

ABSTRACT
Recent reports have revealed diverse and abundant fungal communities in the deep-sea biosphere, while their composition, distribution, and variations in seamount zones are poorly understood. Using a metabarcoding approach targeting the ITS2 regions, we present the structure of the fungal community in 18 sediment samples from the Magellan seamount area of the northwest Pacific.
A total of 1,979 fungal OTUs was obtained, which were taxonomically assigned to seven phyla, 17 classes, 43 orders, 7 families, and 98 genera. The majority of these OTUs were affiliated to Basidiomycota (873 OTUs, 44.11% of total OTUs) and Ascomycota (486 OTUs, 24.56% of total OTUs), followed by other five minor phyla (Mortierellomycota, Chytridiomycota, Mucoromycota, Glomeromycota, and Monoblepharidomycota). Sordriomycetes is the most abundant class, followed by Eurotiomycetes, and Dothideomycetes. Five genera were common in most of the samples, including worldwide reported genera Aspergillus, Cladosporium, Fusarium, Chaetomium, and Penicillium. The environmental data we collected (sampling depth, sampling location latitude and longitude, organic carbon content, and organic nitrogen content in the sediment) had no significant influence on the composition and distribution of fungal communities. Our findings provide valuable information for understanding the distribution and potential ecological functions of fungi in the deep-sea sediments of the Magellan seamounts.
KEYWORDS: Deep-sea sediment, environmental factors, fungal diversity, high-throughput sequencing, seamount area
Introduction
Deep-sea (> 1000 m) covers more than 65% of the Earth’s surface and fulfils a range of key ecosystem functions (Danovaro 2012). Although the deep-sea environment is characterised by the absence of sunlight irradiation, predominantly low temperature, and high hydrostatic pressure, the fungal community is diverse in this extreme environment where fungi are major components of microeukaryotes and play critical roles (Nagano and Nagahama 2012). Since the first report of deep-sea fungi isolated from the Atlantic Ocean at a depth of 4,450 m (Roth et al. 1964), an increasing number of fungal species was found in several deep-sea environments, e.g.: sediments from the Mariana Trench (Takami et al. 1997), the Yap Trench (Xu et al. 2019), the hydrothermal site of South Mid-Atlantic Ridge (Xu et al. 2017), calcareous sediments (Raghukumar and Raghukumar 1998), the Chagos Trench (RaghuKumar et al. 2004), the Central Indian Basin (Damare et al. 2006; Singh et al. 2010), and the deep-sea coral (Galkievicz et al. 2012). These studies clearly illustrate the increasing attention being paid to fungal abundance and diversity in deep-sea environments.
Microorganisms have also been found in the deep-sea area of the Pacific Ocean, such as the deep-sea volcano (Akerman et al. 2013), hydrothermal vent (Fortunato and Huber 2016), and water column (Li et al. 2019). The fungal community in deep-sea sediments from the different Pacific area has been reported before (Zhou et al. 2007; Burgaud et al. 2009, 2010; Nagano et al. 2010; Nagahama et al. 2011; Rédou et al. 2015; Zhang et al. 2015; Xu et al. 2014, 2016, 2018a, 2019). However, it is still insufficient when comparing Bacteria and Archaea communities (Wu et al. 2013; Luo et al. 2015; Zhang et al. 2015, 2018; Walsh et al. 2016; Bienhold et al. 2016; Peoples et al. 2019). Especially, there is a lack of information on fungal richness, diversity, and potential ecological roles in the Pacific seamount area.
Seamounts are undersea mountains that rise steeply from the sea bottom to below sea level, defined as having an elevation of more than 1000 m with a limited extent across the summit (Menard 1964). Seamounts in the world’s oceans are numerous, especially in the Pacific Ocean. The seamounts of the Pacific Ocean are old in history and undulating in terrain, giving birth to a unique deep-sea ecosystem (Geotimes, 1993; Kvile et al. 2014). Magellan seamounts chain locates in the western Pacific and consists of top flat seamounts (1500 m to 6000 m water depth) (Kellogg et al. 1987; Mel’nikov et al. 2009). The top flat seamount is characterised by a large flat roof and steep slope with a listric shape. The flat roofs are covered by Quaternary foraminifer sand and calcium ooze (Zhu et al. 2011). Up to now, previous studies have demonstrated that high productivity is a distinctive characteristic of seamounts because of a large number of organic matters providing sufficient matrix for growth of organisms (Genin and Boehlert 1985; Tseytlin 1985; Boehlert and Genin 2013). Previous studies have shown that seamounts are highly biologically diverse and have an abundance of biomes (Morato et al. 2010; Quattrini et al. 2015; Preez et al. 2016), discovering diverse microbial communities including bacteria (Ettoumi et al. 2010, 2013, 2016), archaea (Liao et al. 2011; Esther et al. 2015; Fortunato and Huber 2016) and fungi (Magnus et al. 2015). So far, however, there has been little concern about fungal diversity in sediments from the Magellan seamounts.
Several studies have shown that the potential drivers of the distribution of marine fungi could be specific environmental parameters, such as temperature, sample depth, and available nutrients (Booth and Kenkel 1986; Jones 2000; Jeffries et al. 2016; Tisthammer et al. 2016; Li et al. 2018). Globally, the distribution of marine fungi is related to temperature and salinity (Booth and Kenkel 1986). In marine sediments, environmental factors, especially sample depth, oxygen, and nitrate, have been found closely related to fungal community composition (Tisthammer et al. 2016). In the Arctic sediments, the diversity of fungi is mainly affected by salinity, organic carbon, silicate, and phosphate content (Zhang et al. 2015). In sediments of the margins of Peru, fungal communities and activities are associated with dissolved and total organic carbon and sulphide (Orsi et al. 2013). In deep-sea sediments of the Gulf of Mexico, the physical and chemical properties of sediments (water content, carbonate, nitrogen, and terrigenous content) and geographic location (region, latitude, longitude, and geographical distance) affect fungal community structure (Lluvia et al. 2019). These studies indicated that there is a relationship between fungal community structure and environmental factors. However, what is not yet understood is the relative importance of the various factors that function in different environments.
Our present knowledge of deep-sea fungal diversity is largely based on the identification of the fruiting body, culturing surveys and conventional sequencing of the internal transcribed spacer (ITS) of rRNA gene clones (RaghuKumar et al. 2004; Bass et al. 2007; Nagano et al. 2010; Singh et al. 2011, 2012; Xu et al. 2014, 2016; Zhang et al. 2016). High-throughput sequencing (HTS) of DNA amplification from marine environments is a powerful approach for screening fungal communities with better capacity for detecting rare species, the taxa that present only as vegetative mycelia and cannot be cultured (Zhang et al. 2016; Nagano et al. 2017; Wang et al. 2018; Xu et al. 2018b). A few of studies have been conducted to detect fungal assemblages present in bathypelagic and abyssopelagic zones and other specialised deep environments including hydrothermal systems, methane-dominated regions, and deep subsurface sediments (Bass et al. 2007; Lai et al. 2007; Takeshita et al. 2007; Jebaraj and Raghukumar 2009; Le Calvez et al. 2009; Nagano et al. 2010; Nagahama et al. 2011; Singh et al. 2011, 2012; Thaler et al. 2012; Xu et al. 2014, 2016, 2017).
To better understand the fungal community in the deep-sea sediment of the Magellan seamounts, the nuclear internal transcribed spacer 2 (ITS2) region was used as a barcode and Illumina MiSeq as sequencing platform. The results of this study will allow us to determine the diversity distribution and composition of the fungal communities, providing new details of fungal communities in the deep-sea seamounts. Besides, we also evaluated the influences of geographic location and physicochemical parameters on the distribution of fungal communities.
Materials and methods
Sampling
Using the Chinese scientific research vessel “Dayang No. 1”, sediment samples were collected from the northwest Pacific during the implementation of the Chinese Ocean 48 cruise from August 12 to 7 September 2018. The sampling location is in the centre of the Magellan Seamount chain (Zhao et al. 2010). Details of the collected samples were shown in Figure 1 and Table 1. Each sediment sample was divided into two fractions and stored at 4°C and −80°C, respectively, for subsequent separation and molecular analysis. Organic nitrogen and carbon were measured using CHNSO Elemental Analyser (Model FLASH2000, Thermo Scientific, USA) (Aoyagi et al. 2015).
Figure 1.
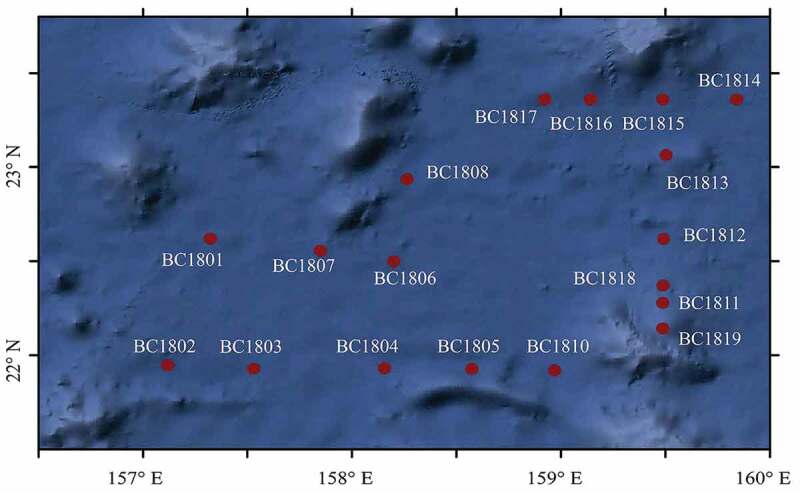
Table 1.
