

The induction and maintenance of robust immune tolerance has been the holy grail of immunology for decades. In the absence of robust unresponsiveness of the immune system to self antigens (immune tolerance), uncontrolled reactivity can lead to disorders like food allergies and autoimmune diseases. Although the first seminal experiments in tolerance were conducted in the 1950s,1 the development of novel tolerance-inducing therapeutic drugs has been fraught with clinical challenges and few durable successes, in spite of advances in our understanding of the fundamental aspects of the immune system. However, in recent years, new breakthroughs in our understanding of these basic mechanisms and maintenance of immune tolerance have led to clinical successes in the fields of organ transplantation and allergic and autoimmune diseases. Moreover, novel peptide therapeutic drugs, anti–T-cell antibodies, and cell therapies have set the stage for short-term treatments of autoimmune diseases that have long-term efficacy and eliminate the need for continuous therapy.
During the past few decades, a more detailed understanding of the molecular events associated with T-cell recognition and activation has advanced various approaches to tolerance, such as reprogramming, costimulatory blockade, checkpoint inhibition, and antigen-specific immune regulation. The term “unresponsiveness” that has been associated with immune tolerance refers to a lack of pathogenic immunity characterized by immune-cell inactivation or deletion or to the diversion of pathogenic immunity to protective immunity through the engagement of regulatory cells, deviation in cell differentiation, or development of immune barriers. Thus, implicit in current approaches to the development of tolerogenic drugs is the assumption that successful therapies would treat and prevent allergic and autoimmune diseases, as well as lead to immunosuppression-free organ and stem cell–derived tissue transplantation and protein-replacement approaches in congenital diseases such as hemophilia. Many of the most recent therapeutic successes involve novel drugs that have been designed to break tolerance to cancers that are dependent on tumor-specific and microenvironment-mediated tolerogenic signals. However, these treatments can lead to autoimmune syndromes, underscoring the delicate balance between breaking tolerance to treat tumors and altering immune homeostasis systemically.
MECHANISMS OF IMMUNE TOLERANCE
THE THYMUS AND CENTRAL TOLERANCE
In the early 1960s, Jacques Miller and Max Cooper independently defined two distinct types of immune cells, T and B cells, that are the hallmark of the antigen-specific adaptive immune system.2,3 T cells orchestrate immune responses both indirectly, by providing soluble and membrane-associated signals that promote the survival, expansion, and differentiation of B cells (which produce antibodies that support productive humoral immunity), and directly, by killing foreign and infected tissues through cellular and soluble mediators.3 T cells recognize foreign antigens through a unique, highly diverse set of T-cell receptors (TCRs) designed to mediate immunity without the collateral damage of destroying native tissues. In a parallel system, regulatory T cells (Tregs) recognize self-peptides and, when activated, control self-reactive pathogenic T cells. This complex, dynamic process of self–nonself discrimination is the basis of immune tolerance. In this review, we focus on TCR αβ cells in immune tolerance, given their dual role in pathogenicity and immune suppression, and on the wealth of new approaches being developed to induce or break tolerance by targeting TCR αβ cells and the antigen-presenting cells that drive their function.
The thymus is the birthplace of T cells. Bone marrow–derived CD34+ stem cells migrate to the thymus, where they differentiate and acquire the expression of the TCR. Each T cell expresses its own unique receptor, which is composed of a heterodimer of two chains (TCRα and TCRβ), each of which is generated through somatic recombination of multiple genetic elements, including the addition of a few nucleotides at the site of recombination. This combinatorial diversity results in a broad TCR repertoire with more than 1010 distinct receptor combinations and is key to the ability of the T-cell population as a whole to recognize the vast array of potential targets (see the Glossary). However, in each person, this repertoire is restricted and skewed as each T cell is selected for its ability to bind a self-peptide in the context of a polymorphic and polygenic component of its ligand, molecules encoded by the major histocompatibility complex (MHC). This selection step, termed positive selection, ensures that T cells are able to recognize a foreign peptide antigen bound to proteins encoded by specific self MHC alleles, most commonly MHC class I and class II molecules for CD8+ and CD4+ T cells, respectively.4 (Details are provided in note 1 in the Supplementary Appendix, available with the full text of this article at NEJM.org.)
After positive selection, T cells pass through an important second selection step, called negative selection. This is a filtering step to remove T cells that have a strong binding affinity to self-peptides that are bound to the same MHC molecules and are consequently autoreactive. Such T cells arise by chance through the stochastic gene-rearrangement process that generated the TCR and, thus, could potentially lead to autoimmunity. To accomplish negative selection, a small, resident cell subset, termed medullary thymic epithelial cells (mTECs), and bone marrow–derived dendritic cells5 interact with the maturing T cells. The mTECs express a transcriptional activator called the autoimmune regulator (AIRE), which enables the expression of thousands of otherwise tissue-restricted proteins, so that peptides, derived from these proteins, are displayed to developing T cells; this process results in robust elimination of self-reactive CD4+ and CD8+ conventional T cells.6 The key role of AIRE-positive mTECs in central tolerance is indicated by the clinical manifestation of a profound, multiorgan autoimmune syndrome, called autoimmune polyglandular syndrome type 1 (APS1), in patients with mutations in AIRE.7
PERIPHERAL TOLERANCE
As efficient as the thymus is in eliminating self-reactive cells, many self-reactive T cells escape thymic negative selection, which leads to the need for peripheral mechanisms to ensure that self-tolerance is maintained.8 A variety of cell types and processes control peripheral tolerance, including cells of the adaptive and innate immune system and signaling components within T cells and antigen-presenting cells themselves.9
Effective T-cell signaling requires both engagement of the primary antigen-specific receptor and a second, costimulatory signal to induce proliferation, differentiation, and survival.10 A constitutive T-cell–surface molecule, CD28, was the first T-cell costimulatory receptor to be identified (Fig. 1).11 The ligands CD80 and CD86 are expressed selectively on antigen-presenting cells, especially after activation through innate agonists of toll-like receptors (TLRs) and soluble factors. Only cells that recognize nominal antigen — often termed “signal one” to reflect the first signal delivered to T cells during an activation event — respond to a second costimulatory signal essential for complete T-cell activation. Blockade of costimulatory pathways leads to an antigen-specific apoptotic cell death, clonal inactivation, and tolerance induction.12 Studies in animal models have shown that costimulatory blockade with monoclonal antibodies and soluble forms of the CD80 and CD86 high-affinity receptor, cytotoxic T-lymphocyte–associated protein 4 (CTLA-4), induces tolerance in the context of autoimmune disorders or organ transplantation.12 Identification of additional costimulatory pathways, such as CD154–CD40, CD11A–CD54, CD18–CD54, and CD2–CD58, has validated and extended the two-signal model of T-cell activation. These findings have provided new therapeutic opportunities for blocking autoreactive T-cell activity and inducing long-term tolerance without the need for continuous therapy.13
Figure 1. Two-Signal Models of Costimulatory and Inhibitory Pathways.
