromoter primers.
Table S3. List of converged parts (BioBricks).
Table S4. List of oligonucleotides used in this study.
Table S5. Plasmids used and constructed in this study.
Fig. S1. PCR amplified SEVA modules.
Fig. S2. Graphical representation of XylS/Pm domestication primers.
Fig. S3. Graphical representation of SevaBrick primers attached on MCS 2.0
Fig. S4. DNA sequencing summary of predicted RBSs + promoter BBa_J23105 + sfGFP variants from Figure 7.
Fig. S6. DNA sequencing summary of random RBSs + sfGFP variants from the last plate of Figure 7G.
Fig. S7. DNA sequencing summary of constitutive promoters + RBS BBa_0034 + sfGFP variants from Fig. 7.
Appendix S1. Calculation of DNA parts concentrations for SevaBrick assembly.