The information of the sample station.
| Samples | Location |
Depth (m) | Sediment type | Organic N% | Organic C% | |
|---|---|---|---|---|---|---|
| Longitude (°E) |
Latitude (°N) |
|||||
| BC1801 | 157.32 | 22.62 | 5437 | Surface clay | 0.06 | 0.34 |
| BC1802 | 157.12 | 21.95 | 5412 | Surface siliceous mud | 0.10 | 0.42 |
| BC1803 | 157.53 | 21.93 | 5426 | Surface clay | 0.09 | 0.40 |
| BC1804 | 158.16 | 21.93 | 5474 | Surface clay | 0.09 | 0.39 |
| BC1805 | 158.58 | 21.93 | 5496 | Surface clay | 0.09 | 0.37 |
| BC1806 | 158.2 | 22.5 | 5442 | Surface clay | 0.09 | 0.47 |
| BC1807 | 157.85 | 22.56 | 5396 | Surface clay | 0.08 | 0.45 |
| BC1808 | 158.25 | 22.94 | 5269 | Surface clay | 0.08 | 0.38 |
| BC1810 | 158.97 | 21.92 | 5255 | Surface clay | 0.10 | 0.71 |
| BC1811 | 159.49 | 22.28 | 4132 | Surface calcareous slime | 0.07 | 0.25 |
| BC1812 | 159.49 | 22.62 | 5087 | Surface clay | 0.08 | 0.34 |
| BC1813 | 159.5 | 23.06 | 5270 | Surface clay | 0.08 | 0.32 |
| BC1814 | 159.84 | 23.36 | 5488 | Surface clay | 0.09 | 0.52 |
| BC1815 | 159.48 | 23.36 | 5417 | Surface clay | 0.08 | 0.38 |
| BC1816 | 159.12 | 23.36 | 5240.38 | Surface clay | 0.09 | 0.40 |
| BC1817 | 158.9 | 23.36 | 5421.31 | Surface clay | 0.09 | 0.44 |
| BC1818 | 159.49 | 22.37 | 4508.03 | Surface clay | 0.08 | 0.41 |
| BC1819 | 159.48 | 22.14 | 3530.98 | Surface calcareous slime | 0.09 | 0.39 |
DNA isolation
The environmental genomic DNA of the sediment samples was extracted by FastDNA®Spin Kit for Soil (MP bio, Santa Ana, USA), according to the manufacturer’s instruction. The ITS region of the fungal ribosomal RNA gene was amplified by PCR in 50-μl reactions (95°C for 2 min, followed by 27 cycles at 98°C for 10 s, 62°C for 30 s, and 68°C for 30 s and a final extension at 68°C for 10 min) (Xu et al. 2019). The primer sequences were: ITS3- KYO2 F: GATGAAGAACGYAGYRAA; ITS4-R: TCCTCCGCTTATTGATATGC (Toju et al. 2012). The PCR amplified product was then recovered and quantified using the QIAquick PCR purification kit (Qiagen) and Qubit 3.0 (Thermo Scientific). Sequencing libraries were generated using NEB Next R Ultra TM DNA Library Prep Kit for Illumina (NEB, USA) and added index codes. The library quality was assessed by the QuantiFluorTM-ST Blue fluorescence quantitative system (Promega) and sequenced by paired-end (2 × 250 bp) Illumina HiSeq 2500 platform at Genedenovo Inc. Guangzhou, China. The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (Accession Number: SAMN14543412-SAMN14543447)
Quality control and reads assembly
Raw sequencing data obtained included dirty reads containing adapters or low-quality bases which would affect sequence assembly and analysis. To get high-quality clean reads, raw reads were filtered according to the following rules: 1) removing reads containing more than 10% of unknown nucleotides (N); and/or 2) removing reads containing less than 80% of bases with quality (Q-value)>20 (Lu et al. 2014; Li et al. 2017; Xu et al. 2018b). Paired-end clean reads were merged as raw tags using FLASH (version 1.2.11) with a minimum overlap of 10 bp and mismatch error rates of 2%. Noisy sequences of raw tags were filtered by QIIME (version 1.9.1) (Caporaso et al. 2010) pipeline under specific filtering conditions (Bokulich et al. 2013) to obtain high-quality clean tags. Clean tags were searched against the reference database (http://drive5.com/uchime/uchime_download.html) to perform reference-based chimera checking using the UCHIME algorithm (Edgar et al. 2011). This analysis was performed on USEARCH (http://www.drive5.com/usearch/manual/uchime_algo.html) (Alloui et al. 2015). All chimeric tags were removed and effective tags were obtained for further analysis (Haas et al. 2011). The software MOTHUR (version 1.39.1) (Schloss et al. 2009) was used to remove redundant tags to get unique tags.
Diversity analysis
The clean reads were clustered into operational taxonomic units (OTUs) of ≥97% similarity using the UPARSE (version 9.2.64) (Edgar 2013). The sequence with the highest abundance was selected as a representative sequence within each cluster. Venn analysis was performed in R to identify unique and common OTUs between-groups using Venn Diagram (version 1.6.17) and UpSet R (version 1.3.3) (Lex and Gehlenborg 2014; Lex et al. 2014). The representative sequences were classified into organisms by a naive Bayesian model using the RDP classifier (version 2.2) (Wang et al. 2007) based on the UNITE database (https://unite.ut.ee/, version 2016.11.20) (Kõljalg et al. 2005). Diversity indices including Chao1value, ACE value, Shannon index, and Simpson index were calculated in QIIME (Paul and Josephine 2010). Rarefaction curves were generated based on the Chao1 value, Shannon index, and Simpson index. KRONA (version 2.6) was then used to interactively visualise the species annotation results (Ondov et al. 2011).
The composition of microbial communities in different samples was studies based on beta diversity analysis. First, use the software Muscle (version 3.8.31) (Edgar 2004) to perform multiple sequence alignment based on the OTU sequences of all samples. Combined with abundance information of the OTU, the GUniFrac (version 1.0) package in the R language was used to calculate the Unweighted Unifrac and Weighted Unifrac distance between pairs of samples (Catherine and Knight 2005). We visualised patterns of variation in community composition using principle coordinates analysis (PCoA) by the cmdscale function with the stats package in R (version 3.2.1) (Cox and Cox 2008).
To further explore the differences in microbial community structure between samples, the Unweighted Pair-group Method with Arithmetic Means (UPGMA) was generated using the MOTHUR (version 1.39.1) (Schloss et al. 2009). To examine the relationship between microbial community structure and environmental factors, canonical correspondence analyses (CCA) were conducted using CANOCO software (Dang et al. 2010). The FUNGuild (version 1.0) database (https://github.com/UMNFuN/FUNGuild) was used to assign ecological functions (trophic modes) to all OTUs (Nguyen et al. 2016).
Results
Sequence analysis and OTU classification
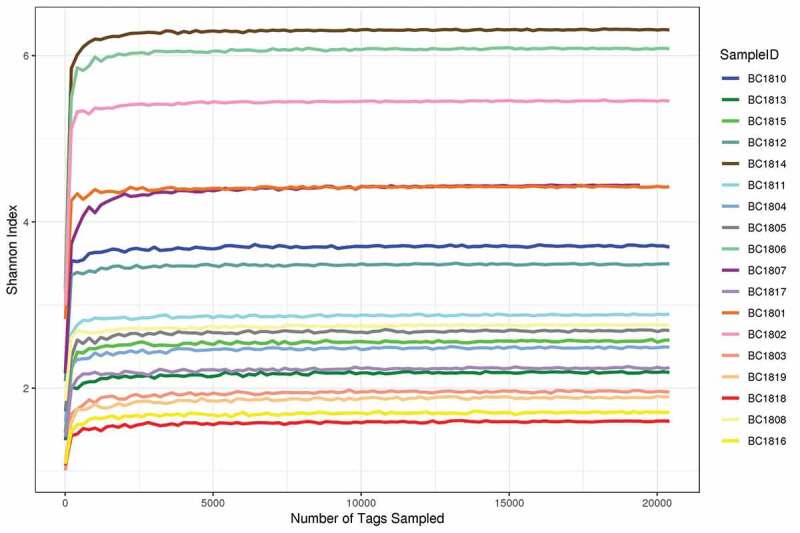
A total of 4548, 928 raw tags with 4536, 922 quality-filtered fungal ITS reads were obtained from the Illumina HiSeq 2500 platform sequencing. Previous studies have shown that removal of low-frequency sequences can reduce error rates and improve microbiota assessment (Tedersoo et al. 2010; ; Li et al. 2016). After filtration and denoising, a total of 1,979 OTUs at ≥ 97% similarity level was obtained from 18 sediment samples. The observed OTU richness and Shannon index were used for further analyses. The OTU richness of 18 samples differed from each other, ranging from 452–741 (Table 2). Shannon diversity ranged from 1.62–6.32 (Figure 2).
Table 2.