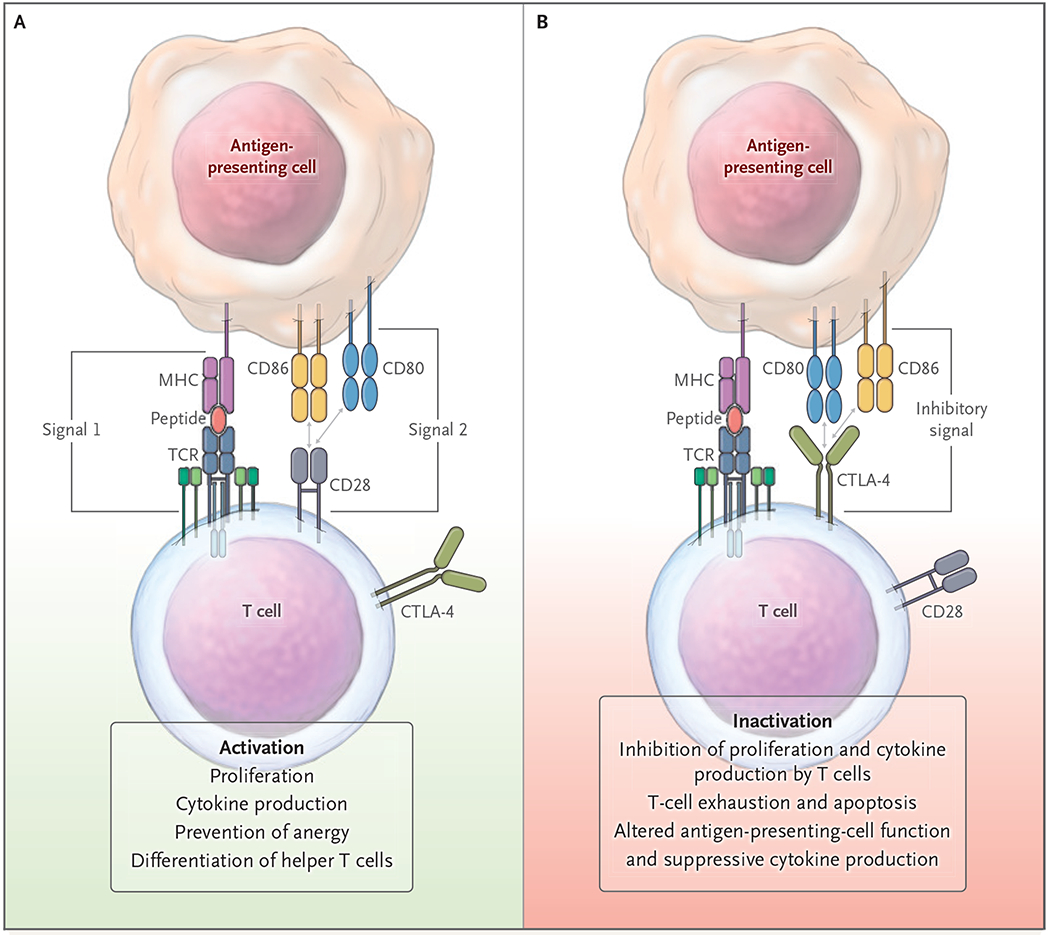
Initiation of a productive T-cell response involves integration of a primary signal delivered through the T-cell receptor (TCR) and major histocompatibility complex (MHC)–peptide, followed by a second signal delivered through the CD28–CD80 or CD28–CD86 pathway (Panel A). After initiation of T-cell activation, other inhibitory checkpoint interactions can shut down T-cell activity (Panel B). Pathways that may be affected as a consequence of both positive and negative second signals are listed at the bottom of the figure. CTLA-4 denotes cytotoxic T-lymphocyte–associated protein 4.
Additional controls on the surface of activated T cells — so-called negative regulators, or checkpoints — are as important as costimulatory pathways in controlling T-cell activation. However, unlike costimulatory pathways, checkpoints, including CTLA-4 and programmed death 1 (PD-1),14 shut down immune activation when engaged by their ligands, which leads to active tolerance induction (Fig. 1). Direct evidence of a role for checkpoints in tolerance has been shown in cases in which checkpoint pathways have been inhibited. For instance, gene ablation or treatment with checkpoint inhibitors can exacerbate autoimmunity and, in some cases, break tolerance induced by other therapies.15 Thus, targeting of checkpoint pathways has resulted in a new generation of tolerance-manipulating drugs (Fig. 2). Inhibition of checkpoint pathways has revolutionized cancer immunotherapy by turning once deadly cancers such as melanoma and non–small-cell lung cancer into treatable diseases.14 Checkpoint agonists are being used in the treatment of autoimmune disease and organ-transplant rejection, as described below.
