

Abstract
Epithelial-to-mesenchymal transition (EMT) in nasal epithelial cells is involved with tissue remodeling of nasal polyps. The present study investigated the molecular mechanisms through which miR-155-5p regulated EMT in chronic rhinosinusitis (CRS). Patients were divided into the following groups: CRSsNP, CRS without nasal polyposis group, CRSwNP, CRS with nasal polyposis and controls. The expression of transforming growth factor (TGF)-β1, EMT markers, sirtuin 1 (SIRT1) and miR-155-5p were determined by western blotting and reverse transcription-quantitative PCR. Cell morphology following TGF-β1 treatment in the presence of miR-155-5p inhibitors or controls was observed under a microscope. Target genes and potential binding sites between miR-155-5p and SIRT1 were predicted by TargetScan and confirmed using dual-luciferase reporter assay. In patients with CRS, the expression levels of E-cadherin were downregulated and the expression levels of TGF-β1, mesenchymal markers and miR-155-5p were upregulated. Additionally, these changes in expression levels were reduced or increased to a greater extent in the CRSwNP group compared with the CRSsNP group. Furthermore, TGF-β1 expression promoted EMT in human nasal epithelial cells (HNEpCs) and upregulated miR-155-5p expression. These effects were reversed by miR-155-5p inhibitors. Additionally, SIRT1 was predicted as a target gene of miR-155-5p. Downregulation of miR-155-5p upregulated epithelial marker expression and downregulated mesenchymal marker expression by regulating SIRT1. Therefore, the downregulation of miR-155-5p inhibited EMT in HNEpCs by targeting SIRT1.
Keywords: microR-155-5p, chronic rhinosinusitis, epithelial-to-mesenchymal transition inhibition, sirtuin 1
Introduction
Chronic rhinosinusitis (CRS) is a multifunctional inflammatory disease of the nasal cavity and sinus mucosa (1) that often lasts >12 weeks (2). CRS is often accompanied by chronic sinus inflammation (3). Some forms of the disease are driven by allergy, often in association with asthma (4). According to nasal endoscopy and whether nasal polyps (NPs) are present, CRS can be divided into two heterogeneous phenotypes: i) CRS with nasal polyposis (CRSwNP); and ii) CRS without nasal polyposis (CRSsNP) (5). CRSsNP is mainly characterized by nasal obstruction, anosmia, drainage, headaches and facial pain or pressure (6). CRSwNP is more closely associated with obstruction of the nasal cavity and anosmia (7). Clinically, CRSwNP is frequently resistant to medical therapy (8). Recently, it has been demonstrated that epithelial-to-mesenchymal transition (EMT) and its effects on nasal epithelial cells may result in the tissue remodeling of nasal polyps (9). However, the regulation of EMT by microRNAs (miRNAs or miRs) in CRS has not been investigated to the same extent and requires further investigation.
miRNAs are single-strand and non-coding RNAs 18–25 nucleotides in length that regulate related gene expression by binding to the 3′-untranslated regions (3′-UTR) of mRNA targets and induce mRNA destruction or translation inhibition (10). A previous study revealed that miRNAs may serve a vital role in the progression of inflammation and CRSwNP remodeling (11). Furthermore, miR-142-3p may regulate inflammatory responses by regulating tumor necrosis factor α (11). Interactions between miR-4492 and interleukin 10 associated with Janus kinase/signal transducers and activators of the transcription signaling pathway may be one of the key mechanisms in CRSwNP (8). miR-155-5p has also been reported to be targeted by circular RNA nuclear receptor subfamily 1 group H member 4 in the pathological process of renal injury regulation in salt-sensitive hypertensive mice (12). Moreover, miR-155-5p has been revealed to inhibit vascular smooth muscle cell viability by targeting proto-oncogene c-Fos and Zic family member 3 to promote aneurysm formation (13).
As the underlying biological functions and molecular mechanisms of miR-155-5p in CRS development and progression remain unclear, the present study investigated the role of miR-155-5p on EMT in CRS related to sirtuin 1 (SIRT1) to establish the molecular mechanisms of miR-155-5p in CRS development.
Materials and methods
Ethical statement
The Ethics Committee of Zhujiang Hospital, Southern Medical University, Guangzhou, P.R. China approved the present study (approval no. EBYHK20190503) and written informed consent was obtained from all patients.
Study population
A total of 35 patients who attended Zhujiang Hospital, Southern Medical University for treatment from June 2019 to December 2019 were enrolled in the present study and were divided into the following groups: i) The CRSsNP group (n=14), from whom mucosa samples originating from the middle turbinates were collected; ii) The CRSwNP group (n=11), from whom polyp tissue samples were collected from the middle meatus; and iii) The control group (n=10), which was comprised of patients who had received septoplasty or rhinoseptoplasty with anatomical variations without sinus disease and who had inferior turbinate samples collected.
Patients with CRS were diagnosed based on clinical symptoms, examinations, nasal endoscopy and sinus-computed tomography (CT) scans in accordance with the criteria from the European Position Paper on Rhinosinusitis and Nasal Polyps (EPOS; 2012) (5). Generally, patients included in the study were examined for: i) Cystic fibrosis; ii) Gross immunodeficiency; iii) Congenital mucociliary problems; iv) Non-Invasive fungal sinusitis; v) Invasive fungal disease; vi) Systemic vasculitis; and vii) Granulomatous diseases as listed by EPOS. None of the patients received antihistamines, antileukotrienes, oral or intranasal decongestants, or intranasal anticholinergics 2 weeks prior to the present study. Moreover, drug treatments, including steroids, antibiotics and antihistamines, were terminated for >1 month prior to the present study. Paranasal sinus CT scans were scored using the Lund-Mackay scoring method (14) and nasal endoscopic quantitative evaluation was performed using the Lund-Kennedy scoring method (15). Moreover, patients were scored using a visual analog scale (16). Patients were excluded if they had immune deficiencies, primary ciliary dyskinesia, fungal sinusitis, cystic fibrosis, antrochoanal polyps, vasomotor rhinitis, asthma or a history of sinus surgery.
