

ABSTRACT
In late-onset Alzheimer disease (AD) pathogenesis, genes, infections and immunity could be significant factors. We have reviewed if the keystone periodontal pathogen Porphyromonas gingivalis may affect genes and microglia (primary immune cells in the brain) to promote AD development. Genes for apolipoprotein, clusterin, CD33, triggering receptor expressed on myeloid cells-2 (TREM-2), tyrosine kinase binding protein (TYR-OBP), and complement receptors can affect microglia. Most of these genes can also be affected by P. gingivalis via its mastering of immune suppression. Besides, P. gingivalis can affect microglia directly in several ways. Taken together, genetic predisposition, P. gingivalis infection and microglia could promote neurodegeneration typical of that reported for AD.
KEYWORDS: Microglia, immune cells, inflammation, brain, training, tolerance, hyperactivity, P. gingivalis
Introduction
Amyloid-beta (Aβ) plaque and phosphorylated tau (p-Tau) binding to neurofibrillary tangles (NFTs) are important neuropathological and diagnostic markers of Alzheimer’s disease (AD). Both lesions in their diverse peptide sizes (Aβ and p-Tau) can act as toxins in and outside cells in vitro and in vivo animal models [1–4]. A second factor of AD is brain infection/inflammation where the keystone pathogen in ‘chronic’ periodontitis Porphyromonas gingivalis, seems to play an important role. Cerebral inflammation in the form of activated microglia is a third major histopathological feature, but without a role in the neuropathological diagnosis of AD.
Aim
The aim of the present review is to discuss how genetic factors, P. gingivalis periodontal infection and microglia can interact to promote AD. Potential mechanisms for microglia affliction by P. gingivalis are listed in Table 1.
Table 1.
Potential mechanisms for microglia affection by Porphyromonas gingivalis (P.g.).
| Factor | Mechanism | Ref |
|---|---|---|
| Gingipains | Inhibitors of gingipains are being tested for reducing p-Tau toxicity in man | [4] |
| Persistent expression of gingipains in P.g.infected neu-rons gave AD-like neurodegeneration | [8] | |
| Secreted gingipains from P.g. induced microglia migration | [11] | |
| Inhibitors of Rgp and Kgp suppressed P.g.induced micro-glia migration | [10] | |
| Matrix metallo-proteinases (MMPs) | ||
| P.g. can induce synthesis of MMPs in host tissues and cells | [16] | |
| P.g. may contribute to the brain pool of MMPs | [4,19] | |
| Inhibitors of MMPs | ||
| (TIMPs) | P.g. inactivated TIMP-1 and TIMP-2 causing destruction of connective tissue | [20] |
| Clusterin gene | Clusterin is a complement cascade regulatory plasma protein. P.g. phosphorylated its ser396 in mice | [8] |
| CR1 gene | CR1 regulates complement cascade. P.g.causes immune subversion in relation to CR1 | [52] |
| CD33 | Belongs to an Ig-like family of receptors expressed on microglia.CR1 and is highly expressed on CD33+ cells to which P. g. binds | [57] |
| TREM-2 gene | Codes for a protein expressed on microglia. P.g. down-regu-lates TREM-2 expression on microglia which may accele-rate AD | [58,59] |
| TYR-OBP gene | Key signaling molecule for TREM-2 | [64] |
| Complement | P.g. initiated AD inflammation involving the comple-ment cascade of ApoE−/- brains | [25] |
| LPS | Initiates neuroinflammation through microglia activation | [68] |
| Migroglia were ‘primed’ inducing increased responses to subsequent challenges | [68] | |
| When located in brains microglia can be activated by P.g. LPS | [4,76] | |
| P.g. causes imbalance in the M1/M2 phenotype of microglia | [78] | |
| Leptomeningeal cells | P.g. LPS stimulated transfer of inflammatory signals from peripheral macrophages to brain-resident microglia | [83,84] |
| Administration of P.g. to mice caused P.g. and its prote-ases to be detected intra- and perinuclear in microglia | [8] | |
Relationship between ‘chronic’ periodontitis and Alzheimer’s disease
Important in the relationship between ‘chronic’ periodontitis and AD is infection where P. gingivalis is a suspect pathogen, for details, see references [3–5]. An infectious episode inevitably gives rise to inflammation (albeit acute and/or longstanding) with a degree of tissue atrophy. P. gingivalis has several virulence factors to promote brain inflammation and associated damage.
Gingipains
Institutionalized AD subjects show all forms of dental diseases (amongst them caries and periodontal disease), to co-exist in their dentition, and good oral health practices are unlikely to be a priority in their daily lives [6]. Recent knowledge of gingipains as virulence factors of P. gingivalis has initiated therapy towards reducing p-Tau peptide-related toxicity [4]. This is being tested clinically by neutralizing P. gingivalis virulence with inhibitors of gingipains (GAIN Trial: Phase 2/3 Study of COR388 in Subjects with Alzheimer’s Disease. ClinicalTrials.gov Identifier: NCT03823404) [4,7].
Gingipains of P. gingivalis are reported to digest the normal tau protein into nine fragments [4], and some of these peptides are from tau residues prone to phosphorylation and some are from two of the four microtubule-binding domains that also lie within peptides that form paired/straight helical filaments constituting NFTs [4]. This may be one pathway to releasing fragments of the tau protein into the brain’s parenchymal tissues. The extracellular phosphotau fragments generated by gingipains may be directly toxic to other neurons or the tau fragments may be of the size that neurons are able to take up at synaptic clefts during neurotransmitter uptake, thereby causing its spread from neuron to neuron. Further research is needed to clarify the role of gingipains fragmented tau peptides in the pathogenesis of AD.