Figure 2. Additional Costimulatory and Checkpoint Pathways.

In addition to the two-signal models of costimulatory and checkpoint pathways, additional stimulatory and inhibitory pathways (indicated by plus and minus signs, respectively) influence the immune response, including molecules of the tumor necrosis factor (TNF)–related family, other members of the CD28 family, adhesion molecules, and T-cell immunoglobulin and mucin (TIM) molecules. The various stimulatory and inhibitory pathways can influence and be influenced by cytokines. Pep denotes peptide, and TGF-β transforming growth factor β.
Another key mechanism of peripheral tolerance is the presence of specialized cell populations designed to suppress pathogenic immune responses that inadvertently target self-tissue. These include naturally occurring Treg cells and in vitro–induced Treg (iTreg) cells, as well as interleukin-10–producing type 1 regulatory T (Tr1) cells and transforming growth factor β (TGF-β)–producing type 3 helper T (Th3) cells.16 Of these subsets, the most extensively studied are Tregs, a subset of the self-reactive CD4+ T cells that develop as a consequence of the expression of a master transcriptional repressor, forkhead box P3 (FOXP3). FOXP3 alters the differentiation of the mature CD4+ T-cell population into this regulatory T-cell subset, which plays a fundamental role in immune homeostasis.17 Functional disruption of FOXP3, either genetically (in Scurfy mice and patients with the IPEX [immune dysregulation, polyendocrinopathy, enteropathy, X-linked] syndrome) or pharmacologically, leads to severe autoimmune disorders that cause death at a very young age unless the patient is given a bone marrow transplant.18
The primary source of Tregs is the thymus, where the cells are generated by an alternative developmental pathway. During negative selection, a subset of developing T cells expressing TCRs with high affinity develop into Treg precursors that up-regulate FOXP3, leading to a stable epigenetic state and resulting in a mature, self-reactive population of thymus-derived Tregs (tTregs) that populate lymphoid and nonlymphoid tissues in the periphery.19,20 The role of Tregs in the periphery is to halt self-reactivity and promote tissue repair and regeneration (Fig. 3).21,22 AIRE-expressing mTECs play a role in the generation of tTregs through presentation of tissue-specific antigens. Moreover, AIRE is expressed in a subset of bone marrow–derived cells in peripheral lymphoid organs and, thus, may influence tolerance in peripheral tissues.23 This finding highlights the connection between peripheral tolerance and T-cell development in the thymus.
Figure 3. Tolerance-Inducing Pathways in the Thymus and Periphery.

The majority of cells interacting with autoimmune regulator (AIRE)–expressing medullary thymic epithelial cells (mTECs) during thymic development undergo negative selection and die (Panel A). A subset of high-affinity, self-reactive CD4+ T cells interact with the mTECs, leading to the development of regulatory T cells (Tregs). The remaining mature naive T cells migrate into the immune periphery, where they have either a pathogenic role in mediating immunity (Panel B, left) or a protective role as peripherally derived Tregs (through interaction with tolerogenic antigen-presenting cells and cytokines) that control potential autoreactive responses (Panel B, right). Additional cell types, such as extrathymic AIRE–expressing cells (eTACs), can also modify potentially autoreactive T cells.
Tregs are generated in the immune periphery as well, under conditions in which naive CD4+ T cells encounter antigen in the context of suppressive factors such as TGF-β, interleukin-10, bacterially derived metabolic products, or altered stimulatory pathways. Under certain conditions, antigen presentation leads to the induction of stable FOXP3 and transformation of conventional T cells into peripherally derived Tregs (pTregs).24 Thus, unlike tTregs, which develop in the thymus from T cells undergoing negative selection based on high self-reactivity, pTregs develop from a conventional peripheral T-cell repertoire selected for low self-reactivity. A growing number of examples show that Tregs recognize commensal bacteria, which have many characteristics normally attributed to self-antigens. This process diversifies the Treg repertoire, which may be most effective in shutting down inflammatory responses. Moreover, pTregs may recognize modified proteins, such as citrullinated peptides, hybrid peptides, and phosphorylated proteins, which are often present in autoimmune states but not in the thymus. Together, the combination of tTregs and pTregs, as well as interluekin-10–producing Tr1 cells and TGF-β–producing Th3 cells, may provide the broadest antigen-recognition repertoire for controlling pathogenic self-reactivity. Moreover, because of thymic involution in adulthood, the development and maintenance of tolerance may differ according to age, with peripheral pathways having a more important regulatory role in autoimmunity in adults than in children25 (note 2 in the Supplementary Appendix).
Antigen-presenting cells, including tolerogenic dendritic cells, immature macrophages, suppressor antigen-presenting cells of the myeloid lineage, and even certain B-cell subsets, function in conjunction with Tregs and other suppressor T cells to control immunity. These cells develop in response to a variety of cell-surface and soluble factors, including those controlled by Tregs.26–28 For instance, Tregs express high levels of the checkpoint CTLA-4, which can block CD28-mediated costimulation and deliver inhibitory signals that change antigen-presenting cells into tolerogenic cells.29,30 Tregs, as well as other regulatory cells, produce cytokines, such as interleukin-10, interleukin-35, TGF-β, and other soluble factors involved in metabolism, such as indoleamine 2,3-dioxygenase. The Tregs can induce myeloid-derived suppressor cells and alter antigen presentation.27,31 Tregs express CD39 and CD73, which affect the duration, magnitude, and chemical nature of purinergic signals delivered to immune cells through the conversion of ADP or ATP to adenosine.32 Finally, metabolic products of the gut microbiome, including the production of short-chain fatty acids such as butyrate, can have a profound effect on immune function. These and other regulatory factors can act on antigen-presenting cells to promote the generation of Tr1 cells and Tregs or directly affect pathogenic T cells, altering differentiation, trafficking, and function. Oral tolerance studies with short-chain fatty acids support a strong interface between the immune system and resident microbiota33 (Fig. 4).