Summary for pyrosequencing data and Alpha diversity index statistics from the 18 deep-sea sediment samples from the Magellan seamounts.
| Sample name | Total Tags | Unique Tags | Taxon Tags | OTUs | ACE | Chao1 | Simpson | Shannon | Coverage % |
|---|---|---|---|---|---|---|---|---|---|
| BC1801 | 333905 | 56051 | 333295 | 741 | 956.91 | 972.26 | 0.91 | 4.44 | 99.93 |
| BC1802 | 442513 | 102539 | 441546 | 705 | 896.43 | 876.55 | 0.96 | 5.47 | 99.95 |
| BC1803 | 173533 | 44072 | 172772 | 652 | 863.96 | 958.01 | 0.45 | 1.98 | 99.88 |
| BC1804 | 173592 | 34598 | 172635 | 672 | 876.36 | 847.01 | 0.60 | 2.51 | 99.88 |
| BC1805 | 303868 | 67530 | 302940 | 762 | 977.65 | 975.13 | 0.62 | 2.71 | 99.93 |
| BC1806 | 431943 | 97404 | 427136 | 571 | 733.46 | 737.44 | 0.96 | 6.10 | 99.97 |
| BC1807 | 20627 | 9074 | 19423 | 506 | 645.12 | 643.04 | 0.75 | 4.44 | 99.28 |
| BC1808 | 237478 | 50021 | 236866 | 621 | 817.78 | 814.16 | 0.76 | 2.78 | 99.92 |
| BC1810 | 227200 | 46678 | 226543 | 515 | 785.68 | 743.29 | 0.75 | 3.72 | 99.92 |
| BC1811 | 262340 | 55193 | 261692 | 525 | 724.13 | 700.18 | 0.75 | 2.90 | 99.94 |
| BC1812 | 314425 | 70658 | 313633 | 589 | 833.36 | 814.84 | 0.81 | 3.51 | 99.94 |
| BC1813 | 197608 | 44125 | 196852 | 692 | 928.03 | 973.97 | 0.54 | 2.21 | 99.89 |
| BC1814 | 338348 | 75770 | 335092 | 452 | 604.59 | 539.00 | 0.98 | 6.32 | 99.97 |
| BC1815 | 294145 | 58515 | 293498 | 717 | 921.94 | 954.25 | 0.58 | 2.59 | 99.93 |
| BC1816 | 202164 | 42840 | 201654 | 661 | 901.62 | 935.38 | 0.35 | 1.73 | 99.89 |
| BC1817 | 197918 | 39058 | 197306 | 668 | 930.91 | 982.22 | 0.49 | 2.26 | 99.88 |
| BC1818 | 213231 | 42427 | 212657 | 709 | 977.53 | 1015.96 | 0.44 | 1.62 | 99.89 |
| BC1819 | 172084 | 38251 | 171392 | 738 | 963.21 | 1000.11 | 0.39 | 1.91 | 99.87 |
Figure 2.
The taxonomical assigned OTUs belonged to seven phyla, 20 classes, 46 orders, 88 families, and 106 genera. Of the 1,979 fungal OTUs with 4536, 922 sequences, 873 (44.11%) were affiliated with Basidiomycota, followed by 486 (37.42%) with Ascomycota, 59 (2.98%) with Mortierellomycota, 17 (0.86%) with Chytridiomycota, 4 (0.20%) with Mucoromycota, 4 with Glomeromycota (0.20%), 1 (0.05%) with Monoblepharidomycota and 535 (27.03%) with unidentified fungi (Figure 3).
Figure 3.
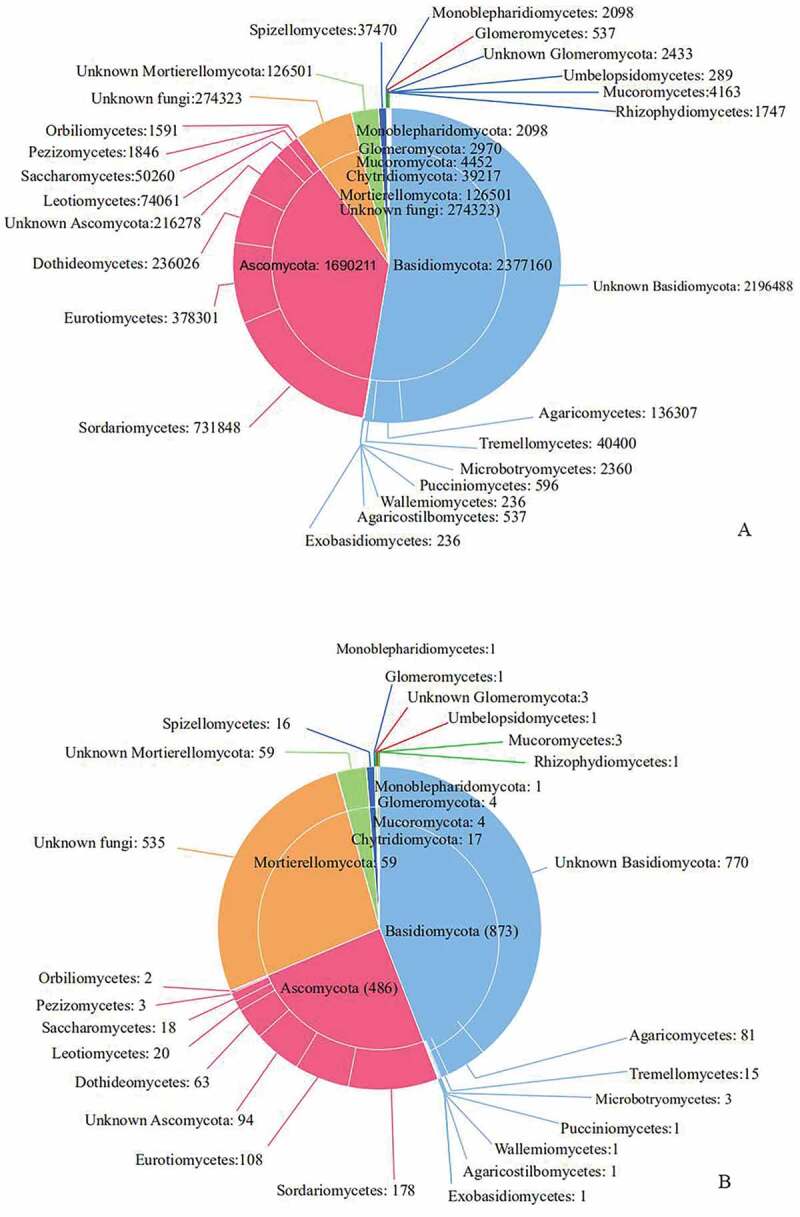
The sequences of Basidiomycota can be referenced in seven known classes, of which Agaricomycetes are the most abundant (136, 307 reads) and moderately diverse (81 OTUs), followed by Tremellomycetes (40, 400 reads and 15 OTUs), Microbotryomycetes (2, 360 and 3 OTUs), Pucciniomycetes (596 reads and 1 OTU), Agaricostilbomycetes (537 reads and 1 OTU), Exobasidiomycetes (236 reads and 1 OTUs) and Wallemiomycetes (236 reads and 1 OTU) (Figure 3).
Sordariomycetes (731,848 reads and 178 OTUs) in Ascomycota was the most abundant classes, followed by Eurotiomycetes (378,301 reads and 108 OTUs), Dothideomycetes (236, 026 reads and 63 OTUs), and Leotiomycetes (74, 061 reads and 20 OTUs), Saccharomycetes (50, 260 reads and 18 OTUs), Pezizomycetes (1, 846 reads and 3 OTUs), Orbiliomycetes (1, 591 reads and 2 OTUs) (Figure 3).
The 59 OTUs (1, 26501 reads) belonging to Mortierellomycota have no more detailed classification information. Spizellomycetes (37470 reads and 16 OTUs) is the most abundant in Chytridiomycota, and the other 1747 reads and 1 OTUs in Chytridiomycota were assigned to Rhizophydiomycetes. In Glomeromycota, only Glomeromycetes (537 reads 1 OTUs) and 3 OTUs (2, 433 reads) were identified. Mucoromycota includes Mucoromycetes (4, 163 reads, and 3 OTUs), Umbelopsidomycetes (289 reads, and 1 OTU). Only Monoblepharidiomycetes (2, 098 reads, and 1 OTU) can be recognised in Monoblepharidomycota (Figure 3).
At the genus level, 106 fungal genera were identified, and 5 genera had great differences in sample richness, namely Aspergillus, Cladosporium, Fusarium, Chaetomium, and Penicillium, which accounted for 4.44%, 1.94%, 3.16%, 2.92% and 2.56% of the total sequences, respectively (Figure 4c). Aspergillus, Cladosporium, Fusarium, and Penicillium were detected in all samples.
Figure 4.
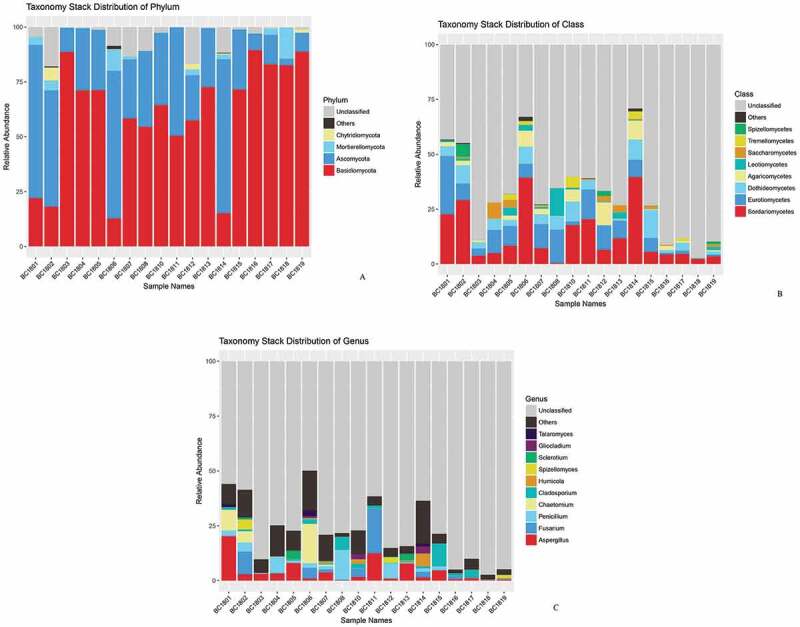
Fungal community composition in different samples
The relative abundance of fungal gates, classes, and genera were found to be different in different samples. In samples BC1801, BC1802, BC1806, and BC1814, Ascomycota accounted for more than 50% of the total sequences. In samples BC1804, BC1805, BC1813, BC1807, BC1808, BC1810, BC1811and BC1812, Basidiomycota accounted for more than 50% less than 75% of the total sequences. In the samples BC1803, BC1816, BC1817, BC1818, and BC1819, Basidiomycota accounted for more than 75% of the total sequences (Figure 4a).
The relative abundance of fungal classes and genera in the different samples were found to be different. Although diverse classes were recognised from samples, the Sordariomycetes, Eurotiomycete, and Dothideomycetes were the most abundant classes in the samples (Figure 4b). However, there are some fungal classes more abundant in some samples. To illustrate, Spizellomycetes was abundant in sample BC1802; Saccharomycetes was plentiful in sample BC1804; Leotiomycetes was abundant in sample BC1808; Agaricomycetes was ample in sample BC1812 (Figure 4b).
