

Significance
The goal of genome-wide association studies is to identify meaningful relationships between genotypes and outcomes of interest. One challenge in the analysis of genetic data is that not all true statistical associations represent relevant biological activity; irrelevant but true associations can arise from the confounding effect of environmental conditions or other factors. We propose a method to analyze such data that is immune to this problem because it uses the variation in inheritance as a randomized experiment. The method can leverage any machine-learning algorithm as well as findings from other studies.
Keywords: transmission disequilibrium test (TDT), family-based association test (FBAT), causal discovery, false discovery rate (FDR), conditional independence testing
Abstract
We introduce a method to draw causal inferences—inferences immune to all possible confounding—from genetic data that include parents and offspring. Causal conclusions are possible with these data because the natural randomness in meiosis can be viewed as a high-dimensional randomized experiment. We make this observation actionable by developing a conditional independence test that identifies regions of the genome containing distinct causal variants. The proposed digital twin test compares an observed offspring to carefully constructed synthetic offspring from the same parents to determine statistical significance, and it can leverage any black-box multivariate model and additional nontrio genetic data to increase power. Crucially, our inferences are based only on a well-established mathematical model of recombination and make no assumptions about the relationship between the genotypes and phenotypes. We compare our method to the widely used transmission disequilibrium test and demonstrate enhanced power and localization.
The ultimate aim of genome-wide association studies (GWAS) is to identify regions of the genome containing variants that causally affect a phenotype of interest (1). This paper works toward this goal by developing a test of a well-chosen conditional independence hypothesis. Specifically, we consider the hypothesis that a phenotype is independent of a group of genetic variants after conditioning on all other observed genetic variation and the genetic information of the subjects’ parents (Eq. 1). This allows us to evaluate the potential causal role of the variants in the group.
Our method addresses a key difficulty arising in the analysis of genetic datasets of increasing size: In this regime, any statistical association between a genetic variant and phenotype will be detectable, including many irrelevant associations arising from nongenetic factors, such as differing environmental conditions. While there are existing methods to mitigate this problem (2–5), such methods are not guaranteed to remove it entirely; the severity of the problem increases with large sample sizes, and current methodology may result in many detectable associations that do not represent interesting biological activity. Therefore, it is critical to move from detecting promising associations to rigorously establishing causality. The most trustworthy way to ascertain that a statistical association is causal is to use a randomized experiment (6), and parent–offspring duo or trio data record such an experiment in the sense that the locations of the recombination points during meiosis are randomized by nature. Building on this, our proposed method analyzes the placement of such sites to provably report only biologically meaningful regions of the genome.
Related Work.
Geneticists have long exploited the randomness in inheritance to identify meaningful associations (7–10). The launching point for this work is the transmission disequilibrium test (TDT) (11, 12), which checks whether a given allele is inherited more or less frequently in affected progeny than expected by chance. If the transmission frequency deviates from the baseline frequency, the TDT reports an association. Beyond the original TDT, additional techniques for analyzing more complex, partially observed pedigrees (13–16) and using multiple markers (17–19) have been developed; these are known as family-based association tests. Moreover, these techniques can be extended to study quantitative traits (20–23). To address the multiple-comparisons problem arising from looking at many variants at once ref. 24 shows how to decouple the selection of promising markers from the final construction of a P value from family-based association tests. These methods are robust both to modeling assumptions about the relationship between the trait and the genotypes and to population structure, namely, the presence of subpopulations with different allele frequencies (e.g., ref. 25). These existing methods, however, restrict the choice of test statistic and do not resolve associations due to linkage disequilibrium (LD)—correlations among sites along the genome.
Turning to the statistics literature, causal inference is concerned with correcting for confounders: variables that create statistical associations between quantities of interest even when there is no causal relationship. The problem of learning the structure of the true underlying causal model from data is known as causal discovery (e.g., refs. 26–28). General methods for causal discovery exist, although they typically require a large number of conditional independence tests and the assumption that such tests can be carried out without any statistical error asymptotically (29, 30). As a result, finite-sample results are rare. This work also uses conditional independence testing as the foundation for causal discovery but builds upon the conditional randomization test (31) to give finite-sample statistical guarantees. Our approach is also related to that of knockoffs (31, 32), which provides finite-sample statistical guarantees and has been successfully deployed to analyze GWAS data (33–35), although the connection with causal discovery was not previously developed.
Our Contribution.
We introduce the digital twin test: an approach for finding causal regions in the genome that generalizes the TDT and related methods. Our contribution has four components:
-
1)
Leveraging black-box models and subject matter knowledge. The digital twin test increases power by incorporating any multivariate model and subject matter information. Critically, the error rate guarantees of the method do not rely whatsoever on the correctness of the prior information or of the phenotype model.
-
2)
Identifying distinct causal regions. The digital twin test provably localizes causal variants within explicit windows along the genome, clearly showing the user when there are distinct causal effects. By contrast, although it is not widely known, the TDT is testing a less exact global null, so spurious findings arise from correlations among variants—see Linkage Disequilibrium in the Trio Design for an example.
-
3)
Testing multiple hypotheses. The digital twin test deals with multiple comparisons in a precise way, controlling either the family-wise error rate or the false discovery rate (FDR) without the need for a conservative Bonferroni correction. The heart of our solution is the creation of independent P values for disjoint regions, which can then be used with more powerful multiple-testing procedures.
-
4)
Establishing causality in the trio design. We formalize the existing notion that family studies are immune to population structure, showing how to leverage the trio design to make causal inferences in a rigorous statistical sense. Our results allow us to describe the properties of the TDT and some of its variations for quantitative traits.
While our inferences are based on trio data, our method can take advantage of additional case–control or population GWAS data to greatly increase power while retaining the certified causal inferences. Since trio samples are harder to collect than case–control or population samples, traditional GWAS designs remain critical, and accordingly we show how joint analysis of population and trio samples can rigorously establish that detected associations are due to causal variants. Finally, we highlight that our approach is flexible and naturally applies to binary, quantitative, or time-to-onset phenotypes.