Demographic and clinical characteristics of all the patients are presented in Table I. Fresh samples of nasal tissues were collected during surgery and stored at −80°C for subsequent experiments.
Table I.
Clinical characteristics of the patients with CRS.
| Characteristics | Controls | CRSsNP | CRSwNP |
|---|---|---|---|
| No. of patients | 10 | 14 | 11 |
| Median age (IQR) | 32 (13–61) | 42 (18–63) | 38 (21–59) |
| Sex | |||
| Male | 4 | 5 | 4 |
| Female | 6 | 9 | 6 |
| Median VAS score (IQR) | 0 | 4 (2–9) | 8 (6–12) |
| Median CT score (IQR) | 0 | 5 (3–8) | 8 (4–9) |
| Median endoscopy score (IQR) | 0 | 10.5 (8–14) | 13 (10–15) |
| Positive skin prick test | 0 | 2 | 1 |
CRS, chronic rhinosinusitis; CRSsNP, CRS without nasal polyposis; CRSwNP, CRS with nasal polyposis; IQR, interquartile range; VAS, visual analog scale; CT, computed tomography.
Skin prick tests
The patients underwent skin prick tests by specially trained nurses with a panel of 13 common allergen, including alder pollen, birch pollen, Timothy grass pollen, smooth meadow grass pollen, Festuca pratensis pollen, mugwort pollen, dandelion pollen, dog dander, cat dander, horse dander, cow dander and house dust mites Dermatophagoides farinae and Dermatophagoides pteronyssinus. Saline was used as negative control and histamine as positive control. Skin prick test responses were considered positive if the allergen caused a wheal with a diameter >3 mm.
Cell culture and treatment
Primary human nasal epithelial cells (HNEpCs) were obtained from PromoCell GmbH. Cells were grown in a RPMI-1640 medium (cat. no. 31870082, Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (Invitrogen; Thermo Fisher Scientific, Inc.) in a humidified incubator at 37°C with 5% CO2.
HNEpCs were cultured in a 96-well plate (3×105 cells/ml) for 24 h. miR-155-5p inhibitor (5′-ACCCCUAUCACGAUUAGCAUUAA-3′) and inhibitor control (5′-CAGUACUUUUGUGUAGUACAA-3′) were purchased from Sigma-Aldrich, Merck KGaA. The medium in the plates was replaced with a fresh one following culturing for 24 h and the HNEpCs were transfected with 50 nM miR-155-5p inhibitor and inhibitor control using Lipofectamine® 2000 transfection reagent (Thermo Fisher Scientific, Inc.). The cells were harvested at 48 h post-transfection for subsequent studies.
To explore the effect of TGF-β1 on HNEpC morphology and EMT, HNEpCs were stimulated with 5 ng/ml transforming growth factor (TGF)-β1 (cat. no. 34-8348-82; Invitrogen; Thermo Fisher Scientific, Inc.) in the presence of miR-155-5p inhibitor or inhibitor control. HNEpC morphology was observed under a DM500 light microscope (Leica Microsystems, Inc.). Furthermore, to explore the function of SIRT1, an SIRT1 inhibitor, Splitomicin (cat. no. S4068; Sigma-Aldrich; Merck KGaA) at final concentration of 100 µM was incubated with the cells for 48 h at 37°C.
Cell Counting Kit-8 (CCK-8) assay
Cell viability was determined using a CCK-8 kit (Beyotime Institute of Biotechnology) according to the manufacturer's protocol. The cells were cultured at 37°C with 95% O2 and 5% CO2 in an incubator for 24, 48 or 72 h. The CCK-8 solution was then added and cultured for another 2 h at 37°C. Optical density values were measured at 450 nm using a microplate reader (model 680; Bio-Rad Laboratories, Inc.).
Western blotting
Protein expression was detected by western blotting as previously described (17). Briefly, HNEpCs were cultured in a 24-well plate (3×105 cells/wells) for 24 h. Total proteins from HNEpCs and nasal samples (CRSsNP, CRSwNP and control groups) were lysed and extracted using RIPA buffer (cat. no. P0013C; Beyotime Institute of Biotechnology) and protein concentrations were measured using a BCA protein kit (Sigma-Aldrich; Merck KGaA). Subsequently, 30 µg protein sample lysates were electrophoresed by 12% SDS-PAGE (cat. no. P0012A; Beyotime Institute of Biotechnology) and transferred to PVDF membranes (cat. no. FFP28; Beyotime Institute of Biotechnology). Membranes were then blocked with 5% non-fat milk for 2 h at room temperature and then incubated with the primary antibodies anti-TGF-β1 (mouse; 1:2,000; cat. no. ab27969), anti-E-cadherin (mouse; 1:1,000; product code ab1416), anti-α-smooth muscle actin (SMA; rabbit; 1:1,000; cat. no. ab5694), anti-fibronectin (rabbit; 1:1,000; cat. no. ab2413), anti-vimentin (rabbit; 1:1,000; cat. no. ab92547), anti-SIRT1 (mouse; 1:1,000; cat. no. ab110304) and anti-β-actin (mouse; 1:1,000; cat. no. ab8226) at 4°C overnight. β-actin served as an internal reference. Membranes were subsequently incubated with secondary horseradish peroxidase (HRP)-combined antibodies goat anti-rabbit immunoglobulin (Ig) G H&L (goat; 1:2,000; cat. no. ab205718) and goat anti-mouse IgG H&L (HRP; goat; 1:2,000; cat. no. ab205719) at room temperature for 1 h and then washed with TBST three times. All antibodies were purchased from Abcam. Protein bands were obtained and developed using an ECL kit (EMD Millipore). Grey values were further collected and calculated using ImageJ software (version 5.0; Bio-Rad Laboratories, Inc.).