The most abundant genera recovered include Aspergillus, Fusarium, Chaetomium, Penicillium, Cladosporium, Spizellomyces, Humicola, Sclerotium, Talaromyces, Gliocladium. Among them, Aspergillus, Cladosporium, Fusarium, Chaetomium, Penicillium showed higher relative abundance in most sediment samples. Meanwhile, some fungal genera (eg, Humicola, Sclerotium, and Gliocladium) are plentiful in partial samples (Figure 4c).
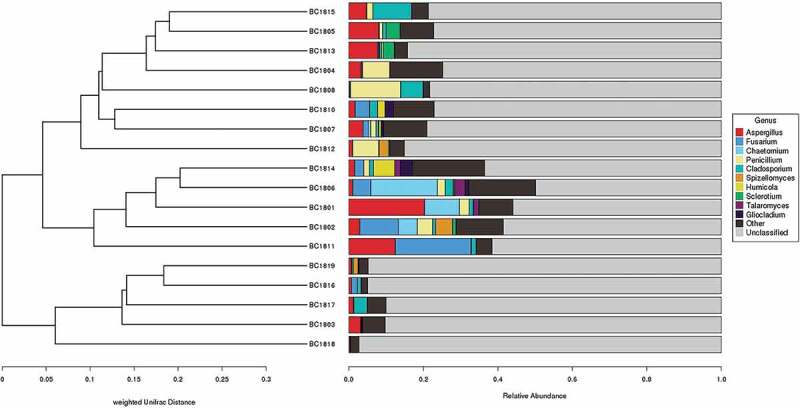
Principal coordinates analysis (PCoA) (Figure 5) based on the distribution of fungal OTUs exhibited a clear separation of fungal community structure between the 18 sediment samples, with the first principal component representing 29% of the total variation. Cluster analysis based on the 10 most abundant fungal genera using weighted unifrac distance analysis (UPGMA) (Figure 6) also provided a similar result with the fungal community structure between the 18 sediments. This difference in fungal communities between different sediment samples may be related to the differences between sediments, such as the spatial differences of sampling stations.
Figure 5.
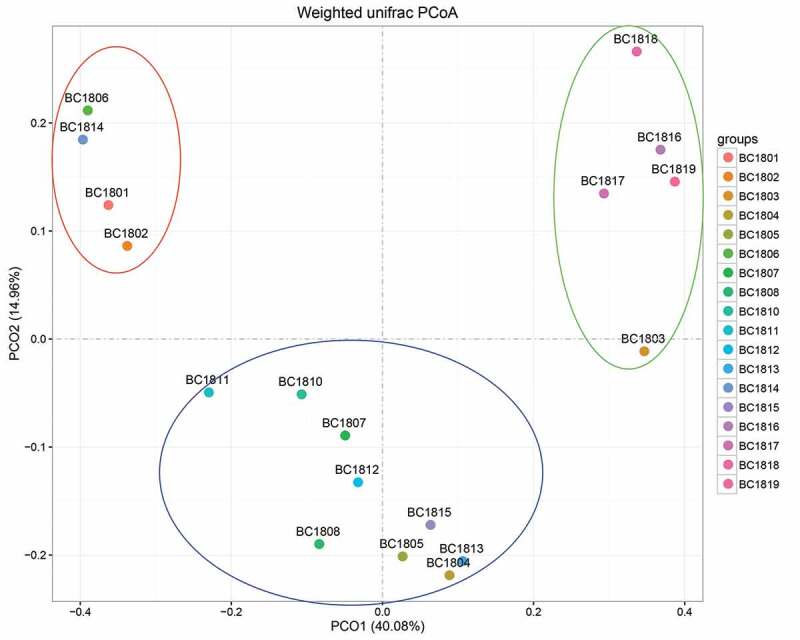
Figure 6.
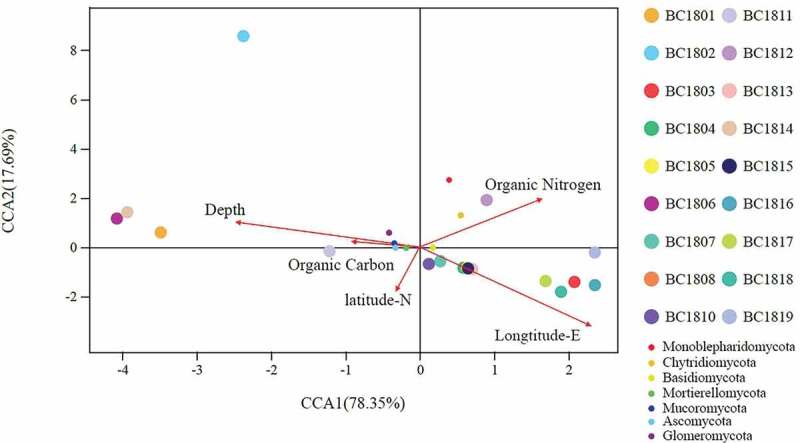
A further CCA analysis was performed to examine the relationship between microbial community structure and environmental factors (Figure 7). Among all environmental factors examined, organic Nitrogen (r2 = 0.1826, P = 0.205), organic Carbon (r2 = 0.0146, P = 0.859), depth (r2 = 0.0933, P = 0.401), Longitude-E (r2 = 0.1412, P = 0.288), Latitude-N (r2 = 0.0655, P = 0.616), no significant correlations were found between these environmental factors and fungi community structure (in all cases, P > 0.05). The present results show that among the analysed environmental factors, none of them has the significant effect on the composition and structure of the microbial communities.
Figure 7.
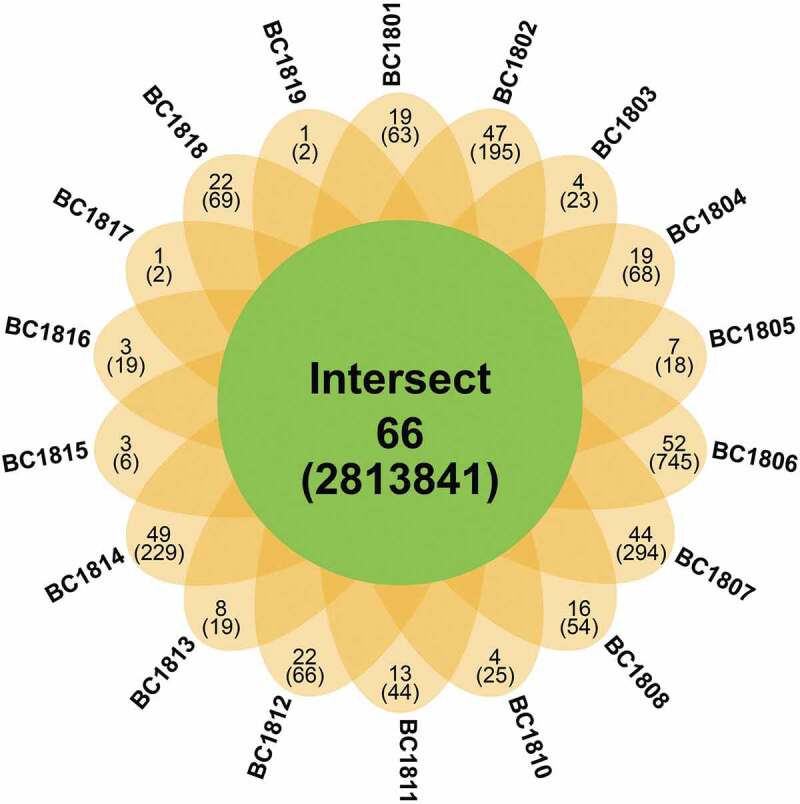
The core fungal taxa were referred to the OTUs shared by all samples, of which symbolise by the overlapping areas in the Venn diagram analysis. 66 fungal (281,384 reads) were shared by all 18 sediment samples and recognised as the core taxonomic group (Figure 8), which accounts for 3.33% of all fungal OTUs and 62.02% of total sequences. Some of these OTUs showed a high relative abundance in deep-sea sediments. For instance, OTU2 assigned to Fusarium and OTU6 assigned to Aspergillus account for 2.84% and 1.51% of the total fungal sequence, respectively. The samples BC1817 and BC1819 contained the lowest number of OTUs, while BC1806, BC1807, and BC1814 contained the highest number of OTUs. Besides, the sample-specific OTUs for each site ranges from 1 to 52. Most unique fungal OTUs were rare, less than 0.34% of total fungal sequences. The coexistence of 66 OTUs only accounts for 3.33% of the total 1979 OTUs. The core taxonomic group indicated the similarity of fungal communities among the 18 samples.
Figure 8.
FunGuild analysis
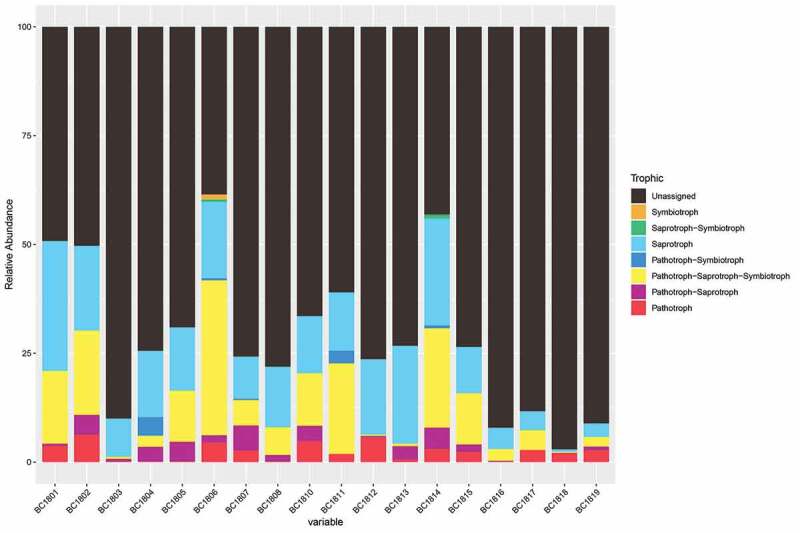
Based on the information of OTU taxonomic assignment and relative published articles, FUNGuild was used to predict the putative living strategies of fungi. Fungi recovered in this study can be characterised as Pathotroph-Saprotroph-Symbiotroph, Pathotroph-Saprotroph, Saprotroph-Symbiotroph, Pathotroph-Symbiotroph, Pathotroph, Saprotroph, Symbiotroph (Figure 9). Among them, Saprotroph was the most common life strategy.