1. The Digital Twin Test
A. Setting.
Human cells have 46 chromosomes organized into 23 pairs; one element in each pair is inherited from the mother and one is from the father. In this work, we consider the case where we measure single-nucleotide polymorphisms (SNPs), sites on the genome where two possible alleles occur in the population, encoded as 0 or 1. The set of observed alleles for one entire chromosome is known as a haplotype. We consider the case where the haplotypes of subjects and their biological parents at sites are known, denoted as follows:
For convenience we define the matrix of all offspring haplotypes as , the matrix of offspring genotypes as , and the set of all ancestral haplotypes as . For any matrix , we let be column of and be row of , with the exception that is defined as . Finally, for any we let .
Our method takes the haplotypes as given, even though typically only the genotypes are directly measured in a GWAS study. Haplotypes are then reconstructed algorithmically through phasing (36). While experimental techniques are being developed to directly measure haplotypes, these are not yet widespread. We instead take the phased haplotypes as a reasonable approximation, since phasing is known to be accurate with family data (36–38). In a simulation using a synthetic population with known ground-truth haplotypes, we find that our method performs identically with known haplotypes and computationally phased haplotypes (SI Appendix, section S.5).
Crucially, the distribution of the offspring genotypes conditional on the parental haplotypes is known. Informally, the model for a single offspring is this: For the haplotype inherited from the mother, the SNP is inherited either from or from , with equal probability. Furthermore, long continuous blocks of are jointly inherited either from or , with occasional switches at recombination sites; see Fig. 1 for an illustration. This process was formalized as a hidden Markov model (HMM) by Haldane (39); see The Haldane HMM for a formal description. Throughout, we will leverage our knowledge of the distribution of the offspring to carry out hypothesis tests.
Fig. 1.

A visualization of the process of recombination on a single chromosome.
B. The Hypothesis and Its Test.
We now introduce a randomization test to find regions of the genome that contain distinct causal variants. Our method partitions the genome into disjoint regions and then constructs a P value for the hypothesis that a given region contains no causal SNPs. The special case where the group is the entire genome corresponds to a test of the global null: whether the trait is heritable or not.
Formally, let denote the distribution of a random variable given the observed value of a random variable , and let denote that is conditionally independent of given . Let be a partition of . For each group of SNPs , we consider the hypothesis
| [1] |
In words, this is the hypothesis that knowing the SNPs in group is not informative about the response once we know the remaining SNPs and the parental haplotypes. Conditioning on the SNPs outside ensures that any rejections reflect the existence of causal SNPs in the region rather than elsewhere on the chromosome. Conditioning on the parental haplotypes, , guarantees that the test yields valid causal inferences; we discuss this at length in Causal Inference in the Trio Design. While any partition of the SNPs is permitted by the theory, we recommend taking continuous blocks of equal genetic length; see The Haldane HMM. The size of the groups will affect the power; larger group sizes correspond to weaker statistical statements and hence the corresponding tests have higher power (e.g., refs. 35 and 40). For simplicity, this work assumes a prespecified partition; see ref. 35 for a proposal for jointly analyzing multiple resolutions in a closely related setting and ref. 41 for a discussion of hierarchical testing in GWAS.
The digital twin test—presented in Algorithm 1—tests the null hypothesis in Eq. 1 by creating synthetic offspring (the “digital twins”) from a subject’s parents, with the constraint that they match outside the region . That is, the synthetic offspring are sampled from the distribution of
| [2] |
See Fig. 2 for an illustration.
Fig. 2.

A visualization of a digital twin. The gray shaded region represents the group ; the digital twin always matches the true offspring outside this region.
Algorithm 1: The digital twin test.
| Compute the test statistic on the true data: |
| for do |
| Sample the digital twins from the |
| distribution in Eq. 2, independent from and |
| (see SI Appendix, section S.2 for an explicit sampler). |
| Compute the test statistic using the digital twins: |
| Compute the quantile of the true statistic among the |
| digital twin statistics : |
The digital twin test is a special case of the conditional randomization test (31), so it is a valid test:
Proposition 1.
Suppose that the distribution of given follows the distribution in The Haldane HMM. Then, under the hypothesis in Eq. 1, the distribution of the output of Algorithm 1 stochastically dominates the uniform distribution.
Many existing tests fall into this family. Notably, the TDT is a special case of the digital twin test with the test statistic
| [3] |
where denotes the indicator of event , and the region is the entire chromosome containing site (Linkage Disequilibrium in the Trio Design). Note that to calculate the P value, the digital twin test uses an exact, finite-sample rejection threshold, whereas the TDT uses an asymptotic approximation. Similarly, the quantitative TDTs (21, 22) are also special cases of the above procedure. Moreover, the digital twin test can exploit arbitrary black-box machine-learning models, such as deep neural networks, gradient boosting, random forests, and penalized regression to form a test statistic that incorporates information from multiple sites in a data-driven way; see Eq. 4 below for a concrete example. This is useful because more sophisticated models can explain away more of the variation in the phenotype, leading to more sensitive tests.
In sum, the digital twin test framework unifies many existing procedures while incorporating varying disease models, subject matter knowledge, fitting algorithms, principal component corrections, screening and replication, and so on, without requiring a new mathematical analysis for each case. While well-chosen models will lead to more powerful tests, we emphasize that the validity of the automatic, finite-sample inference does not depend on the correctness of the chosen model.
C. Incorporating External GWAS Data.
The digital twin test can also leverage large external GWAS datasets that do not contain trio observations to increase power. This is important because such datasets are common and endowed with large sample sizes. Specifically, we can use the external GWAS to find a powerful test statistic , as suggested by ref. 42. For example, suppose we fit a penalized linear or logistic regression model on the external GWAS data to obtain an estimated coefficient vector . Then, on the trio data, we can use the digital twin test with test statistic
(this is the negative squared loss) for real-valued , or
| [4] |
(this is the negative logistic loss) for binary . In words, the digital twin test with this test statistic is asking, “Are the residuals smaller when I use the real genotypes to predict the response, compared to when I use the digital twin genotypes?” If the residuals are systematically smaller, it must be because of a causal effect, and the digital twin test rejects the null hypothesis.
To further increase power, the external GWAS data should be used to prioritize the most promising regions; ref. 43 gives a general discussion of incorporating weighted testing in GWAS and ref. 44 shows how to use side information to improve the ordering for knockoff testing. As a concrete example, when using the test statistic in Eq. 4, one could order the hypothesis by a decreasing value of
| [5] |
and then use the Selective SeqStep procedure (32) or an accumulation test (45) to give a final selection set with guaranteed FDR control. We numerically explore this approach in Simulation Experiments.