RNA isolation and reverse transcription-quantitative PCR (RT-qPCR)
HNEpCs were cultured in a 96-well plate (3×105 cells/ml) for 24 h at 37°C. Total RNA was extracted from HNEpCs and nasal samples (CRSsNP, CRSwNP and control groups) using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.), preserved in a refrigerator at −80°C. Total RNA concentration was detected and quantified using a biological spectrometer (Nano Drop 2000; Thermo Fisher Scientific, Inc.). cDNA was synthesized from 1 µg of total RNA using a First-strand cDNA Synthesis kit (cat. no. E6300L; New England BioLabs, Inc.), according to the manufacturer's protocol. RT-qPCR was conducted using a SYBR Premix Ex Taq II kit (cat. no. RR820L; Takara, Bio, Inc.) and a Touch real-time PCR Detection system (cat. no. CFX96; Bio-Rad Laboratories, Inc.), according to the manufacturer's protocol. The thermocycling conditions were as follows: Denaturation at 95°C for 30 sec, 95°C for 5 sec and 60°C for 1 min for a total of 40 cycles. Primer sequences are presented in Table II. β-actin and U6 were used as internal references. Relative gene expression was quantified using the 2−ΔΔCq method (18).
Table II.
Primers used for reverse transcription-quantitative PCR.
| Genes | Primers |
|---|---|
| miR-155-5p | |
| Forward | 5′-GGGTGTCGTATCCAGTGCAA-3′ |
| Reverse | 5′-GTCGTATCCAGTGCGTGTCG-3′ |
| SIRT1 | |
| Forward | 5′-AAATGCTGGCCTAATAGAGTGG-3′ |
| Reverse | 5′-TGGCAAAAACAGATACTGATTACC-3′ |
| U6 | |
| Forward | 5′-GAGAAAGTTAGCACGGCTTCTG-3′ |
| Reverse | 5′-CAAAATATGGAATGCTTCAAAGAG-3′ |
| β-actin | |
| Forward | 5′-ATTGGCAATGAGCGGTTC-3′ |
| Reverse | 5′-GGATGCCACAGGACTCCA-3′ |
miR, microRNA; SIRT1, sirtuin 1.
Target gene prediction and dual-luciferase reporter assay
Target genes and potential binding sites of miR-155-5p and SIRT1 were predicted using TargetScan software (version 7.2; targetscan.org) and subsequently confirmed by a dual-luciferase reporter assay.
Wild-type (WT) or mutant (MUT) SIRT1 sequences were cloned into pMirGLO reporter vectors (Promega Corporation) to generate SIRT1-WT and SIRT1-MUT vectors. HNEpCs were cultured in 96-well plates (5×103 cells/well) for 24 h at 37°C and co-transfected with SIRT1-WT or SIRT1-MUT and miR-155-5p inhibitor (I group) or inhibitor control using Lipofectamine® 2000 Transfection reagent (Thermo Fisher Scientific, Inc.) at 37°C. Cells were harvested at 48 h post-transfection and subjected to a dual-luciferase reporter assay system (cat. no. E1910; Promega Corporation). Luciferase activity was normalized to Renilla luciferase.
Statistical analysis
All experiments were performed in triplicate. Data are expressed as the mean ± standard deviation. Statistical analysis was performed using SPSS software (version 21.0; IBM Corp.). Data were analyzed using an unpaired Student's t-test and one-way ANOVA followed by Dunnett's post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Epithelial markers are downregulated and TGF-β1, mesenchymal markers and miR-155-5p are upregulated in CRSwNP
To determine whether EMT occurs in CRS, the protein expression of TGF-β1 and EMT-associated markers (E-cadherin, vimentin, α-SMA, and fibronectin) in the CRSwNP, CRSsNP and control groups was determined by western blotting. The protein expression of E-cadherin was downregulated and TGF-β1, vimentin, α-SMA, and fibronectin were upregulated in the CRSsNP and CRSwNP groups compared with the control (Fig. 1A; P<0.001), indicating that epithelial marker expression was downregulated and the expression of TGF-β1 and mesenchymal markers were upregulated in the CRSsNP and CRSwNP group. Furthermore, changes in E-cadherin, TGF-β1, vimentin, α-SMA, and fibronectin expression were greater in the CRSwNP group than in the CRSsNP group.
Figure 1.

Epithelial markers are downregulated and TGF-β1, mesenchymal markers and miR-155-5p are upregulated in patients with CRSwNP. (A) Relative protein/β-actin expression levels of TGF-β1 and epithelial-to-mesenchymal transition-associated proteins (E-cadherin, α-SMA, fibronectin and vimentin) in the control, CRSsNP and CRSwNP groups were detected by western blotting. β-actin served as the internal control. (B) Relative miR-155-5p expression in the control, CRSsNP and CRSwNP groups was assessed by reverse transcription-quantitative PCR. U6 served as the internal control. Experiments were performed in triplicate. Data are presented as the mean ± standard deviation. *P<0.05, **P<0.01 and ***P<0.001 vs. control. TGF-β1, transforming growth factor β1; α-SMA, α-smooth muscle actin; CRSsNP, chronic rhinosinusitis without nasal polyps; CRSwNP, chronic rhinosinusitis with nasal polyps; miR, microRNA.
Additionally, the expression of miR-155-5p was determined to investigate the role of miR-155 in CRS. The expression of miR-155-5p was significantly increased in the CRSwNP and CRSsNP groups compared with the control (P<0.001; Fig. 1B), indicating that miR-155-5p may be a therapeutic target for CRS.
TGF-β1 upregulates miR-155-5p expression and induces EMT in HNEpCs, which is reversed by miR-155-5p inhibitors
To determine the function of miR-155-5p in HNEpCs, HNEpCs were transfected with miR-155-5p inhibitors to knockdown miR-155-5p expression. Control and NC groups were established and the transfection rate of miR-155-5p was measured by RT-qPCR. The results demonstrated that miR-155-5p expression in the I group was downregulated compared with the control (Fig. 2A; P<0.05 vs. NC), indicating that miR-155-5p inhibitors reduced miR-155-5p expression.