P. gingivalis infection promotes tau phosphorylation
Gingipains have been found to be neurotoxic in vivo and in vitro, having detrimental effects on tau [4]. The capsular serotype K1 P. gingivalis W83 strain has shown the potential to contribute to tau phosphorylation at Ser396 in the in vivo wild-type mouse model [8]. Furthermore, an in vitro neuronal cell line model reported by Haditsch et al. [9] demonstrated an increased tau phosphorylation at Thr231 following P. gingivalis infection with persistent gingipain expression. Liu et al. [10] observed in their P. gingivalis--infected microglial cells towards the site of infection, activation of the phosphoinositide 3-kinase/Akt (PI3K/AKT) pathway. Our own in house data show that purified P. gingivalis lipopolysaccharide (LPS) application to a neuroblastoma cell line, in vitro cell model also activated the PI3K/AKT pathway in which glycogen synthase kinases-3 beta (GSK-3β) mRNA expression increased. The importance here is that GSK-3β is one of the enzymes that phosphorylates tau suggesting that P. gingivalis plays an important role in the NFT lesion formation and subsequent pathophysiology of AD.
P. gingivalis infection promotes neurodegeneration
As mentioned, Haditsch et al. [9] reported AD-like neurodegeneration in P. gingivalis infected neurons in an in-vitro culture system with persistent expression of active gingipains. Following infection with live P. gingivalis (ATCC 33277) 25% of the neurons were lost in a time-dependent manner. Full-length tau was reduced in surviving cells with an increase in phosphorylation over time. This finding was related to loss of neuronal synapses and was comparable to features of associated neurodegeneration together with the presence of gingipains in AD autopsy brains. Accordingly, P. gingivalis can invade and survive in neurons and generate intra-neuronal gingipains that are proteolytically active, leading to neurodegeneration associated with AD.
Nonata and Nakanishi [11] found in an in vitro study that secreted gingipains from P. gingivalis induced microglial cell migration. This was likely achieved through endosomal signaling by protease-activated receptor 2 (PAR 2).
Liu et al. [10], attempting to clarify the potential effects of the gingipains – Rgp and Kgp on the cellular activation of brain-resident microglia in mice, found that Rgp and Kgp cooperated thereby contributing to the migration of P. gingivalis-infected microglial cells towards the site of infection, and initiated expression of proinflammatory mediators by activating PAR 2. The mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase/ERK pathway contributed to both cell migration and invoked an inflammatory response in microglia. Furthermore, PI3K/AKT pathway mRNA expression increased together with pro-inflammatory mediators such as IL-6, TNF-α and inducible nitric oxide synthase. The mRNA expression of the anti-inflammatory mediators interleukin 10 (IL-10), arginase-1 and IL-4 was not affected. The authors proposed that microglial cell migration was likely to have been associated with actin polymerization and may be necessary for invoking inflammatory responses in microglia following activation of PAR 2. Further observations by Liu et al. [10] suggest and that Rgp and Kgp gingipains may be responsible for degrading components of the epithelial cell basal membrane, which may be facilitating the invasion of P. gingivalis into the brain. Liu et al. [10] experimentally tested their hypothesis by incorporating inhibitors of Rgp – KYT1 and Kgp – KYT36 and found the P. gingivalis-induced microglia cell migration was suppressed in the presence of the activated PAR 2 pathway. This provided proof of principle indicating that Rgp and Kgp were largely responsible for inducing migration of microglia in the brain. In the study by Dominy et al. [4] synthesized small-molecule inhibitors targeting gingipains were tested and this resulted in a reduction in the bacterial load. Furthermore, the small-molecule inhibitors of P. gingivalis reduced the extent of the brain infection established in mice. In addition, the small-molecule inhibitors blocked Aβ1-42 production, diminished neuroinflammation, and rescued neurons in the hippocampus.
Matrix metalloproteinases
Matrix metalloproteinases (MMPs) have an important role in neuroinflammatory disorders including AD [12,13]. Increase in the expression of MMPs in the brain tissue and blood of demented patients is reported to be part of the overall inflammatory process in AD [14]. Mroczko et al. [15] detected MMP-3 and MMP-9 localized around NFTs and Aβ plaques in AD brains. Healthy elderly with increased risk of developing AD had increased levels of MMP-3 and MMP-9 protein levels in the cerebrospinal fluid. Mroczko et al. [15] proposed that increased protein levels of these MMPs may be related to neuronal degeneration and/or formation of NFTs prior to clinical cognitive deterioration [13]. Further research is required to clarify this observation.
It is accepted that P. gingivalis can induce the synthesis of MMPs in tissues and cells of the host. For example, cytokine and MMP expression in fibroblasts from peri-implantitis lesions were reported to be induced by P. gingivalis [16]. In addition, the sustained upregulation of inflammatory mediators and MMP-1 was suggested to play a role in the pathogenesis of peri-implantitis [16]. In oral squamous cell carcinoma, P. gingivalis promoted invasion by induction of proMMP-9 and its activation [17]. It was suggested that P. gingivalis activated protease-activated receptor 4 (PAR4) signaling pathways, causing proMMP-9 over-expression and invasion of oral squamous carcinoma cells [18]. Since P. gingivalis does spread to the AD brain as shown experimentally in mice and in humans [4,19] it is plausible to suggest that P. gingivalis could contribute to the pool of MMPs in the brain.