D. Looking Everywhere with Independent P Values.
Testing the conditional nulls in Eq. 1 correctly addresses the scientific question; indeed, the conditional nulls both provide localization information and are guaranteed to detect only causal variants (Causal Inference in the Trio Design). With these hypotheses in hand, the analyst is likely to evaluate many separate regions of the genome with a single study, so we must take care to control the number of false positives. Note that the digital twin test can only yield P values as small as where is the number of iterations of the digital twin sampling, unlike parametric methods (e.g., ref. 46) which can yield very small P values. While small P values are generally needed to account for multiple comparisons when one looks at each variant individually, other multiple-testing corrections are available when partitioning the genome into regions and using conditional testing (33, 35). With this end in mind, independent P values for different regions are desirable for at least two reasons: First, they can be used with powerful error-controlling procedures, such as SeqStep (32) and accumulation tests (45); and second, with algorithms such as the Benjamini–Hochberg procedure (47), it is well known that dependent P values can lead to a high number of false positives for a given dataset (48).
Motivated by these advantageous statistical properties, we next develop a technical modification of the digital twin test that yields independent P values. Loosely speaking, the idea is to additionally condition on the boundary of each group. Because of the Markovian structure, this makes the remaining behavior within each group independent of all others. The details require substantial additional notation, however, so they are deferred to Constructing Independent P Values; see Algorithm 2 therein.
Theorem 1 (Independence of Null P Values).
Suppose that given follows the distribution in The Haldane HMM. Then Algorithm 2 (in Constructing Independent P Values) produces P values satisfying the following:
-
1)
For null groups —according to Eq. 1—the distribution of stochastically dominates the uniform distribution; i.e., is a valid P value.
-
2)
The P values for null groups are jointly independent of each other and are independent of the nonnull P values.
The proof of Theorem 1 is provided in SI Appendix, section S.1.
E. Parent–Offspring Duos and Other Pedigrees.
The digital twin test can also be applied to offspring for whom only one parent is genotyped, with a small adjustment. The modification is simple: Whenever a parent is unknown, the algorithm fixes the offspring’s haplotype from that parent (recall the offspring’s haplotypes are observed). For example, if and are unknown, then in Algorithm 1 we set in each iteration of the loop. An analogous version of Theorem 2 continues to hold in this setting, and so we still detect only causal regions.† Furthermore, the digital twin test can be applied to data with a variety of pedigree structures. One can select any set of duos or trios from the pedigree, with the restriction that no offspring in a duo or trio is an ancestor of any other offspring in another duo or trio.
F. Linkage Disequilibrium in the Trio Design.
The TDT is testing the null hypothesis
| [6] |
Because all sites on a chromosome are dependent, this null is technically equivalent to the null in Eq. 1 when is taken to be the entire chromosome. By contrast, the digital twin test can explicitly localize the causal signals into regions by basing the test on smaller groups in Eq. 1. Depending on the levels of LD, interpreting the TDT discoveries as localizing important genetic variation can lead to confusion. We now highlight this limitation of the TDT with a practical example.
We create a synthetic population of 2,500 second-generation admixed individuals whose parents are the children of one ethnically British individual and one ethnically African individual. The haplotypes of the parents are real in the sense that they are phased haplotypes from the UK Biobank dataset (49). For simplicity, we create a binary synthetic response from a logistic regression model with a single causal SNP. We choose a signal strength such that the heritability of the trait is 18% and an intercept such that 20% of the observations have . The causal SNP is chosen at a site with a large difference in allele frequency between the British and African populations. Then, we carry out the TDT at each site and report the P values in a Manhattan plot in Fig. 3. Note that even if we demand that the P values are smaller than the genome-wide significance threshold of , the TDT reports discoveries all across the chromosome. We compare this to an identical simulation composed only of British individuals in Fig. 3, Center. Here, the TDT reports discoveries only near the true causal SNP.
Fig. 3.

Results of the TDT in two populations. (Left and Center) Manhattan plots on chromosome 22, which contains the one true causal SNP, indicated with a dashed vertical line. The genome-wide significance threshold is shown with a gray horizontal line. Left panel shows an admixed population, whereas Center panel shows a British population. (Right) A plot of the absolute correlations between the causal SNP and the other SNPs, conditional on the parental haplotypes. The red solid and blue dotted-dashed curves indicate a smoothed 90% quantile of the absolute correlation with the causal SNP across the chromosome, for the admixed and British populations, respectively.
The TDT behaves differently in these two populations because of the different correlation structure after conditioning on the parental haplotypes. In the admixed population, there are large correlations between sites far away, but not in the British population; see Fig. 3, Right. Because of the large LD in the admixed population, testing the null in Eq. 6 cannot give reliable information about the location of the causal SNP. This weakness of the TDT has been noted before in in the admixed setting (50). That work developed an analytical correction based on population-genetic quantities, whereas here we address this problem using conditional independence testing. Although the TDT can be confused by linkage disequilibrium, both the TDT and the digital twin test are robust to confounding variables that can invalidate GWAS, which we turn to next.
2. Causal Inference in the Trio Design
A. Establishing Causality.
We now explain why it is possible to draw causal inference from trio data by formulating the inheritance process as a high-dimensional randomized experiment. The main idea is to condition on the parental haplotypes: Once these are fixed the remaining randomness in meiosis is independent of possible confounders, so the resulting inferences are immune to these factors.
We begin with a concrete example of confounding in GWAS. Suppose we have a genetic study involving individuals with either French or German ancestries, and we wish to study whether a set of SNPs affects the cholesterol level . Next, suppose that both the distribution of cholesterol levels and the distribution of the SNPs differ in the two populations. As a result, there is a valid statistical association between and , but this statistical association may or may not represent a causal effect. That is, if we manipulated the SNPs , it may or may not change the cholesterol levels of the subjects. The association could instead be the result of a confounder; for example, if the consumption of beer leads to higher cholesterol and Germans consume more beer on average, then the SNPs will be associated with , even if they have no causal effect. Since randomized experiments detect only causal effects (51), to circumvent the above problem we could, in principle, flip fair coins, set the values of accordingly immediately after conception, and then check for an association with . While we of course do not carry out such experiments on people, we can exploit a similar experiment occurring in nature.