Figure 2.

TGF-β1 upregulates miR-155-5p expression and induces epithelial-to-mesenchymal transition of HNEpCs, which is reversed by miR-155-5p inhibitors. (A) Relative miR-155-5p expression in the control, NC and I groups were measured using RT-qPCR. U6 served as the internal control. *P<0.05 vs. NC. (B) Relative miR-155-5p expression levels in the control, TGF-β1 and TGF-β1+I groups were determined by RT-qPCR. U6 served as the internal control. ###P<0.001 vs. control; ^P<0.05 vs. TGF-β1. (C) Cell viability in the HNEpC control, TGF-β1 and TGF-β1+I groups was assessed using a Cell Counting Kit-8 assay. ##P<0.01 and ###P<0.001 vs. control; ^P<0.05 and ^^^P<0.001 vs. TGF-β1. (D) Morphological changes on HNEpCs were observed under a microscope. Experiments were performed in triplicate. Data are presented as the mean ± standard deviation. TGF-β1, transforming growth factor β1; miR, microRNA; HNEpCs, human nasal epithelial cells; NC, negative control; I, miR-155-5p inhibitor; RT-qPCR, reverse transcription-quantitative PCR.
miR-155-5p expression has been reported to be upregulated by TGF-β1 (19). Therefore, miR-155-5p expression in HNEpCs prior to or following 5 ng/ml TGF-β1 treatment was examined. The results demonstrated that miR-155-5p expression was significantly upregulated following 5 ng/ml TGF-β1 treatment (P<0.001 vs. the control; Fig. 2B); however, this expression was inhibited by miR-155-5p inhibitors (P<0.05 vs. TGF-β1). Furthermore, cell viability was determined via CCK-8 assay and the results demonstrated that viability in HNEpCs was promoted by TGF-β1 (P<0.001 vs. the control) and inhibited by miR-155-5p inhibitors (P<0.05 vs. TGF-β1; Fig. 2C).
A previous study revealed that TGF-β1 induced EMT in primary airway epithelial cells (20). In the present study, HNEpCs were stimulated by TGF-β1 to observe whether TGF-β1 induced EMT in HNEpCs. TGF-β1 treatment in HNEpCs induced transition HNEpCs from normal epithelial morphology with cobblestone-like appearance into migratory mesenchymal morphology with an abnormally elongated appearance (Fig. 2D). However, these changes were inhibited following the transfection of miR-155-5p inhibitors. These results indicated that TGF-β1 treatment induced EMT in HNEpCs. Immunohistochemical staining and transepithelial resistance could be performed in future studies to examine cell-cell contact adhesion.
During EMT development, cell-to-cell adhesion, polarity of epithelial cells and transformation of epithelial cells into mesenchymal cells gradually decline (21). The protein expression levels of EMT-related proteins (E-cadherin, α-SMA, fibronectin, and vimentin) were assessed using western blotting. In accordance with changes in cell morphology, TGF-β1 treatment significantly downregulated E-cadherin protein expression and significantly upregulated α-SMA, fibronectin and vimentin protein expression in HNEpCs (P<0.01 and P<0.001 vs. the control; Fig. 3). These expression levels were reversed by miR-155-5p inhibitors (P<0.01 and P<0.001 vs. TGF-β1+IC), indicating that TGF-β1 induced EMT in HNEpCs.
Figure 3.

TGF-β1 regulates the expression of EMT-related proteins in human nasal epithelial cells. These expression levels were reversed by miR-155-5p inhibitors. The expression of EMT-related proteins E-cadherin, α-SMA, fibronectin and vimentin were detected by western blotting. β-actin served as the internal control. **P<0.01 and ***P<0.001 vs. control; ##P<0.01 and ###P<0.001 vs. TGF-β1 + IC. Experiments were performed in triplicate. Data are presented as the mean ± standard deviation. TGF-β1, transforming growth factor β1; EMT, epithelial-to-mesenchymal transition; miR, microRNA; α-SMA, α-smooth muscle actin; IC, inhibitor control; I, miR-155-5p inhibitor.
miR-155-5p mediates TGF-β1-induced EMT in HNEpCs by regulating SIRT1 expression
miRNAs regulate gene expression by binding to the 3′-UTR of target mRNAs (22). The online database TargetScan was used to successfully predict that SIRT1 was a possible target of miR-155-5p (Fig. 4A). This prediction was confirmed by dual-luciferase reporter assays (Fig. 4B). The results demonstrated that the relative luciferase activity of the SIRT1-WT group was significantly increased compared with the IC group (P<0.001 vs. IC). miR-155-5p inhibitors did not affect the luciferase activity in the SIRT1-MUT group. These results demonstrated that SIRT1 was a target of miR-155-5p.
Figure 4.
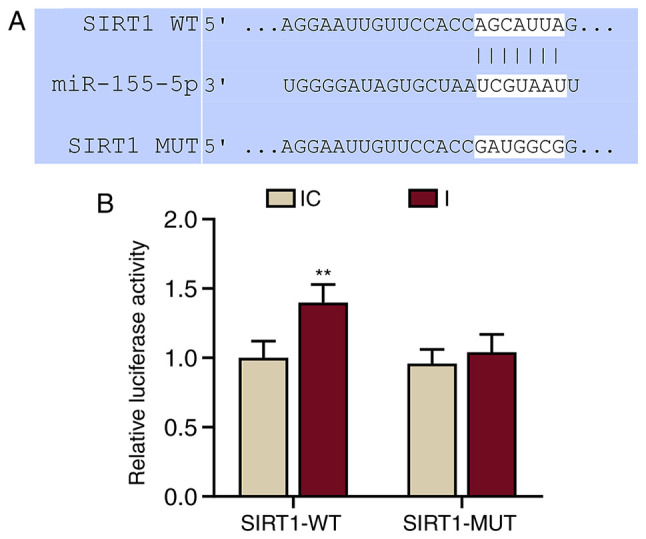
miR-155-5p targets SIRT1 in human nasal epithelial cells. (A) Sequences of SIRT1-WT (top), miR-155-5p (middle) and SIRT1-MUT (bottom) used to predict that SIRT1 may be a potential target of miR-155-5p. (B) Dual-luciferase reporter assays confirmed that SIRT1 was a target of miR-155-5p. **P<0.01, vs. IC. Experiments were performed in triplicate. Data are presented as the mean ± standard deviation. miR, microRNA; SIRT1, sirtuin 1; WT, wild-type; MUT, mutant; IC, inhibitor control; I, miR-155-5p inhibitor.