Inhibitors of matrix metalloproteinases
Tissue inhibitors of metalloproteinases (TIMPS) can modulate the activity of MMPs [15]. This is important because dysregulation of MMPs can lead to several disorders. In a study by Sato et al. [20], sonicated P. gingivalis extracts caused the destruction of connective tissue, contributing to the process of periapical disease by activating pro-MMP-2 as well as by inactivating TIMP-1 and TIMP-2. In another study using human gingival fibroblasts, P. gingivalis LPS differentially modulated the expression of MMP-1, −2, and −3 and TIMP-1 [21]. Alternatively, there is a possibility that P. gingivalis suppresses TIMPs activity in the brain, to dysregulate the MMP pool in AD patients. Again further research is warranted to clarify this possibility.
Relationship between microglia and Alzheimer’s disease
Microglia comprise 10% of the total brain cells. They are resident macrophages and the brain’s primary innate immune cells responding to diverse stimuli (Figure 1(a,b)). They also act as inflammatory cells by rapidly changing morphology, proliferating and migrating to the site of infection/injury where they phagocytize and destroy invaders and remove damaged cells. Microglia secrete cytokines, chemokines, prostaglandins, nitric oxide and reactive oxygen species [22]. During aging, they develop a more inflammatory (activated) phenotype possibly due to having previously confronted diverse antigens [23], following which they may fail to return to their original resting (non-activated) state. Some microglia can survive for long periods, even more than two decades [24]. Thus, the microglial cell population in the human adult brain is characterized by a slow turnover. Their efforts to resolve any inflammatory response involves the production of anti-inflammatory cytokines such as IL-10. In the case of experimental oral infection with P. gingivalis in apolipoprotein E−/- (ApoE−/-) mice, the host releases copious amount of IL-10 in the serum. However, the bacterium itself still spreads to the brain and encounters microglia, which as a result become activated [25]. It appears that peripheral IL-10 mediated immune resolution in the ApoE−/- mice remains inadequate for microglia to return to the resting state [25,26]. Recent research in mice has shown that microglia are able to ‘remember’ a previous inflammatory challenge and become ‘trained’ or ‘tolerant’ to toxins like LPS [27]. This may prolong the existence of the endotoxin in infected brains. Thus, immune training can inadvertently increase cerebral β-amyloidosis while tolerance may decrease it. The immune memory affects the reaction of microglia to new stimuli and the way in which they deal with toxic Aβ plaque in the brain (Figure 1(b)), thereby modifying neuropathology.
Figure 1.
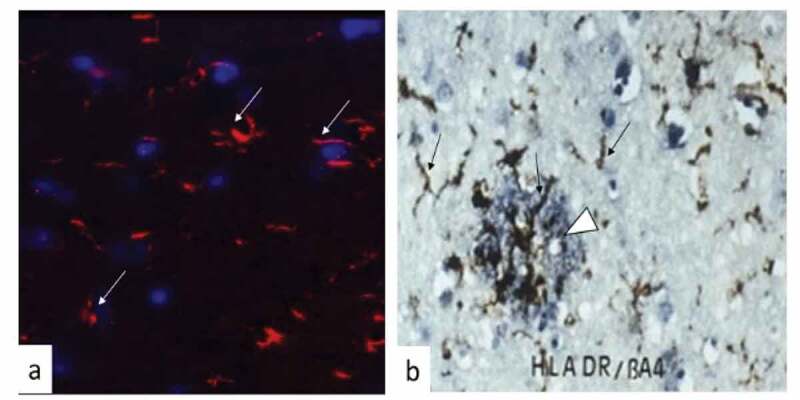
Brain tissue showing microglia responding to infection in a mouse model and to Aβ plaque in a brain tissue section from Alzheimer’s disease.
(a) Confocal image. Brain tissue showing microglia (white arrows) following mono-P. gingivalis infection (24 weeks) from an apolipoprotein E−/- mouse brain immunolabelled to demonstrate microglia (anti-1ba1); Blue = DAPI; Red = TRITC label for immunopositive microglia. (b) Immunohistochemistry. Double labelling of a cortical human AD brain tissue section showing activated microglia brown (anti-HLADR) demarcated by black arrows, and Aβ plaque (anti-Aβ) blue (white arrow head) to demonstrate their relationship.
It seems the severity of AD and its progression may be linked to chronic inflammation [28,29]. It is therefore plausible to suggest that immunological memory in long-living microglia can represent a risk for not only protracting but also initiating clinical AD at the appropriate age, particularly if they become tolerant to inflammation [27]. Singhrao et al. [30] proposed that the long-term effect of inflammatory mediators, pathogens, and/or their virulence factors could, over time, prime the brain’s microglia in individuals with inherent susceptibility traits.
Microglia are not uniform cells, and this is why only fragments of microglia are seen in tissue sections following their immuno/fluorescence/histochemistry reactivities (Figure 1(a,b)). Activation of microglia in the central nervous system involves two opposing phenotypes denoted M1 and M2. Depending on the trigger that activates these two phenotypes, microglia (M1) can exert cytotoxic (proinflammatory cytokine release) or neuroprotective (M2) (immune resolution) effects [31]. Hammond et al. [32] performed single-cell RNA sequencing and in situ brain mapping, and detected nine transcriptionally distinct stages of >76,000 microglial cells in mice expressing unique sets of genes. Some of these genes were upregulated in microglia-surrounding Aβ plaques [33]. Microglial cell phenotypes were most diverse in the developing brain and following aging and injury. Furthermore, RNA sequencing revealed that the expression of genes from microglial cell activation increased in several neurological diseases including AD [34].