Figure 4. Activation and Functional Consequences of Suppressive Cells.
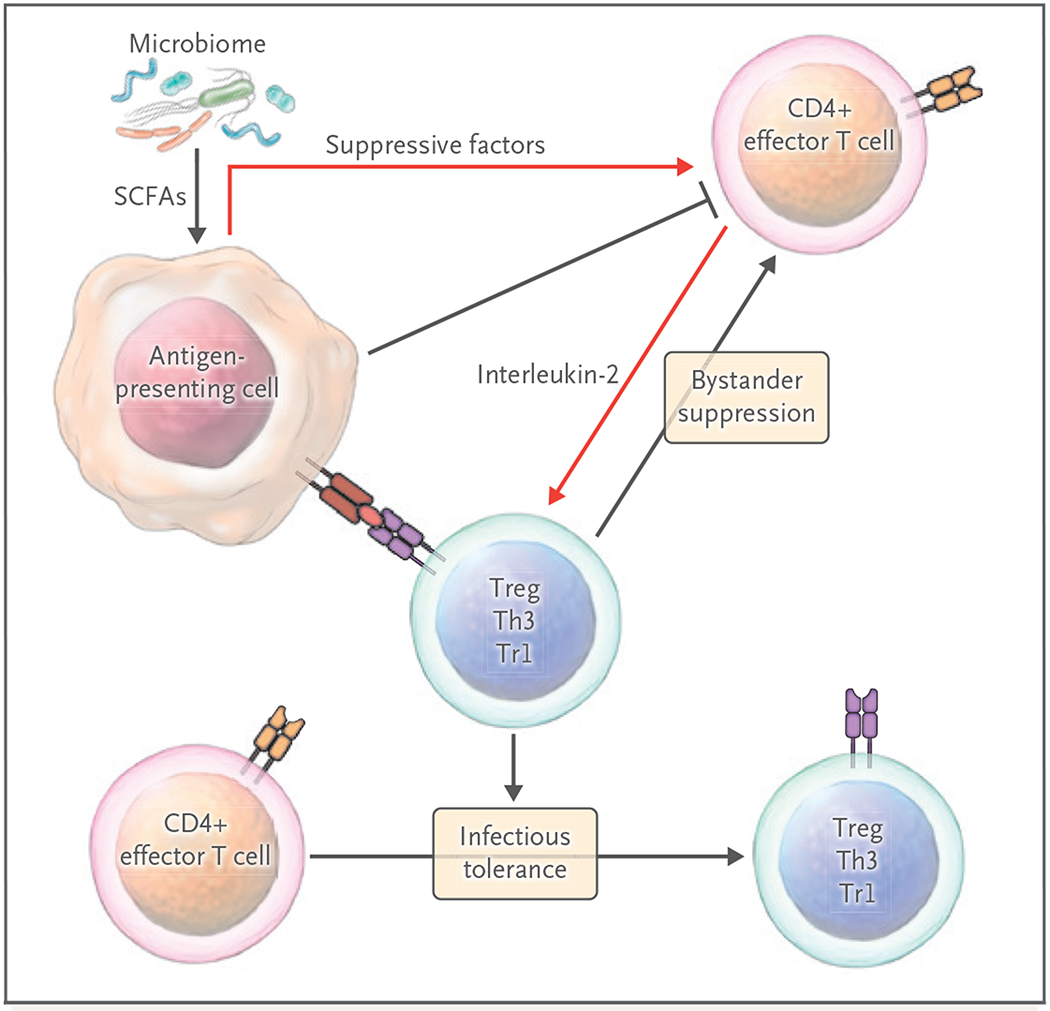
Tregs and other suppressive cells circulate and reside in lymphoid and somatic tissues to control unwanted autoimmune and inflammatory responses. Multiple cell–cell contacts, as well as soluble molecules (including the roduction of metabolites by microbiota), are generated by Tregs or antigen-presenting cells after Treg interactions to control immunity. Tregs can also act through bystander suppression, leading to dominant local immunosuppression and tolerance induction. IDO denotes indoleamine 2,3-dioxygenase, SCFA short-chain fatty acid, Th3 type 3 helper T cell, and Tr1 type 1 regulatory T cell.
Thus, it is clear that a dynamic, homeostatic immune system has evolved to deal with endogenous and exogenous insults. There is a balancing act between the need to develop potent effector cells in order to combat foreign pathogens and the need for homeostatic control of the immune system to shut down unwanted autoinflammation, as has been reported in some patients with coronavirus disease 2019 (Covid-19)34 and autoimmunity.
INDUCING TOLERANCE
Historically, the only option for patients with autoimmune diseases or organ-transplant rejection has been the use of broadly immunosuppressive drugs, which carry substantial risks of serious side effects. Approved therapies, such as calcineurin inhibitors, glucocorticoids, and tumor necrosis factor α and interleukin-1 antagonists, require continuous treatment and target the end stage of the immune dysfunction rather than the induction of tolerance. An improved understanding of the molecular and functional basis for immune tolerance has driven the development of tolerogenic drugs that may fundamentally change the therapeutic landscape.
RESETTING THE IMMUNE SYSTEM
The most effective way to generate immune tolerance would be to reset and rebalance the immune system in order to prevent the development and progression of the autoimmune response. In this regard, two approaches — autologous hematopoietic stem-cell transplantation (HST) and massive immune-cell depletion — have been pioneered. Both of these methods are intended to induce tolerance through exposure to the tolerogenic antigen during regeneration and recovery of the immune system. Autologous HST has been reported to halt the progression of certain forms of autoimmunity, such as multiple sclerosis.35 Combining autologous and donor-derived HST can result in lasting tolerance to the donor MHC while allowing effective immune reconstitution (i.e., mixed chimerism).36,37 The immune-cell depletion approach to rebooting the immune system has been enabled by the use of broad immune-depleting agents such as alemtuzumab (anti-CD52 antibody), B-cell-depleting agents (rituximab, ocrelizumab, and obinutuzumab), and antilymphocyte antibody therapies.38–41 These drugs have been successful in slowing the progression of disease, in some cases through the elimination of antigen presentation by the autoreactive B cells, which are very efficient in capturing and presenting autoantigens.38,42 However, both autologous HST and the broad cell-depleting strategies lead to the elimination of immune-cell subsets, including those involved in protection from infection and, potentially, cancer.