To further determine the role of SIRT1 in CRS, the protein and mRNA expression levels of SIRT1 in CRSsNP, CRSwNP and control groups were detected by western blotting and RT-qPCR. The results demonstrated that the protein and mRNA expression levels of SIRT1 were downregulated in the CRSsNP and CRSwNP groups compared with the control (Fig. 5A and B; P<0.01 and P<0.001 vs. the control).
Figure 5.

miR-155-5p mediates TGF-β1-induced EMT of human nasal epithelial cells by regulating SIRT1 expression. (A) Relative SIRT1/β-actin expression in the control, CRSsNP and CRSwNP groups was measured by western blotting. β-actin served as the internal control. (B) Relative SIRT1 mRNA expression in the control, CRSsNP and CRSwNP groups was measured by reverse transcription-quantitative PCR. β-actin served as the internal control. (C) Relative protein/β-actin expressions of EMT-related proteins (E-cadherin, α-SMA, fibronectin and vimentin) were assessed by western blotting. β-actin served as the internal control. **P<0.01 and ***P<0.001 vs. the control; #P<0.05, ##P<0.01 and ###P<0.001 vs. TGF-β1; ^P<0.05 and ^^P<0.01 vs. TGF-β1+spl. Experiments were performed in triplicate. Data are presented as the mean ± standard deviation. miR, microRNA; TGF-β1, transforming growth factor β1; EMT, epithelial-to-mesenchymal transition; SIRT1, sirtuin 1; CRSsNP, chronic rhinosinusitis without nasal polyps; CRSwNP, chronic rhinosinusitis with nasal polyps; α-SMA, α-smooth muscle actin; spl, splitomicin; I, miR-155-5p inhibitor.
Following treatment with 5 ng/ml TGF-β1 and 100 µM splitomicin treatment, the protein expression of EMT-associated markers (E-cadherin, α-SMA, fibronectin and vimentin) in HNEpCs was assessed by western blotting to detect the association between miR-155-5p and SIRT1, and their effects on EMT. Consistent with previous results, following TGF-β1 treatment, the protein expression of E-cadherin was downregulated and the expression levels of α-SMA, fibronectin and vimentin were upregulated compared with the control (Fig. 5C; P<0.01 and P<0.001 vs. the control). Splitomicin treatment further enhanced the effect of TGF-β1 on protein expression (P<0.01 and P<0.001, vs. TGF-β1), indicating that SIRT1 inhibition promoted EMT. Furthermore, these changes in protein expression were reversed by miR-155-5p inhibitors (P<0.05 and P<0.01 vs. TGF-β1+spl). This suggested that miR-155-5p downregulation mediated TGF-β1-induced EMT in HNEpCs by regulating SIRT1 expression.
Discussion
The present study revealed that miR-155-5p mediated EMT in HNEpCs by regulating SIRT1 expression in response to TGF-β1. To the best of knowledge, the present study is the first to report that miR-155-5p served an essential role in EMT in CRS. Although miRNA dysfunction has been widely discussed in various pathophysiological processes, such as coronary heart disease (23), atherosclerosis (24) and cancer (25), their biological roles and molecular mechanisms in CRS development and progression remain unclear. miR-155 upregulation and TGF-β1 overexpression accelerate EMT and promote cell migration and invasion, thereby inducing hepatocellular carcinoma progression (26). Furthermore, TGF-β1 has been reported to upregulate miR-155 expression to alter osteoclast differentiation (27). Based on these findings, the present study hypothesized that miR-155 induction by TGF-β1 may serve a potential role in CRS pathogenesis.
CRS is an inflammatory upper respiratory tract disease characterized by a persistent inflammatory state and paranasal sinuses, with a minimum duration of 12 weeks (28,29). Based on the presence or absence of polyposis, CRS can be divided into CRSwNP and CRSsNP (5). Though the occurrence of CRSwNP is infrequent, medical intervention is unsuccessful for a large proportion of patients with CRSwNP (8). In addition to inflammation, CRSwNP and CRSsNP are characterized by remodeling of structural components, particularly in the epithelium (30). Remodeling under normal conditions is defined as an essential reconstruction process in response to minor inflammatory conditions; however, dysregulated remodeling, which may be induced by severe or chronic inflammation, can result in pathological reconstruction and formation of pathological tissues during healing and repairing (31). Depending on the disease and its severity, inflammation often causes different degrees of tissue injury, indicating that remodeling may occur during inflammatory diseases, including CRSwNP (17). Recently, the role of remodeling in upper airway diseases, such as CRS, has received considerable attention, partly since remodeling begins at an early disease stage and contributes to later development and progression (31).
To date, the mechanisms of tissue remodeling have not been fully elucidated, however, accumulating evidence has indicated that EMT may be involved (32–34). In multiple progressive fibrotic diseases, EMT is seen as a point of convergence between inflammation and pathological remodeling (31). EMT is a process by which differentiated epithelial cells undergo phenotypic transformation and acquire the mesenchymal cell phenotype (35). A previous study reported that ongoing EMT may result in the progression of numerous diseases, such as cancer (36) and chronic kidney disease (37), particularly in CRSwNP (1). Following injury, the expression of junctional proteins, including E-cadherin, are downregulated and the expression of mesenchymal proteins, such as α-SMA, vimentin and fibronectin, are upregulated in epithelial cells, which then undergo EMT and alter morphology (38). In accordance with previous studies, the present study suggested that E-cadherin expression was reduced, while expression of α-SMA, vimentin and fibronectin were increased in CRSwNP and CRSsNP tissues compared with control groups. CRSsNP is characterized by fibrosis, a thickened membrane of basement and excessive goblet cells (39). TGF-β1, a significant regulator of fibrosis promotion and airway remodeling, was demonstrated to be upregulated in CRSsNP compared with control. This result was consistent with a previous study (40).