Consider a potential confounder , such as beer consumption above. The critical observation is that for essentially all possible confounders of concern in genetic studies, the distribution of a set of SNPs given the parental haplotypes does not change given knowledge of , since the randomness in inheritance is a result only of random biological processes independent of . To make this precise, we define the following set of possible confounders:
Definition 1 (External Confounder).
We say that a random variable is an external confounder if the distribution of the offspring’s haplotypes given the parental haplotypes does not change given knowledge of :
| [7] |
The relation in Eq. 7 is true for the offspring’s beer consumption, for example, as well as for all environmental conditions occurring after conception. Importantly, this implies that is independent of the offspring’s SNPs given the parental haplotypes and remaining SNPs:
This then implies that if there is an association between and after conditioning on the parental haplotypes and remaining SNPs , then the association is not due to the confounder :
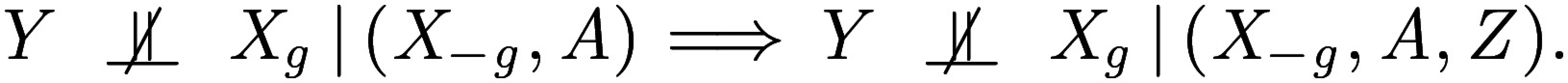 |
Returning to the language of hypothesis testing, this proves that if we test the null hypothesis in Eq. 1, we automatically account for the random variable . We record this fact formally next:
Theorem 2 (Conditioning on Parents Accounts for External Confounders).
Let be an external confounder; i.e., satisfies Eq. 7. Then, any valid test of the null hypothesis in Eq. 1 is also a valid test of the stronger null hypothesis
| [8] |
that accounts for the confounder .
In words, if we test the hypothesis in Eq. 1, which is possible based on observed trio data, then we have perfectly adjusted for the confounder , even if it is not specified or measured in the data. Thus, if we reject the null in Eq. 1, it cannot be the case that and are dependent due to an external confounder . The digital twin test is such a test, so it is immune to external confounders:
Corollary 1.
Suppose that given follows the distribution in The Haldane HMM. Then the digital twin test is a valid test of the hypothesis in Eq. 8 that accounts for the (possibly unmeasured) external confounder . That is, if the null in Eq. 8 holds, then the distribution of the output of Algorithm 1 stochastically dominates the uniform distribution.
Note that this implies that the TDT is immune to external confounders, since it is a special case of the digital twin test. This is a formal statement of the existing notion that the TDT is robust to population structure (e.g., ref. 25).
B. Connection to Structural Equation Modeling.
We next frame these results within a structural equation model to make the connection with the causal inference literature explicit, and we similarly formulate our results in the potential outcomes framework in SI Appendix, section S.3.
Consider a structural equation model involving the variables and the external confounder . For a response , we assume that can only cause and not the reverse, which is reasonable because a subject’s genotype is fixed after conception. We further know that the parental haplotypes cause and not the reverse. We also assume that causes since the reverse case does not result in confounding. Finally, by definition the external confounder is conditionally independent of given , which implies that there is no causal effect from to . The corresponding structural equation model is
where , and are fixed functions and , and are independent uniform random variables; see Fig. 4 for a graphical representation. Within this model, rejecting the hypothesis in Eq. 1 implies that there is a causal effect from to . The digital twin test makes formal causal inferences in this sense, and crucially, it does not require the analyst to specify or restrict or .
Fig. 4.

A graphical depiction of the causal argument in Causal Inference in the Trio Design. A shows that the random variable can create an association between and , even if there is no causal effect. B shows that conditional on the parental haplotypes , the external confounder is independent of the offspring’s genotype . As a result, cannot be responsible for the remaining association between the genotype and the trait . Note that in our hypothesis test we also condition on , which is omitted from the figure for simplicity.
C. Discussion of Possible Confounders.
Virtually all confounders of concern in genetic studies do not affect the transmission of the genetic information from parents to offspring and are thus external confounders which are correctly accounted for in the trio design by Theorem 2. We list the most important examples below:
-
•
Environmental conditions after conception. The mechanism for producing from is unaffected by anything occurring after conception.
-
•
Population structure, ethnic composition, and geographic location. The mechanism for producing from does not change with subpopulation information, ethnicity, or geographic location.
-
•
Cryptic relatedness. The presence of distantly related individuals in a sample does not change the distribution of given , even if this relatedness is unknown and unspecified.
-
•
Family effects, altruistic genes. Information about the quality of the environment caused by parental behavior does not impact the distribution of given .
-
•
Assortive mating. Tests of the null in Eq. 8 condition on the observed mating pattern, making them immune to this form of confounding.
By contrast, the following are not external confounders:
-
•
Germline mutations. A few environmental factors of the parents can affect the inheritance process, such as the exposure of a parent to radiation, which changes the distribution of the offspring by increasing the frequency of mutations. While this does affect the model for inheritance in principle, we do not expect this to practically invalidate tests of the null in Eq. 8. In any case, this is a narrow set of possible confounders.
-
•
Unmeasured SNPs. In typical studies, only a subset of SNPs is sequenced. Knowledge of a subject’s unmeasured SNPs gives additional information about the distribution of given , so the unmeasured SNPs are not external confounders. Since we condition on and recombination events are rare, however, our method is effectively independent of the unmeasured SNPs outside the region .
Finally, we note that there is potential for selection bias in all genetic studies, since some individuals are more likely to be included in a sample. Tests of the null in Eq. 8 are more robust to this bias, as any potential selection bias due to external confounders, such as geographic location, is automatically accounted for by Theorem 2. However, if a SNP causally influences the probability of inclusion in a study, then it is not null according to our null in Eq. 8, so it may be detected.
3. Simulation Experiments
In this section, we examine the performance of the digital twin test in semisynthetic examples, focusing on the binary response case so that the standard TDT can serve as a benchmark. We form our parent–offspring population by taking real haplotypes from the UK Biobank dataset and sample offspring according to the recombination model in The Haldane HMM. In each experiment, we sample the offspring once and then repeat the generation of the synthetic phenotype multiple times, indicating the SE with error bars. When presenting the results, we index the signal strength by heritability: a -valued scale defined in SI Appendix, section S.5. An R package implementing the methods below together with notebook tutorials is available at https://github.com/stephenbates19/digitaltwins (52).