Figure 9.
Discussion
Ascomycota and Basidiomycota have been recovered from other deep-sea habitats frequently (Nagano and Nagahama 2012; Xu et al. 2014, 2016, 2017, 2018b; Rédou et al. 2015; Nagano et al. 2017). High levels of Basidiomycota found in this study were different from most previous studies, of which described Ascomycota as the dominant phylum in the fungal community in other deep-sea habitats (Nagano and Nagahama 2012; Xu et al. 2014, 2016, 2017, 2018b; Rédou et al. 2015; Zhang et al. 2016; Nagano et al. 2017), indicating that the composition of fungal communities in this area may be unique. Similarly, Xu et al. (2019) presented high levels of Basidiomycota in several deep-sea sediment samples from Yap Trench by using high throughput sequencing. Ascomycota has been reported dominating the fungal community in deep-sea sediments worldwide, for instances: in the Southwest India Ridge of the Indian Ocean (Xu et al. 2018b), in the Okinawa Trough (Zhang et al. 2016) and the Sao Paulo Plateau of the Atlantic Ocean (Nagano et al. 2017). This deviance is probably due to the differences in the source of samples. Rämä et al. (2017) found that the dominant fungal phyla may be substratum-specific in the marine environment, Chytridiomycota, and Basidiomycota prevails in sea ice and seawater and Ascomycota overabundant on driftwood and sediments.
Analyses of environmental samples by molecular techniques recovered unknown clades from diverse marine ecosystems, especially in the deep-sea environment (Le Calvez et al. 2009; Nagano and Nagahama 2012; Xu et al. 2018b, 2019). Some phyla discovered in the deep-sea environment formerly were also recovered by a metabarcoding method in this study: Mortierellomycota (Xu et al. 2019), Chytridiomycota (Le Calvez et al. 2009; Nagano et al. 2010; Nagahama et al. 2011; Singh et al. 2011; Zhang et al. 2014, 2016; Xu et al. 2014, 2019), Mucoromycota (Xu et al. 2019), Glomeromycota (Le Calvez et al. 2009; Nagano et al. 2010; Nagahama et al. 2011). Monoblepharidomycota has been recovered from marine surface sediments previously but was firstly recovered from the deep-sea environment (Guo et al. 2015). Consistently with the previous study mentioned above, members of these phyla represented only small proportions of the sedimentary fungal communities. Nevertheless, Chytrids was found to dominate Arctic marine fungal communities and might change primary production patterns rapidly with increased light penetration through the Arctic Ocean (Hassett and Gradinger 2016). In marine habitats, Chytridiomycota, Mortierellomycota, and Mucoromycota have been characterised as decomposers of pollens and leaves (Phuphumirat et al. 2016) or pathogens of marine algae and animals (Scholz et al. 2016; Wang et al. 2018). These discoveries suggest that these fungi played a wide array of ecological roles potentially in the marine environment.
In this study, the most abundant class was the Sordariomycetes of Ascomycota, which is abundant in the marine environment. Previous studies have found that many obligate marine mycelium fungi belong to Sordariomycetes (Raghukumar 2017). Most classes recognised in this study were widely distributed in the deep-sea environment, as they were recovered from other regions of the deep-sea environment by molecular approach previously: Tremellomycetes, Microbotryomycetes, Agaricostilbomycetes, Exobasidiomycetes, Wallemiomycetes, Sordariomycetes, Eurotiomycetes, Dothideomycetes, and Leotiomycetes, Saccharomycetes, Pezizomycetes, Orbiliomycetes, Umbelopsidomycetes, Spizellomycetes, Pucciniomycetes and Glomeromycetes (Bass etal. 2007; Nagahama etal. 2011; Zhang etal. 2016; Nagano etal. 2017; Xu etal. 2018a, 2019). By ametabarcoding approach, we recovered Mucoromycetes, Rhizophydiomycetes, and Monoblepharidiomycetes in the deep-sea environments for the first time, updated the ecological distribution of these fungi. Some classes have been discovered from other environments, may be adapted to wide range habitats, such as Pucciniomycetes found from (Gao etal. 2010), Mucoromycetes found in the White Sea Sediments (Khusnullina etal. 2018), Monoblepharidiomycetes recovered from freshwater phytoplankton and lake samples (Ishida etal. 2015), Rhizophydiomycetes recovered in freshwater sites and high alpine exposed soils (Powell and Letcher 2014; Tedersoo et al. 2018).
Aspergillus, Cladosporium, Fusarium, Penicillium were detected in all deep-sea sediment samples. These genera have been widely recovered in deep-sea sediments around the world and were considered to be ubiquitous in the deep-sea environment (Nagano et al. 2010; Singh et al. 2012; Zhang et al. 2014; Rédou et al. 2015; Nagahama et al. 2011; Vargas-Gastelum et al. 2019; Xu et al. 2017, 2018a, 2018b, 2019). Penicillium and Aspergillus were amongst the most common genera in deep-sea ecosystems (Burgaud et al. 2009; Singh et al. 2010; Nagano and Nagahama 2012; Zhang et al. 2013; Xu et al. 2017, 2018b), as well as the most widely terrestrial forms of fungi in the sea and proved to be active in the marine environment owing to their physiological versatility (Raghukumar 2017).
Furthermore, the Venn diagram analysis revealed that the unique OTUs existed in every sample, reflecting the divergence among all the stations. These unique OTUs in each station may represent a rapidly changing community that is associated with the unique physicochemical properties of that location. While the common OTUs represent a more stable fungal community well adapted to habitat dynamics (Vargas-Gastelum et al. 2019).
Our study revealed a diverse fungal group in the deep-sea sediments of the Magellan Seamount by a metabarcoding approach, supplementing the fungal diversity information in this area. Compared with previous studies on fungal diversity of deep-sea sediments from other sites of the Pacific Ocean by using culture-dependent methods (Burgaud et al. 2009; Rédou et al. 2015; Xu et al. 2018a), clone libraries (Zhou et al. 2007; Nagano et al. 2010; Xu et al. 2014, 2016), studies by high throughput sequencing (Rédou et al. 2014; Zhang et al. 2015; Xu et al. 2019) notably extended our knowledge of the marine mycobiota in this area. Combining different methods for research could help us to understand the fungal diversity in the deep-sea environment more comprehensively.
However, 27.03% of the fungal OTUs could not be assigned to any fungal phyla based on the available databases. The high percentage of unidentified fungi was also detected in other deep-sea sediments, suggesting that there are largely unknown fungal taxa inhabiting in the deep-sea sediments, which probably includes indigenous fungi and species of potential biotechnological importance (Zhang et al. 2015; Barone et al. 2018; Vargas-Gastelum et al. 2019). This discovery of unidentified fungi may result from the insufficient coverage of ITS sequences in databases (Khomich et al. 2017). Currently, molecular studies have revealed a high diversity of ascomycetes and basidiomycetes in deep-sea with many novel lineages (Nagahama and Nagano 2012), while the low detection of taxonomic groups other than Ascomycota and Basidiomycota possibly results from the same reason (Tedersoo et al. 2015). The universal sequencing method targeting ITS2 regions are more likely to amplify ITS regions from Ascomycota and Basidiomycota, instead of other fungal groups (Op De Beeck et al. 2014; Amend et al. 2019). This limitation could mask the presence of other fungal groups. The use of multiple or group-specific primers might solve this obstacle (Singh et al. 2012a). Furthermore, a concordance between the rDNA and mRNA taxonomic diversity should be applied to estimate whether the fungi detected from the deep-sea environment by the sequencing approach are metabolically active.
Surprisingly, the canonical correspondence analysis revealed that all the environmental factors considered in this study (including organic Carbon, organic Nitrate, depth, longitude, and latitude) are not significantly related to fungal community composition. This result is different from other studies which showed that depth (Roth et al. 1964; Gong et al. 2015; Zhang et al. 2015), carbon content (Orsi et al. 2013; Zhang et al. 2015; Vargas-Gastelum et al. 2019) and sampling station latitude (Lluvia et al. 2019) were correlated with microbial community structure. These differences might result from the difference in sample source or insufficient environmental data. However, the organic nitrate, depth, longitude had more obvious influence on fungal community composition relatively. Global analysis of marine fungal community structure from water columns and sediments revealed that environmental factors, especially sample depth, oxygen, and nitrate are closely related to the fungal community composition (Tisthammer et al. 2016). This consistency suggested that the nitrate component of the sediments and sample depth are crucial factors to the community structure of marine fungal. Therefore, the distribution of marine fungi may depend on their interaction with a variety of environmental factors, which needs more environmental factors to do further analysis in subsequent studies.
Based on FUNGuild database analysis, saprotroph tends to be the most abundant life strategy. The higher abundance of saprotrophs in the deep-sea environment may be due to their key roles in decomposition processes. Saprotrophs are essential for nutrient turnover and sediment C storage. The saprotroph fungi in the deep-sea environment probably contributed to the maintenance of the sediment structure and nutrient cycling as to their great capability at producing extracellular enzymes in soil (Treseder and Lennon 2015). Agaricomycetes and Eurotiomycetes assigned as saprophytic fungi, which had been the most abundant and common classes in this study. (Cannon and Kirk 2007; Sterkenburg et al. 2015). Aspergillus and Penicillium are the major species of fungi in this study, also assigned as saprotrophs (Baldrian 2010). Some members of Dothideomycetes and Sordariomycetes had been discovered as plant pathogenic fungi, which contains Fusarium, Chaetomium, and Cladosporium (; Tedersoo et al. 2014; Lawrey and Diederich 2018). Another common life strategy is Pathotroph. Pathogenic fungi might accelerate the leaching out of Dissolved Organic Matter (DOM) from the host, which is then available to other microorganisms for their growth in the marine environment (Raghukumar 2017). Fungal functional groups may have important implications for the functions of various fungi which may reflect their function in the deep-sea environment.