TGF-β1 is a multifunctional peptide regulating various cellular functions, including cell growth, differentiation, apoptosis and migration (41). TGF-β1 is implicated in the pathogenesis of airway diseases, including chronic obstructive pulmonary disease (42) and CRS (1). TGF-β1 serves a key role in fibrosis and EMT, and induces epithelial cell transition, contributes to tissue remodeling and CRSwNP pathogenesis (43). The present results revealed that TGF-β1 expression was upregulated in the CRSwNP group compared with the control. Furthermore, TGF-β1 altered HNEpCs morphology and regulated the expression of EMT-related proteins. These expression levels were reversed by downregulating the expression of miR-155-5p.
miR-155-5p is associated with various diseases. For example, miR-155-5p affects Wilms' tumor cell proliferation and apoptosis by targeting CREB1 (44). Moreover, p53/miR-155-5p/SIRT1-mediated autophagic processes have been implicated in diabetic kidney injury (45). miR-155-5p has also been demonstrated to mediate the TGF-β1 signaling pathway and EMT (26). Zhang et al (46) reported that miR-155-5p regulated cardiac fibrosis induced by glucose via the TGF-β1 signaling pathway. Wang et al (47) demonstrated that miR-155-5p regulated TGF-β-induced EMT in human coronary artery endothelial cells by targeting c-Ski to affect cardiac fibrosis (47). The results of the present study revealed that miR-155-5p expression was increased in the CRSwNP group compared with the control and that upregulated miR-155-5p expression induced by TGF-β1 caused EMT in HNEpCs. Furthermore, these results were reversed by miR-155-5p downregulation.
A previous study reported that SIRT1 attenuates nasal polypogenesis through EMT suppression (48). Furthermore, it has been demonstrated that SIRT1 is a target gene of miR-155-5p (49). Huang et al (49) reported that miR-155 downregulation stimulated cardioprotection against myocardial ischemia/reperfusion injury by binding to SIRT1. However, the role of miR-155-5p in inhibiting EMT has not been extensively reported. The present study demonstrated that miR-155 downregulation attenuated CRS by inhibiting EMT via targeting of SIRT1.
However, the present study still had some limitations. Firstly, the number of samples in the experimental groups were not equal and an animal model was not established. Secondly, there was a lack of immunohistochemical data. Further studies could perform Transwell assays to confirm the ETM state of the HNEpCs cells.
In conclusion, the results of the present study revealed that miR-155 may be a potential therapeutic target for CRS treatment by suppressing EMT. Furthermore, miR-155 inhibitors may be a novel anti-polyp drug in the regulation of SIRT1 expression in HNEpCs and miR-155 downregulation may be a potential method for treating CRS.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Clinical Medical Technology Innovation Guide Project of Hunan Province (grant no. 2017SK51405).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
NY and HC conceptualized and designed the present study, and drafted and critically revised the manuscript for intellectual content. QM, XZ and MX performed data acquisition, analysis and interpretation. All authors agreed to be accountable in ensuring that enquiries related to the accuracy and/or integrity of the present work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The Ethics Committee of Zhujiang Hospital, Southern Medical University, Guangzhou, China approved the present study (approval no. EBYHK20190503) and written informed consent was obtained from all patients. Furthermore, all procedures involving human participants were in accordance with the ethical standards of the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Schleimer RP. Immunopathogenesis of chronic rhinosinusitis and nasal polyposis. Annu Rev Pathol. 2017;12:331–357. doi: 10.1146/annurev-pathol-052016-100401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee S, Lane AP. Chronic rhinosinusitis as a multifactorial inflammatory disorder. Curr Infect Dis Rep. 2011;13:159–168. doi: 10.1007/s11908-011-0166-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savovic S, Buljcik-Cupic M, Jovancevic L, Kljajic V, Lemajic-Komazec S, Dragicevic D. Frequency and intensity of symptoms in patients with chronic rhinosinusitis. Srpski Arhiv Za Celokupno Lekarstvo. 2018;147:13. [Google Scholar]
- 4.Hull BP, Chandra RK. Refractory chronic rhinosinusitis with nasal polyposis. Otolaryngol Clin North Am. 2017;50:61–81. doi: 10.1016/j.otc.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Fokkens WJ, Lund VJ, Mullol J, Bachert C, Alobid I, Baroody F, Cohen N, Cervin A, Douglas R, Gevaert P, et al. European position paper on rhinosinusitis and nasal polyps 2012. Rhinol Suppl. 2012;23:3. [PubMed] [Google Scholar]