A. Testing the Global Causal Null.
We first examine the ability of the digital twin test to test the global causal null. In this simulation, we test only one hypothesis, so we seek to control the usual type I error rate at the level. We create a synthetic population of parent–child trios and generate a binary valued response coming from a sparse logistic regression model
| [9] |
with 10 nonzero entries of of equal value, chosen uniformly at random. The intercept is chosen so that the fraction of cases is , or . We use SNPs from chromosome 20, which has width 63 Mb. We emphasize that the above gives a well-defined structural equation model on , and the nonzero entries of the coefficient vector correspond to the SNPs that have a causal effect.
To illustrate how the digital twin test can be used for confirmatory analysis, we make an external GWAS dataset using nontrio observations from the UK Biobank (not included in our previous sample) and generating phenotypes with the same rule as above. We use these GWAS data to fit an -penalized logistic regression model (with regularization parameter chosen by cross-validation) to obtain a predictive model for the trait. We denote as the resulting coefficient estimate. Then, we apply the digital twin test on the trio data with the feature importance statistic in Eq. 4 to produce a single P value, rejecting when it falls below .
We take the TDT as a natural benchmark, interpreting its output in two alternative ways. First, we take the minimum P value after applying the TDT at every SNP and then Bonferroni correct it (this does not use the nontrio GWAS data). Second, we compute the Bonferroni-adjusted minimum P value only on the coordinates with nonzero coefficients in the lasso fit on the external GWAS data. Because is sparse, this method has a less severe Bonferroni correction and may be more powerful than the other TDT procedure.
Since all three methods are valid tests of the null hypothesis that there is no causal SNP on the chromosome, we directly compare their power in Fig. 5, using 20 independent realizations for each data point. We find that the digital twin test has higher power than the TDT, even when the latter attempts to leverage the external GWAS as a screening step. The leftmost point in each panel is the null case with zero heritability; the empirical error of the digital twin test does not exceed the nominal level of in any of the three cases.
Fig. 5.

Power of the digital twin test compared to TDT benchmarks for testing the full-chromosome causal null.
B. Localization.
We now examine the ability of the digital twin test to identify causal regions. Here, we use SNPs on chromosomes 1 to 22, split into 532 predetermined groups of size approximately 5 Mb. The response is again generated from the logistic regression model in Eq. 9, and the number of nonzero coefficients in the true causal model is varied as a control parameter. We consider a sample of trios with an external GWAS of size used to fit a logistic regression model as in the previous section. The fitted coefficients are used to form the test statistic in Eq. 4. Here, we take the nominal level for the FDR to be . Each experiment is repeated 10 times. Additional technical details about these simulations can be found in SI Appendix, section S.5.
We compare the following procedures:
-
•
Digital twin test–accum. We apply the digital twin test at each of the groups to obtain one P value per group. We also use the external GWAS data to order the regions from most to least promising as in Eq. 5 and use an accumulation test (45) to produce a final set of discoveries. This method is guaranteed to control the FDR.§
-
•
TDT–Screen–BH. For each group, we apply the TDT to the SNPs with nonzero entries of , the model fit on the external GWAS. Then, we report the minimum P value after adjusting it with Bonferroni. Finally, we apply the Benjamini–Hochberg procedure to report a set of groups. This method assumes the TDT P values are valid for the group null hypothesis in Eq. 1, which is not fully correct, so this method does not have formal guarantees.
-
•
TDT–BH. We proceed as above, except that we apply the TDT to all SNPs. This method also incorrectly assumes the TDT P values are valid for the group null hypothesis in Eq. 1, so it does not have formal guarantees.
We report the results in Fig. 6. The digital twin test has generally comparable power to the screened TDT method with Benjamini–Hochberg, with moderate power improvements for traits caused by many SNPs. We report on a similar experiment with a continuous response in SI Appendix, section S.5.
Fig. 6.

Performance of the digital twin test and TDT in the binary-response full-genome simulations from Localization. Here, error bars give one SD and the dashed horizontal line indicates the nominal FDR level.
Although the TDT benchmarks empirically control the FDR here, we emphasize that they test the full-chromosome null in Eq. 6, so they are not valid for our goal of localizing the causal SNPs into the given groups. The group hypothesis in Eq. 1 can be rigorously tested by a test of the null in Eq. 6 only if the SNPs in different groups are independent. If the TDT-based group P values were valid, these two benchmarks would control the FDR. This is a reasonable approximation within this experiment, because the groups are wide and the population is homogeneous, so the LD decays rapidly. However, this assumption fails in other cases, as we turn to next.
C. Spurious Discoveries with the TDT.
We have seen in Linkage Disequilibrium in the Trio Design an example where the TDT makes false discoveries throughout the chromosome because it does not account for LD. Here, we revisit this example more carefully, demonstrating that the TDT-based benchmarks above can dramatically fail to control type I errors for the group hypotheses.
For simplicity, we perform the TDT at each SNP and report a SNP as significant if the P value is below the genome-wide significance level, . We use this demanding threshold to emphasize that even the most conservative existing methods violate type I error control here. Proceeding as in the previous experiment, we split the chromosome into 25 groups of size 2 Mb and consider the procedure that reports a group as significant if any SNP therein has a P value below the genome-wide significance threshold. Each experimental setting is repeated 20 times. Fig. 7 shows that this TDT benchmark badly violates FDR control, suggesting all of the other less conservative TDT benchmarks will also fail to control the FDR in this setting. We report additional metrics about the TDT in SI Appendix, Fig. S.4. By contrast, the digital twin test controls the FDR because its P values are valid for the conditional null hypotheses in Eq. 1.
Fig. 7.

Performance of the digital twin test and TDT in an admixed population. The dashed horizontal line (Top row) indicates the nominal FDR level for the digital twin test. Because the TDT is using the genome-wide significance level, the nominal FDR level for the TDT is less than 0.05.
4. Analysis of Autism Spectrum Disorders
We apply the digital twin test to study the genetic basis of autism spectrum disorder (ASD) using a dataset of 2,565 parent–child trios from the Autism Sequencing Consortium (ASC) (53), accessed through dbGaP (54, 55); see SI Appendix, section S.4 for details about the sample and data processing. ASD has complex genetic roots, with variability arising from common SNPs, copy number variation, and de novo gene-disrupting mutations (56, 57). It has been theoretically conjectured (58) and empirically observed (59) that common variants have only small effects on ASD. As a result, only recently have individual SNPs been implicated by GWAS, and the first individual SNPs appear in ref. 60, where five SNPs were reported at the genome-wide significance level. One such SNP, rs910805 (an intergenic variant on chromosome 20), was also genotyped in the ASC trio data, and we test its causal validity with the digital twin test. Note, however, that the ASC trio data were used as part of ref. 60, so our results here must be viewed as a practical demonstration and not as an independent replication of these findings.