Microglia can also develop functional defects as seen in other neurodegenerative disorders [35]. During the early stages of AD, they play a key role in the clearance of Aβ and reducing the plaque burden [36]. However, Aβ plaques and extracellular tau peptides can eventually become surrounded by glial cells with dysfunctional homeostatic control and as a result acquire a proinflammatory phenotype amplifying neuronal damage [37]. Similarly, cytokines and proinflammatory molecules secreted by microglia that initially have a neuroprotective role can subsequently become the cause of further neurodegeneration [37]. If microglia become overactive, they can initiate the biosynthesis of complement proteins and with the appropriate trigger, activate the complement cascade [38]. This can lead to their aberrant digestion of nerve synapses [39]. This is observed in mice lacking the TAR DNA-binding protein 43 (TPD-43) [40]. A complement-microglial axis has been found to drive synapse loss in AD [41] and is a plausible issue for deteriorating memory.
Relationship between P. gingivalis and microglia
It is noteworthy that gingipains have been detected in microglia [8], and in the capillaries of the hippocampus in ApoE−/- mice brains that were from a mono-P. gingivalis-infected group [42]. Mice brains have shown a potent microglial activation response to mono-P. gingivalis infection [25]. However, there are also several other effects that P. gingivalis may exert on microglia, which are less appreciated.
Affliction by genetic factors
Genome-Wide-Association Studies (GWAS) have identified several susceptibility genes expressed by microglia in AD. Genome-wide meta-analysis of clinically diagnosed AD and AD-by-proxy (71,880 cases, 383,378 controls) found associated genes to be strongly expressed in immune-related tissues (spleen and liver), and cell types such as microglia [43]. Other GWAS and integrated network studies identified immune-related pathways, as risk factors for AD, with microglia as central players. These studies strongly support the idea that genes, pathogens and the immune system act together in the eventual development of AD [5,44]. Among the genes related to microglia and AD were clusterin (apolipoprotein J), complement receptor 1 (CR1), CD33, triggering receptor expressed on myeloid cells-2 (TREM-2) and tyrosine kinase binding protein (TYR-OBP) [45]. These genes play a role in the clearance of cellular debris from the brain. However, in the context of P. gingivalis infection of the brain, the impressive immune subversion of this bacterium in cleaving receptors challenges this very function as discussed below.
Clusterin gene
The clusterin gene was identified as an important risk locus for AD with the three SNPs (rs 11136000, rs 2279590 and rs9331888) showing a statistically significant relationship with the disease [46,47]. Clusterin (CLU) is one of the complement cascade regulatory plasma proteins that significantly increases during AD [48]. It is present in Aβ plaques where it binds to insoluble amyloid peptides and interacts with Aβ40 and Aβ42 [49]. Due to its well-accepted role in the complement cascade, CLU is likely to affect Aβ clearance, amyloid deposition and subsequent neurotoxicity [50]. CLU is also said to stimulate expression and secretion of various chemotactic cytokines such as TNF-α, which has a critical role in promoting macrophage chemotaxis via the Pi3K/Akt mitogen-activated protein ERK and JNK pathways [51].
CR1 gene
It has been reported that SNPs rs3818361 and rs6656401 of the CR1 gene are associated with increased likelihood of AD [49]. This supports a CR1 gene defect in AD [5]. CR1 helps with regulating the complement cascade and promotes phagocytosis of cellular debris and Aβ plaques, and adherence of immune complexes to erythrocytes [5]. Interestingly, P. gingivalis mediates immune subversion in relation to CR1 [52]. This may suggest that a vulnerability-axis exists, within a protein region (e.g. CR1), which is exploited by both genetic defects and pathogens like P. gingivalis.
CD33
CD33 appears to have an important role in Aβ clearance and other neuroinflammatory pathways in the brain aided by microglia [50]. CD33 belongs to an immunoglobulin (Ig)-like family of receptors that are expressed on myeloid cells including microglia [53,54]. CD33 binds to alpha2-3- or alpha2-6-linked sialic acids (N-acetyl neuraminic acid) to which P. gingivalis also binds [55]. Sialylation of P. gingivalis cell surface components such as LPS may provide additional benefits to this prominent periodontal pathogen in biofilm formation and in escaping complement-mediated killing [56]. CR1 is highly expressed on CD33+ cells which facilitate P. gingivalis binding to them and is also a general clearance receptor for pathogens [57]. However, P. gingivalis is either able to cleave CD33 from the surface membrane of cells or down-regulate functional cell surface receptors on myeloid cells. If this was the case, then the CD33 receptor expression would be affected in a similar way on microglia.
TREM-2 gene
The TREM-2 gene codes for a protein in the brain that is expressed on microglial cells and is also involved in removing degenerated tissue, including remnants from neuroinflammation [58]. The TREM-2 protein has been found to increase the susceptibility to AD, with an odds ratio similar to that of the apolipoprotein ε4 allele [59,60]. TREM-2 deficiency inhibited Aβ degradation in a primary microglial culture and in a mouse brain model [61]. Interestingly, P. gingivalis significantly down-regulated TREM-2 expression in microglia [62]. Lack of TREM-2 protein may accelerate aging processes, neuronal cell loss and reduce microglial activity leading to neuroinflammation [63].
TYR-OBP gene
TYR-OBP has been identified as a key regulator among genes involved in phagocytosis [64]. It is a key signaling molecule for TREM-2, as determined from networks involved in immune and microglia-specific modules disrupted in AD brains [64]. The association of this gene defect with P. gingivalis activity is little understood. Further research is needed to clarify if P. gingivalis can affect TREM-2 signaling through TYR-OBP.
