

Hereditary spastic paraplegias (HSP) are a group of genetically and phenotypically heterogeneous disorders resulting from progressive degeneration of the corticospinal tract. 1 They may have autosomal dominant, autosomal recessive, x‐linked, or mitochondrial inheritance, and the main clinical features derive from lower limb spasticity. There are now more than 80 genes or loci associated to this condition, globally identified as spastic paraplegia genes (SPG). 1 , 2
The SPG15 subtype represents 2%–4% of the autosomal recessive inherited HSP and are associated to mutations in the ZFYVE26 gene that code for spastizin. It belongs to the group of autosomal recessive HSP with thin corpus callosum (AR HSP‐TCC), which includes a diverse genetic basis covering SPG11, SPG15, SPG21, SPG32, and SPG 47. 3 , 4
SPG15 is clinically characterized by progressive lower limbs spasticity that may be associated to a variable number of other manifestations such as cognitive impairment, retinal degeneration, motor neuropathy, dysarthria, and rarely parkinsonism, although the knowledge about its phenotypic spectrum is still limited. 5 Here, we report a case of a juvenile‐onset levodopa‐responsive parkinsonism associated to SPG 15 that developed motor complications under chronic levodopa therapy.
Case Report
A young Brazilian man, born from non‐consanguineous parents, with an apparent normal neuropsychomotor development, presented school difficulties initiating at childhood. In addition, at the age of 10, he developed progressive dysarthria, stiffness, and bradykinesia of the right limbs that gradually progressed to involve all four limbs, associated to gait slowing. At the age of 14, he was referred to our service for evaluation, being diagnosed as having juvenile‐onset asymmetric parkinsonism. He started levodopa as the first dopaminergic treatment with excellent response. However, there was a gradual need for dosage increase and pramipexole was associated to the treatment. The brain magnetic resonance imaging (MRI) showed thinning of the corpus callosum and the “sign of the ears of the lynx” (Fig. 1A,B), which directed the genetic tests. Analysis of the ZFYVE26 gene revealed the presence of a known homozygous deletion (c.918delA), confirming the diagnosis of SPG15. Other complementary tests were normal (complete blood count, iron metabolism analysis, copper metabolism analysis, and serum ceruloplasmin). Nerve conduction studies and electroencephalography were also within the normal range. At the age of 17, he started to present motor complications related to dopaminergic treatment with wearing‐off, off‐dystonia, and peak‐dose dyskinesia. On clinical examination in the “off” state, there was marked global bradykinesia and dystonic posture of the right arm, Unified Parkinson's disease rating scale motor scoring was 78 out of 108 points (video segment 2). The levodopa challenge test after administration of 250 mg of levodopa led to a 72% improvement in motor score (22 out of 108 points) and to the appearance of generalized severe choreic movements in the “on state” (video segment 1). At this time, the mini mental state examination (MMSE) revealed a score of 23 (lost 5 points in calculation, 1 point in day of the month, and 1 point in drawing). He was independent for basic and instrumental activities of daily living. Eye movement's examination was normal, and there was mild spasticity of the lower limbs, no cerebellar signs, or other abnormal findings on neurological examination.
FIG. 1.
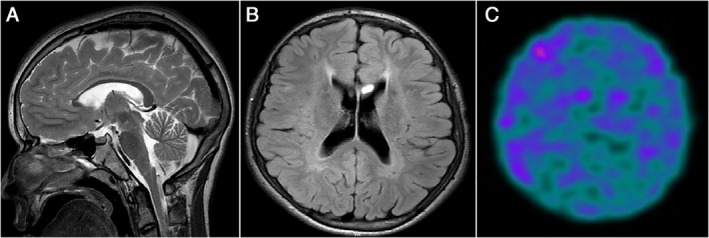
(A) Sagittal T2‐weighted brain MRI showing marked thinning of corpus callosum. (B) Axial FLAIR brain MRI showing hypersignal of the anterior periventricular white matter, “ears of the lynx sign”. (C) [99mTc] TRODAT‐1 SPECT showed markedly reduced dopamine transporter levels in the striatum.
[99mTc]TRODAT‐1 SPECT (Fig. 1C) demonstrated a severe symmetric reduction of striatal dopaminergic terminals. Amantadine was started for better control of dyskinesia.
Discussion
Parkinsonism has already been reported in AR HSP‐TCC patients, initially SPG11 was proposed as a cause of atypical juvenile parkinsonism and later confirmed by other studies. 6 , 7 Shortly thereafter, mutations in Spastizin gene were found in three Turkish descent brothers and in a patient of Portuguese origin, characterizing the SPG15 phenotype. 8 , 9 They all had parkinsonism responsive to levodopa but the authors did not describe motor complications due to chronic dopaminergic treatment. More recently a 30‐year‐old woman with SPG15 presented a similar phenotype. 10
Our case strengthens the previous evidence that levodopa‐responsive parkinsonism, especially of juvenile‐onset, should be included in the SPG15 phenotype. It also describes, for the first time, the development of motor fluctuations and dyskinesia induced by chronic levodopa treatment in a patient with juvenile parkinsonism associated to SPG15. These findings not only extend the phenotypic spectrum of SPG15 but also provide support for phenotypic overlap between SPG15 and SPG11. We conclude that the benefit of dopaminergic therapy emphasizes the importance of identifying parkinsonian characteristics in HSP patients.