Since we do not have a corresponding cohort of nontrio observations, we use the digital twin test with the test statistic in Eq. 3 and report the results in Table 1. We test groups centered around SNP rs910805 of increasing size, ranging from 1 Mb to the whole chromosome (in which case this digital twin test is equivalent to the TDT). For large enough groups, we find significance at the level—we are testing only one SNP so we do not need to achieve the genome-wide significance level. We are unable to reject at finer resolutions; smaller groups correspond to fundamentally more demanding statistical hypotheses, and the effect size here is small. Our analysis suggests that the observed association with rs910805 is not due to confounding and that there is instead a causal variant in its vicinity.
Table 1.
Analysis of ASD with digital twin test at different resolutions
| Resolution | 1 Mb | 2 Mb | 3 Mb | 4 Mb | Full chromosome |
| P value | 0.237 | 0.146 | 0.100 | 0.0168 | 0.011 |
Discussion
We have developed a statistical test to rigorously establish that a genomic region contains causal variants; the inferences are based only on variation arising in meiosis, a randomized experiment performed by nature. Next, we highlight the two most important limitations of this work. First, our method currently relies on computationally phased haplotypes. Although phasing is typically accurate for family data and numerical experiments show that the digital twin test works well with computationally phased data (SI Appendix, section S.5), it would be interesting to thoroughly understand the robustness to phasing errors. Second, to operate at practical resolutions, our method requires many parent–child trios or duos, which are more challenging to collect than unrelated individuals. As such, contemporary studies genotyping entire populations [e.g., in Iceland (61) and Finland (62)] are particularly promising because they simultaneously address the challenges of accurate phasing and obtaining data from both parents and offspring. Here, drawing conclusions from GWAS that are provably immune to confounding variables is appealing, making the digital twin test a tantalizing way to confirm candidate GWAS discoveries.
Turning to the statistical aspect of this work, our finite-sample causal conclusions come from the fact that 1) we measure a random variable that blocks the relevant confounders and 2) we exactly know the distribution of given . This statistical form may appear in other settings. Concretely, for any random variables , and such that the relationship in Definition 1 holds, Theorem 2 holds as well. More generally, for a structural equation model on vectors , if satisfies the back-door criterion (63) with respect to and , then tests of the hypothesis Eq. 1 detect only causal effects. One can then leverage conditional testing techniques, such as the conditional randomization test or knockoffs, to make causal inferences with finite-sample guarantees.
Supplementary Material
Acknowledgments
S.B. is partly supported by a Ric Weiland Fellowship. S.B., M.S., C.S., and E.C. are supported by NSF Grant DMS 1712800 (M.S. was in the Department of Statistics at Stanford University). C.S. and E.C. are also supported by a Math+X grant (Simons Foundation) and by NSF Grant 1934578. E.C. is also supported by Office of Naval Research Grant N000142012157 and a generous gift from TwoSigma. We thank Trevor Hastie, Manuel Rivas, Robert Tibshirani, and Wing Wong for discussions of an early version of this manuscript and the Stanford Research Computing Center for computational resources and support.
A. Constructing Independent P Values
Here, we present a technical refinement of the digital twin test (still testing the null hypothesis in Eq. 1) that produces independent P values for disjoint null groups. The core idea is that for each individual, we determine which groups contain a recombination event; then, we generate the digital twins only in those groups, sampling from a modified version of the distribution Eq. 2. By construction, these digital twins will have recombination events in the same groups as the true observations, but the recombination events will be randomly perturbed. This separates the randomness used to test each region. We then show how to create feature importance statistics that ignore the randomness in other regions. As a result, the tests in different regions are fully decoupled.
To begin, let denote the collection of disjoint groups of SNPs that we wish to test. We will assume that each group is a continuous block of the form for endpoints in . Also, let be the set of SNPs that form the boundary of some group: .
For simplicity, we consider a single observation and define and to be the underlying ancestral states for the two haplotypes and . We first sample from the posterior distribution conditional on the data based on the HMM described in The Haldane HMM and then condition on and . This conditioning splits the distribution of and into independent HMMs across the groups ; see ref. 64 for a general discussion of this conditioning idea. In the modified digital twin test, each digital twin will be sampled from the distribution of
| [10] |
This is essentially the same as in the original digital twin test, which instead samples from Eq. 2, because the values are almost perfectly determined by the data and , since the measured SNPs are dense and recombination is rare. Sampling from the distribution in Eq. 10 is straightforward: We simply sample and , an HMM sampling operation, and then from this we sample and from the emission distribution.
We now introduce additional notation that will be needed to describe the digital twin sampling. Given and , for each observation , let be the set of groups where individual has an odd number of recombinations (for small groups, there will typically be exactly one recombination). Define in the analogous way. Now, let be the set of observations and sites in groups with an even number of recombinations. Define in the analogous way. The upcoming algorithm will hold for fixed when generating the digital twins.
Next, we define the masked version of which is safe to use in the feature importance statistics; feature importance statistics using the masked version of will operate independently in different groups. The masked version is defined as
| [11] |
for each observation . The entries of in groups with no recombination events will remain unchanged in both the digital twins and . On the other hand, for groups with observed recombinations, the digital twins will vary, so, for these entries, we set to be a constant: the average imputation based on the parental haplotypes and . These conditional expectations can be computed easily, due to the Markov property. Note that, since there are very few groups with recombination events, most entries of will be equal to those of , and most entries of will be equal to those of . For instance, with groups of size 1 Mb we will have that over of the entries match.
With the notation in place, we can finally present the procedure in Algorithm 2.