TARGETING PATHOGENIC T CELLS
In the past several years, treatments have been developed to selectively target the highly activated, pathogenic T cells involved in mediating tissue-reactive immunity. These treatments include an anti-CD3 monoclonal antibody (teplizumab), a soluble form of the CD2 costimulatory receptor LFA-3 (alefacept), and low-dose antithymocyte globulin.40,43,44 These biologics have been shown to induce apoptosis and functional inactivation of highly activated effector cells, leaving naive T cells and Tregs intact or even expanding the regulatory pathways. This class of therapeutic drugs has been shown to be efficacious in a variety of autoimmune diseases, even when patients were treated for only a short period of time. In the case of teplizumab, Herold and colleagues reported that the median time until disease onset was prolonged by more than 3 years and the median disease-free survival was 5 or more years among patients who were at risk for type 1 diabetes and who were treated for only 2 weeks.45,46 The therapy not only delays the development of disease but also may induce more sustained tolerance in some patients. In trials conducted by the Immune Tolerance Network, one or two courses of teplizumab given over a 3-month period to patients with newly diagnosed type 1 diabetes led to prolonged preservation of c-peptide (insulin) production, with no increase in rates of infection or cancer. A significant percentage of residual CD8+ effector T cells expressed an exhausted or anergic T-cell phenotype that has been shown to be the major marker of tumor tolerance.47 These results highlight the importance of targeting pathogenic T cells to induce tolerance while maintaining naive T cells and regulatory pathways to preserve immune competence and homeostasis. Moreover, a combination of these therapies, as well as the pro-Treg drugs described below, provides an opportunity to rebalance the immune system, since it is likely that control of autoimmunity requires both a decreased number of pathogenic T cells and increased regulatory activity.
The second approach to tolerance induction with more selective tolerogenic therapeutic drugs has been the development of inhibitors of the costimulatory pathway, as noted above and shown in Figures 1 and 2. Two soluble CD28 antagonists, abatacept and belatacept (CTLA-4–IgG1 and mutant CTLA-4–IgG1, respectively), have been approved by the Food and Drug Administration for the treatment of rheumatoid arthritis and kidney-transplant rejection, respectively.48,49 Unfortunately, the ability of these drugs to induce protracted tolerance in humans is unclear. In a clinical trial involving patients with newly diagnosed type 1 diabetes, treatment with abatacept for 6 months resulted in significant prolongation of insulin production, as compared with insulin production in the control group, but this protection diminished over time.50 One reason for the decline in the efficacy of the drug may have been its depletion of Tregs, which may be essential for the development of tolerance.12,51 Treg development and survival are dependent on CD28 signaling in vivo.52 A second generation of costimulatory antagonists (including those that block CD154–CD40 interactions) that may synergize with CD28 costimulatory blockade are currently being evaluated in a number of clinical settings.53 The substantial effect of antibodies directed at negative regulators in cancer immune therapy, including multiple antibodies against PD-1 and its ligand, PD-L1, and antibodies against CTLA-4 to promote activation signals to T-cells, opens up a new area for tolerance induction.54 New treatments are being developed to activate (rather than block) the key checkpoints and inactivate pathogenic T cells.
The third approach, the induction of antigen-specific tolerance with the use of autoantigen therapeutic drugs, has been the most challenging.55–58 These therapies are potentially the most selective in eliminating autoreactive pathogenic T cells while reducing the risks of infections and cancer, which make broadly immunosuppressive agents problematic. However, the requirement for knowledge of the autoantigen, the large number of pathogenic epitopes, and the consequence of epitope spreading (which expands the number of pathogenic epitopes) complicate these deletional therapies. Such therapies have nevertheless been successful in the treatment of immune responses to allergens, such as peanuts, dust mites, and certain grasses, and metabolic deficiencies, such as hemophilia,59,60 in which the uncontrolled inflammatory responses to the proteins after exposure can prevent the use of these lifesaving treatments. In a seminal proof-of-principle study, newborn children at high risk for atopic allergies were randomly assigned to be exposed to peanuts or to have no exposure over a 5-year period. A majority of the children who were exposed to peanuts had a higher sustained reduction in the incidence of peanut allergy than those who had not been exposed.61 At a mechanistic level, tolerance was due in part to immune deviation from a pathogenic IgE response to a nonpathogenic IgG response.