In summary, our study characterised the fungal communities in deep-sea sediments of the Magellan Seamounts. We also explored the reasons for the divergence of the fungal community composition between different samples, which need to be further studied and anatomised in combination with more environmental factors. Nevertheless, our findings provide valuable information for understanding the distribution and potential ecological effects of fungi in the deep sea of the Magellan seamount area.
Acknowledgements
This research work was financially supported by the China Ocean Mineral Resources R&D Association (COMRA) Program (DY135-B-09), National Natural Science Foundation of China (41776170 and 41606145), and the Startup Foundation for Introducing Talent of NUIST (2020r028). We thank Dr. Jichao Yang for sample sites map in National Deep-sea Center, State Ocean Administration, Qingdao, China. We would like to thank the crew and scientific team of R/V Xiangyanghong 09, the pilots and the supporting team of Jiaolong manned submersible in 48th Dayang Cruise for the sampling.
Funding Statement
This work was supported by the China Ocean Mineral Resources Research and Development Association [(DY135-B-01)]; National Natural Science Foundation of China [(41776170 and 41606145)]; Scientific Research Foundation of Third Institute of Oceanography [SOA (2016039)].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Akerman NH, Butterfield DA, Huber JA.. 2013. Phylogenetic diversity and functional gene patterns of sulfur-oxidizing subseafloor epsilonproteo bacteria in diffuse hydrothermal vent fluids. Front Microbiol. 4:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloui T, Boussebough I, Chaoui A, Nouar A, Chettah M. 2015. Usearch: A meta search engine based on a new result merging strategy. Proceedings of the 7th International Joint Conference on Knowledge Discovery, Knowledge Engineering and Knowledge Management (IC3K). Lisbon (LIS): IEEE Computer Society. p. 531–536. [Google Scholar]
- Amend A, Burgaud G, Cunliffe M, Edgcomb VP, Ettinger CL, Gutiérrez MH, Heitman J, Hom EFY, Laniri G, Jones AC, et al. 2019. Fungi in the marine environment: open questions and unsolved problems. Mbio. 10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi T, Kimura M, Yamada N, Navarro RR, Itoh H, Ogata A, Sakoda A, Katayama Y, Takasaki M, Hori T.. 2015. Dynamic transition of chemolithotrophic sulfur-oxidizing bacteria in response to amendment with nitrate in deposited marine sediments. Front Microbiol. 6:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldrian P. 2010. Chapter 12. Effect of heavy metals on saprotrophic soil fungi. In: Sherameti I, Varma A, editors. Soil heavy metals. Berlin (BE): Springer; p. 263–279. [Google Scholar]
- Barone G, Rastelli E, Corinaldesi C, Tangherlini M, Danovaro R, Dell’Anno A. 2018. Benthic deep-sea fungi in submarine canyons of the Mediterranean Sea. Prog Oceanogr. 168:57–64. [Google Scholar]
- Bass D, Howe A, Brown N, Barton H, Demidova M, Michelle H, Li L, Sanders H, Watkinson SC, Willcock S, et al. 2007. Yeast forms dominate fungal diversity in the deep oceans. Proc Royal Soc B. 274(1629):3069–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienhold C, Zinger L, Boetius A, Ramette A. 2016. Diversity and biogeography of bathyal and abyssal seafloor bacteria. PloS One. 11(1):e0148016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehlert GW, Genin A. 2013. A review of the effects of seamounts on biological processes. In: Barbara HK, Patricia F, Rodey B, George WB, editors. American geophysical union geophysical monograph series. Vol. 43. Seamounts (Islands, and Atolls. Washington (WA)): American Geophysical Union; p. 319-334. [Google Scholar]
- Bokulich NA, Subramanian S, Faith JJ, Gordon JI, Knight R, Mills DA, Caporaso JG. 2013. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 10:57–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth T, Kenkel N. 1986. Ecological distribution of lignicolous marine fungi: a distribution model based on ordination and classification. In: Moss ST, editor. The biology of marine fungi. Cambridge (UK): Cambridge University Press; p. 297–309. [Google Scholar]
- Burgaud G, Arzur D., Durand L., Cambon-Bonavita M-A., Barbier G.. 2010. Marine culturable yeasts in deep-sea hydrothermal vents: species richness and association with fauna. FEMS Microbiology Ecology. 73(1):121-133. doi: 10.1111/j.1574-6941.2010.00881.x [DOI] [PubMed] [Google Scholar]
- Burgaud G, Le Calvez T, Arzur D, Vandenkoornhuyse P, Barbier G. 2009. Diversity of culturable marine filamentous fungi from deep-sea hydrothermal vents. Environ Microbiol. 11(6):1588–1600. [DOI] [PubMed] [Google Scholar]
- Cannon PF, Kirk PM. 2007. Fungal families of the world. Cambridge (UK): CABI. [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JA, et al. 2010. Qiime allows analysis of high-throughput community sequencing data. Nat Methods. 7:335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherine L, Knight R. 2005. Unifrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 71(12):8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox M, Cox T. 2008. Multidimensional scaling. In: Chen CH, Härdle W, Unwin A, editors.Handbook of data visualization. Berlin (BE): Springer; p. 338–341. [Google Scholar]
- Damare S, Raghukumar C, Raghukumar S. 2006. Fungi in deep-sea sediments of the Central Indian Basin. Deep Sea Res Part I Oceanogr Res Pap. 53(1):14–27. [Google Scholar]
- Dang HY, Li J, Chen RP, Wang L, Guo LZ, Zhang ZN, Klotz MG. 2010. Diversity, abundance, and spatial distribution of sediment ammonia-oxidizing Betaproteobacteria in response to environmental gradients and coastal eutrophication in Jiaozhou Bay, China. Appl Environ Microbiol. 76(14):4691–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danovaro R. 2012. Marine biodiversity and ecosystem functioning: frameworks, methodologies, and integration. Chapter 9. Oxford (UK): Oxford University Press; p. 26–115. [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 10(10):996–998. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 27(16):2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esther S, Chong LS, Heidelberg JF, Edwards KJ. 2015. Similar microbial communities found on two distant seafloor basalts. Front Microbiol. 6:1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettoumi B, Bouhajja E, Borin S, Daffonchio D, Boudabous A, Cherif A. 2010. Gammaproteo bacteria occurrence and micro diversity in Tyrrhenian sea sediments as revealed by cultivation-dependent and -independent approaches. Syst Appl Microbiol. 33(4):222–231. [DOI] [PubMed] [Google Scholar]
- Ettoumi B, Chouchane H, Guesmi A, Mahjoubi M, Brusetti L, Neifar M, Borin S, Daffonchio D, Cherif A. 2016. Diversity, ecological distribution, and biotechnological potential of actinobacteria inhabiting seamounts and non-seamounts in the Tyrrhenian sea. Microbiol Res. 186–187:71–80. [DOI] [PubMed] [Google Scholar]
- Ettoumi B, Guesmi A, Brusetti L, Borin S, Najjari A, Boudabous A, Cherif A. 2013. Microdiversity of deep-sea bacillales isolated from Tyrrhenian sea sediments as revealed by Arisa, 16s rRNA gene sequencing and boxPCR fingerprinting. Microbes Environ. 28(3):361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortunato CS, Huber JA. 2016. Coupled RNA-sip and metatranscriptomics of active chemolithoautotrophic communities at a deep-sea hydrothermal vent. Isme J. 10:1925–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkievicz JP, Stellick SH, Gray MA, Kellogg CA. 2012. Cultured fungal associates from the deep-sea coral Lophelia pertusa. Deep Sea Res Part I Oceanogr Res Pap. 67:12–20. [Google Scholar]
- Gao Z, Johnson ZI, Wang G.. 2010. Molecular characterization of the spatial diversity and novel lineages of mycoplankton in Hawaiian coastal waters. ISME J. 4(1):111–120. doi: 10.1038/ismej.2009.87 [DOI] [PubMed] [Google Scholar]
- Genin A, Boehlert GW. 1985. Dynamics of temperature and chlorophyll structures above a seamount: an oceanic experiment. J Mar Res. 43(4):907–924. [Google Scholar]
- Gong J, Shi F, Ma B, Pachiadaki M, Zhang XL, Edgcomb VP. 2015. Depth shapes α- and β-diversities of microbial eukaryotes in surficial sediments of coastal ecosystems. Environ Microbiol. 17(10):3722–3737. [DOI] [PubMed] [Google Scholar]
- Guo X, Zhang Q, Zhang X, Zhang J, Gong J. 2015. Marine fungal communities in water and surface sediment of a sea cucumber farming system: habitat-differentiated distribution and nutrients driving succession. Fungal Ecol. 14:87–98. [Google Scholar]
- Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, et al. 2011. Chimeric 16s rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21(3):494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassett BT, Gradinger R. 2016. Chytrids dominate Arctic marine fungal communities. Environ Microbiol. 18:2001–2009. [DOI] [PubMed] [Google Scholar]
- Ishida S, Nozaki D, Grossart HP, Kagami M. 2015. Novel basal, fungal lineages from freshwater phytoplankton and lake samples. Environ Microbiol Rep. 7(3):435–441. [DOI] [PubMed] [Google Scholar]
- Jebaraj CS, Raghukumar C. 2009. Anaerobic denitrification in fungi from the coastal marine sediments off Goa, India. Mycol Res. 113(1):100–109. [DOI] [PubMed] [Google Scholar]
- Jeffries TC, Curlevski NJ, Brown MV, Harrison DP, Doblin MA, Petrou K, Ralph PJ, Seymour JR. 2016. Partitioning of fungal assemblages across different marine habitats. Environ Microbiol Rep. 8:235–238. [DOI] [PubMed] [Google Scholar]
- Jones EBG. 2000. Marine fungi: some factors influencing biodiversity. Fungal Divers. 4:53–73. [Google Scholar]
- Kellogg JN, Wedgeworth BS, Freymueller JT. 1987. Isostatic compensation and conduit structures of Western Pacific seamounts: results of three-dimensional gravity modeling. Seamounts, Islands, and Atolls. In: Keating BH, Fryer P, Batiza R, Boehlert GW, editors. Geophysical monograph series. Vol. 43. New York (NY): American Geophysical Union; p. 85-96. [Google Scholar]
- Khomich M, Davey ML, Kauserud H, Rasconi S, Andersen T. 2017. Fungal communities in Scandinavian lakes along a longitudinal gradient. Fungal Ecol. 27:36–46. [Google Scholar]
- Khusnullina AI, Bilanenko EN, Kurakov AV. 2018. Microscopic fungi of White Sea sediments. Contemp Probl Ecol. 11:503–513. [Google Scholar]
- Kõljalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, et al. 2005. UNITE: a database providing web‐based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 166(3):1063–1068. [DOI] [PubMed] [Google Scholar]
- Kvile K, Taranto GH, Pitcher TJ, Morato T. 2014. A global assessment of seamount ecosystems knowledge using an ecosystem evaluation framework. Biol Conserv. 173:108–120. [Google Scholar]
- Lai X, Cao L, Tan H, Fang S, Huang Y, Zhou S. 2007. Fungal communities from methane hydrate-bearing deep-sea marine sediments in the South China Sea. Isme J. 1:756–762. [DOI] [PubMed] [Google Scholar]
- Lawrey JD, Diederich P. 2018. Lichenicolous fungi-worldwide checklist, including isolated cultures and sequences. [accessed 2016 May27] http://www.lichenicolous.net.