Algorithm 2: The digital twin test with independent P values
| Sample and given according to the |
| HMM in The Haldane HMM (see SI Appendix, section S.2 |
| for an explicit sampler). |
| Define and according to Eq. 11. |
| for do |
| Compute the test statistic on the true data: |
| for do |
| For observations such that , sample |
| from the distribution in Eq. 10, independent of |
| and (see SI Appendix, section S.2 |
| for an explicit sampler). Otherwise, set . |
| Sample analogously. |
| Compute the test statistic on the digital twins: |
| Compute the quantile of the true statistic among the |
| digital twin statistics : |
| randomly breaking any ties. |
The null P values produced by this algorithm are jointly independent, which we record in Theorem 1 in The Digital Twin Test. The proof is given in SI Appendix, section S.1.
B. The Haldane HMM
In service of our tests of the hypothesis in Eq. 1, we formally describe the distribution of offspring’s genotype given the parental haplotypes. In particular, the process by which a subject’s two haplotypes arise from the parental haplotypes was formalized by Haldane as an HMM (39). Without loss of generality, we describe the model for a single observation on one chromosome; in the general case, each chromosome of each observation is an independent instance of this model. For concreteness, we focus on .
Let the random vector indicate the following:
Our model is that is distributed as a Markov chain, where and
Here, is the genetic distance between SNPs and , which is fixed and known. Note that the genetic distance is not always proportional to the physical distance due to recombination hotspots: regions that have more frequent recombination events (65, 66). Conditional on , each is independently sampled from
Here, is the probability of a de novo mutation, which for humans is about (67). The analogous HMM describes the distribution of given and , which is taken to be independent of given .
C. Sampling Full-Chromosome Digital Twins
In this section, we explicitly describe how to sample the digital twins in Algorithm 1 for the simple case where is an entire chromosome. Explicit samplers for other cases require substantial additional notation, so we instead present them in SI Appendix, section S.2. For convenience, we consider a single observation, i.e., . Without loss of generality, we consider one chromosome; different chromosomes are independent, so this is sufficient. In addition, for we define when and when , i.e., the complementary ancestral strand. A digital twin is simply an independent draw from the model in The Haldane HMM. This is sampled as follows:
-
1)
(Hidden ancestral states) Sample uniformly from .
-
2)
For , independently sample as
-
3)
(De novo mutations) For each , independently sample as
The digital twin is sampled analogously.
Footnotes
The authors declare no competing interest.
†We simply condition on rather than and for each unit where the father’s haplotypes are unknown, and so on.
The reader may wonder about the identifiability of this model. Note that both and are random variables, so provided that the distribution of is not contained in a subspace of rank less than , then this model is identified.
§Strictly speaking, this procedure controls a modified version of the FDR (45), but the difference will be unimportant in settings with a large number of discoveries. This is a property of the accumulation test, not the digital twin test, and other procedures can be used for standard FDR control.
See online for related content such as Commentaries.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2007743117/-/DCSupplemental.
Data Availability.
Some study data are available. An R package implementing the methods below together with notebook tutorials is available at https://github.com/stephenbates19/digitaltwins (52).
Change History
October 6, 2020: The license for this article has been updated.
References
- 1.Visscher P. M., et al. , 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Devlin B., Roeder K., Genomic control for association studies. Biometrics 55, 997–1004 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Cordell H. J., Clayton D. G., Genetic association studies. Lancet 366, 1121–1131 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Price A. L., et al. , Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Kang H. M., et al. , Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubin D. B., Estimating causal effects of treatments in randomized and nonrandomized studies. J. Educ. Psychol. 66, 688–701 (1974). [Google Scholar]
- 7.Rubinstein P., et al. , Genetics of HLA-disease associations. the use of the haplotype relative risk (HRR) and the ‘haplo-delta’ (Dh) estimates in juvenile diabetes from three racial groups. Hum. Immunol. 3, 384 (1981). [Google Scholar]
- 8.Falk C. T., Rubinstein P., Haplotype relative risks: An easy reliable way to construct a proper control sample for risk calculations. Ann. Hum. Genet. 51, 227–233 (1987). [DOI] [PubMed] [Google Scholar]
- 9.Terwilliger J., Ott J., A haplotype-based haplotype relative risk’ approach to detecting allelic associations. Hum. Hered. 42, 337–346 (1992). [DOI] [PubMed] [Google Scholar]
- 10.Ott J., Kamatani Y., Lathrop M., Family-based designs for genome-wide association studies. Nat. Rev. Genet. 12, 465–474 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Spielman R. S., McGinnis R. E., Ewens W. J., Transmission test for linkage disequilibrium: The insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet. 52, 506–516 (1993). [PMC free article] [PubMed] [Google Scholar]
- 12.Spielman R. S., Ewens W. J., The TDT and other family-based tests for linkage disequilibrium and association. Am. J. Hum. Genet. 59, 983–989 (1996). [PMC free article] [PubMed] [Google Scholar]
- 13.Thomson G., Mapping disease genes: Family-based association studies. Am. J. Hum. Genet. 57, 487–498 (1995). [PMC free article] [PubMed] [Google Scholar]
- 14.Sinsheimer J. S., Blangero J., Lange K., Gamete-competition models. Am. J. Hum. Genet. 66, 1168–1172 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabinowitz D., Laird N., A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum. Hered. 50, 211–223 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Lange C., Laird N. M., Power calculations for a general class of family-based association tests: Dichotomous traits. Am. J. Hum. Genet. 71, 575–584 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazzeroni L. C., Lange K., A conditional inference framework for extending the transmission/disequilibrium test. Hum. Hered. 48, 67–81 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Xu X., Rakovski C. S., Xu X., Laird N. M., An efficient family-based association test using multiple markers. Genet. Epidemiol. 31, 789–796 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Rakovski C. S., Weiss S. T., Laird N. M., Lange C., FBAT-SNP-PC: An approach for multiple markers and single trait in family-based association tests. Hum. Hered. 66, 122–126 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Allison D. B., Transmission-disequilibrium tests for quantitative traits. Am. J. Hum. Genet. 60, 676–690 (1997). [PMC free article] [PubMed] [Google Scholar]