- 6.Piromchai P, Kasemsiri P, Laohasiriwong S, Thanaviratananich S. Chronic rhinosinusitis and emerging treatment options. Int J Gen Med. 2013;6:453–464. doi: 10.2147/IJGM.S29977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koskinen A, Numminen J, Markkola A, Karjalainen J, Karstila T, Seppälä M, Julkunen A, Lemmetyinen R, Pekkanen J, Rautiainen M, et al. Diagnostic accuracy of symptoms, endoscopy, and imaging signs of chronic rhinosinusitis without nasal polyps compared to allergic rhinitis. Am J Rhinol Allergy. 2018;32:121–131. doi: 10.1177/1945892418762891. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Feng J, Zhang D, Yong J, Wang Y, Yao J, Huang R. Differential expression of miR-4492 and IL-10 is involved in chronic rhinosinusitis with nasal polyps. Exp Ther Med. 2019;18:3968–3976. doi: 10.3892/etm.2019.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Konnecke M, Burmeister M, Pries R, Boscke R, Bruchhage KL, Ungefroren H, Klimek L, Wollenberg B. Epithelial-mesenchymal transition in chronic rhinosinusitis: Differences revealed between epithelial cells from nasal polyps and inferior turbinates. Arch Immunol Ther Exp (Warsz) 2017;65:157–173. doi: 10.1007/s00005-016-0409-7. [DOI] [PubMed] [Google Scholar]
- 10.Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141:1202–1207. doi: 10.1016/j.jaci.2017.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qing X, Zhang Y, Peng Y, He G, Liu A, Liu H. miR-142-3p regulates inflammatory response by contributing to increased TNF-α in chronic rhinosinusitis with nasal polyposis. Ear Nose Throat J. 2019;9:145561319847972. doi: 10.1177/0145561319847972. [DOI] [PubMed] [Google Scholar]
- 12.Lu C, Chen B, Chen C, Li H, Wang D, Tan Y, Weng H. CircNr1h4 regulates the pathological process of renal injury in salt-sensitive hypertensive mice by targeting miR-155-5p. J Cell Mol Med. 2019;24:1700–1712. doi: 10.1111/jcmm.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao L, Ouyang Y, Bai Y, Gong J, Liao H. miR-155-5p inhibits the viability of vascular smooth muscle cell via targeting FOS and ZIC3 to promote aneurysm formation. Eur J Pharmacol. 2019;853:145–152. doi: 10.1016/j.ejps.2019.03.021. [DOI] [PubMed] [Google Scholar]
- 14.Thwin M, Weitzel EK, McMains KC, Athanasiadis T, Psaltis A, Field J, Wormald PJ. Validating the use of report-derived lund-macKay scores. Am J Rhinol Allergy. 2009;23:33–35. doi: 10.2500/ajra.2009.23.3255. [DOI] [PubMed] [Google Scholar]
- 15.Lund VJ, Kennedy DW. Quantification for staging sinusitis. The staging and therapy group. Ann Otol Rhinol Laryngol Suppl. 1995;167:17–21. doi: 10.1177/000348949510410s02. [DOI] [PubMed] [Google Scholar]
- 16.Linder A. Symptom scores as measures of the severity of rhinitis. Clin Allergy. 1988;18:29–37. doi: 10.1111/j.1365-2222.1988.tb02840.x. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Li C, Zhu G, Yuan W, Xiao ZA. TGF-β1 induces epithelial-mesenchymal transition of chronic sinusitis with nasal polyps through MicroRNA-21. Int Arch Allergy Immunol. 2019;179:304–319. doi: 10.1159/000497829. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Liu F, Kong X, Lv L, Gao J. TGF-β1 acts through miR-155 to down-regulate TP53INP1 in promoting epithelial-mesenchymal transition and cancer stem cell phenotypes. Cancer Lett. 2015;359:288–298. doi: 10.1016/j.canlet.2015.01.030. [DOI] [PubMed] [Google Scholar]
- 20.Park IH, Kang JH, Shin JM, Lee HM. Trichostatin A inhibits epithelial mesenchymal transition induced by TGF-β1 in airway epithelium. PLoS One. 2016;11:e0162058. doi: 10.1371/journal.pone.0162058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35:645–654. doi: 10.1007/s10555-016-9648-7. [DOI] [PubMed] [Google Scholar]
- 22.Ma Z, Shen Y, Zeng Q, Liu J, Yang L, Fu R, Hu G. MiR-150-5p regulates EGR2 to promote the development of chronic rhinosinusitis via the DC-Th axis. Int Immunopharmacol. 2018;54:188–197. doi: 10.1016/j.intimp.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 23.Wu ZW, Liu YF, Wang S, Li B. miRNA-146a induces vascular smooth muscle cell apoptosis in a rat model of coronary heart disease via NF-κB pathway. Genet Mol Res. 2015;14:18703–18712. doi: 10.4238/2015.December.28.19. [DOI] [PubMed] [Google Scholar]
- 24.Canfrán-Duque A, Rotllan N, Zhang X, Fernández-Fuertes M, Ramírez-Hidalgo C, Araldi E, Daimiel L, Busto R, Fernández-Hernando C, Suárez Y. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol Med. 2017;9:1244–1262. doi: 10.15252/emmm.201607492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Haidari AA, Syk I, Thorlacius H. miR-155-5p positively regulates CCL17-induced colon cancer cell migration by targeting rhoA. Oncotarget. 2017;8:14887–14896. doi: 10.18632/oncotarget.14841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li DP, Fan J, Wu YJ, Xie YF, Zha JM, Zhou XM. MiR-155 up-regulated by TGF-β promotes epithelial-mesenchymal transition, invasion and metastasis of human hepatocellular carcinoma cells in vitro. Am J Transl Res. 2017;9:2956–2965. [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao H, Zhang J, Shao H, Liu J, Jin M, Chen J, Huang Y. Transforming growth factor β1/Smad4 signaling affects osteoclast differentiation via regulation of miR-155 expression. Mol Cells. 2017;40:211–221. doi: 10.14348/molcells.2017.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koennecke M, Benecke F, Masche A, Linke R, Bruchhage KL, Pries R, Klimek L, Wollenberg B. Increased phosphorylation of eNOS in nasal polyps of chronic rhinosinusitis patients can be diminished by 1,8-cineol. Nitric Oxide. 2018;78:89–94. doi: 10.1016/j.niox.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 29.DeConde AS, Soler ZM. Chronic rhinosinusitis: Epidemiology and burden of disease. Am J Rhinol Allergy. 2016;30:134–139. doi: 10.2500/ajra.2016.30.4297. [DOI] [PubMed] [Google Scholar]