In an equally dramatic demonstration of the power of antigens to induce tolerance under the right conditions, allograft tolerance has been maintained after withdrawal of immunosuppressive drugs in a subset of liver-transplant recipients, presumably because of the constant exposure of alloantigen in a protolerogenic hepatic environment.62 These successes have expanded to the use of peptides, either administered alone intravenously or coupled to cells, nanoparticles, or other multimeric scaffolds; DNA vaccines, which incorporate both antigenic peptides and tolerogenic therapeutics to modify TCR recognition; immobilized HLA–peptide complexes; and tolerogenic dendritic cells pulsed with multiple peptides.63 Peptide-induced tolerance has often been associated with the induction of Tregs or tolerogenic dendritic cells. Uptake of antigen through scavenger receptors, such as DEC-205, SR-A, or MARCO, may alter the cell phenotype and function and, in some cases, down-regulate costimulatory ligands. Thus, peptide therapies tap into some of the most basic aspects of T-cell activation. The safety of antigen-specific therapeutic approaches has been shown in multiple phase 1 clinical trials using several autoantigenic peptides from multiple proteins, and controlled phase 2 trials of efficacy are now under way.64,65
TARGETING REGULATORY PATHWAYS
Defects in or defective regulation of key immune cells such as tolerogenic FOXP3-positive Treg cells has been documented in several types of human autoimmunity, which suggests that enhanced Treg numbers, increased Treg functioning, or both might stop autoimmunity. Several approaches have been undertaken to increase or enhance Treg numbers and activity in patients with autoimmune disorders. These approaches include treatment with low-dose interleukin-2 and interleukin-2 mutants to selectively increase Treg numbers and functioning, short-term combination therapies that promote the development of regulatory cells, such as rapamycin (sirolimus), and the coadministration of tolerogenic peptides, as described above.66–69 One of the more active research areas has been the administration of ex vivo–generated, tolerogenic cellular therapies, which can result in a shutdown of autoimmunity, potentially lasting for years.15,70 Treg therapy takes advantage of two distinct factors that enhance tolerance: bystander suppression and infectious tolerance. These mechanisms enable Tregs to suppress the immune response widely within a local environment and create a tolerogenic environment in which other local cells take on a tolerogenic phenotype, which leads to broadening of the number and specificities of the cells involved in the control of unwanted immunity. More than 50 active and completed clinical trials are testing the safety and efficacy of Treg cell therapy for indications such as kidney or liver transplantation, pemphigus vulgaris, systemic lupus erythematosus, inflammatory bowel disease, autoimmune hepatitis, allergy, and asthma. In patients with chronic graft-versus-host disease, Treg-cell therapy alleviated symptoms, and pharmacologic immunosuppression could be reduced.71 In addition to Tregs, a second suppressive T-cell population, Tr1 cells, is being used therapeutically to induce tolerance.72 Efforts are under way to increase the potency and life span of these regulatory cells by inserting specific TCRs or chimeric antigen receptors and using gene editing to modify durability and stability, alter trafficking, and enhance tissue repair of the adoptively transferred cells.
Additional potentially tolerogenic approaches using antigen-presenting cells are also being tested in the clinic, including the use of mesenchymal stromal cells73 and dendritic cells.74,75 Each approach has advantages and disadvantages with respect to the potential for “off-the-shelf” treatments, the ability to modify the cell therapy with current genetic approaches, and the mechanism (direct vs. bystander) of suppression. Clinical-grade tolerogenic dendritic cells, generated in vitro, are being tested in autoimmune and organ-transplant settings to induce clonal deletion of pathogenic T cells in vivo or to induce the generation of antigen-specific regulatory T and B cells.76–78 All these approaches are focused on the induction of active, dominant, antigen-specific tolerance and thus avoid the need to delete all T cells against potential pathogenic specificities.
Finally, other cell therapies are being tested to treat autoantibody-mediated autoimmunity, including the use of genetically engineered effector T cells transduced with a chimeric antigen receptor that is directed at the cell-surface CD19 molecule. On adoptive transfer, the T cells are triggered by CD19 on B cells, which induces cytolytic activity and B-cell destruction.79,80 Another approach is the expression of autoantigen (target protein) chimerized to the signal domains of a typical chimeric antigen receptor (a so-called chimeric autoantigen receptor), which when put into CD8+ T cells, leads to the destruction of B cells and plasma cells expressing a receptor that recognizes the autoantigen.79 These cells are being introduced into clinical practice and will soon be assessed for their safety and efficacy in a variety of autoantibody-mediated diseases.
CONCLUSIONS
Despite the genetic predisposition observed in the majority of autoimmune diseases, the inability to predict the development of autoimmunity, coupled with the likelihood that disease has already developed by the time patients come to clinical attention, means that treatment is usually initiated after the onset of disease. Efforts are under way to predict the onset of diseases such as rheumatoid arthritis and type 1 diabetes. Individual tolerogenic therapies may be more effective if they can be used before sufficient tissue has been damaged to develop a disease phenotype.45 Achieving durable tolerance is likely to require eliminating or regulating disease effectors and repairing damaged tissue. The complexity of the process suggests that the use of combination therapies will be required to establish long-term tolerance.
Supplementary Material
Acknowledgments
We thank Dr. Gerald Nepom at the Benaroya Institute for kindly reading and advising on an earlier version of the manuscript and the members of the Bluestone and Anderson laboratory for their work, which has resulted in many of the ideas highlighted in this review, and for their ongoing advice and commitment to the discovery effort.
Glossary
- Autoimmune regulator (AIRE)
A protein that binds to chromatin and regulates the process of gene transcription such that a plethora of self-proteins are ectopically expressed in medullary thymic epithelial cells (mTECs) involved in thymic selection.
- Bystander suppression
Immunosuppression in a local environment through direct cell-to-cell contacts or short-range cytokines that are independent of specific antigen reactivity.
- Infectious tolerance
Tolerance that results when forkhead box P3 (FOXP3)–positive regulatory T cells (Tregs) convert conventional T cells into peripherally derived Tregs through secretion of the suppressive cytokines transforming growth factor β (TGF-β), interleukin-10, or interleukin-35 or indirectly through the alteration of antigen-presenting cells.
- In vitro–induced Tregs (iTregs)
Tregs that can clearly be distinguished from Treg populations generated in vivo; iTregs can be induced by cytokines such as TGF-β, altered antigen-presenting cells, or costimulatory blockade.
- Medullary thymic epithelial cells (mTECs)
An epithelial-cell population present in the thymic medulla that expresses AIRE protein and is responsible for negative selection and Treg development.
- Peripherally derived Tregs (pTregs)
Tregs that develop in tissue sites from conventional CD4+ T cells as a consequence of exposure to certain cytokines, microbial products, or altered antigen-presenting cells.