- Le Calvez T, Burgaud G, Mahe S, Barbier G, Vandenkoornhuyse P. 2009. Fungal diversity in deep-sea hydrothermal ecosystems. Appl Environ Microbiol. 75(20):6415–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lex A, Gehlenborg N. 2014. Sets & intersections. Nat Methods. 11:779. [DOI] [PubMed] [Google Scholar]
- Lex A, Gehlenborg N, Strobelt H, Vuillemot R, Pfister H. 2014. Upset: Visualization of intersecting sets. IEEE Trans Vis Comput Graph. 20(12):1983–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhou H, Pan X, Xu T, Zhang Z, Zi X, Jiang Y. 2017. Cassava foliage affects the microbial diversity of Chinese indigenous geese caecum using 16S rRNA sequencing. Sci Rep. 7:45697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wang M, Burgaud G, Yu H, Cai L. 2019. Fungal community composition and potential depth-related driving factors impacting distribution pattern and trophic modes from epi- to abyssopelagic zones of the Western Pacific Ocean. Microb Ecol. 78:820–831. [DOI] [PubMed] [Google Scholar]
- Li W, Wang MM, Pan HQ, Burgaud G, Liang SK, Guo JJ, Luo T, Li ZX, Zhang SM, Cai L. 2018. Highlighting patterns of fungal diversity and composition shaped by ocean currents using the East China Sea as a model. Mol Ecol. 27:564–576. [DOI] [PubMed] [Google Scholar]
- Li W, Wang MM, Wang XG, Cheng XL, Guo JJ, Bian XM, Cai L. 2016. Fungal communities in sediments of subtropical Chinese seas as estimated by DNA metabarcoding. Sci Rep. 6(1):26528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, Xu XW, Jiang XW, Wang CS, Zhang DS, Ni JY, Wu M. 2011. Microbial diversity in deep-sea sediment from the cobalt-rich crust deposit region in the Pacific ocean. FEMS Microbiol Ecol. 78(3):565–585. [DOI] [PubMed] [Google Scholar]
- Lluvia VG, Jennyfers CR, Asunción LL, John LD, Anthony SA, Meritxell R. 2019. Targeted ITS1 sequencing unravels the mycodiversity of deep-sea sediments from the Gulf of Mexico. Environ Microbiol. 21(11):4046–4061. [DOI] [PubMed] [Google Scholar]
- Lu X, Kim H, Zhong S, Chen H, Hu Z, Zhou B. 2014. . De novo transcriptome assembly for rudimentary leaves in Litchi Chinensis Sonn. and identification of differentially expressed genes in response to reactive oxygen species. BMC Genom. 15(1):805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZH, Xu W, Li M, Gu JD, Zhong TH. 2015. Spatial distribution and abundance of ammonia-oxidizing microorganisms in deep-sea sediments of the Pacific Ocean. Antonie Van Leeuwenhoek. 108(2):329–342. [DOI] [PubMed] [Google Scholar]
- Magnus I, Jörn P, Anders T, Curt B, Wolfgang B, Katharina B, Böttcher ME, Ivarsson LN. 2015. Zygomycetes in vesicular basanites from vestries seamount, Greenland basin a new type of cryptoendolithic fungi. PloS One. 10(7):e0133368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mel’nikov ME, Pletnev SP, Basov IA, Sedysheva TE. 2009. New data on the morphology and geological structure of the Gramberg Guyot (Magellan Seamounts, Pacific Ocean). Russ J Pacific Geol. 3:401–410. [Google Scholar]
- Menard HW. 1964. Marine geology of the Pacific. In: Mc Graw-Hill, editor. International series in the earth sciences. New York (NY): Mc Graw-Hill; p. 271. [Google Scholar]
- Morato T, Hoyle SD, Allain V, Nicol SJ. 2010. Seamounts are hotspots of pelagic biodiversity in the open ocean. Proc Natl Acad Sci USA. 107(21):9707–9711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahama T, Nagano Y. 2012. Biology of Marine Fungi. In: Raghukumar C, editor. Progress in molecular and subcellular biology. Vol. 53. Berlin (BE): Springer; p.173-189. [Google Scholar]
- Nagahama T, Takahashi E, Nagano Y, Abdel-Wahab MA, Miyazaki M. 2011. Molecular evidence that deep-branching fungi are major fungal components in deep-sea methane cold-seep sediments. Environ Microbiol. 13(8):2359–2370. [DOI] [PubMed] [Google Scholar]
- Nagano Y, Miura T, Nishi S, Lima AO, Nakayama C, Pellizari VH, Fujikura K. 2017. Fungal diversity in deep-sea sediments associated with asphalt seeps at the Sao Paulo Plateau. Deep Sea Res Part II Top Stud Oceanogr. 146:59–67. [Google Scholar]
- Nagano Y, Nagahama T. 2012. Fungal diversity in deep-sea extreme environments. Fungal Ecol. 5(4):463–471. [DOI] [PubMed] [Google Scholar]
- Nagano Y, Nagahama T, Hatada Y, Nunoura T, Takami H, Miyazaki J, Takai K, Horikoshi K. 2010. Fungal diversity in deep-sea sediments—the presence of novel fungal groups. Fungal Ecol. 3(4):316–325. [Google Scholar]
- Nguyen NH, Song Z, Bates ST, Branco S, Tedersoo L, Menke J, Kennedy PG. 2016. FUNGuild an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20:241–248. [Google Scholar]
- Ondov BD, Nicholas HB, Adam MP. 2011. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 12(1):385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Op De Beeck M, Lievens B, Busschaert P, Declerck S, Vangronsveld J, JV C. 2014. Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PloS One. 9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi W, Biddle JF, Edgcomb V. 2013. Deep sequencing of subseafloor eukaryotic rRNA reveals active Fungi across marine subsurface provinces. PloS One. 8(2):e56335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul FK, Josephine YA. 2010. Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microbiol Ecol. 47:161–177. [DOI] [PubMed] [Google Scholar]
- Peoples LM, Grammatopoulou E, Pombrol M, Xu X, Osuntokun O, Blanton J, Allen EE, Nunnally CC, Drazen JC, Mayor DJ, et al. 2019. Microbial community diversity within sediments from two geographically separated hadal trenches. Front Microbiol. 10:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuphumirat W, Ferguson DK, Gleason FH. 2016. The colonization of palynomorphs by chytrids and thraustochytrids during pre-depositional taphonomic processes in tropical mangrove ecosystems. Fungal Ecol. 23:11–19. [Google Scholar]
- Powell MJ, Letcher PM. 2014. Systematics and Evolution. Berlin (BE): Springer. (McLaughlin D, Spatafora J, editors. The Mycota; vol.7A.). [Google Scholar]
- Preez CD, Curtis JMR, Clarke ME. 2016. The structure and distribution of benthic communities on a shallow seamount (cobb seamount, northeast Pacific ocean). PloS One. 11(10):e0165513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrini AM, Nizinski MS, Chaytor JD, Demopoulos AWJ, Brendan RE, France SC, Moore JA, Heyl T, Auster PJ, Kinlan B, et al. 2015. Exploration of the canyon-incised continental margin of the northeastern united states reveals dynamic habitats and diverse communities. PloS One. 10(10):e0139904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghukumar C, Raghukumar S. 1998. Barotolerance of fungi isolated from deep-sea sediments of the Indian Ocean. Aquat Microb Ecol. 15(2):153–163. [Google Scholar]
- RaghuKumar C, Raghukumar S, Sheelu G, Gupta SM, Nath BN, Rao BR. 2004. Buried in time culturable fungi in a deep-sea sediment core from the Chagos Trench. The Indian Ocean. Deep Sea Res Part I Oceanogr Res Pap. 51(11):1759–1768. [Google Scholar]
- Raghukumar S. 2017. Chapter 2, The marine environment and the role of fungi. In: Raghukumar S, editor.Fungi in coastal and oceanic marine ecosystems. Berlin (BE): Springer International Publishing; p. 15–36. [Google Scholar]
- Rämä T, Hassett BT, Bubnova E. 2017. Arctic marine fungi: from filaments and flagella to operation taxonomic units and beyond. Bot Mar. 60:433–452. [Google Scholar]
- RédouV, Ciobanu MC, Pachiadaki MG, Edgcomb V, Alain K, Barbier G, Burgaud G. 2014. In-depth analyses of deep subsurface sediments using 454-pyrosequencing reveals a reservoir of buried fungal communities at record-breaking depths. FEMS Microbiol Ecol. 90(3):908–921. [DOI] [PubMed] [Google Scholar]