Complement activation
P. gingivalis has been proposed to exploit complement receptors 1 and 3 for evading innate immune clearance [65,66]. Active invasion of P. gingivalis-induced complement activation in ApoE−/- mice brains has been investigated [25]. Microglia in both infected (P. gingivalis, oral infection) and control groups exhibited strong intracellular labeling with complement components/opsonins from C3 and C9, due to ongoing biosynthesis and activation. Further, Poole et al. [25] showed that P. gingivalis was able to access the ApoE−/- brain and contribute to the development of AD inflammatory pathology through mechanisms involving acute-phase proteins, cytokines and the complement cascade where neurons would be attacked. It has since been shown that ApoE binds to activated C1q and that the resulting C1q-ApoE complex becomes a common player to affect brain inflammation [67]. Thus, inappropriate complement activity plays a significant role in AD pathophysiology. Interestingly, treatment with small interfering RNA (siRNA) against C5, which is formed in all complement pathways, attenuated Aβ-associated microglia accumulation [67]. As mentioned, microglia and the complement-dependent pathway can over-prune functional synapses and lead to memory loss [45].
Activation by lipopolysaccharide
LPS is one of the major virulence factors of P. gingivalis. Several animal studies have shown that LPS administered directly into the peritoneum of the brain initiates neuroinflammation in the form of microglial cell activation [e.g. 68]. Researchers measured the inflammatory response following LPS administration in experimental mice and this demonstrated learning and memory impairment in test mice [69,70]. In the Cunningham study [68] the microglial cells were ‘primed’ so that they induced increased inflammatory responses to subsequent LPS challenges.
In a study by Henry et al. [71] peripheral LPS challenge in aged mice induced a hyperactive microglial response together with a higher induction of inflammatory IL-1β and anti-inflammatory IL-10. Injection of LPS caused a marked induction of mRNA expression of both IL-1β and IL-10 in the cortex of aged mice as compared to adults. An age-dependent increase in the major histocompatibility complex (MHC) class II mRNA and protein expression was also seen in microglia, suggesting their activated status. Other studies have indicated that peripheral injection of P. gingivalis LPS also causes a higher increase in IL-1β. Interestingly, the most prominent induction of IL-1β was detected in MHC II (+) microglia from aged mice [72]. In another study, Zhang et al. [73] found that P. gingivalis LPS induced cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. Both microglia and astrocytes in the cortex and hippocampus were activated. Accordingly, age-associated priming of microglia seems to have a central role in exaggerated inflammation induced by activation of the peripheral immune system. IL-1β is also implicated in synaptic loss [74,75], promoting deterioration in cognition [45] by stimulating Aβ cleavage indirectly from the action of cathepsin B on the APP with its cognate receptor (IL-1 R) on neurons [72]. Last, but not least, P. gingivalis LPS has been reported in the human brain, thus suggesting it might activate brain microglia participating in brain inflammation [4]. This idea was supported in an 18-h in vitro stimulation study with ultrapure P. gingivalis LPS in rats that resulted in classical and alternative activation of rat brain microglia and the concomitant release of cytokines and chemokines [76].
Microglia, being influenced by their environment, can assume a diversity of phenotypes and can change functions aimed to maintain homeostasis. Like their macrophage ‘cousins’, microglia show unique features with regard to phenotype polarization. As mentioned, they can be stimulated by LPS and IFN-ɣ to develop into an M1 phenotype for expression of proinflammatory cytokines, or by IL-4/IL-13 to an M2 phenotype for resolution of inflammation and tissue repair [77]. Whether P. gingivalis-LPS has this capacity is not known. P. gingivalis causes an imbalance in M1/M2 activation in macrophages, resulting in a hyperinflammatory environment that promotes the pathogenesis of periodontitis [78]. These authors reported that P. gingivalis or P. gingivalis-derived LPS-induced inflammatory responses enhanced M1 macrophages and suppressed M2 macrophages, even in the presence of IL-4. Interestingly, resveratrol has been found to reduce inflammatory damage and promote microglia polarization to the M2 phenotype in LPS-induced neuroinflammation [79].
Transduction of inflammatory signals to microglia by leptomeningeal cells
The leptomeninges (pia mater and the arachnoid together housing the brain and spinal cord) plays a role as secretory cells, which transduce systemic inflammatory signals into the CNS [80–82]. In studies by Liu et al. [83] and Wu and Nakanishi [84] leptomeningeal cells transduced inflammatory signals from peripheral macrophages to brain-resident microglia exposed to P. gingivalis LPS. The mean amount of TNF-α and IL-1β after exposure to conditioned medium from P. gingivalis LPS-stimulated macrophages were significantly higher than after treatment with P. gingivalis LPS alone. This indicated that leptomeningeal cells could transduce inflammatory signals to microglia in the deeper brain areas, which in turn initiated neuroinflammation.
Porphyromonas gingivalis DNA in brain microglia
Repeated chronic oral administration of P. gingivalis in wild-type mice transferred P. gingivalis to the brain where the bacterium and its proteases (gingipains) were detected within intra-nuclear and peri-nuclear locations of microglia, astrocytes, neurons, and extracellular spaces [8]. Microgliosis and astrogliosis were found in the experimental but not in the control group, and significantly higher levels of expression of IL6, TNF-α and IL-1β were detected in the experimental group. Also, neurodegeneration was more evident in the experimental group. Extracellular Aβ42 was detected in the parenchyma of the experimental group but not in controls. This was the first report of p-Tau (Ser396) and NFT formation. Ilievski et al. [8] have proven the concept that chronic periodontal infection can result in the formation of the diagnostic neuropathology lesions consistent with AD. Haditsch et al. [9] confirmed the findings of Ilievski et al. [8] for p-Tau on Ser396 and additionally demonstrated an increased tau phosphorylation at Thr231 following P. gingivalis infection with persistent gingipain expression with ongoing neurodegeneration.