- T-cell receptor (TCR) repertoire
The diverse use of unique TCR alpha and beta chains to recognize individual antigen–major histocompatibility complexes.
- Thymus-derived Tregs (tTregs)
Tregs induced as a consequence of negative selection, in which high-affinity, self-reactive T cells develop into FoXP3-positive Tregs.
- Toll-like receptors (TLRs)
A class of proteins, usually expressed on macrophages and dendritic cells, that recognize structurally conserved molecules derived from microbes to activate the innate immune system.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
REFERENCES
- 1.Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature 1953; 172: 603–6. [DOI] [PubMed] [Google Scholar]
- 2.Cooper MD, Peterson RD, Good RA. Delineation of the thymic and bursal lymphoid systems in the chicken. Nature 1965; 205: 143–6. [DOI] [PubMed] [Google Scholar]
- 3.Miller JFAP. The golden anniversary of the thymus. Nat Rev Immunol 2011; 11: 489–95. [DOI] [PubMed] [Google Scholar]
- 4.Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol 2014; 14: 377–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Meerwijk JPM, Marguerat S, Lees RK, Germain RN, Fowlkes BJ, MacDonald HR. Quantitative impact of thymic clonal deletion on the T cell repertoire. J Exp Med 1997; 185: 377–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson MS, Venanzi ES, Klein L, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002; 298: 1395–401. [DOI] [PubMed] [Google Scholar]
- 7.Husebye ES, Anderson MS, Kämpe O. Autoimmune polyendocrine syndromes. N Engl J Med 2018; 378: 1132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Theofilopoulos AN, Kono DH, Baccala R. The multiple pathways to autoimmunity. Nat Immunol 2017; 18: 716–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bluestone JA, Bour-Jordan H, Cheng M, Anderson M. T cells in the control of organ-specific autoimmunity. J Clin Invest 2015; 125: 2250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol 2001; 19: 225–52. [DOI] [PubMed] [Google Scholar]
- 11.Weiss A, Manger B, Imboden J. Synergy between the T3/antigen receptor complex and Tp44 in the activation of human T cells. J Immunol 1986; 137: 819–25. [PubMed] [Google Scholar]
- 12.Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity 2016; 44: 973–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharpe AH. Mechanisms of costimulation. Immunol Rev 2009; 229: 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature 2017; 541: 321–30. [DOI] [PubMed] [Google Scholar]
- 15.Fife BT, Guleria I, Gubbels Bupp M, et al. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1–PD-L1 pathway. J Exp Med 2006; 203: 2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discov 2019; 18: 749–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev 2011; 241: 260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ochs HD, Gambineri E, Torgerson TR. IPEX, FOXP3 and regulatory T-cells: a model for autoimmunity. Immunol Res 2007; 38: 112–21. [DOI] [PubMed] [Google Scholar]
- 19.Aschenbrenner K, D’Cruz LM, Vollmann EH, et al. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat Immunol 2007; 8: 351–8. [DOI] [PubMed] [Google Scholar]
- 20.Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA. Aire enforces immunetolerance by directing autoreactive T cells into the regulatory T cell lineage. Immunity 2016; 44: 1102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Josefowicz SZ, Niec RE, Kim HY, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012; 482: 395–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arpaia N, Green JA, Moltedo B, et al. A distinct function of regulatory T cells in tissue protection. Cell 2015; 162: 1078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardner JM, Devoss JJ, Friedman RS, et al. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 2008; 321: 843–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abbas AK, Benoist C, Bluestone JA, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol 2013; 14: 307–8. [DOI] [PubMed] [Google Scholar]
- 25.Ander SE, Diamond MS, Coyne CB. Immune responses at the maternal-fetal interface. Sci Immunol 2019; 4(31): eaat6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity 2015; 42: 607–12. [DOI] [PubMed] [Google Scholar]
- 27.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer 2013; 13: 739–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell 2014; 157: 121–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008; 322: 271–5. [DOI] [PubMed] [Google Scholar]
- 30.Walker LSK, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol 2015; 36: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Josefowicz SZ, Lu L-F, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30: 531–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med 2013; 19: 355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pandiyan P, Bhaskaran N, Zou M, Schneider E, Jayaraman S, Huehn J. Microbiome dependent regulation of Tregs and Th17 cells in mucosa. Front Immunol 2019; 10: 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 2020; 395: 1033–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nash RA, Hutton GJ, Racke MK, et al. High-dose immunosuppressive therapy and autologous HCT for relapsing-remitting MS. Neurology 2017; 88: 842–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Busque S, Scandling JD, Lowsky R, et al. Mixed chimerism and acceptance of kidney transplants after immunosuppressive drug withdrawal. Sci Transl Med 2020; 12(528):eaax8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spitzer TR, Sykes M, Tolkoff-Rubin N, et al. Long-term follow-up of recipients of combined human leukocyte antigenmatched bone marrow and kidney transplantation for multiple myeloma with end-stage renal disease. Transplantation 2011; 91: 672–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabatino JJ Jr, Zamvil SS, Hauser SL. B-cell therapies in multiple sclerosis. Cold Spring Harb Perspect Med 2019; 9(2): a032037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hersh CM, Cohen JA. Alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Immunotherapy 2014; 6: 249–59. [DOI] [PubMed] [Google Scholar]