- Rédou V, Navarri M, Meslet-Cladière L, Barbier G, Burgaud G. 2015. Species richness and adaptation of marine fungi from deep-subseafloor sediments. Appl Environ Microbiol. 81(10):3571–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth FJ, Orpurt PA, Ahearn DJ. 1964. Occurrence and distribution of fungi in a subtropical marine environment. Can J Bot. 42(4):375–383. [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ. 2009. Introducing mothur: open source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 75:7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz B, Guillou L, Marano AV, Neuhauser S, Sullivan BK, Karsten U, Küpper FC, Gleason FH. 2016. Zoosporic parasites infecting marine diatoms - a black box that needs to be opened. Fungal Ecol. 19:59–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Raghukumar C, Verma P, Shouche Y. 2010. Phylogenetic diversity of culturable fungi from the deep-sea sediments of the Central Indian Basin and their growth characteristics. Fungal Divers. 40:89–102. [Google Scholar]
- Singh P, Raghukumar C, Verma P, Shouche Y. 2011. Fungal community analysis in the deep-sea sediments of the Central Indian Basin by culture-independent approach. Microb Ecol. 61(13):507–517. [DOI] [PubMed] [Google Scholar]
- Singh P, Raghukumar C, Verma P, Shouche Y. 2012. Assessment of fungal diversity in deep-sea sediments by multiple primer approach. World J Microbiol Biotechnol. 28(2):659–667. [DOI] [PubMed] [Google Scholar]
- Sterkenburg E, Bahr A, Mikael BD, Karina EC, Björn DL. 2015. Changes in fungal communities along a boreal forest soil fertility gradient. New Phytol. 207(4):1145–1158. [DOI] [PubMed] [Google Scholar]
- Takami H, Inoue A, Fuji F, Horikoshi K. 1997. Microbial flora in the deepest sea mud of the Mariana Trench. FEMS Microbiol Lett. 152(2):279–285. [DOI] [PubMed] [Google Scholar]
- Takeshita K, Yubuki N, Kakizoe N, Inagaki Y, Maruyama T. 2007. Diversity of microbial eukaryotes in sediment at a deep-sea methane cold seep, surveys of ribosomal DNA libraries from raw sediment samples and two enrichment cultures. Extremophiles. 11(4):563–576. [DOI] [PubMed] [Google Scholar]
- Tedersoo L, Anslan S, Bahram M, Põlme S, Riit T, Liiv I, Kõljalg U, Kisand V, Nilsson H, Hildebrand F, et al. 2015. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys. 10(10):1–43. [Google Scholar]
- Tedersoo L, Bahram M, Põlme S, Kõljalg U, Yorou NS, Wijesundera R, Ruiz LV, Vasco-Palacios AM, Thu PQ, Suija A, et al. 2014. Global diversity and geography of soil fungi. Science. 346(6213):1256688. [DOI] [PubMed] [Google Scholar]
- Tedersoo L, Nilsson RH, Abarenkov K, Jairus T, Sadam A, Saar I, Bahram M, Bechem E, Chuyong G, Kõljalg U.. 2010. 454 Pyrosequencing and Sanger sequencing of tropical mycorrhizal fungi provide similar results but reveal substantial methodological biases. New Phytologist. 188(1):291-301. doi: 10.1111/j.1469-8137.2010.03373.x [DOI] [PubMed] [Google Scholar]
- Tedersoo L, Sánchez-Ramírez S, Kõljalg U, Bahram M, Döring M, Schigel D, May T, Ryberg M, Abarenkov K. 2018. High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Divers. 90:135–159. [Google Scholar]
- Ü, ODP. . 1993. History of Pacific Guyots uncovered. Geotimes. 38(1):18–19. [Google Scholar]
- Thaler AD, van Dover CL, Vilgalys R. 2012. Ascomycete phylotypes recovered from a Gulf of Mexico methane seep are identical to an uncultured deep-sea fungal clade from the Pacific. Fungal Ecol. 5(2):270–273. [Google Scholar]
- Tisthammer KH, Cobian GM, Amend AS. 2016. Global biogeography of marine fungi is shaped by the environment. Fungal Ecol. 19:39–46. [Google Scholar]
- Toju H, Tanabe AS, Yamamoto S, Sato H. 2012. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PloS One. 7(7):e40863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treseder KK, Lennon JT. 2015. Fungal traits that drive ecosystem dynamics on land. Microbiol Mol Biol Rev. 79(2):243–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseytlin VB. 1985. Energetics of fish populations inhabiting seamounts. Oceanology (Wash DC). 25:237–239. [Google Scholar]
- Vargas-Gastelum L, Chong-Robles J, Lago-Leston A, Darcy JL, Amend AS, Riquelme M. 2019. Targeted ITS1 sequencing unravels the mycodiversity of deep-sea sediments from the Gulf of Mexico. Environ Microbiol. 21(11):4046–4061. [DOI] [PubMed] [Google Scholar]
- Walsh EA, Kirkpatrick JB, Rutherford SD, Smith DC, Sogin M, D’Hondt S. 2016. Bacterial diversity and community composition from the sea surface to subseafloor. Isme J. 10:979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 73:5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sen B, He Y, Xie N, Wang G. 2018. Spatiotemporal Distribution and Assemblages of Planktonic Fungi in the Coastal Waters of the Bohai Sea. Front Microbiol. 9:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Gao W, Johnson RH, Zhang W, Meldrum DR. 2013. Integrated metagenomic and metatranscriptomic analyses of microbial communities in the meso- and bathypelagic realm of the North Pacific ocean. Mar Drugs. 11(10):3777–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Gao YH, Gong LF, Li M, Pang KL, Luo ZH. 2019. Fungal Diversity in the Deep Sea Hadal Sediments of the Yap Trench by Cultivation and High Throughput Sequencing Methods based on the ITS rRNA gene. Deep Sea Res Part I Oceanogr Res. Pap.145:125–136. [Google Scholar]
- Xu W, Gong LF, Pang KL, Luo ZH. 2018b. Fungal diversity in deep-sea sediments of a hydrothermal vent system in the Southwest Indian Ridge. Deep Sea Res Part I Oceanogr Res Pap. 131:16–26. [Google Scholar]
- Xu W, Guo SS, Gong LF, He GY, Pang KL, Luo ZH. 2018a. Cultivable fungal diversity in deep-sea sediment of the East Pacific Ocean. Geomicrobiol J. 35(9):1–8. [Google Scholar]
- Xu W, Guo SS, Pang KL, Luo ZH. 2017. Fungi associated with chimney and sulfide samples from a South Mid-Atlantic Ridge hydrothermal site: distribution, diversity, and abundance. Deep Sea Res Part I Oceanogr Res Pap. 123:48–55. [Google Scholar]
- Xu W, Luo ZH, Guo SS, Pang KL. 2016. Fungal community analysis in the deep-sea sediments of the Pacific Ocean assessed by comparison of ITS, 18S and 28S ribosomal DNA regions. Deep Sea Res Part I Ocanogr Res ePap. 109:51–60. [Google Scholar]
- Xu W, Pang KL, Luo ZH. 2014. High fungal diversity and abundance recovered in the deep-sea sediments of the Pacific Ocean. Microb Ecol. 68:688–698. [DOI] [PubMed] [Google Scholar]
- Zhang J, Sun QL, Zeng ZG, Chen S, Sun L. 2015. Microbial diversity in the deep-sea sediments of the north and the ridge, Okinawa trough. Microbiol Res. 177:43–52. [DOI] [PubMed] [Google Scholar]
- Zhang X, Xu W, Liu Y, Cai M, Luo Z, Li M. 2018. Metagenomics reveals microbial diversity and metabolic potentials of seawater and surface sediment from a hadal biosphere at the Yap Trench. Front Microbiol.9:2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XY, Tang GL, Xu XY, Nong XH, Qi SH. 2014. Insights into deep-sea sediment fungal communities from the East Indian Ocean using targeted environmental sequencing combined with traditional cultivation. PloS One. 9(10):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XY, Wang GH, Xu XY, Nong XH, Wang J, Amin M, Qi SH. 2016. Exploring fungal diversity in deep-sea sediments from Okinawa Trough using high-throughput Illumina sequencing. Deep Sea Res Part I Oceanogr Res Pap. 116:99–105. [Google Scholar]
- Zhang X-Y., Zhang Y, Xu X-Y., Qi S-H... 2013. Diverse Deep-Sea Fungi from the South China Sea and Their Antimicrobial Activity. Curr Microbiol. 67(5):525-530. doi: 10.1007/s00284-013-0394-6 [DOI] [PubMed] [Google Scholar]
- Zhao LH, Jin XL, Gin JY, Li JB. 2010. 麦哲伦海山链漂移史及可能的来源. [The research on the drifting history and possible origin of the Magellan seamount trail]. Hai Yang Xue Bao. 32: 3. Chinese. [Google Scholar]
- Zhou S, Huang Y, Lai X, Cao L, Tan H, Fang S. 2007. Fungal communities from methane hydrate-bearing deep-sea marine sediments in south china sea. Isme J. 1:756–762. [DOI] [PubMed] [Google Scholar]
- Zhu BD, Liang DH, Cui ZG. 2011. 西太平洋麦哲伦海山链的海山地貌及成因[Geomorphologic characteristics and genesis of the Magellan seamount chain in the western Pacific]. Cent South Univ. 42: 2. Chinese. [Google Scholar]