In conclusion, SPG15 should be suspected in patients with juvenile‐onset atypical parkinsonism responsive to levodopa presenting signs of spasticity, motor neuropathy, pigmentary maculopathy, cognitive impairment, family history suggestive of HSP, or the typical MRI brain findings (thin corpus callosum and the “sign of the ears of the lynx”).
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Manuscript: A. Writing of the First Draft, B. Review and Critique.
F.M.M.A.: 1A, 1B, 1C, 2A, 2B
W.M.:1A, 1B, 2B
P.J.T.: 1A, 1B, 2B
Â.V.P.: 1A, 1B, 2A
M.M.C.M.B.: 1B, 1C, 2A
V.T.: 1A, 1B, 1C, 2A, 2B
Disclosures
Ethical Compliance Statement
This study was submitted to and approved by the Ethics Committee of Ribeirão Preto Medical School Hospital, University of São Paulo‐USP. Reference number: 3.892.743. Submitted via Platform Brasil (https://platformbrasil.saude.gov.br). A written and verbalized consent form (“free and informed consent form”) was presented to the patient, obtained according to the guidelines and guidelines of the local ethics committee, the Wiley Online Library, the Movement Disorders Clinical Practice, and the Law United States Health Insurance Portability and Liability Act 1996 (“HIPAA”), including all necessary information, including all. key elements in the patient's consent form, in accordance with HIPAA. Permission was obtained and signed by the patient himself, in accordance with any laws regarding patient authorizations related to the use or disclosure of health information. This declaration of consent can be provided to the magazine or to its readers, if requested. Finally, we confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with these guidelines.
Funding Sources and Conflicts of Interest
No specific funding was received for this work. The authors declare that there are no relevant conflicts of interest for this work.
Financial Disclosures for the Previous 12 Months
The authors declare that there are no additional disclosures to be reported.
Supporting information
Video S1. Segment 1: patient in “on state” presenting significant improvement in speech, facial expression, bradykinesia, and stiffness. He is able to get up from the chair and walk without help. There are generalized choreic movements (peak‐dose dyskinesia). Segment 2: patient in “off state” presenting dysarthria, hypomimia, and severe hypokinesia, associated to dystonic right arm posture. He is unable to get up from the chair and walk without help
References
- 1. Burguez D, Polese‐Bonatto M, Scudeiro LAJ, et al. Clinical and molecular characterization of hereditary spastic paraplegias: a nextgeneration sequencing panel approach. J Neurol Sci 2017;383:18–25. [DOI] [PubMed] [Google Scholar]
- 2. Pensato V, Castellotti B, Gellera C, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 2014;137(Pt. 7):1907–1920. [DOI] [PubMed] [Google Scholar]
- 3. Fink JK. Hereditary spastic paraplegia: clinico‐pathologic features and emerging molecular mechanisms. Acta Neuropathol 2013;126(3):307–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brockmann K, Simpson MA, Faber A, Bönnemann C, Crosby AH, Gärtner J. Complicated hereditary spastic paraplegia with thin corpus callosum (HSP‐TCC) and childhood onset. Neuropediatrics 2005;36:274–278. [DOI] [PubMed] [Google Scholar]
- 5. Hanein S, Martin E, Boukhris A, et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal‐recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet 2008;82:992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anheim M, Lagier‐Tourenne C, Stevanin G, et al. SPG11 spastic paraplegia. A new cause of juvenile parkinsonism. J Neurol 2009;256:104–108. [DOI] [PubMed] [Google Scholar]
- 7. Guidubaldi A, Piano C, Santorelli FM, Silvestri G, Petracca M, Tessa A, Bentivoglio AR. Novel mutations in SPG11 cause hereditary spastic paraplegia associated with early‐onset levodopa‐responsive parkinsonism. Mov Disord 2011;26:553–556. [DOI] [PubMed] [Google Scholar]
- 8. Schincks J, Synofzik M, Petursson H, et al. Atypical juvenile parkinsonism in a consanguineous SPG 15 family. Mov Disord 2011;26:564–566. [DOI] [PubMed] [Google Scholar]
- 9. Mallaret M, Lagha‐Boukbizza O, Biskup S, et al. SPG 15: a cause of juvenile atypical levodopa responsive parkinsonism. J Neurol 2014;261:435–437. [DOI] [PubMed] [Google Scholar]
- 10. Picillo M, Munhoz R, Fasano A, et al. When shaking during standing points to hereditary spastic paraplegias. Parkinsonism Relat Disord 2018;46:92–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Segment 1: patient in “on state” presenting significant improvement in speech, facial expression, bradykinesia, and stiffness. He is able to get up from the chair and walk without help. There are generalized choreic movements (peak‐dose dyskinesia). Segment 2: patient in “off state” presenting dysarthria, hypomimia, and severe hypokinesia, associated to dystonic right arm posture. He is unable to get up from the chair and walk without help