- 40.Haller MJ, Long SA, Blanchfield JL, et al. Low-dose anti-thymocyte globulin preserves C-peptide, reduces HbA1c, and increases regulatory to conventional T-cell ratios in new-onset type 1 diabetes: two-year clinical trial data. Diabetes 2019; 68: 1267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gitelman SE, Gottlieb PA, Felner EI, et al. Antithymocyte globulin therapy for patients with recent-onset type 1 diabetes: 2 year results of a randomised trial. Diabetologia 2016; 59: 1153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arneth BM. Impact of B cells to the pathophysiology of multiple sclerosis. J Neuroinflammation 2019; 16: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rigby MR, Harris KM, Pinckney A, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest 2015; 125: 3285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herold KC, Gitelman SE, Ehlers MR, et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013; 62: 3766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herold KC, Bundy BN, Long SA, et al. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med 2019; 381: 603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sims EK, Bundy BN, Stier KD, et al. 277-OR: Teplizumab reverses the loss of C-peptide in relatives at risk for type 1 diabetes (T1D). Diabetes 2020; 69: Suppl 1 ( 10.2337/db20-277-OR). [DOI] [Google Scholar]
- 47.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15: 486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vincenti F, Rostaing L, Grinyo J, et al. Belatacept and long-term outcomes in kidney transplantation. N Engl J Med 2016; 374: 333–43. [DOI] [PubMed] [Google Scholar]
- 49.Bathon J, Robles M, Ximenes AC, et al. Sustained disease remission and inhibition of radiographic progression in methotrexate-naive patients with rheumatoid arthritis and poor prognostic factors treated with abatacept: 2-year outcomes. Ann Rheum Dis 2011; 70: 1949–56. [DOI] [PubMed] [Google Scholar]
- 50.Orban T, Bundy B, Becker DJ, et al. Costimulation modulation with abatacept in patients with recent-onset type 1 diabetes: follow-up 1 year after cessation of treatment. Diabetes Care 2014; 37: 1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glatigny S, Höllbacher B, Motley SJ, et al. Abatacept targets T follicular helper and regulatory T cells, disrupting molecular pathways that regulate their proliferation and maintenance. J Immunol 2019; 202: 1373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang Q, Henriksen KJ, Boden EK, et al. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol 2003; 171: 3348–52. [DOI] [PubMed] [Google Scholar]
- 53.Pinelli DF, Ford ML. Novel insights into anti-CD40/CD154 immunotherapy in transplant tolerance. Immunotherapy 2015; 7: 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paluch C, Santos AM, Anzilotti C, Cornall RJ, Davis SJ. Immune checkpoints as therapeutic targets in autoimmunity. Front Immunol 2018; 9: 2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol 2007; 7: 665–77. [DOI] [PubMed] [Google Scholar]
- 56.Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat Biotechnol 2019; 37: 238–51. [DOI] [PubMed] [Google Scholar]
- 57.Roep BO, Wheeler DCS, Peakman M. Antigen-based immune modulation therapy for type 1 diabetes: the era of precision medicine. Lancet Diabetes Endocrinol 2019; 7: 65–74. [DOI] [PubMed] [Google Scholar]
- 58.Wraith DC. The future of immunotherapy: a 20-year perspective. Front Immunol 2017; 8: 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maldonado RA, LaMothe RA, Ferrari JD, et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci U S A 2015; 112(2): E156–E165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adair P, Su Y, Scott DW. Tolerance induction in hemophilia A animal models: battling inhibitors with antigen-specific immunotherapies. Discov Med 2013; 15: 275–82. [PubMed] [Google Scholar]
- 61.Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med 2015; 372: 803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feng S, Bucuvalas J. Tolerance after liver transplantation: where are we? Liver Transpl 2017; 23: 1601–14. [DOI] [PubMed] [Google Scholar]
- 63.Luo X, Miller SD, Shea LD. Immune tolerance for autoimmune disease and cell transplantation. Annu Rev Biomed Eng 2016; 18: 181–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steinman L, Ho PP, Robinson WH, Utz PJ, Villoslada P. Antigen-specific tolerance to self-antigens in protein replacement therapy, gene therapy and autoimmunity. Curr Opin Immunol 2019; 61: 46–53. [DOI] [PubMed] [Google Scholar]
- 65.Wraith DC. Designing antigens for the prevention and treatment of autoimmune diseases. Curr Opin Chem Eng 2018; 19: 35–42. [Google Scholar]
- 66.Peterson LB, Bell CJM, Howlett SK, et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun 2018; 95: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 2015; 15: 283–94. [DOI] [PubMed] [Google Scholar]
- 68.Saadoun D, Rosenzwajg M, Joly F, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med 2011; 365: 2067–77. [DOI] [PubMed] [Google Scholar]
- 69.Koreth J, Matsuoka K, Kim HT, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med 2011; 365: 2055–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bluestone JA, Tang Q. Treg cells — the next frontier of cell therapy. Science 2018; 362: 154–5. [DOI] [PubMed] [Google Scholar]
- 71.Koreth J, Kim HT, Jones KT, et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. Blood 2016; 128: 130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gregori S, Roncarolo MG. Engineered T tegulatory type 1 cells for clinical application. Front Immunol 2018; 9: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rasmusson I Immune modulation by mesenchymal stem cells. Exp Cell Res 2006; 312: 2169–79. [DOI] [PubMed] [Google Scholar]
- 74.Takenaka MC, Quintana FJ. Tolerogenic dendritic cells. Semin Immunopathol 2017; 39: 113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maldonado RA, von Andrian UH. How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol 2010; 108: 111–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Naranjo-Gómez M, Raïch-Regué D, Oñate C, et al. Comparative study of clinical grade human tolerogenic dendritic cells. J Transl Med 2011; 9: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beriou G, Moreau A, Cuturi MC. Tolerogenic dendritic cells: applications for solid organ transplantation. Curr Opin Organ Transplant 2012; 17: 42–7. [DOI] [PubMed] [Google Scholar]
- 78.Flórez-Grau G, Zubizarreta I, Cabezón R, Villoslada P, Benitez-Ribas D. Tolerogenic dendritic cells as a promising antigen-specific therapy in the treatment of multiple sclerosis and neuromyelitis optica from preclinical to clinical trials. Front Immunol 2018; 9: 1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ellebrecht CT, Bhoj VG, Nace A, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016; 353: 179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kansal R, Richardson N, Neeli I, et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Transl Med 2019; 11(482): eaav1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.