- 21.Rabinowitz D., A transmission disequilibrium test for quantitative trait loci. Hum. Hered. 47, 342–350 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Monks S. A., Kaplan N. L., Removing the sampling restrictions from family-based tests of association for a quantitative-trait locus. Am. J. Hum. Genet. 66, 576–592 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ewens W. J., Li M., Spielman R. S., A review of family-based tests for linkage disequilibrium between a quantitative trait and a genetic marker. PLoS Genet. 4, 1–6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Steen K., et al. , Genomic screening and replication using the same data set in family-based association testing. Nat. Genet. 37, 683–691 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Laird N. M., Lange C., The role of family-based designs in genome-wide association studies. Stat. Sci. 24, 388–397 (2010). [Google Scholar]
- 26.Spirtes P., Glymour C., Scheines R., Causation, Prediction, and Search (MIT Press, ed. 2, 2000). [Google Scholar]
- 27.Kalisch M., Bühlmann P., Estimating high-dimensional directed acyclic graphs with the PC-algorithm. J. Mach. Learn. Res. 8, 613–636 (2007). [Google Scholar]
- 28.Maathuis M. H., Kalisch M., Bühlmann P., Estimating high-dimensional intervention effects from observational data. Ann. Stat. 37, 3133–3164 (2009). [Google Scholar]
- 29.Chickering D. Optimal structure identification with greedy search. J. Mach. Learn. Res. 3, 507–554, 2002. . [Google Scholar]
- 30.He Y., Geng Z., Active learning of causal networks with intervention experiments and optimal designs. J. Mach. Learn. Res. 9, 2523–2547 (2008). [Google Scholar]
- 31.Candès E., Fan Y., Janson L., Lv J., Panning for gold: Model-X knockoffs for high dimensional controlled variable selection. J. R. Stat. Soc. Ser. B Stat. Methodol. 80, 551–577 (2018). [Google Scholar]
- 32.Barber R. F., Candès E., Controlling the false discovery rate via knockoffs. Ann. Stat. 43, 2055–2085 (2015). [Google Scholar]
- 33.Sesia M., Sabatti C., Candès E., Gene hunting with hidden Markov model knockoffs. Biometrika 106, 1–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sesia M., Sabatti C., Candès E., Rejoinder: ‘Gene hunting with hidden Markov model knockoffs’. Biometrika 106, 35–45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sesia M., Katsevich E., Bates S., Candès E., Sabatti C., Multi-resolution localization of causal variants across the genome. Nat. Commun. 11, 1093 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Browning S. R., Browning B. L., Haplotype phasing: Existing methods and new developments. Nat. Rev. Genet. 12, 703–714 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marchini J., et al. , A comparison of phasing algorithms for trios and unrelated individuals. Am. J. Hum. Genet. 78, 437–450 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Connell J., et al. , A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 10, e1004234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haldane J. B. S., The combination of linkage values and the calculation of distances between the loci of linked factors. J. Genet. 8, 299–309 (1919). [Google Scholar]
- 40.Dai R., Barber R., “The knockoff filter for FDR control in group-sparse and multitask regression”in International Conference on Machine Learning, Balcan M. F., Weinberger K. Q., Eds. (PMLR, 2016), pp. 1851–1859. [Google Scholar]
- 41.Renaux C., Buzdugan L., Kalisch M., Bühlmann P., Hierarchical inference for genome-wide association studies: A view on methodology with software. Comput. Stat. 35, 1–40 (2020). [Google Scholar]
- 42.Tansey W., Veitch V., Zhang H., Rabadan R., Blei D. M.. The holdout randomization test: Principled and easy black box feature selection. arXiv:1811.00645 (1 November 2018).
- 43.Roeder K., Wasserman L., Genome-wide significance levels and weighted hypothesis testing. Stat. Sci. 24, 398–413 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ren Z., Candès E., Knockoffs with side information. arXiv:2001.07835 (22 January 2020).
- 45.Li A., Barber R. F., Accumulation tests for FDR control in ordered hypothesis testing. J. Am. Stat. Assoc. 112, 837–849 (2017). [Google Scholar]
- 46.Loh P.-R., Kichaev G., Gazal S., Schoech A. P., Price A. L., Mixed-model association for biobank-scale datasets. Nat. Genet. 50, 906–908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57, 289–300 (1995). [Google Scholar]
- 48.Efron B., Large-Scale Inference: Empirical Bayes Methods for Estimation, Testing, and Prediction (Institute of Mathematical Statistics Monographs, Cambridge University Press, Cambridge, UK, 2012). [Google Scholar]
- 49.Bycroft C., et al. , The UK biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X., Xiao R., Zhu X., Li M., Gene mapping in admixed families: A cautionary note on the interpretation of the transmission disequilibrium test and a possible solution. Hum. Hered. 81, 106–116 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Fisher R. A., Statistical Methods for Research Workers (Oliver & Boyd, Edinburgh, Scotland, 1925). [Google Scholar]
- 52.Bates S., Sesia M., digitaltwins R package. GitHub. https://github.com/stephenbates19/digitaltwins 24 February 2020.
- 53.Buxbaum J. D., et al. , The autism sequencing consortium: Large-scale, high-throughput sequencing in autism spectrum disorders. Neuron 76, 1052–1056 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mailman M. D., et al. , The NCBI dbGaP database of genotypes and phenotypes. Nat. Genet. 39, 1181–1186 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tryka K. A., et al. NCBI’s database of genotypes and phenotypes: dbGaP. Nucleic Acids Res. 42, 975–979 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iossifov I., et al. , The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gaugler T., et al. , Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Devlin B., Melhem N., Roeder K., Do common variants play a role in risk for autism? Evidence and theoretical musings. Brain Res. 1380, 78–84 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anney R., et al. , Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum. Mol. Genet. 21, 4781–4792 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grove J., et al. , Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.deCODE Genetics , https://www.decode.com/. Accessed 6 December 2019.
- 62.FinnGen , https://www.finngen.fi/en. Accessed 6 December 2019.
- 63.J. Pearl, Causality: Models, Reasoning and Inference (Cambridge University Press, ed. 2, 2009). [Google Scholar]
- 64.Bates S., Candès E., Janson L., Wang W., Metropolized knockoff sampling. J. Am. Stat. Assoc., 10.1080/01621459.2020.1729163 (2020). [DOI] [Google Scholar]
- 65.Altshuler D., Donnelly P., International HapMap Consortium , A haplotype map of the human genome. Nature 437, 1299–1320 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bherer C., Campbell C. L., Auton A., Refined genetic maps reveal sexual dimorphism in human meiotic recombination at multiple scales. Nat. Commun. 8, 14994 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Acuna-Hidalgo R., Veltman J. A., Hoischen A., New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 17, 241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Some study data are available. An R package implementing the methods below together with notebook tutorials is available at https://github.com/stephenbates19/digitaltwins (52).