Concluding remarks
GWAS have indicated that genes, pathogens and the immune system act together to generate AD. In addition, neuroinflammation plays a pivotal role and this has made scientists ask the question if AD is an infectious disease. In this complex interaction of different players, microglia seem to be important in the host defense against invasion of the keystone periodontopathogen P. gingivalis. The latter may affect microglia in both direct and indirect ways. Whether other putative periopathogens and even intestinal bacteria also affect microglia of the AD brain remain to be tested. Astrocytes, which are macroglia, can also be activated by P. gingivalis. Such activation may have toxic effects on neurons. The chronic nature of low-level infections such as ‘chronic’ periodontitis and associated byproducts, e.g. endo/exotoxins and cytokines could affect susceptible brains’ defense capacity to a point where microglia involved in brain protection become adversely affected. Whether microglia will ‘remember’ inflammation caused by P. gingivalis and develop ‘tolerance’ to it, requires further research. However, it is plausible to suggest that once microglia are primed by P. gingivalis exposure, there remains the possibility of developing tolerance through the mastery of innate immunity manipulation by this bacterium, which may be the result of inadequate clearance of cellular debris (Aβ) from the AD brain.
Acknowledgments
We acknowledge the Newcastle Brain Tissue Resource, UK for the human brain specimens for the approved study formerly published by Poole et al. [19] and later by Siddiqui et al. [85] from which the immunohistochemistry image was prepared.
Funding Statement
SKS has received a PreViser award from the Oral and Dental Research Trust, 2018 and continued financial support from the School of Dentistry, University of Central Lancashire, UK.
Disclosure statement
No conflict of interest was reported by the authors.
References
- [1].Soscia SJ, Kirby E, Washicosky KJ, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):09505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kumar DKV, Choi SH, Washicosky KJ, et al. Amyloid-ß peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8(340):340ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Singhrao SK, Olsen I.. Assessing the role of Porphyromonas gingivalis in periodontitis to determine a causative relationship with Alzheimer’s disease. J Oral Microbiol. 2019;11(1):1563405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dominy SS, Lynch C, Ermini F, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5:eaaa3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Olsen I, Singhrao SK. Is there a link between genetic defects in the complement cascade and Porphyromonas gingivalis in Alzheimer’s disease? J Oral Microbiol. 2019;12(1):1676486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chalmers JM, Hodge C, Fuss JM, et al. The prevalence and experience of oral diseases in Adelaide nursing home residents. Aust Dent J. 2002;47(2):123–10. [DOI] [PubMed] [Google Scholar]
- [7].Olsen I, Potempa J. Strategies for the inhibition of gingipains for the potential treatment of periodontitis and associated systemic diseases. J Oral Microbiol. 2014;6. DOI: 10.3402/jom.v6.24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ilievski V, Zuchowska PK, Green SJ, et al. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One. 2018;13(10):e0204941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Haditsch U, Roth T, Rodriguez L, et al. Alzheimer’s disease-like neurodegeneration in Porphyromonas gingivalis infected neurons with persistent expression of active gingipains. J Alzheimer’s Dis. 2020;75:1361–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liu Y, Wu Z, Nakanishi Y, et al. Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci Rep. 2017;7(1):11759. Liu Y, Wu Z, Nakanishi Y, et al. Author Correction: Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci Rep. 2018; 8: 10304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nonaka S, Nakanishi H. Secreted gingipains from Porphyromonas gingivalis induce microglia migration through endosomal signaling by protease-activated receptor 2. Neurochem Int. 2020;104840. DOI: 10.1016/j.neuint.2020.104840. [DOI] [PubMed] [Google Scholar]
- [12].Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8(2):205–216. [DOI] [PubMed] [Google Scholar]
- [13].Stomrud E, Björkqvist M, Janciauskiene S, et al. Alterations of matrix metalloproteinases in the healthy elderly with increased risk of prodromal Alzheimer’s disease. Alzheimers Res Ther. 2010;2(3):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sochocka M, Diniz BS, Leszek J, et al. Inflammatory response in the CNS: friend or foe? Mol Neurobiol. 2017;54(10):8071–8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mroczko B, Groblewska M, Barcikowska M. The role of matrix metalloproteinases and tissue inhibitors of metalloproteinases in the pathophysiology of neurodegeneration: a literature study. J Alzheimers Dis. 2013;37(2):273–283. [DOI] [PubMed] [Google Scholar]
- [16].Irshad M, Scheres N, Anssari Moin D, et al. Cytokine and matrix metalloproteinase expression in fibroblasts from peri-implantitis lesions in response to viable Porphyromonas gingivalis. J Periodontal Res. 2013;48(5):647–656. [DOI] [PubMed] [Google Scholar]
- [17].Inaba H, Sugita H, Kuboniwa M, et al. Porphyromonas gingivalis promotes invasion of oral squamous cell carcinoma through induction of proMMP9 and its activation. Cell Microbiol. 2014;16(1):131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Inaba H, Amano A, Lamont RJ, et al. Involvement of protease-activated receptor 4 in over-expression of matrix metalloproteinase 9 induced by Porphyromonas gingivalis. Med Microbiol Immunol. 2015;204(5):605–612. [DOI] [PubMed] [Google Scholar]