- 30.Hupin C, Gohy S, Bouzin C, Lecocq M, Polette M, Pilette C. Features of mesenchymal transition in the airway epithelium from chronic rhinosinusitis. Allergy. 2014;69:1540–1549. doi: 10.1111/all.12503. [DOI] [PubMed] [Google Scholar]
- 31.Lee HM, Kang JH, Shin JM, Lee SA, Park IH. Chemical chaperone of endoplasmic reticulum stress inhibits epithelial-mesenchymal transition induced by TGF-β1 in airway epithelium via the c-src pathway. Mediators Inflamm. 2017;2017:8123281. doi: 10.1155/2017/8123281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pu Y, Liu Y, Liao S, Miao S, Zhou L, Wan L. Azithromycin ameliorates OVA-induced airway remodeling in Balb/c mice via suppression of epithelial-to-mesenchymal transition. Int Immunopharmacol. 2018;58:87–93. doi: 10.1016/j.intimp.2018.03.016. [DOI] [PubMed] [Google Scholar]
- 33.Hirota N, Martin JG. Mechanisms of airway remodeling. Chest. 2013;144:1026–1032. doi: 10.1378/chest.12-3073. [DOI] [PubMed] [Google Scholar]
- 34.Tian X, Tian X, Huo R, Chang Q, Zheng G, Du Y, Chen Y, Niu B. Bacillus calmette-guerin alleviates airway inflammation and remodeling by preventing TGF-β1 induced epithelial-mesenchymal transition. Hum Vaccin Immunother. 2017;13:1758–1764. doi: 10.1080/21645515.2017.1313366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cruz-Solbes AS, Youker K. Epithelial to mesenchymal transition (EMT) and endothelial to mesenchymal transition (EndMT): Role and implications in kidney fibrosis. Results Probl Cell Differ. 2017;60:345–372. doi: 10.1007/978-3-319-51436-9_13. [DOI] [PubMed] [Google Scholar]
- 36.Title AC, Hong SJ, Pires ND, Hasenöhrl L, Godbersen S, Stokar-Regenscheit N, Bartel DP, Stoffel M. Genetic dissection of the miR-200-Zeb1 axis reveals its importance in tumor differentiation and invasion. Nat Commun. 2018;9:4671. doi: 10.1038/s41467-018-07130-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu L, Zhu J, Zhang Y, Wang Y, Zhang S, Xia A. Febuxostat inhibits TGF-β1-induced epithelial-mesenchymal transition via downregulation of USAG-1 expression in madin-darby canine kidney cells in vitro. Mol Med Rep. 2019;19:1694–1704. doi: 10.3892/mmr.2019.9806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sisto M, Lisi S, Ribatti D. The role of the epithelial-to-mesenchymal transition (EMT) in diseases of the salivary glands. Histochem Cell Biol. 2018;150:133–147. doi: 10.1007/s00418-018-1680-y. [DOI] [PubMed] [Google Scholar]
- 39.Khalmuratova R, Park JW, Shin HW. Immune cell responses and mucosal barrier disruptions in chronic rhinosinusitis. Immune Netw. 2017;17:60–67. doi: 10.4110/in.2017.17.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, Xiao L, Liu J, Shen Y, Ke X, Huang J, Hu G, Yang Y. Differential expression of the aryl hydrocarbon receptor and transforming growth factor beta 1 in chronic rhinosinusitis with nasal polyps with allergic rhinitis. ORL J Otorhinolaryngol Relat Spec. 2017;79:295–305. doi: 10.1159/000481510. [DOI] [PubMed] [Google Scholar]
- 41.Kim SI, Choi ME. TGF-β-activated kinase-1: New insights into the mechanism of TGF-β signaling and kidney disease. Kidney Res Clin Pract. 2012;31:94–105. doi: 10.1016/j.krcp.2012.04.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahmood MQ, Reid D, Ward C, Muller HK, Knight DA, Sohal SS, Walters EH. Transforming growth factor (TGF) β1 and Smad signalling pathways: A likely key to EMT-associated COPD pathogenesis. Respirology. 2017;22:133–140. doi: 10.1111/resp.12882. [DOI] [PubMed] [Google Scholar]
- 43.Ryu NH, Shin JM, Um JY, Park IH, Lee HM. Wogonin inhibits transforming growth factor β1-induced extracellular matrix production via the p38/activator protein 1 signaling pathway in nasal polyp-derived fibroblasts. Am J Rhinol Allergy. 2016;30:128–133. doi: 10.2500/ajra.2016.30.4329. [DOI] [PubMed] [Google Scholar]
- 44.Zhao XS, Han B, Zhao JX, Tao N, Dong CY. miR-155-5p affects wilms' tumor cell proliferation and apoptosis via targeting CREB1. Eur Rev Med Pharmacol Sci. 2019;23:1030–1037. doi: 10.26355/eurrev_201902_16990. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Zheng ZJ, Jia YJ, Yang YL, Xue YM. Role of p53/miR-155-5p/sirt1 loop in renal tubular injury of diabetic kidney disease. J Transl Med. 2018;16:146. doi: 10.1186/s12967-018-1486-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang D, Cui Y, Li B, Luo X, Li B, Tang Y. miR-155 regulates high glucose-induced cardiac fibrosis via the TGF-β signaling pathway. Mol Biosyst. 2016;13:215–224. doi: 10.1039/C6MB00649C. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, He W, Xu X, Guo L, Zhang Y, Han S, Shen D. The mechanism of TGF-β/miR-155/c-Ski regulates endothelial-mesenchymal transition in human coronary artery endothelial cells. Biosci Rep. 2017;37:BSR20160603. doi: 10.1042/BSR20160603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee M, Kim DW, Yoon H, So D, Khalmuratova R, Rhee CS, Park JW, Shin HW. Sirtuin 1 attenuates nasal polypogenesis by suppressing epithelial-to-mesenchymal transition. J Allergy Clin Immunol. 2016;137:87–98. doi: 10.1016/j.jaci.2015.07.026. [DOI] [PubMed] [Google Scholar]
- 49.Huang G, Hao F, Hu X. Downregulation of microRNA-155 stimulates sevoflurane-mediated cardioprotection against myocardial ischemia/reperfusion injury by binding to SIRT1 in mice. J Cell Biochem. 2019;120:15494–15505. doi: 10.1002/jcb.28816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.