- [19].Poole S, Singhrao SK, Kesavalu L, et al. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis. 2013;36(4):665–677. [DOI] [PubMed] [Google Scholar]
- [20].Sato Y, Kishi J, Suzuki K, et al. Sonic extracts from a bacterium related to periapical disease activate gelatinase A and inactivate tissue inhibitor of metalloproteinases TIMP-1 and TIMP-2. Int Endod J. 2009;42(12):1104–1111. [DOI] [PubMed] [Google Scholar]
- [21].Bozkurt SB, Hakki SS, Hakki EE, et al. Porphyromonas gingivalis lipopolysaccharide induces a pro-inflammatory human gingival fibroblast phenotype. Inflammation. 2017;40(1):144–153. [DOI] [PubMed] [Google Scholar]
- [22].Green K. Microbial function in the healthy brain. [cited 2019 January8]. https:faculty.sites.uci.edu/kimgreen/bio/microglia-in-the-healthy-brain
- [23].Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. 2013;39:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Réu P, Khosravi A, Bernard S, et al. The lifespan and turnover of microglia in the human brain. Cell Rep. 2017;20(4):779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Poole S, Singhrao SK, Chukkapalli S, et al. Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE−/- mice brains. J Alzheimers Dis. 2015;43(1):67–80. [DOI] [PubMed] [Google Scholar]
- [26].Velsko IM. Periodontal pathogen-induced atherosclerosis in ApoE−/- and integrin β 6−/- mice (PhD thesis). University of Florida; 2014. [Google Scholar]
- [27].Wendeln AC, Degenhardt K, Kaurani L, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556(7701):332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sparks Stein P, Steffen MJ, Smith C, et al. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimers Dement. 2012;8(3):196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen C-K, Wu Y-T, Chang Y-C. Association between chronic periodontitis and the risk of Alzheimer’s disease: a retrospective, population-based, matched cohort study. Alzheimers Res Ther. 2017;9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Singhrao SK, Harding A, Poole S, et al. Porphyromonas gingivalis periodontal infection and its putative links with Alzheimer’s disease. Mediators Inflammation. 2015;2015:137357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53(2):1181–1194. [DOI] [PubMed] [Google Scholar]
- [32].Hammond TR, Dufort C, Dissing-Olesen L, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Keren-Schaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- [34].Bennett JP Jr, Keeney PM, Brohawn DG. RNA sequencing reveals small and variable contributions of infectious agents to transcriptomes of postmortem nervous tissues from amyotrophic lateral sclerosis, Alzheimer’s disease and Parkinson’s disease subjects, and increased expression of genes from disease-activated microglia. Front Neurosci. 2019;13:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013;39(1):3–18. [DOI] [PubMed] [Google Scholar]
- [36].Wang WY, Tan MS, Yu JT, et al. Role of pro- inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. 2015;3(10):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bronzuoli MR, Iacomino A, Steardo L, et al. Targeting neuroinflammation in Alzheimer’s disease. J Inflamm Res. 2016;9:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Singhrao SK, Neal JW, Morgan BP, et al. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp Neurol. 1999;159(2):362–376. [DOI] [PubMed] [Google Scholar]
- [39].Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Paolicelli RC, Jawaid A, Henstridge CM, et al. TDP-43 depletion in microglia promotes amyloid clearance but also induces synapse loss. Neuron. 2017;95(2):297–308.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vasek MJ, Garber C, Dorsey D, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534(7608):538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Singhrao SK, Chukkapalli S, Poole S, et al. Chronic Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE−/- mice brains. J Oral Microbiol. 2017;9(1):1270602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jansen IE, Svage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Carter C. Alzheimer’s disease: APP, gamma secretase, APOE, Clu, CR1, PICALM, ABCA7, BINI1, CD2AP, CD33, EPHA1, and MS4A2, and their relationships with Herpes simplex, C. pneumoniae, other suspect pathogens, and the immune system. Int J Alzheimer’s Dis. 2011;2011:501862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at GLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–1099. [DOI] [PubMed] [Google Scholar]
- [48].Hakobyan S, Harding K, Aiyaz M, et al. Complement biomarkers as predictors of disease progression in Alzheimer’s disease. J Alzheimers Dis. 2016;54(2):707–716. [DOI] [PubMed] [Google Scholar]
- [49].Giri M, Zhang M, Lü Y, et al. Genes associated with Alzheimer’s disease: an overview and current status. Clin Interv Aging. 2016;11:665–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of gene pathogenesis. Biol Psychiatry. 2015;77(1):43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shim YJ, Kang BH, Choi BK, et al. Clusterin induces the secretion of TNF-α and the chemotactic migration of macrophages. Biochim Biophys Res Commun. 2012;422(1):200–205. [DOI] [PubMed] [Google Scholar]
- [52].Hajischengallis G, Liang S, Payne MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10(5):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Malik M, Simpson JF, Parikh I, et al. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci. 2013;33(33):13320–13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Griciuc A, Serrano-Pozo A, Parrado AR, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hallén U, Björkner AE, Hallberg EC. Binding of the periodontitis associated bacterium Porphyromonas gingivalis to glycoproteins from human epithelial cells. Oral Microbiol Immunol. 2008;23(5):367–371. [DOI] [PubMed] [Google Scholar]
- [56].Zaric SS, Lappin MJ, Fulton CR, et al. Sialylation of Porphyromonas gingivalis LPS and its effect on bacterial-host interactions. Innate Immun. 2017;23(3):319–326. [DOI] [PubMed] [Google Scholar]
- [57].Repik A, Pincus SE, Ghiran I, et al. A transgenic mouse model for studying the clearance of blood-borne pathogens via human complement receptor 1 (CR1). Clin Experiment Immunol. 2005;140(2):230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Neumann H, Daly MJ. Variant TREM2 as risk factor for Alzheimer’s disease. N Engl J Med. 2013;368(2):182–184. [DOI] [PubMed] [Google Scholar]
- [59].Jiang T, Yu J-T, Zhu X-C, et al. TREM2 in Alzheimer’s disease. Mol Neurobiol. 2013;48(1):180–185. [DOI] [PubMed] [Google Scholar]
- [60].Chen X, Zhong L. The merging roles and therapeutic potential of soluble TREM2 in Alzheimer’s disease. Front Aging Neurosci. 2019;11:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhao N, Liu CC, Qiao W, et al. Apolipoprotein E, receptors, and modulation of Alzheimer’s Disease. Biol Psychiatry. 2018;83(4):347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liang S, Domon H, Hosur KB, et al. Age-related alterations in innate immune receptor expression and ability of macrophages to respond to pathogen challenge in vitro. Mech Ageing Dev. 2009;130(8):538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Raha AA, Henderson JW, Stott SR, et al. Neuroprotective effect of TREM-2 in aging and Alzheimer’s disease model. J Alzheimers Dis. 2017;55(1):199–217. [DOI] [PubMed] [Google Scholar]
- [64].Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hajishengallis G, Harokopakis E. Porphyromonas gingivalis interactions with complement receptor 3 (CR3): innate immunity or immune evasion? Front Biosci. 2007;12:4547–4557. [DOI] [PubMed] [Google Scholar]
- [66].Hajishengallis G. Immune evasion strategies of Porphyromonas gingivalis. J Oral Biosc. 2011;53(3):233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yin C, Ackermann S, Ma Z, et al. ApoE attenuates unresolved inflammation by complex formation with activated C1q. Nat Med. 2019. Ackermann S, Ma Z, et al. Publisher Correction: ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019; 25(3):496–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Cunningham C, Wilcockson DC, Campion S, et al. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25(40):9275–9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Tanaka S, Ide M, Shibutani T, et al. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J Neurosci Res. 2006;83(4):557–566. [DOI] [PubMed] [Google Scholar]
- [70].Chen J, Buchanan JB, Sparkman NL, et al. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brian Behav Immun. 2008;18:223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Henry CJ, Huang Y, Wynne AM, et al. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23(3):309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wu Z, Ni J, Liu Y, et al. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun. 2017;65:350–361. [DOI] [PubMed] [Google Scholar]
- [73].Zhang J, Yu C, Zhang X, et al. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J Neuroinflammation. 2018;15(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bellinger FP, Madamba S, Siggins GR. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. [DOI] [PubMed] [Google Scholar]
- [75].Mishra A, Kim HJ, Shin AH, et al. Synapse loss induced by interleukin-1beta requires pre- and post-synaptic mechanisms. J Neuroimmune Pharmacol. 2012;7(3):571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Memedovski Z, Czerwonka E, Han J, et al. Classical and alternative activation of rat microglia treated with ultrapure Porphyromonas gingivalis lipopolysaccharide in vitro. Toxins (Basel). 2020;12(5):333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173(4):649–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yu S, Ding L, Liang D, et al. Porphyromonas gingivalis inhibits M2 activation of macrophages by suppressing α-ketoglutarate production in mice. Mol Oral Microbiol. 2018;33(5):388–395. [DOI] [PubMed] [Google Scholar]
- [79].Yang X, Xu S, Qian Y, et al. Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav Immun. 2017;64:162–172. [DOI] [PubMed] [Google Scholar]
- [80].Wu Z, Zhang J, Nakanishi H. Leptomeningeal cells activate microglia and astrocytes to induce IL-10 production by releasing pro-inflammatory cytokines during systemic inflammation. J Neuroimmunol. 2005;167(1–2):90–98. [DOI] [PubMed] [Google Scholar]
- [81].Wu Z, Hayashi Y, Zhang J, et al. Involvement of prostaglandin E2 released from leptomeningeal cells in increased expression of transforming growth factor-β in glial cells and cortical neurons during systemic inflammation. J Neurosci Res. 2007;85(1):184–192. [DOI] [PubMed] [Google Scholar]
- [82].Wu Z, Tokuda Y, Zhang XW, et al. Age-dependent responses of glial cells and leptomeninges during systemic inflammation. Neurobiol Dis. 2008;32(3):543–551. [DOI] [PubMed] [Google Scholar]
- [83].Liu Y, Wu Z, Zhang X, et al. Leptomeningeal cells transduce peripheral macrophages inflammatory signal to microglia in response to Porphyromonas gingivalis LPS. Mediators Inflammation. 2013;2013:407562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wu Z, Nakanishi H. Connection between periodontitis and Alzheimer’s disease: possible roles of microglia and leptomeningeal cells. J Pharmacol Sci. 2014;126(1):8–13. [DOI] [PubMed] [Google Scholar]
- [85].Siddiqui H, Eribe ERK, Singhrao SK, et al. High throughput sequencing detects gingivitis and periodontal oral bacteria in Alzheimer’s disease autopsy brains. Neuro Res. 2019;1(1):3. [Google Scholar]